

Abstract
One quarter of the world’s population is infected with Mycobacterium tuberculosis (Mtb), the causative agent of tuberculosis (TB). Although most infected individuals successfully control or clear the infection, some individuals will progress to TB disease. Immune correlates identified using animal models are not always effectively translated to human TB, thus resulting in a slow pace of translational discoveries from animal models to human TB for many platforms including vaccines, therapeutics, biomarkers and diagnostic discovery. Therefore, it is critical to improve our poor understanding of immune correlates of disease and protection that are shared across animal TB models and human TB. In this study, we have provided an in-depth identification of the conserved and diversified gene/immune pathways in TB models of non-human primate and diversity outbred mouse, and human TB. Our results show that prominent differentially expressed genes/pathways induced during TB disease progression are conserved in genetically diverse mice, macaques and humans. Additionally, using gene-deficient inbred mouse models, we have addressed the functional role of individual genes comprising the gene signature of disease progression seen in humans with Mtb infection. We show that genes representing specific immune pathways can be protective, detrimental or redundant in controlling Mtb infection, and translate into identifying immune pathways that mediate TB immunopathology in humans. Together, our cross-species findings provide insights into modeling TB disease and the immunological basis of TB disease progression.
One sentence summary:
Comparison of human TB gene signature to mouse and macaque models reveal common immune correlates of TB disease and risk.
INTRODUCTION
The immune mechanisms that govern risk of progression from infection to TB disease remain poorly defined. Animal models have been used to characterize immune mechanisms of Mtb infection outcomes (1). However, not all of the immune correlates identified using animal models have been translated to human studies (2). Similarly, several human blood transcriptomic signatures that differentiate asymptomatic Mtb infection and active TB disease have been reported (3–5). Using transcriptional profiling approaches, we recently described genes that were differentially expressed between progressors and non-progressors in a well-characterized longitudinal adolescent cohort study (ACS) of South African (SA) individuals with Mtb infection, some of whom progressed to pulmonary disease (6, 7). We also identified a correlate of risk (COR) of TB signature (7), comprising a set of 16 genes (hereafter referred to as the “16-gene ACS signature”), which predicted onset of TB disease more than a year before disease was diagnosed. Most of these 16 genes were also differentially expressed between individuals with asymptomatic Mtb infection and TB cases (3–5), suggesting conserved biology during progression and clinical disease. However, a mechanistic understanding of the role of the immune pathways identified in the human transcriptomic studies has not been functionally probed using animal models.
Although animal models, especially mice, are used commonly to study mechanisms of immunity or inflammation in TB (1), disease outcomes seen in inbred mouse models do not fully reflect the heterogeneity observed in human TB (1). We and others have established a mouse model of TB in diversity outbred (DO) mice which demonstrate considerable heterogeneity in inflammatory and protective responses following Mtb infection, reflecting some of the genetic diversity found in humans (8, 9). In addition, the non-human primate (NHP) model of TB allows pre-clinical studies of Mtb infection and TB disease progression, as granulomas in rhesus macaques with TB disease recapitulate the morphology and physiology observed in human TB (10). Thus, using a combination of the DO Mtb-mouse model and the macaque model of TB, we recently identified that lung inflammation during TB correlated with increased accumulation of neutrophils and associated production of neutrophilic proteins S100A8/A9 (8). Importantly, increased S100A8/A9 protein concentrations in serum of patients with TB also correlated with increased disease severity (8). Thus, a combination of animal models and human clinical samples allowed us to functionally implicate neutrophils in mediating inflammation and disease severity in TB. However, the pace of such translational discoveries could be enhanced by a comprehensive and in-depth understanding of common immune correlates of protection and inflammation across relevant animal models and human TB. To address this gap, we evaluated whether relevant animal models of TB exhibit similar transcriptional profiles or expressed the 16-gene ACS signature identified in humans. By comparing transcriptional signatures in lung samples from a spectrum of DO mice infected with Mtb, macaques with TB and blood transcriptomic profiles in patients with TB, we identified immune correlates of TB disease that are prominently shared across animal models and humans. Additionally, using gene-deficient mice, we have addressed the functional role of individual genes comprising the 16-gene ACS signature (7).
RESULTS
Common immune correlates of TB disease identified using analysis across animal models and human TB
We compared our existing ACS human blood global transcriptomic profiles dataset (6, 7) to lung transcriptional profiles from animal models to identify common patterns of gene expression and biological pathways, or mechanisms of immune control or disease of Mtb infection. Between 2005 and 2007, 6,363 adolescents were enrolled in Western Cape, SA, a setting with a TB incidence of ~1%, and followed for 2 years (11, 12). Among those with evidence of Mtb infection (Tuberculin Skin Test - positive and/or Quantiferon Gold In-Tube positive), 46 adolescents developed culture and/or sputum smear-positive, intrathoracic TB (termed progressors), and we selected 107 matched adolescents by age at enrolment, sex, ethnic origin, school of attendance, and presence or absence of previous episodes of tuberculosis disease and remained healthy (termed controllers). We measured mRNA expression by RNA-Seq in whole blood ex vivo from progressors and controllers (table S1) and investigated the biological pathways associated with progression to TB disease (table S2). For animal studies, mouse lung samples from DO mice that were left uninfected (naive), individual DO mice that controlled Mtb infection (controllers) or individual DO mice that progressed to severe TB infection (progressors; Fig. 1A) were included. In the DO infected mice, phenotypic clinical observations were used to determine the severity of the infection. The samples were divided into controller and progressor groups based on a “TB severity score” assessed at euthanasia (log10 bacterial burden of the lung tissue sample) × (log10 percentage lung inflammation, by area), using thresholds of < 6.5 and > 8.5, respectively (Fig. 1B). These conservative thresholds excluded 15 samples as not classifying as progressor or controller types (“Both” in Fig. 1B), with the rest clustering as either progressors or controllers. Thus, the naive, controller and progressor mouse samples were subjected to RNA-Seq analysis and the distinct clustering is shown in Fig. 1C.
Fig.1. Overview of the samples used and the computational analysis.
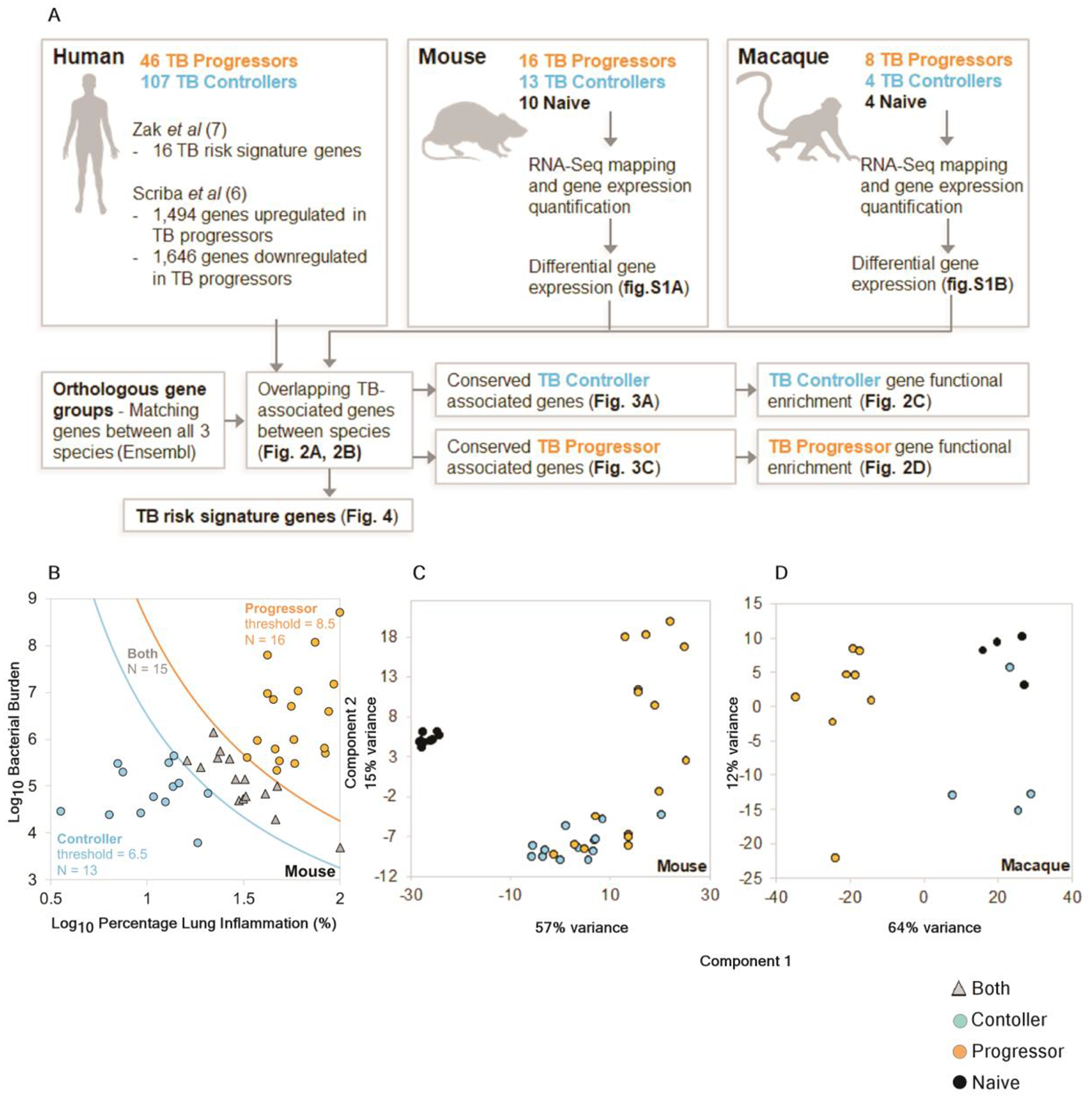
(A) Workflow of cross-species immune correlates of TB disease. (B) Mouse samples were divided into progressor and controller sample groups based on (Log10 bacterial burden) × (Log10 percentage lung inflammation), using thresholds of < 6.5 and > 8.5, respectively, (C) PCA clustering of the mouse RNA-Seq samples based on the default DESeq2 method (top 500 most variable genes) and (D) PCA clustering of the macaque RNA-Seq samples.
Lung samples from uninfected macaques (naive), macaques that were classified as asymptomatic TB (controllers) or macaques that exhibited clinical symptoms and TB disease (progressors; Fig. 1A) were also analyzed by RNA-Seq and showed distinct clustering (Fig. 1D). Macaque lungs from progressors were collected 5–8 weeks after Mtb-infection, while those from controllers were collected 22–24 weeks after Mtb-infection.
We performed pairwise differential gene expression analysis between each species group including mouse and macaques (Fig. 2 A and B). Adequate power and consistent expression patterns allowed for the identification of significantly differentially expressed genes (using DEseq2 (13) with default settings, P ≤ 0.05 after FDR correction), in DO mice (fig. S1A), and in macaques (fig. S1B). The key difference between mice and macaques was that in the latter, the controllers and the naive groups expressed relatively similar transcriptional profiles. As a result, a smaller number of differentially expressed genes (n=184) were identified between the controllers and the naive group in macaques (fig. S1B). Genes were then matched between mouse, macaque, and human using Ensembl (14) orthology matches; 17,044 macaque protein coding genes and 18,658 human protein coding genes matched to the 22,005 protein coding mouse genes. Differences in expression values of genes between human progressors and controllers from the human progressor study (7) were also aligned to the dataset, facilitating the identification of overlapping, differentially expressed orthologous genes across the three species. The results are presented in Fig. 2A (identifying genes consistently higher in controller versus both progressors and naïve), fig.S1C (lower gene expression in controllers versus both progressors and naïve groups), fig. 2B (higher expressed in progressor groups versus both controllers and naïve) and fig. S1D (lower expression in progressors versus both controllers and naïve). We further defined genes expressed higher and lower in progressors compared to both naive and controllers in mice and macaque with relation to human gene expression. These results together define transcriptional profiles from Mtb-infected DO mice and macaques that demonstrate conserved biology in disease processes that overlap with human TB disease. We observed significant overlaps of differentially expressed genes in each of these comparisons across species (P < 10−10, FDR-corrected negative binomial distribution tests; table S3), with the exception of the macaque comparisons involving controller vs naive (due to the minimal differences observed for this, as discussed above), and a significant overlap for macaque controller vs progressor and the human negative controller-associated comparison (P = 2.0×10−4).
Fig.2. Conserved differentially expressed genes in controllers and progressors.
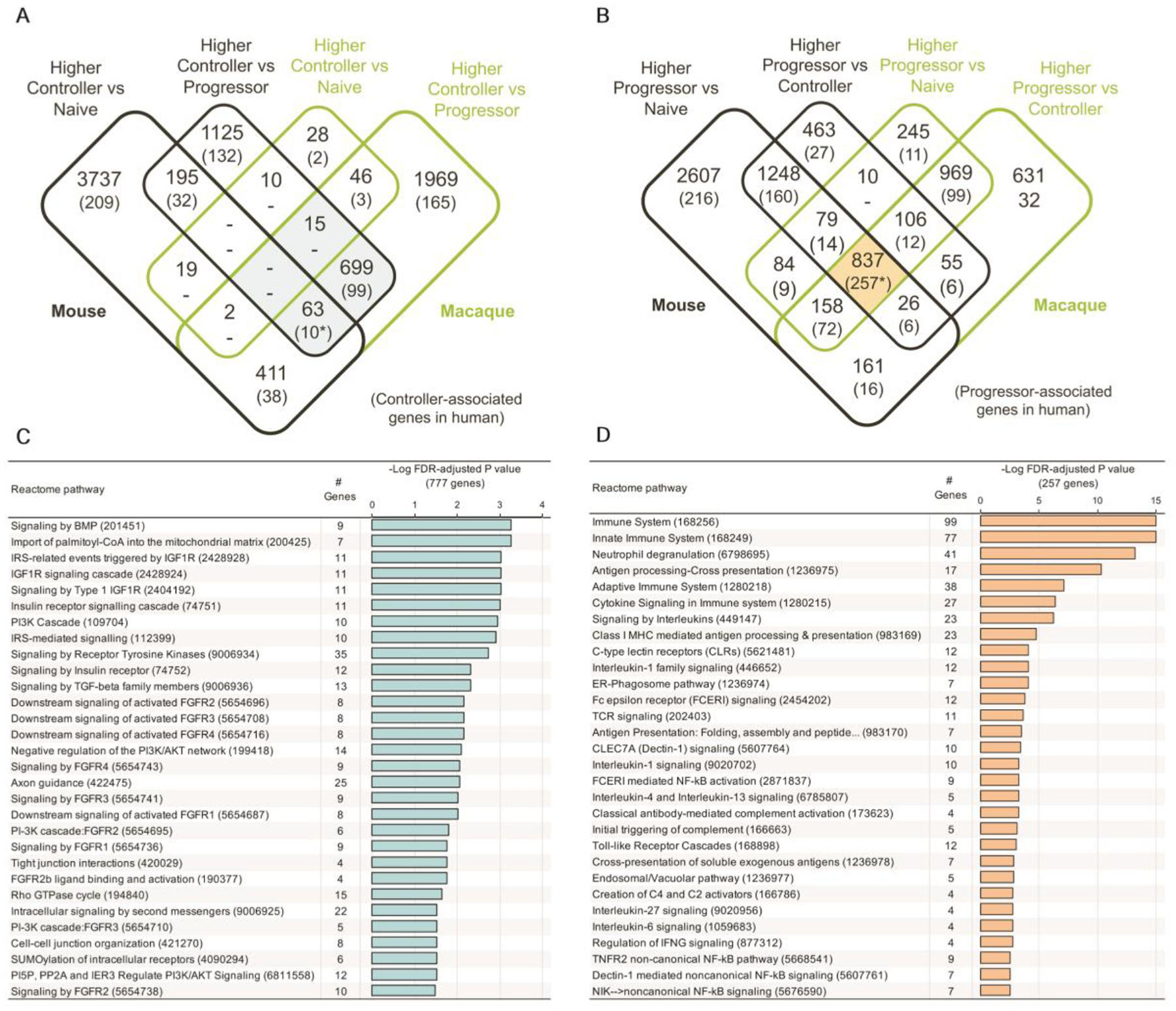
The counts of overlapping significantly differentially expressed genes between mouse genes, macaque genes and human genes (7) are shown for (A) genes higher in controller and (B) genes higher in progressor. Shaded areas represent gene sets used for REACTOME enrichment testing, as shown in panels (C) for genes higher in controller and (D) for genes higher in progressor. *Gene sets visualized in Fig.3.
Many of the most significant pathways that were expressed more abundantly in controller versus progressor samples in animal models were associated with lung tissue repair following injury (Fig. 2C) such as Bone morphogenetic Protein (BMP) signaling pathways (15), Insulin Like Growth Factor 1 Receptor (IGF1R) (16), and fibroblast growth factor receptor (FGFR) (17). Although no enriched pathways were identified among the 109 of 777 genes that are higher in controller versus progressor in all three species (Fig. 2C, table S2), there are several genes related to cellular repair including: (i) Latent Transforming Growth Factor Beta Binding Protein 4 (LTBP4) (18); (ii) myosin IXA (MYO9a) (19) and (iii) v-erb-b2 erythroblastic leukemia viral oncogene homolog 2 (ERB2), required for epithelial cell repair following neutrophil elastase exposure (20). These data suggest that lung tissue damage is being actively repaired in controllers, without triggering a substantial inflammatory response.
Many general immune response pathways including those linked to inflammation and lung damage were associated with disease progression in all three species (Fig. 2D). Among the top progression-associated pathways is “neutrophil degranulation”, which is consistent with the role of neutrophils in TB pathology (8, 21–23) and the prominence of neutrophils in published TB disease gene signatures (3). There also are enriched pathways related to cytokines and chemokines, which initiate and coordinate immune cell recruitment and activation in Mtb-infected lungs (24). Overall, the pathways associated with progressors suggest an active and broad inflammatory response, with the most significantly enriched pathway (P = 3.9 × 10−11) being neutrophil pathways.
Upon further analysis, we identified 10 genes with significantly higher expression in controllers across species, (P ≤ 0.05 for the comparisons in each species), which suggests possible immune parameters that may limit TB disease. This did not take into consideration controllers versus naive groups in macaque, since there was minimal difference in their gene signatures; Fig. 3A. Among this set are the genes, PH Domain And Leucine Rich Repeat Protein Phosphatase 2 (PHLPP2), which dephosphorylates AKT1 (25), and HECT And RLD Domain Containing E3 Ubiquitin Protein Ligase Family Member 1(HERC-1) which regulates the ERK signaling pathway(26), suggesting a potential mechanism by which ERK signaling is altered during TB infection to promote Mtb control. In comparison, there were 13 genes that were consistently lower in controllers across species (Fig. 3B). Among these are SCARF-1, one of the genes in the 16-gene ACS signature previously identified to be upregulated during TB infection in humans (7), and Intercellular Adhesion Molecule 1(ICAM-1).
Fig.3. Differentially expressed genes that are significantly different in TB controllers across species.
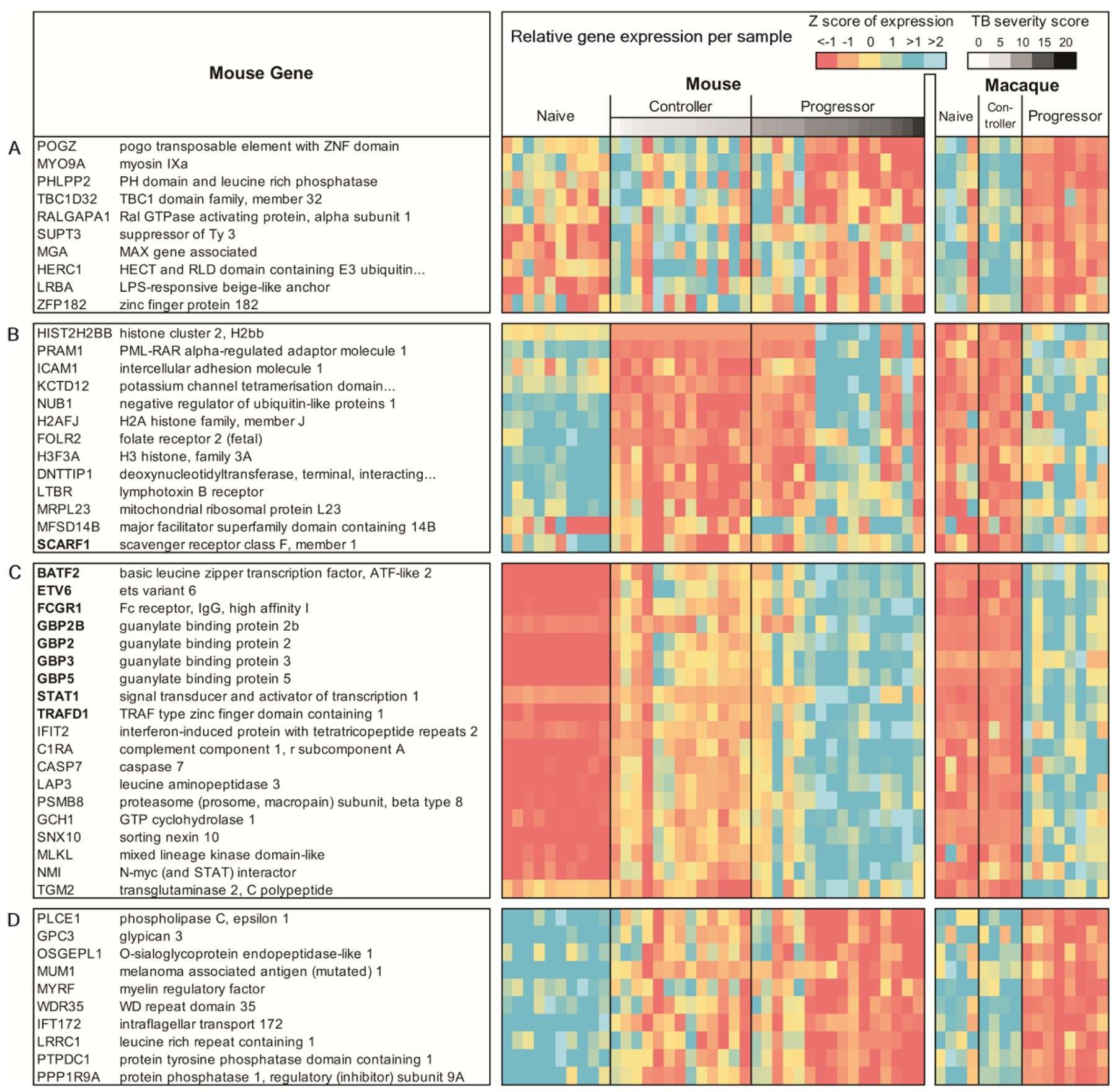
(A) Higher in controller vs progressor and naive in mouse, and higher in controller vs progressor in macaque and human; (B) lower in controller vs progressor and naive in both mouse and macaque; (C) higher in progressor vs controller and naive in both mouse and macaque, and higher in progressor vs controller in mouse (includes ACS signature genes and top 10 most significant other genes); (D) lower in progressor vs controller and naive in both mouse and macaque and lower in progressor vs controller in mouse (top 10 most significant shown).
Of the 837 genes more highly expressed in progressor versus controller and naive groups in both mouse and macaque, the top 19 genes also included 9 of the 16 genes in the 16-gene ACS signature, including BATF2, Etv6 (the mouse ortholog of human ETV7), FCRG1, GBP2, GBP2B, GBP3, GBP5, STAT1, and TRAFD1 (Fig. 3C). Among the remaining 10 of the 19 top genes were several in the interferon-stimulated genes (ISGs) namely interferon induced protein with tetratricopeptide repeats 2 (IFIT2). A total of 777 genes had lower expression in progressors in both animal models versus both naive and controller samples (top 10 lower expressed genes are listed in Fig. 3D). The most significantly expressed genes among these include: (i) Phospholipase C (PLCE1) and (ii) Glypican 3 (GPC3), which regulates macrophage recruitment into tissues (27) (table S3). Collectively, our results suggest that highly expressed genes in Mtb-infected DO mouse and macaque recapitulate the IFN-inducible gene expression pattern previously reported in human TB progressors (3).
The previously identified human 16-gene ACS signature is enriched in progressors across animal models
We analyzed if genes in the previously identified human 16-gene ACS signature were enriched in mouse and macaque progressors. Of the 16 genes, 13, including BATF2, Etv6 (the mouse ortholog of human ETV7), FCGR1, SERPING1, GBP family members (p65 guanylate-binding proteins-GBP1, GBP2, GBP4, GBP5), STAT1, TRAFD1, TAP1 (macaque ortholog absent), APOL1 (mouse ortholog absent), and ANKRD22 (mouse ortholog absent), were expressed more highly in progressors than naive groups in both mouse and macaque DESeq2 (13) results (Fig. 4A). Additionally, expression of these 12 genes in mice also was increased in controllers compared to the naïve group, suggesting a potential role in controlling Mtb infection (Fig. 4A). Expression of 14 genes was increased in macaque progressors, but not in controllers, relative to the naive group (Fig. 4A–C). Of interest was SCARF1, which is involved in apoptotic cell clearance (28). Scarf1 gene expression in mice was lower in controllers than naïve but induced in progressors (Fig. 4B). Similarly, the expression of SCARF1 in macaques was higher in progressors than naive and controllers (Fig. 4B), as also observed in humans (6, 7). Another gene of interest was SEPT4 which belongs to a family of nucleotide binding proteins involved in cytoskeletal organization (29). Whereas SEPT4 expression was induced in macaque progressors when compared with naive or controllers as observed in human TB progressors, the reverse was true in mice where Sept4 expression was lower in controllers and further decreased in progressors (Fig. 4C).
Fig.4. Expression of the 16-gene ACS signature genes across species.
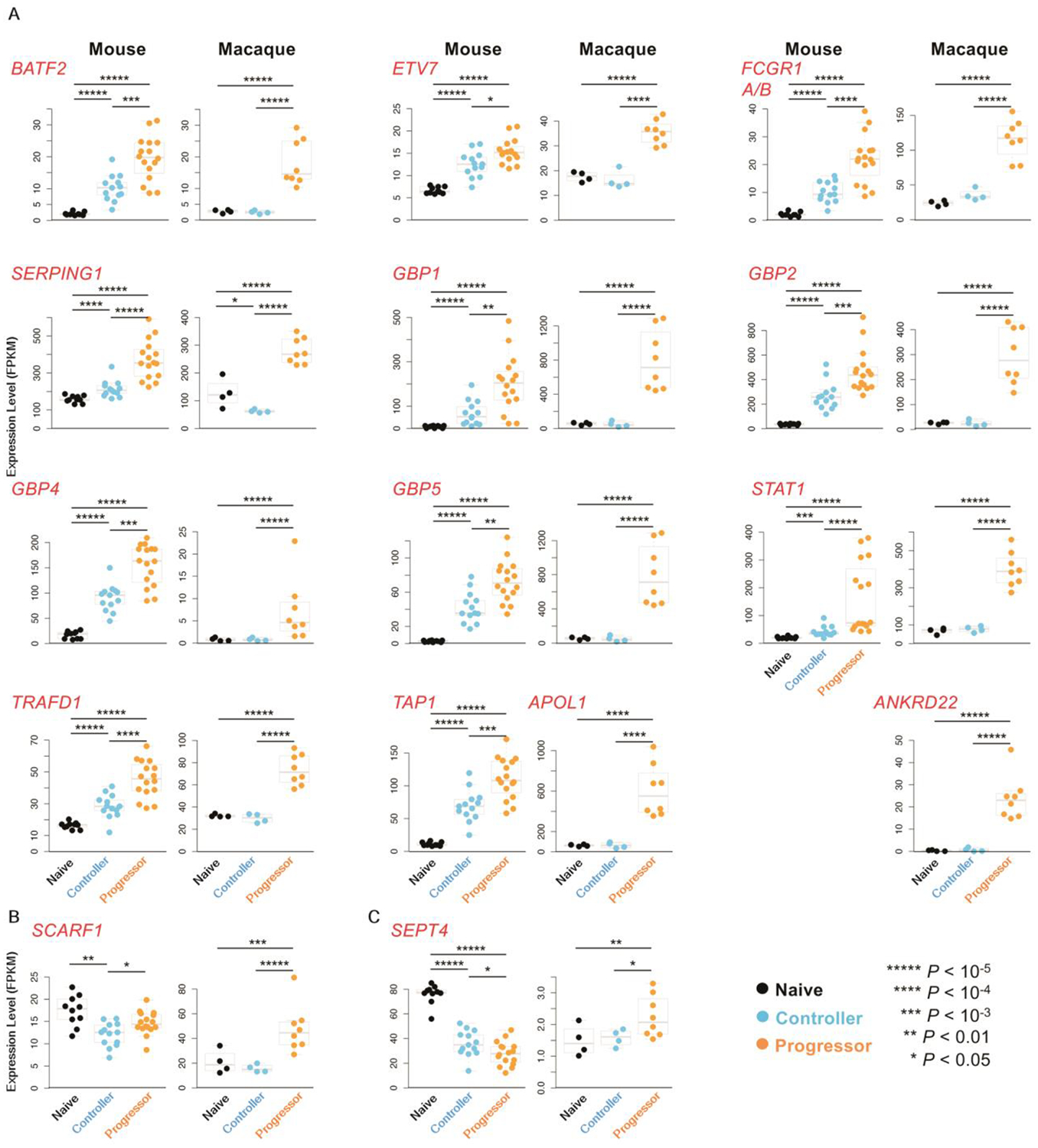
(A) Ortholog gene expression of 13 out of 16 human ACS signature genes (7) in progressor versus controller, progressor versus naive and controller versus naive, in mouse and macaque. Note that both FCGR1A and FCGR1B are represented by a single gene in mice and macaques, and human ETVTs ortholog in mouse is called Etv6. (B) SCARF1 expression in progressor, controller versus naive in mouse and macaque is shown. (C) SEPT4 expression in progressor, controller and naive is shown in mouse and macaque. All P values shown on the expression swarm plots represent FDR-corrected significance values for differential expression calculated by DESeq2.
In addition to analyzing the differences between the groups, the correlation of the gene expression of these ACS signature genes was compared to the percentage lung inflammation and the bacterial burden (table S4) across the controller and progressor samples (N = 29 for mouse, N = 12 for macaque). Significant correlations (P ≤ 0.05, after FDR correction) were observed for all ACS signature genes in macaque, and in all genes except for SCARF1 and bacterial burden in mouse (P = 0.07), although it was significantly correlated with lung inflammation (P = 0.002). In both mouse and macaque, the ortholog of FCGR1a / FCGR1b was the most significantly associated with the metadata (P = 4.9×10−5 with lung inflammation and P = 8.3×10−5 with bacterial burden in mouse, P = 1.2×10−6 with lung inflammation and P = 1.8×10−14 with bacterial burden in macaque). In macaques, correlations were higher with bacterial burden than with lung inflammation in all genes except GBP4 and SEPT4, while this was only true for half of the mouse genes. These correlation results suggest that the expression of these ACS signature genes correlate with disease severity even within the controller and progressor groups. Collectively, our results suggest that the majority of human immune pathways induced in blood during TB disease progression are similarly induced in the lungs of the animal models in this study. However, pathways involving Sept4 may function differentially in TB disease in mice when compared with macaque or humans. Thus, our studies provide an in-depth understanding of the conserved and diversified immune pathways in animal models and human TB.
Delineating the functional relevance of genes in the 16-gene ACS signature using gene-deficient mouse models
Using the well-established inbred mouse model of TB, we conducted mechanistic studies to delineate the functional relevance of genes in the 16-gene ACS signature that were increased in progressors in mice, macaque and humans. Of the 16 human genes, APOL1 and ANKRD22 lack a mouse ortholog, and FCGR1a and FCGR1b only have one mouse ortholog (Fcgr1), leaving 13 potential genes for study. Etvfr−/− mice are not viable and die between days 10–11 of embryonic development (30) and could not be evaluated. Therefore, we focused on the remaining 12 genes in this study. Accordingly, Scarft−/−, Statt−/−, Sept4−/−, Tap1−/−, Trafdt−/−, Batf2−/−, Serpingt−/−, Fcgrt−/− mice, and mice deficient for 5 murine GBP genes located on chromosome 3 (Gbp1, 2, 3, 5 and 7−/−), namely GbpChr3−/−, were inoculated with low doses of aerosolized Mtb clinical W-Beijing strain, HN878. At acute (30 dpi) and chronic stages (60 dpi or more) of Mtb infection, lungs were harvested and accumulation of immune cells, disease inflammation in the lungs as well as Mtb burden were measured. In these studies, lung cellular immune responses such as CD4+, CD8+, accumulation of myeloid dendritic cells, alveolar macrophages, monocytes, recruited macrophages, and neutrophils were all enumerated and are shown in table S5. Only cellular responses in gene deficient mice that were significantly different from control Mtb-infected control mice are included in the figures below. Based on the Mtb burden in the lung during either acute, chronic or both stages of infection, we classified the gene-deficient mice into three groups: (a) gene-deficient mice that did not exhibit any effects on Mtb control, suggesting these genes do not contribute to protective outcomes in TB, at least in mice; (b) gene-deficient mice that were more susceptible to Mtb infection and sustained higher Mtb burden, suggesting that these genes are important for protection; and (c) gene-deficient mice that were resistant to Mtb infection and had decreased burden in the lung, suggesting that these genes were involved in mediating disease progression and TB susceptibility.
Serping 1 of the 16 gene ACS signature mediates no effect on Mtb control in mice
SERPING1 is a highly glycosylated plasma protein that inhibits activated C1r and C1s, and thus, negatively regulates complement activation (31). Although SERPING1 mRNA expression is upregulated in Mtb-infected lungs (32, 33), its contribution to Mtb control and inflammation has not been assessed. Mtb-infected wild-type C57BL/6 mice expressed Serping1 mRNA within granulomas (fig. S2A). Upon Mtb infection, no differences in lung bacterial burden were observed between Serping1−/− and wild-type mice with no major differences in myeloid or T cell populations, except recruited macrophages at 30 dpi (fig. S2B, table S5). In contrast, we observed more inflammation in the lungs of Serpingt−/− mice than wild-type mice at multiple time points (fig. S2B), which may be due to the activation of the complement cascade (34, 35) in the absence of Serping1. Thus, Serping1 had no effect on Mtb control yet still regulated inflammation. Although no role in Mtb control was observed, a deficiency of Serping1 led to dysregulation of inflammation. In the absence of Serping1, mice undergo greater activation of the complement pathway and sustain increased inflammation (34, 35). It remains to be determined if this mechanism explains the increased inflammation seen in Serping1 deficiency during Mtb infection.
Genes in the 16-gene ACS signature that mediate protective responses in mice
Of the gene-deficient mice tested those with an absence of Stat1, Tap1, Gbps, Trafd1 and Sept4, displayed increased Mtb susceptibility. STAT1 is critical for type I, II, and III IFN signaling pathways (36), and mice deficient in Stat1 are extremely susceptible to Mtb (37). Accordingly, Statt−/− mice exhibited high lung Mtb burden during acute infection and severe lung inflammation, when compared with control Mtb-infected C57BL/6 mice (Fig. 5A). TAP1 facilitates loading of peptide antigens onto MHC class I molecules for presentation to CD8+ T cells, and Tap1−/− mice exhibit defects in MHC class I antigen presentation (38, 39). We found a significant increase in lung Mtb burden of Tapt−/− mice during the acute and chronic stages of infection (Fig. 5B), and decreased numbers of activated CD8+ T cells compared to infected wild-type mice (Fig. 5B, table S5). However, differences in the cytokine secretion profile of CD8+ but not CD4+ T cells were observed in Tapt−/− mice when compared with controls (fig. S3A and S3B). Tapt−/− mice also had no changes in accumulation of neutrophils and recruited macrophages (fig. S3C) but exhibited decreased accumulation of alveolar macrophages at acute (30 dpi) phases of infection (fig. S3D and S3E). Consistent with the increased lung Mtb burden, greater lung inflammation was observed in Tapt−/− than wild-type mice at all the time-points assessed (Fig. 5B). GBPs are IFN-inducible proteins with documented key antiviral and antibacterial functions (40, 41). The mouse genome has 13 Gbp genes (2 of which are pseudogenes) organized in specific clusters on chromosomes 3 and 5 (42). To elucidate the contribution of the GBPs to host immunity to Mtb, we utilized mice lacking the entire cluster of Gbps on chromosome 3 (GbpChr3−/−). In contrast to results with M. bovis BCG infection in Gbp1−/− mice (41), GbpChr3−/− mice demonstrated increased susceptibility only during chronic infection and not during the acute phase, and this coincided with decreased accumulation of lung neutrophils and recruited macrophages, without impacting inflammation (Fig. 5C). TRAFD1 belongs to the TNF family like genes and is a negative feedback regulator of the TLR signaling pathway, which results in the control of excessive immune responses (43, 44). Similar to GbpChr3−/− mice, Trafdt−/− mice displayed increased susceptibility only during chronic infection and not during the acute phase (Fig. 5D). This susceptibility coincided with a significant decrease (P = 0.001) in neutrophil accumulation at 60 dpi, without affecting recruited macrophage accumulation (Fig. 5D). Surprisingly, we observed a decrease in lung inflammation in Trafdt−/− mice during acute and chronic infections (Fig. 5D). Trafd1 deficiency is known to regulate Type I IFN responses (43), which could have an impact on the reduced inflammation observed.
Fig.5. Genes in the 16-gene ACS signature that mediate protective immune responses in the mouse model.
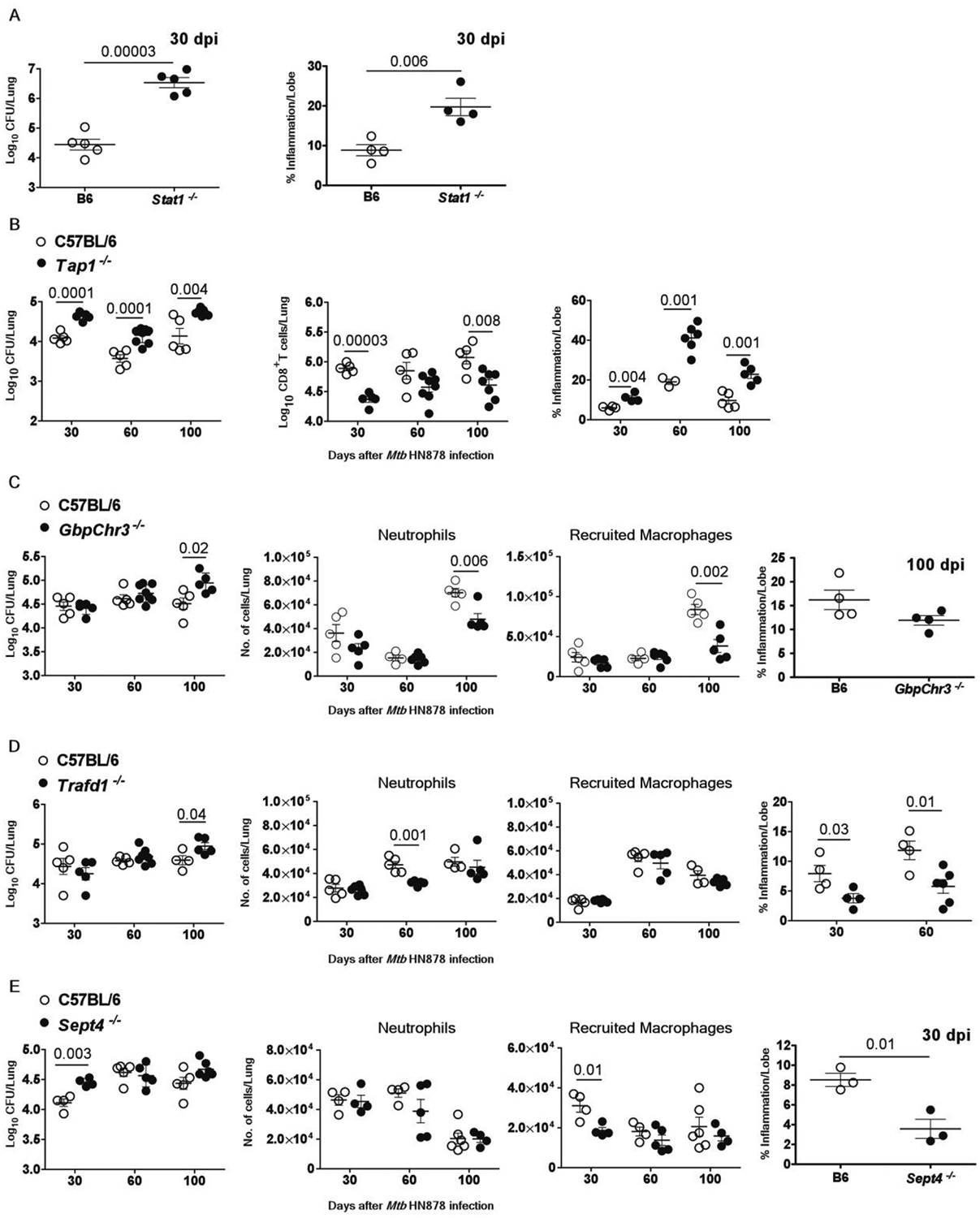
Control or gene deficient mice were infected with Mtb HN878 (100 CFU) by aerosol route. On 30, 60 and 100 days post-infection (dpi), lungs were harvested to assess bacterial burden by plating, or inflammatory cell infiltration by flow cytometry, or histologically by H&E staining in (A) Statt−/− mice (B) Tap1−/− mice, (C) GbpChr3−/− mice (D) Trafdt−/− mice and (E) Sept4−/− mice and relevant controls are shown. The data points represent the means (±SEM) of one out of two separate experiments. Significance for bacterial burden analysis for C57BL/6 and gene deficient mice was determined using unpaired two-tailed Student’s t-test at different dpi.
Sept4 originally was described in yeast as a cell division cycle regulatory protein (45). Upon Mtb infection, Sept4−/− mice showed enhanced susceptibility to Mtb at early stages, which correlated with reduced accumulation of lung recruited macrophages but not neutrophils and increased inflammation, but this was not maintained during chronic stages of infection (Fig. 5E). Together, our results show that Stat1 and Tap1 contribute to protection through acute and chronic stages, whereas Gbps, Trafd1 and Sept4 have disease stage-dependent protective roles during Mtb infection.
Genes in the 16-gene ACS signature that mediate detrimental effects during infection of mice
Of the 16-gene ACS signature genes analyzed for functional relevance in mice, we identified three genes (Batf2, Fcgr1, and Scarf1) which contribute to Mtb susceptibility at distinct phases of infection. BATF2 is a basic leucine zipper transcription factor that is closely related to BATF and BATF3 (46), which together regulate dendritic cell differentiation and development. Batf2/3−/− mice were more resistant to Mtb infection and unexpectedly showed improved Mtb control upon infection during the transition from acute to chronic stages at 60 and 100 dpi (Fig. 6A). Since Batf3−/− mice are more susceptible to Mtb infection at 60 dpi, improved Mtb control likely depends on the loss of Batf2 expression (fig. S4), as also suggested recently (46). The increased resistance to Mtb infection was associated with diminished neutrophil and recruited macrophage accumulation in the lungs at 60 dpi (Fig. 6A). However, the enhanced protection against Mtb infection observed in the lungs of Batf2/3−/− mice was associated with enhanced proportion of CD4+ IL-17+ cells as well as CD4+ IL-17+ IFNγ+ cells (fig. S5). Batf2/3−/− mice demonstrated increased lung inflammation at 60 and 100 dpi (Fig. 6A), suggesting that while Batf2 expression was detrimental to Mtb control, its expression limits inflammatory responses during Mtb infection, possibly mediated by Th17 cells.
Fig.6. Genes in the 16-gene ACS signature that mediate detrimental effects during infection.
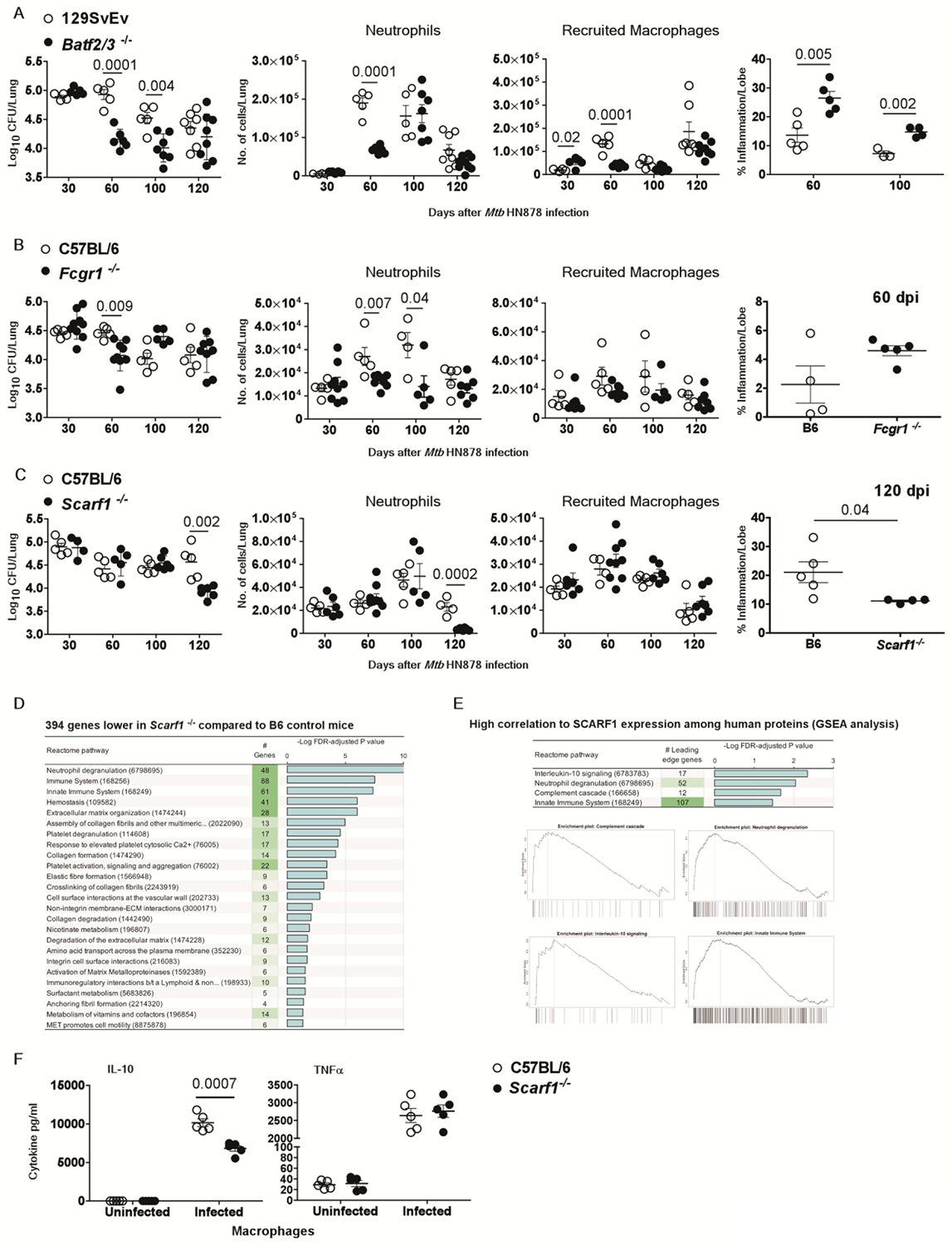
Wild-type or gene-deficient mice strains were infected with Mtb HN878 (100 CFU) by aerosol route. On 30, 60, 100 and 120 days post-infection (dpi), lungs were harvested to assess bacterial burden by plating, or inflammatory cell infiltration by flow cytometry, or histologically by H&E staining in (A) Batf2/3r−/− mice, (B) Fcgrt−/− mice (C) Scarft−/− mice and relevant controls (D) DESeq2 differential gene expression comparisons identified in infected Scarft−/− mice compared to wild-type mice at 120 dpi respectively, (E) Gene set enrichment analysis (GSEA) for Reactome pathways significantly enriched among human proteins with high correlation to SCARF1. Enrichment plots are shown for each of the enriched terms, showing the relative rank of genes from the enriched pathways (across the x-axis) and (F) IL-10 and TNFa production was measured in culture supernatants from uninfected and Mtb HN878-infected bone marrow derived macrophages (MOI 1) from C57BL/6J and ScarfT−/− mice after 2 dpi. The data points represent the means (±SEM) of one out of two separate experiments. Bacterial burden in gene deficient mice compared to control mice and cytokine production from macrophages was done by unpaired two-tailed Student’s t-test at different dpi.
FCGR1 inactivation impacts nitric oxide production by neutrophils (47), cytokine responses and antigen presentation (48, 49), and antibody-dependent killing of pathogens by macrophages (50). We utilized Fcgr1−/− mice to assess the functional significance of this gene in protective immunity to Mtb. We observed improved Mtb control in the lungs of Fcgrt−/− mice compared to the control mice at 60 dpi (Fig. 6B). The decrease in lung Mtb burden in Fcgrt−/− mice was associated with reductions in neutrophils, but not recruited macrophages, in the lung at both 60 and 100 dpi (Fig. 6B) but no major effect on inflammation in the lung tissue at 60 dpi (Fig. 6B), which coincided with increased IFNγ/IL-17 co-producing CD4+ T cell during early disease (fig. S6). Thus, both Batf2 and Fcgr1 are detrimental for Mtb control but likely required to limit inflammation in the lung.
SCARF1 is involved in the uptake of modified low density lipoprotein (LDL) or acetylated LDL (AcLDL) (51, 52). We assessed the functional relevance of this gene for Mtb using Scarft−/− mice. We observed a significant reduction (P = 0.002) in bacterial burden in the lungs of Scarf1−/− mice during the chronic phase (120 dpi), but not earlier (Fig. 6C). This was associated with reduced accumulation of neutrophils but not of recruited macrophages in the lungs of Scarft−/− Mtb-infected mice compared to wild-type Mtb-infected mice (Fig. 6C). Although Scarf1 has been associated with clearance of apoptotic cells by macrophages (28) and impaired apoptotic cell clearance is associated with chronic inflammation (53), we observed reduced inflammation in the lungs of Scarft−/− mice (Fig. 6C). To further characterize the functional role of Scarf1 in TB, we performed RNA-Seq profiling of lung tissue from C57BL/6 mice and Scarft−/− mice at 120 dpi, the time point at which Scarf1−/− mice exhibited better Mtb control and reduced inflammation. Differential gene expression analysis identified 218 higher and 394 lower expressed genes in infected Scarf1−/− compared to infected wild-type mice (table S6). While there was no functional enrichment among the 218 induced genes (Fig. 6D), analysis of the reduced genes suggested that Scarf1 might affect neutrophil-related and other downstream immune signaling pathways. To investigate whether similar biological pathways may be associated with SCARF1 in humans, we sought to identify plasma proteins quantified in a previous study (6, 54) that correlate with SCARF1 mRNA abundance in ACS progressors and controllers. Since soluble mediators of immune responses produced in inflamed tissues more readily enter the blood than intracellular RNAs, we hypothesized that plasma protein concentrations more likely reflect Mtb disease in the lungs than transcriptomic data from blood leukocytes. Pearson correlation coefficients were calculated for SCARF1 mRNA abundance in whole blood (measured by RNA-Seq) and proteomics abundance data for 2872 plasma proteins (measured with SOMAscan, a highly multiplexed proteomic assay) (6, 54) across 311 samples from 30 progressors (65 samples) and 102 non-progressors (209 samples). These Pearson correlations per protein were used as a ranked input list for Gene Set Enrichment Analysis (GSEA) (55); performed using WebGestalt (56), to identify Reactome (57) pathways enriched among the highest or lowest-ranking proteins. Since the GSEA analysis considers the ranked positions of proteins within the list rather than the total number, any functional bias among the 2872 proteins relative to the entire proteome will not affect the results. This analysis identified four significantly enriched pathways with SCARF1 gene expression in humans: neutrophil degranulation, interleukin (IL)-10 signaling (comprising primarily innate cytokines and chemokines), the complement cascade, and the innate immune system (Fig. 6E, table S6). Importantly, genes in the neutrophil degranulation and innate immune system pathways were enriched as downregulated genes in Mtb-infected Scarf1−/− mice when compared to infected wild-type control mice (table S6). This coincided with significantly decreased (P = 0.0007) IL-10 production in Mtb-infected Scarft−/− macrophages when compared with C57BL6 Mtb-infected macrophages, while other cytokines such as TNF- α levels were comparable between B6 and Scarf1−/− macrophages (Fig. 6F). These data align with the human proteomics data, although genes in the IL-10 signaling pathway were not enriched among downregulated genes in the Scarf1−/− mice (table S6). Indeed, compared to the well-defined large granulomas with enhanced collagen expression in the lungs of infected wild-type mice, the lung granulomas in Scarft−/− mice were not well formed, with little to no collagen expression (fig. S7A and S7B). Mtb-infected wild-type C57BL/6 mice expressed Scarf1 mRNA within granulomas (fig. S7C). Furthermore, Scarf1 protein localized to granulomas within the lungs of Mtb-infected wild-type mice and progressor macaques (Fig. S7D). Importantly, 5 of the 394 genes lower in the Scarf1−/− mice also overlapped with the 17 proteins correlated with SCARF1 expression in humans (IL-1 β-interleukin 1 beta, TIMP1-tissue inhibitor of metalloproteinase 1, IL-1RA-interleukin 1 receptor antagonist, TNFRSF1a-tumor necrosis factor receptor superfamily member 1a and PTGS2-prostaglandin-endoperoxide synthase 2) (table S6). Together, these results implicate SCARF1 as a key regulator of the innate immune response that possibly can contribute to immunopathology and tissue destruction during the chronic stages of TB disease in mice and humans.
DISCUSSION
The pace of translational discoveries from animal models to human TB for many platforms including vaccines, therapeutics, biomarkers and diagnostics is suboptimal because of a poor understanding of shared immune correlates of disease and protection across species. In this study, we provide an in-depth identification of the conserved and diversified immune pathways in animal models and human TB. Our results suggest that most of the prominent differentially expressed genes induced during TB disease progression are conserved in mice, macaques, and humans. However, some pathways (e.g., SEPT4) may function differently in TB disease in mice or be regulated differentially when compared with macaques or humans. Using the well-established inbred mouse model of TB, our mechanistic studies begin to define the functional relevance of a suite of genes strongly associated with disease progression (6, 7, 58) and TB (4, 5) in humans. We classified the gene-deficient mice infected with Mtb into groups that were more susceptible to Mtb infection, suggesting that these host genes contribute to protection. Additionally, gene-deficient mice that had increased resistance to Mtb infection suggest that these genes likely mediate disease progression and TB susceptibility. One such pathway is the Scarf1 pathway, which we identified as a driver of TB immunopathogenesis across species. Our studies in mice also identified genes that did not exhibit any direct effects on Mtb control but regulated inflammation. Finally, we identified that genes expressed during control of Mtb infection across species are related to the repair of lung injury. Together, these results identified cross-species correlates of disease and annotated the functional role of specific genes within the 16-gene ACS signature. These findings may allow more effective use of appropriate animal models for translation to human TB.
Animal models are critical for testing vaccines and therapeutics for clinical development for human use. The two most widely used animal models of TB are mice and NHPs. While the former are most extensively utilized, the latter are universally accepted to recapitulate key aspects of the human TB disease. However, some cellular phenotypes and immune signaling events occurring in uninfected hosts are different between mouse, NHPs and humans (59). Animal models have been predictive of some human mechanisms of immunological protection, such as the roles of the cytokines IFNγ and TNFα in protective immunity to TB (60–62). Indeed, mice deficient in IFNγ and TNFα are vulnerable to Mtb infection in inbred mouse models (60–62). In humans, deleterious mutations in the IFN-IFNAR-STAT1 pathway give rise to Mendelian susceptibility to TB (63), and humans with Mtb infection who undergo therapeutic TNFα blockade are at increased risk of TB reactivation (64). However, these examples are limited, as it is not known if immune correlates of TB disease are similar or distinct in mice, macaques, and humans. Additionally, most mouse studies have utilized inbred strains with the major limitation that genetic diversity, a key driver of variability in infection outcome and immune responses in macaques and humans, is not reflected in the inbred mouse model. While the mouse model of TB has been useful in identifying some key immune mechanisms responsible for the control of TB (65), and assessment of drugs and therapeutics, its utility in effectively modeling the progression of Mtb infection and pathology has been questioned (66, 67). Recently, two mouse models with the ability to offer maximal allelic variation, thus mimicking human populations, have been generated. One, the Collaborative Cross (CC) model, is a large panel of inbred mouse strains that display a broad range of susceptibility to Mtb infection (68). The other, the DO mouse model, is produced by an outbreeding strategy such that each DO mouse is genetically unique and their groups approximate the genetic diversity found in human populations. When DO mice were infected with standard doses of Mtb via aerosol, super-susceptible, susceptible and resistant phenotypes were observed (8, 9). In the current study, we show that with the exception of Sept4 and orthologs absent in mice, 12 of the ACS signature (7) genes are upregulated in the lungs of progressor DO mice, when compared with controller DO mice. Therefore, vaccines or therapeutics targeting immune pathways in mice, excluding the Sept4 pathway, may reflect outcomes occurring in human TB. Upon aerosol infection with Mtb, macaques typically either progress to TB disease or, unlike mice but similar to humans, remain asymptomatic. Our study demonstrates that the rhesus macaque progressors faithfully express the 16-gene ACS signature, with the exception of TAP1, for which there is no known macaque ortholog. This is consistent with a study of the cynomolgus macaque model of low-dose Mtb infection, which showed that a subset of genes comprising the 16-gene ACS signature prospectively discriminated progressor from controller animals 3–6 weeks after infection (69). One limitation of the study is that the number of NHP animals per group was lower (naïve and controller) than the number of mice and human samples per group owing to associated expense of experimentation. Additionally, another limitation was in the cross species RNA-seq comparison analysis of the human ACS gene signature, as it was based on pre-defined orthologous protein family groups by Ensembl. And if no ortholog was identified in the other two model species (mice or macaque), then only the top most homologous gene match was accepted (above the set threshold).
In our NHP experiments, the challenge dose of 10 CFU versus 100CFU was utilized as the key determinant of disease outcome as either “controller” or “progressor”. However, the DO mice were all challenged with the same Mtb dose and therefore in the DO mouse model, the dose of Mtb is not the primary determinant of disease progression where the same infectious dose leads to a range of bacterial burdens and inflammation in the lungs. We interpret this spectrum of clinical outcome as a significant advantage of the DO model given that such a spectrum is observed in human Mtb infection outcomes. Bacterial burden and inflammatory responses are interdependent, and some gene expression changes may be a response to heightened bacteria, not the cause. We were not able to rule out from these correlative data that the differentially expressed genes are the consequence of the different bacterial loads, which may very well be the case and is a limitation of the current study.
Consistent with humans who progress to TB, IFN response pathways, innate immune system, cytokine signaling, complementation activation, myeloid cell activation pathways such as TLR cascades and signaling by interleukins, were elevated in progressor mice and macaques. Importantly, when we considered pathways that were upregulated in controllers compared to progressors in macaques and mice, those associated with lung remodeling and repair were prominent, thus providing important insights into protective immune parameters that limit TB disease. The identification of several genes associated with controlled Mtb infection, projects these genes as potential candidates for drug targeting or biological experimentation to better understand their role in Mtb control.
Our work suggests that molecular mechanisms that govern the equilibrium between lung repair and inflammatory immune responses are the key determinants of progression of Mtb infection. This result was remarkable in light of recent work demonstrating that IL-22, a mediator of epithelial repair, was protective against chronic infection of mice with the virulent HN878 strains of Mtb (70). The biological pathways associated with control of Mtb infection thus can engender hypotheses of protective immunity. One limitation of this study is that we were unable to follow up on specific mechanisms uncovered by our findings in DO mice as these studies will require gene knockouts to enable functional studies.
Despite the limitation that genotype-phenotype relationships are complex to infer by studying a single genetic background (71), our studies with gene-deficient mice show that greater susceptibility to Mtb infection is likely due to underlying deficiencies in distinct immunological mechanisms. For example, an absence of Stat1 in mice, and humans with mutations in STAT1, results in greater susceptibility to Mtb infection (37). Accordingly, Statt−/− mice had increased Mtb burden and increased inflammation due to defects in Th1 immunity, which is critical for control of Mtb replication (37). By contrast, the upregulation of STAT1 transcripts in TB progressors reflects Mtb-induced inflammation and type I and II IFN signaling, which is indicative of elevated Mtb replication. In Tapt−/− mice, cell surface expression of class I MHC is impaired and thus CD8+ T cell priming is defective as shown here and before (39). GBPs are GTPases induced in response to IFNs and regulate activation of macrophages in response to many pathogens. GBPs function through induction of autophagy, activation of inflammasomes, and formation of vacuolar compartments (72). GBPs have important roles in controlling several pathogens, including T.gondi and M.bovis (41, 73). In light of this, our finding that increased Mtb lung burden was observed only at late time points after Mtb aerosol infection in GbpChr3−/− mice was surprising. We acknowledge that gene deficiency in multiple GBP proteins (GbpChrS−/− mice lack functional Gbp1, 2, 3, 5 and 7), only some of which may be implicated in immunity to Mtb, may underlie this finding. However, it is also possible that while GBPs are critical for immunity to M.bovis (41, 73), the role of GBPs in Mtb infection may be more redundant. As TRAFD1 is associated with negative regulation of inflammation (43, 44) it would be expected that a genetic deficiency might result in enhanced inflammation in the lungs. Surprisingly, a deficiency of Trafd1 resulted in reduced lung inflammation during late stages of Mtb infection. It has been reported that cells lacking TRAFD1 secrete more IFNβ than wild-type cells (43). As IFNβ has anti-inflammatory effects by promoting a switch from Th1 to Th2 responses in blood cells (74), this might explain the reduced inflammation observed in our study. Although there is as yet no published data on interaction between Trafd1 and Gbp genes, they belong to the category of Interferon stimulated genes whose expression occur in response to Type 1 or Type II IFN stimulation (75). Therefore, it is interesting to observe a similar pattern in the bacterial burden in the lungs of GbpChr3−/− and Trafdt−/− infected mice, which could possibly implicate a role for Stat signaling. SEPT4 has been implicated in diverse cellular functions, including cytokinesis, apoptosis, and tumor suppression (76, 77). Sept4−/− mice have improved wound healing and regeneration of hair follicles, are protected against apoptosis (76), and males are sterile due to immotile and structurally defective sperm (77). In our study, Sept4−/− mice exhibited transient susceptibility due to reduced macrophage recruitment.
In the absence of Serping1, mice undergo greater activation of the complement pathway and sustain increased inflammation (34, 35). It remains to be determined if this mechanism explains the increased inflammation seen in Serping1 deficiency during Mtb infection. In a third group of gene-deficient mice, the absence of Batf2, Scarf1 and Fcgr1 improved Mtb control and were associated with reduced neutrophil recruitment. Neutrophil accumulation is associated with increased disease severity in human TB (3, 8) and in mouse models of TB (21, 78). Although these three genes were directly associated in KEGG (79) or STRING (80) functional protein databases, identification of pathways overlapping in the absence of these genes may allow us to define targets that can limit neutrophil accumulation and improve Mtb control. Our studies show that plasma proteome markers of innate immune activation, neutrophil degranulation, and complement activation pathways correlate with SCARF1 mRNA expression of human TB progressors. These pathways were downregulated in Scarft−/− infected mice, suggesting a previously unknown functional role for SCARF1 in modulating neutrophil mediated immunopathology during TB. A number of mediators of lung matrix destruction, the process that leads to cavitary TB in humans, were among the proteins and genes associated with SCARF1 expression; this suggests a possible role for SCARF1 in regulating pulmonary caseation, necrosis and cavitation, which are associated with poor treatment outcome (81). Moreover the IL-10 signaling pathway was the top enriched reactome in the human proteomics analysis conducted. IL-10-producing macrophages preferentially clear apoptotic cells (82) and Scarf1 has been associated with clearance of apoptotic cells (28). However, IL-10 also aids immune evasion strategies of mycobacteria, and IL-10 overexpression in mice fails to control Mtb infection and mice develop severe lung pathology (83). In summary, we investigated transcriptional signatures in lung samples from DO mice and NHPs infected with Mtb that reflect a spectrum of infection and disease outcomes. Comparison of these transcriptional signatures with those from human TB led to identification of common immune correlates of TB disease that implicate overlapping biology but also highlight distinct processes between animal models and humans. Our results and data provide a significant new resource that will be valuable to others in the field. Furthermore, our functional studies using gene-deficient mouse models have addressed the functional role of TB disease risk signatures and demonstrate that specific immune pathways associated with TB progression can be protective, detrimental, or regulate infection and inflammation during Mtb infection. Together, our cross-species findings provide insights into modeling TB disease and the immunological basis of TB disease progression.
MATERIALS AND METHODS
Study design
Our objective was to compare the common immune correlates of TB disease and risk across species. Whole blood RNA-Seq data from human subjects was aligned to the hg19 human genome using GSNAP as done previously (7). Normalized gene-level expression estimates were derived from mapped read pairs. Briefly, mapped read pairs were assigned to genes by collapsing all transcripts into a single gene model and counting the number of reads that fully overlap the resulting exons using htseq (v. 0.6.0) (84), with strict intersection and including strand information. Gene models for protein-coding genes were downloaded from Ensembl (GRCh37.74). Reads that mapped to multiple locations were only counted once and those mapping to ambiguous regions were excluded. Log2-transformed values of counts normalized by adjusted library counts were computed using the cpm function of the edgeR package (85). All mouse strains were bred in-house, used at 6 to 8 weeks of age, and sex matched and both sexes were used for all experiments. Specific pathogen-free (SPF), adult Indian rhesus macaques of both sexes were obtained from the Tulane National Primate Research Center. Animals were infected with Mtb via the aerosol route and tissues were collected for RNA-Seq analysis. All animal experiments were performed in accordance with National and Institutional guidelines for animal care under approved protocols (Animal Protocol Number: mice-20160129 and macaque-P0295R). Primary data are reported in data file S1.
Mice
C57BL/6J, DO (86) and 129SvEv (87) mice were obtained from Jackson Laboratory and bred at Washington University in St. Louis. Fcgr1−/− (88), Scarft−/− (28) Batf3r−/−(89), Batf2−/−(90), Batf2/3−/− mice, and GbpChr3−/− (73) mice were obtained from Dr. Jeffery Ravetch, (Rockefeller University), Dr. Terry Means (Sanofi), Dr. Ken Murphy (Washington University in St. Louis), Dr. Masahiro Yamamoto (Osaka University, Japan) respectively. 129SvEv mice were used as relevant controls for Batf3−/−, Batf2−/− and Batf2/3−/− mice while C57BL/6J mice were controls for all other gene-deficient mice. CRISPR/Cas9 gene editing was used to generate mice with an out-of-frame 7 nucleotide (nt) deletion in Serping1. The sequence of the Serping1 locus was retrieved from NCBI and a sgRNA targeting the third exon was designed based on the RefSeq annotation. The sgRNA was selected based on its low off-target profile and absence of single nucleotide polymorphisms: 5’-AGCACTGAGCAAGCGGCTCCGGG-3’. The gRNA containing the scaffold was generated by in vitro synthesis (Invitrogen, MEGAscript T7 Transcription Kit) followed by cleanup (Invitrogen, MEGAclear Transcription Clean-Up Kit). Cas9 protein and gRNA were complexed and electroporated into C57BL/6J zygotes. After next-generation sequencing of founders and two generations of mice backcrossed to C57BL6/J mice, a mouse line with a 5’-GCCCGGA-3’ deletion at nt 383–389 in the Serpingl cDNA was generated. After an additional round of backcrossing to C57BL6/J mice, homozygous Serpingl−/− mice were produced and used for experiments. Sanger sequencing of a PCR amplicon (forward primer: 5’ CCTGGCCAACAACCAGTGTAGC-3’, reverse primer: 5’-GGCACAGCCTCGCAGAAG G-3’) was used for genotyping. Serpingl−/− mice were born in normal Mendelian frequencies and showed no apparent defects in development, growth, or fecundity. Lack of expression of Serpingl mRNA expression was confirmed in lungs of Serpingl−/− Mtb-infected mice. Cryopreserved sperm from Trafdl−/− (43) mice was obtained from Riken Resource Center (Tokyo, Japan) and the in vitro fertilization was performed in the Micro-injection Core at Washington University in St. Louis. All mouse strains were bred in-house, used at 6 to 8 weeks of age, and sex-matched for all experiments.
Infection and tissue harvest
Inbred and outbred mice were co-housed but segregated by sex in an ABSL3 facility and infected via aerosol route with ~100 CFU of Mtb strain HN878 using a Glas-Col airborne infection system as described previously (91). At given time points following infection, lungs were collected and homogenized to assess bacterial burden. Indian rhesus macaques were housed in an ABSL3 facility and verified to be free of Mtb infection by tuberculin skin test. Mtb- naive, SPF Indian rhesus macaques were exposed to aerosols of Mtb strain CDC1551 (92). Animals were either exposed to a high (93), or low (94) dose designed to inoculate 100 or 10 CFU in their lungs. While both groups of animals developed positivity to mammalian old tuberculin following experimental aerosol infection, animals exposed to the higher dose developed TB, as characterized by fever, rapid weight-loss, elevated serum C-reactive protein (CRP) levels, high chest X-ray scores, and detection of viable Mtb CFUs in the bronchoalveolar lavage fluid (93). The animals exposed to low dose were deemed to have asymptomatic TB based on the absence of clinical signals but evidence of TB exposure (Tuberculin skin test, PRIMAGAM-IGRA). The animals were subjected to weekly physical examinations by board certified clinician(s), including evaluation of body temperature and weight, and complete blood chemistries, including serum CRP levels (94, 95). Lung tissue from both mice and NHPs were homogenized, snap-frozen in RLT buffer, and DNAse-treated total RNA was extracted using the Qiagen RNeasy Mini kit (Qiagen) for RNA-Seq analysis.
In vitro Mtb infection of bone marrow derived-macrophages
Bone marrow cells from the femur and tibia of C57BL/6J and gene deficient mice were extracted, and 1×107 cells were plated in 10 ml of complete Dulbecco’s modified eagle’s medium (cDMEM) supplemented with 20 ng/ml mouse recombinant mouse (rm) granulocyte-macrophage colony-stimulating factor (GM-CSF) (Peprotech) (8). Cells were then cultured at 37°C in 5% CO2. On day 3, 10 ml of cDMEM containing 20 ng/ml rmGM-CSF was added. On day 7, adherent cells were collected as macrophages (8). Macrophages were infected with Mtb strain HN878 (Multiplicity of infection, MOI 1) in antibiotic-free cDMEM for 2 dpi, after which culture supernatants were assayed for IL-10 and TNF-a using ELISA (R&D Biosystems) according to recommended standard protocols.
Generation of single-cell suspensions from tissues and flow cytometry staining
Single-cell suspensions of lungs from untreated or Mtb-infected mice were isolated as previously described (91). Briefly, mice were euthanized with CO2, the right lower lobe was isolated and perfused with heparin in saline. Lungs were minced and incubated in collagenase/DNAse for 30 minutes at 37°C. Lung tissue was pushed through a 70 μm nylon screen to obtain a single cell suspension. Following lysis of erythrocytes, the cells were washed and resuspended in complete DMEM (DMEM high glucose/10% fetal bovine serum/ 1% Penicillin/Streptomycin) for flow cytometry staining. The following fluorochrome conjugated antibodies were used for cell surface staining CD11b-APC (Tonbo Biosciences) (Clone: M1/70), CD11c-PeCy7 (Clone: HL3, BD Biosciences) and GR-1 PE (Clone: RB6–8C5, Tonbo Biosciences). CD3 AF700 (clone: 500A2, BD Biosciences), CD4 PB (Clone: RM4.5, BD Biosciences), CD44 PeCy7 (Clone: 1M7, Tonbo Biosciences), CD8 APC Cy7 (Clone: 53–6.7, BD Biosciences). For intracellular staining, fixation/permeabilization concentrate and diluent (eBioscience) were used to fix and permeabilize lung cells following manufacturer’s instructions. Intracellular staining with IFNγ APC (clone: XMG1.2, Tonbo Biosciences) and IL-17A PE (BD Pharmingen) or the respective isotype control antibodies (APC rat IgG1 k and PE rat IgG1 k isotype, BD Pharmingen) was performed for 30 min. Samples were acquired on a 4 laser BD Fortessa Flow Cytometer and the analysis was performed using FlowJo (Treestar LLC). As before (70), alveolar macrophages were gated on CD11c+CD11b− cells, neutrophils were defined as CD11b+CD11c−Gr-1hi cells, monocytes were defined as CD11b+CD11c−Gr-1med cells, and recruited macrophages were defined as CD11b+CD11c−Gr-1low cells (fig. S8). Total numbers of cells within each gate were back calculated based on cell counts/ individual lung sample.
Histological analysis
The left upper lobe was collected for histological analysis of inflammation. The lobes were infused with 10% neutral buffered formalin and embedded in paraffin. Lung section (5 μm) were cut using a microtome, stained with hematoxylin and eosin (H&E) and processed for light microscopy. Formalin fixed paraffin-embedded (FFPE) lung sections were stained for collagen and muscle using Masson’s Trichrome 2000 stain procedure kit (American Master Tech) as per the manufacturer’s instructions. Images were captured using the automated Nanozoomer digital whole slide imaging system (Hamamatsu Photonics). Regions of inflammatory cell infiltration were delineated utilizing the NDP view2 software (Hamamatsu Photonics), and the percentage of inflammation was calculated in relation to the total lung area of each section. All scoring was conducted in a blinded manner utilizing n=3 to 5 per group of mice.
In situ hybridization
FFPE lung tissue sections were subjected to in situ hybridization (ISH) with the RNAscope Probes - Scarf1 (Cat#535551) or Serping1 (Cat#535071), using the RNAscope 2.5 HD Detection Reagent-Red kit, or the RNAscope 2.5 HD Detection Reagent-BROWN kit, as per the manufacturer’s recommendations (Advanced Cell Diagnostics). Representative images were acquired with the Leica Application Suite V4.5 microscope and software (Leica Microsystems).
RNA-Seq
Mouse or macaque lung tissues were homogenized, snap-frozen in RLT buffer, and DNAse-treated total RNA was extracted using the Qiagen RNeasy Mini kit (Qiagen). RNA sequencing libraries were generated using Clontech SMART-Seq v4 Ultra Low Input RNA Kit for sequencing and Illumina Nextera XT DNA Library preparation kit following the manufacturer’s protocol. The cDNA libraries were validated using KAPA Biosystems primer premix kit with Illumina-compatible DNA primers and quality was examined using Agilent Tapestation 2200. The cDNA libraries were pooled at a final concentration of 1.8 pM. Cluster generation and 75 bp Paired-read dual-indexed sequencing was performed on Illumina Next Seq 500. After adapter trimming using Trimmomatic v0.36 (96), RNA-seq reads were aligned to their respective genome assemblies (Mus musculus GRCm38 and Macaca mulatta Mmul_8.0.1 - Ensembl release 91.0(14)) using HISAT2 v2.1.0 (97). All raw RNA-Seq fastq files were uploaded to the NCBI Sequence Read Archive [SRA (98)], and complete sample metadata and accession information is provided in table S1. Read fragments (read pairs or single reads) were quantified per gene per sample using feature Counts (version 1.5.1) (99). FPKM (fragments per kilobase of gene length per million reads mapped) normalization was also performed. Gene annotations and matches across species were assigned by retrieving the top hit macaque and human gene to each mouse gene using data retrieved from Ensembl (14) (release 91.0). Human data for the blood RNA signature was retrieved from a previous study (7), and significance values for the association with progressors or controllers were aligned to mouse genes using these human gene matches to the mouse genome. Because Ensembl annotations did not provide a direct ortholog for several key signature genes, these were instead matched by identifying the top mouse hit for the human genes: GBP4 to ENSMUSG00000028268 (reciprocal top hit), GBP-1 to ENSMUSG00000040264 (mouse to human second-best hit; GBP-2 was the top hit) and ETV-7 to ENSMUSG00000030199 (mouse to human second-best hit; ETV-6 was the top hit). Also, note that FCRG1a and FCRG1b both matched to the same mouse gene (ENSMUSG00000015947; mouse to human two top hits are to these two genes. Significantly differentially expressed genes between naive, controller and progressor sample sets were identified using DESeq2 (version 1.4.5) (100) with default settings, and a minimum P value significance threshold of 0.05 (after False Discovery Rate [FDR (101)] correction for the number of tests). Principal components analysis also was calculated using DESeq2 output (default settings, using the top 500 most variable genes.. Read counts, relative gene expression levels, gene annotations, and differential expression data for the mouse genes and their corresponding macaque genes, and the alignment to the human dataset (7) are available in table S2. Pathway enrichment analysis for Reactome (57) pathways among gene sets of interest was performed using the WebGestalt (56) web server (table S6).
Statistical analysis
Where applicable, comparisons between two groups were performed using the unpaired two-tailed Student t-test, using the GraphPad Prism 5 software. In all instances, P values ≤0.05 were considered as significant. DESeq2 (version 1.4.5) (100) with default settings, and a minimum P value significance threshold of 0.05 (after False Discovery Rate [FDR (101)] correction for the number of tests was used for all RNA-Seq comparisons of differential expression. Associations between ACS signature gene expression levels and TB infection metadata (presented as significance P values for Pearson correlations; table S4) were tested using the Student’s ‘t’ distribution, and performing FDR correction for the number of tests performed.
Supplementary Material
Table S1. RNA-Seq sample metadata and accessions.
Table S3. Significance testing for overlaps of differentially expressed genes between species.
Table S4. Correlation analysis between ACS signature genes and tuberculosis metadata (lung inflammation and bacterial bacteria) among mouse controller and progressor samples.
Table S5. Flow cytometric quantification of lung cellular subsets in control and gene-deficient models infected with Mtb.
Table S6. Significantly enriched REACTOME pathways among gene sets of interest.
Table S2. FPKM gene expression values and differential expression statistics for the top 1000 mouse genes, including information about ortholog matches in human and macaque.
Fig.S1. Differential gene expression (DESeq2) across species.
Fig.S2. Serping1 regulates inflammation but not Mtb control in mice.
Fig.S3. T cell cytokine profile and myeloid cell recruitment in Mtb-infected Tap1−/− mice.
Fig.S4. Protection from Mtb HN878 infection is mediated by Batf2.
Fig.S5. IL-17-producing CD4+ T cell cytokines are increased in Mtb-infected Batf2/3−/− mice.
Fig.S6. T cell cytokine profile in Mtb-infected Fcgr1−/− mice.
Fig.S7. SCARF1 expression drives inflammation within TB granulomas.
Fig. S8. Hierarchical gating strategy used to identify myeloid populations in mice lungs.
Data file S1. Primary Data
ACKNOWLEDGEMENTS
This work was supported by Washington University in St. Louis, NIH grant HL105427, AI111914-02 to S.A.K., and AI123780 to S.A.K., D.K., T.S. and M.M., AI134236–02 to S.A.K and D.K. Department of Molecular Microbiology, Washington University, and Stephen I. Morse Fellowship to S.D. J-R.M. was supported by funds of the Department of Medicine, University of Rochester Medical Center, and NIH grant U19 AI91036. Work was supported by NIH grant U19AI106772 to M.S.D. We thank the Genome Engineering and iPSC center and Department of Pathology Micro-Injection Core (Washington University in St. Louis) for their assistance in generating the Serpingt−/− and Trafdt−/− mice. We thank Nadia Golden and Allison Bucsan (Tulane National Primate Research Center) for assistance with the macaque samples.
Footnotes
COMPETING FINANCIAL INTERESTS
T.J.S. is co-inventor of a patent (pending) of the 16-gene ACS signature. All other authors declare no competing financial interests.
DATA AND MATERIALS AVAILABILITY
All data associated with this study are present in the paper or Supplementary Materials. All unprocessed RNA-Seq reads are available for download from the NCBI Sequence Read Archive (SRA), Bioproject PRJNA523820. Accession numbers and metadata per sample are provided in table S1, and processed read counts are provided in table S2. Serpingt−/− mice are available upon execution of MTA with Washington University in St.Louis (http://otminnovate.wustl.edu/).
REFERENCES AND NOTES
- 1.Flynn JL, Lessons from experimental Mycobacterium tuberculosis infections. Microbes and Infection 8, 1179–1188 (2006). [DOI] [PubMed] [Google Scholar]
- 2.Kagina BM, Abel B, Scriba TJ, Hughes EJ, Keyser A, Soares A, Gamieldien H, Sidibana M, Hatherill M, Gelderbloem S, Mahomed H, Hawkridge A, Hussey G, Kaplan G, Hanekom WA, other members of the South African Tuberculosis Vaccine, Specific T cell frequency and cytokine expression profile do not correlate with protection against tuberculosis after bacillus Calmette-Guerin vaccination of newborns. American journal of respiratory and critical care medicine 182, 1073–1079 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Berry MP, Graham CM, McNab FW, Xu Z, Bloch SA, Oni T, Wilkinson KA, Banchereau R, Skinner J, Wilkinson RJ, Quinn C, Blankenship D, Dhawan R, Cush JJ, Mejias A, Ramilo O, Kon OM, Pascual V, Banchereau J, Chaussabel D, O’Garra A, An interferon-inducible neutrophil-driven blood transcriptional signature in human tuberculosis. Nature 466, 973–977 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kaforou M, Wright VJ, Oni T, French N, Anderson ST, Bangani N, Banwell CM, Brent AJ, Crampin AC, Dockrell HM, Eley B, Heyderman RS, Hibberd ML, Kern F, Langford PR, Ling L, Mendelson M, Ottenhoff TH, Zgambo F, Wilkinson RJ, Coin LJ, Levin M, Detection of Tuberculosis in HIV-Infected and -Uninfected African Adults Using Whole Blood RNA Expression Signatures: A Case-Control Study. PLOS Medicine 10, e1001538 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maertzdorf J, Repsilber D, Parida SK, Stanley K, Roberts T, Black G, Walzl G, Kaufmann SH, Human gene expression profiles of susceptibility and resistance in tuberculosis. Genes and immunity 12, 15–22 (2011). [DOI] [PubMed] [Google Scholar]
- 6.Scriba TJ, Penn-Nicholson A, Shankar S, Hraha T, Thompson EG, Sterling D, Nemes E, Darboe F, Suliman S, Amon LM, Mahomed H, Erasmus M, Whatney W, Johnson JL, Boom WH, Hatherill M, Valvo J, De Groote MA, Ochsner UA, Aderem A, Hanekom WA, Zak DE, A. C. S. c. s. t. other members of the, Sequential inflammatory processes define human progression from M. tuberculosis infection to tuberculosis disease. PLOS Pathogens 13, e1006687 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zak DE, Penn-Nicholson A, Scriba TJ, Thompson E, Suliman S, Amon LM, Mahomed H, Erasmus M, Whatney W, Hussey GD, Abrahams D, Kafaar F, Hawkridge T, Verver S, Hughes EJ, Ota M, Sutherland J, Howe R, Dockrell HM, Boom WH, Thiel B, Ottenhoff THM, Mayanja-Kizza H, Crampin AC, Downing K, Hatherill M, Valvo J, Shankar S, Parida SK, Kaufmann SHE, Walzl G, Aderem A, Hanekom WA, Acs GC c. s. groups, A blood RNA signature for tuberculosis disease risk: a prospective cohort study. Lancet 387, 2312–2322 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Gopal R, Monin L, Torres D, Slight S, Mehra S, McKenna KC, Fallert Junecko BA, Reinhart TA, Kolls J, Baez-Saldana R, Cruz-Lagunas A, Rodriguez-Reyna TS, Kumar NP, Tessier P, Roth J, Selman M, Becerril-Villanueva E, Baquera-Heredia J, Cumming B, Kasprowicz VO, Steyn AJ, Babu S, Kaushal D, Zuniga J, Vogl T, Rangel-Moreno J, Khader SA, S100A8/A9 proteins mediate neutrophilic inflammation and lung pathology during tuberculosis. American journal of respiratory and critical care medicine 188, 1137–1146 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niazi MK, Dhulekar N, Schmidt D, Major S, Cooper R, Abeijon C, Gatti DM, Kramnik I, Yener B, Gurcan M, Beamer G, Lung necrosis and neutrophils reflect common pathways of susceptibility to Mycobacterium tuberculosis in genetically diverse, immune-competent mice. Dis Model Mech 8, 1141–1153 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kaushal D, Mehra S, Didier PJ, Lackner AA, The non-human primate model of tuberculosis. Journal of Medical Primatology 41, 191–201 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Machingaidze S, Verver S, Mulenga H, Abrahams DA, Hatherill M, Hanekom W, Hussey GD, Mahomed H, Predictive value of recent QuantiFERON conversion for tuberculosis disease in adolescents. American journal of respiratory and critical care medicine 186, 1051–1056 (2012). [DOI] [PubMed] [Google Scholar]
- 12.Mahomed H, Ehrlich R, Hawkridge T, Hatherill M, Geiter L, Kafaar F, Abrahams DA, Mulenga H, Tameris M, Geldenhuys H, Hanekom WA, Verver S, Hussey GD, TB incidence in an adolescent cohort in South Africa. PloS one 8, e59652 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zerbino DR, Achuthan P, Akanni W, Amode MR, Barrell D, Bhai J, Billis K, Cummins C, Gall A, Giron CG, Gil L, Gordon L, Haggerty L, Haskell E, Hourlier T, Izuogu OG, Janacek SH, Juettemann T, To JK, Laird MR, Lavidas I, Liu Z, Loveland JE, Maurel T, McLaren W, Moore B, Mudge J, Murphy DN, Newman V, Nuhn M, Ogeh D, Ong CK, Parker A, Patricio M, Riat HS, Schuilenburg H, Sheppard D, Sparrow H, Taylor K, Thormann A, Vullo A, Walts B, Zadissa A, Frankish A, Hunt SE, Kostadima M, Langridge N, Martin FJ, Muffato M, Perry E, Ruffier M, Staines DM, Trevanion SJ, Aken BL, Cunningham F, Yates A, Flicek P, Ensembl 2018. Nucleic Acids Research 46, D754–D761 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Weaver M, Yingling JM, Dunn NR, Bellusci S, Hogan BL, Bmp signaling regulates proximal-distal differentiation of endoderm in mouse lung development. Development (Cambridge, England) 126, 4005–4015 (1999). [DOI] [PubMed] [Google Scholar]
- 16.Lopez IP, Pineiro-Hermida S, Pais RS, Torrens R, Hoeflich A, Pichel JG, Involvement of Igf1r in Bronchiolar Epithelial Regeneration: Role during Repair Kinetics after Selective Club Cell Ablation. PloS one 11, e0166388 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Usul Afsar C, Sahin B, Gunaldi M, Kilic Bagir E, Gumurdulu D, Burgut R, Erkisi M, Kara IO, Paydas S, Karaca F, Ercolak V, Expression of fibroblast growth factor receptor 1, fibroblast growth factor 2, phosphatidyl inositol 3 phosphate kinase and their clinical and prognostic significance in early and advanced stage of squamous cell carcinoma of the lung. International journal of clinical and experimental pathology 8, 9760–9771 (2015). [PMC free article] [PubMed] [Google Scholar]
- 18.Tomasovic A, Kurrle N, Wempe F, De-Zolt S, Scheibe S, Koli K, Serchinger M, Schnutgen F, Surun D, Sterner-Kock A, Weissmann N, von Melchner H, Ltbp4 regulates Pdgfrbeta expression via TGFbeta-dependent modulation of Nrf2 transcription factor function. Matrix biology : journal of the International Society for Matrix Biology 59, 109–120 (2017). [DOI] [PubMed] [Google Scholar]
- 19.Omelchenko T, Hall A, Myosin-IXA regulates collective epithelial cell migration by targeting RhoGAP activity to cell-cell junctions. Current biology : CB 22, 278–288 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Fischer BM, Cuellar JG, Byrd AS, Rice AB, Bonner JC, Martin LD, Voynow JA, ErbB2 activity is required for airway epithelial repair following neutrophil elastase exposure. FASEB journal: official publication of the Federation of American Societies for Experimental Biology 19, 1374–1376 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Lyadova IV, Neutrophils in Tuberculosis: Heterogeneity Shapes the Way? Mediators Inflamm 2017, 8619307 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martineau AR, Newton SM, Wilkinson KA, Kampmann B, Hall BM, Nawroly N, Packe GE, Davidson RN, Griffiths CJ, Wilkinson RJ, Neutrophil-mediated innate immune resistance to mycobacteria. J Clin Invest 117, 1988–1994 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nouailles G, Dorhoi A, Koch M, Zerrahn J, Weiner J 3rd, Fae KC, Arrey F, Kuhlmann S, Bandermann S, Loewe D, Mollenkopf HJ, Vogelzang A, Meyer-Schwesinger C, Mittrucker HW, McEwen G, Kaufmann SH, CXCL5-secreting pulmonary epithelial cells drive destructive neutrophilic inflammation in tuberculosis. J Clin Invest 124, 1268–1282 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Domingo-Gonzalez R, Prince O, Cooper A, Khader SA, Cytokines and Chemokines in Mycobacterium tuberculosis Infection. Microbiology spectrum 4, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nitsche C, Edderkaoui M, Moore RM, Eibl G, Kasahara N, Treger J, Grippo PJ, Mayerle J, Lerch MM, Gukovskaya AS, The phosphatase PHLPP1 regulates Akt2, promotes pancreatic cancer cell death, and inhibits tumor formation. Gastroenterology 142, 377–387.e875 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schneider T, Martinez-Martinez A, Cubillos-Rojas M, Bartrons R, Ventura F, Rosa JL, The E3 ubiquitin ligase HERC1 controls the ERK signaling pathway targeting C-RAF for degradation. Oncotarget 9, 31531–31548 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Takai H, Ashihara M, Ishiguro T, Terashima H, Watanabe T, Kato A, Suzuki M, Involvement of glypican-3 in the recruitment of M2-polarized tumor-associated macrophages in hepatocellular carcinoma. Cancer biology & therapy 8, 2329–2338 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Ramirez-Ortiz ZG, Pendergraft WF 3rd, Prasad A, Byrne MH, Iram T, Blanchette CJ, Luster AD, Hacohen N, El Khoury J, Means TK, The scavenger receptor SCARF1 mediates the clearance of apoptotic cells and prevents autoimmunity. Nature immunology 14, 917–926 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ihara M, Kinoshita A, Yamada S, Tanaka H, Tanigaki A, Kitano A, Goto M, Okubo K, Nishiyama H, Ogawa O, Takahashi C, Itohara S, Nishimune Y, Noda M, Kinoshita M, Cortical Organization by the Septin Cytoskeleton Is Essential for Structural and Mechanical Integrity of Mammalian Spermatozoa. Developmental cell 8, 343–352 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Wang LC, Kuo F, Fujiwara Y, Gilliland DG, Golub TR, Orkin SH, Yolk sac angiogenic defect and intra embryonic apoptosis in mice lacking the Ets related factor TEL. The EMBO Journal 16, 4374–4383 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Noris M, Remuzzi G, Overview of complement activation and regulation. Seminars in nephrology 33, 479–492 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Marquis JF, Nantel A, LaCourse R, Ryan L, North RJ, Gros P, Fibrotic response as a distinguishing feature of resistance and susceptibility to pulmonary infection with Mycobacterium tuberculosis in mice. Infection and immunity 76, 78–88 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mehra S, Alvarez X, Didier PJ, Doyle LA, Blanchard JL, Lackner AA, Kaushal D, Granuloma correlates of protection against tuberculosis and mechanisms of immune modulation by Mycobacterium tuberculosis. The Journal of Infectious Diseases 207, 1115–1127 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rathi S, Jalali S, Patnaik S, Shahulhameed S, Musada GR, Balakrishnan D, Rani PK, Kekunnaya R, Chhablani PP, Swain S, Giri L, Chakrabarti S, Kaur I, Abnormal Complement Activation and Inflammation in the Pathogenesis of Retinopathy of Prematurity. Frontiers in immunology 8, 1868–1868 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sprong T, Moller AS, Bjerre A, Wedege E, Kierulf P, van der Meer JW, Brandtzaeg P, van Deuren M, Mollnes TE, Complement activation and complement-dependent inflammation by Neisseria meningitidis are independent of lipopolysaccharide. Infection and immunity 72, 3344–3349 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Durbin JE, Hackenmiller R, Simon MC, Levy DE, Targeted disruption of the mouse Stat1 gene results in compromised innate immunity to viral disease. Cell 84, 443–450 (1996). [DOI] [PubMed] [Google Scholar]
- 37.Sugawara I, Yamada H, Mizuno S, STAT1 knockout mice are highly susceptible to pulmonary mycobacterial infection. The Tohoku journal of experimental medicine 202, 41–50 (2004). [DOI] [PubMed] [Google Scholar]
- 38.Van Kaer L, Ashton-Rickardt PG, Ploegh HL, Tonegawa S, TAP1 mutant mice are deficient in antigen presentation, surface class I molecules, and CD4–8+ T cells. Cell 71, 1205–1214 (1992). [DOI] [PubMed] [Google Scholar]
- 39.Behar SM, Dascher CC, Grusby MJ, Wang C-R, Brenner MB, Susceptibility of Mice Deficient in CD1D or TAP1 to Infection with Mycobacterium tuberculosis. The Journal of experimental medicine 189, 1973–1980 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Anderson SL, Carton JM, Lou J, Xing L, Rubin BY, Interferon-induced guanylate binding protein-1 (GBP-1) mediates an antiviral effect against vesicular stomatitis virus and encephalomyocarditis virus. Virology 256, 8–14 (1999). [DOI] [PubMed] [Google Scholar]
- 41.Kim BH, Shenoy AR, Kumar P, Das R, Tiwari S, MacMicking JD, A family of IFN-gamma-inducible 65-kD GTPases protects against bacterial infection. Science (New York, N.Y.) 332, 717–721 (2011). [Google Scholar]
- 42.Kresse A, Konermann C, Degrandi D, Beuter-Gunia C, Wuerthner J, Pfeffer K, Beer S, Analyses of murine GBP homology clusters based on in silico, in vitro and in vivo studies. BMC Genomics 9, 158 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sanada T, Takaesu G, Mashima R, Yoshida R, Kobayashi T, Yoshimura A, FLN29 deficiency reveals its negative regulatory role in the Toll-like receptor (TLR) and retinoic acid-inducible gene I (RIG-I)-like helicase signaling pathway. The Journal of biological chemistry 283, 33858–33864 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mashima R, Saeki K, Aki D, Minoda Y, Takaki H, Sanada T, Kobayashi T, Aburatani H, Yamanashi Y, Yoshimura A, FLN29, a novel interferon- and LPS-inducible gene acting as a negative regulator of toll-like receptor signaling. The Journal of biological chemistry 280, 41289–41297 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Byers B, Goetsch L, A highly ordered ring of membrane-associated filaments in budding yeast. The Journal of cell biology 69, 717–721 (1976). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Guler R, Mpotje T, Ozturk M, Nono JK, Parihar SP, Chia JE, Abdel Aziz N, Hlaka L, Kumar S, Roy S, Penn-Nicholson A, Hanekom WA, Zak DE, Scriba TJ, Suzuki H, Brombacher F, Batf2 differentially regulates tissue immunopathology in Type 1 and Type 2 diseases. Mucosal Immunology 12, 390–402 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Newbrough SA, Mocsai A, Clemens RA, Wu JN, Silverman MA, Singer AL, Lowell CA, Koretzky GA, SLP-76 regulates Fcgamma receptor and integrin signaling in neutrophils. Immunity 19, 761–769 (2003). [DOI] [PubMed] [Google Scholar]
- 48.Edberg JC, Yee AMF, Rakshit DS, Chang DJ, Gokhale JA, Indik ZK, Schreiber AD, Kimberly RP, The Cytoplasmic Domain of Human FcyRIa Alters the Functional Properties of the FcyRI-y-Chain Receptor Complex. Journal of Biological Chemistry 274, 30328–30333 (1999). [DOI] [PubMed] [Google Scholar]
- 49.van Vugt MJ, Kleijmeer MJ, Keler T, Zeelenberg I, van Dijk MA, Leusen JHW, Geuze HJ, van de Winkel JGJ, The FcyRIa (CD64) Ligand Binding Chain Triggers Major Histocompatibility Complex Class II Antigen Presentation Independently of Its Associated FcR y-Chain. Blood 94, 808–817 (1999). [PubMed] [Google Scholar]
- 50.DiLillo DJ, Ravetch JV, Fc-Receptor Interactions Regulate Both Cytotoxic and Immunomodulatory Therapeutic Antibody Effector Functions. Cancer immunology research 3, 704–713 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Tamura Y, Osuga J.-i., Adachi H, Tozawa R.-i., Takanezawa Y, Ohashi K, Yahagi N, Sekiya M, Okazaki H, Tomita S, Iizuka Y, Koizumi H, Inaba T, Yagyu H, Kamada N, Suzuki H, Shimano H, Kadowaki T, Tsujimoto M, Arai H, Yamada N, Ishibashi S, Scavenger Receptor Expressed by Endothelial Cells I (SREC-I) Mediates the Uptake of Acetylated Low Density Lipoproteins by Macrophages Stimulated with Lipopolysaccharide. Journal of Biological Chemistry 279, 30938–30944 (2004). [DOI] [PubMed] [Google Scholar]
- 52.van Erp EA, Luytjes W, Ferwerda G, van Kasteren PB, Fc-Mediated Antibody Effector Functions During Respiratory Syncytial Virus Infection and Disease. Frontiers in Immunology 10, 548 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Munoz LE, Berens C, Lauber K, Gaipl US, Herrmann M, Apoptotic Cell Clearance and Its Role in the Origin and Resolution of Chronic Inflammation. Frontiers in Immunology 6, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Penn-Nicholson A, Hraha T, Thompson EG, Sterling D, Mbandi SK, Wall KM, Fisher M, Suliman S, Shankar S, Hanekom WA, Janjic N, Hatherill M, Kaufmann SHE, Sutherland J, Walzl G, De Groote MA, Ochsner U, Zak DE, Scriba TJ, Acs GC c. s. groups, Discovery and validation of a prognostic proteomic signature for tuberculosis progression: A prospective cohort study. PLOS Medicine 16, e1002781 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP, Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102, 15545–15550 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang J, Vasaikar S, Shi Z, Greer M, Zhang B, WebGestalt 2017: a more comprehensive, powerful, flexible and interactive gene set enrichment analysis toolkit. Nucleic Acids Research 45, W130–W137 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fabregat A, Jupe S, Matthews L, Sidiropoulos K, Gillespie M, Garapati P, Haw R, Jassal B, Korninger F, May B, Milacic M, Roca CD, Rothfels K, Sevilla C, Shamovsky V, Shorser S, Varusai T, Viteri G, Weiser J, Wu G, Stein L, Hermjakob H, D’Eustachio P, The Reactome Pathway Knowledgebase. Nucleic Acids Research 46, D649–d655 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Joosten SA, Fletcher HA, Ottenhoff THM, A Helicopter Perspective on TB Biomarkers: Pathway and Process Based Analysis of Gene Expression Data Provides New Insight into TB Pathogenesis. PloS one 8, e73230 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bjornson-Hooper ZB, Fragiadakis GK, Spitzer MH, Madhireddy D, McIlwain D, Nolan GP, A comprehensive atlas of immunological differences between humans, mice and non-human primates. bioRxiv, 574160 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Cooper AM, Dalton DK, Stewart TA, Griffin JP, Russell DG, Orme IM, Disseminated tuberculosis in interferon gamma gene-disrupted mice. The Journal of experimental medicine 178, 2243–2247 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Flynn JL, Chan J, Triebold KJ, Dalton DK, Stewart TA, Bloom BR, An essential role for interferon gamma in resistance to Mycobacterium tuberculosis infection. The Journal of experimental medicine 178, 2249–2254 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Flynn JL, Goldstein MM, Chan J, Triebold KJ, Pfeffer K, Lowenstein CJ, Schreiber R, Mak TW, Bloom BR, Tumor necrosis factor-alpha is required in the protective immune response against Mycobacterium tuberculosis in mice. Immunity 2, 561–572 (1995). [DOI] [PubMed] [Google Scholar]
- 63.Bustamante J, Boisson-Dupuis S, Abel L, Casanova JL, Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-gamma immunity. Seminars in immunology 26, 454–470 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Keane J, Gershon S, Wise RP, Mirabile-Levens E, Kasznica J, Schwieterman WD, Siegel JN, Braun MM, Tuberculosis associated with infliximab, a tumor necrosis factor alpha-neutralizing agent. The New England journal of medicine 345, 1098–1104 (2001). [DOI] [PubMed] [Google Scholar]
- 65.Orme IM, The mouse as a useful model of tuberculosis. Tuberculosis (Edinburgh, Scotland) 83, 112–115 (2003). [DOI] [PubMed] [Google Scholar]
- 66.Rhoades ER, Frank AA, Orme IM, Progression of chronic pulmonary tuberculosis in mice aerogenically infected with virulent Mycobacterium tuberculosis. Tubercle and lung disease : the official journal of the International Union against Tuberculosis and Lung Disease 78, 57–66 (1997). [DOI] [PubMed] [Google Scholar]
- 67.Vilaplana C, Cardona P-J, The lack of a big picture in tuberculosis: the clinical point of view, the problems of experimental modeling and immunomodulation. The factors we should consider when designing novel treatment strategies. Frontiers in Microbiology 5, 55 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Smith CM, Proulx MK, Olive AJ, Laddy D, Mishra BB, Moss C, Gutierrez NM, Bellerose MM, Barreira-Silva P, Phuah JY, Baker RE, Behar SM, Kornfeld H, Evans TG, Beamer G, Sassetti CM, Tuberculosis Susceptibility and Vaccine Protection Are Independently Controlled by Host Genotype. mBio 7, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aderem A, Thompson EG, Braun J, Valvo J, Shankar S, Zak DE, Gideon HP, Flynn JL, Lin PL, Skinner JA, Prospective Discrimination of Controllers From Progressors Early After Low-Dose Mycobacterium tuberculosis Infection of Cynomolgus Macaques using Blood RNA Signatures. The Journal of Infectious Diseases 217, 1318–1322 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Treerat P, Prince O, Cruz-Lagunas A, Munoz-Torrico M, Salazar-Lezama MA, Selman M, Fallert-Junecko B, Reinhardt TA, Alcorn JF, Kaushal D, Zuniga J, Rangel-Moreno J, Kolls JK, Khader SA, Novel role for IL-22 in protection during chronic Mycobacterium tuberculosis HN878 infection. Mucosal Immunology 10, 1069–1081 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sittig LJ, Carbonetto P, Engel KA, Krauss KS, Barrios-Camacho CM, Palmer AA, Genetic Background Limits Generalizability of Genotype-Phenotype Relationships. Neuron 91, 1253–1259 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Praefcke GJK, Regulation of innate immune functions by guanylate-binding proteins. International Journal of Medical Microbiology 308, 237–245 (2018). [DOI] [PubMed] [Google Scholar]
- 73.Yamamoto M, Okuyama M, Ma JS, Kimura T, Kamiyama N, Saiga H, Ohshima J, Sasai M, Kayama H, Okamoto T, Huang DC, Soldati-Favre D, Horie K, Takeda J, Takeda K, A cluster of interferon-gamma-inducible p65 GTPases plays a critical role in host defense against Toxoplasma gondii. Immunity 37, 302–313 (2012). [DOI] [PubMed] [Google Scholar]
- 74.Inacio AR, Liu Y, Clausen BH, Svensson M, Kucharz K, Yang Y, Stankovich T, Khorooshi R, Lambertsen KL, Issazadeh-Navikas S, Deierborg T, Endogenous IFN-beta signaling exerts anti-inflammatory actions in experimentally induced focal cerebral ischemia. Journal of neuroinflammation 12, 211 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Schneider WM, Chevillotte MD, Rice CM, Interferon-stimulated genes: a complex web of host defenses. Annual review of immunology 32, 513–545 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fuchs Y, Brown S, Gorenc T, Rodriguez J, Fuchs E, Steller H, Sept4/ARTS regulates stem cell apoptosis and skin regeneration. Science (New York, N.Y.) 341, 286–289 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kissel H, Georgescu MM, Larisch S, Manova K, Hunnicutt GR, Steller H, The Sept4 septin locus is required for sperm terminal differentiation in mice. Developmental cell 8, 353–364 (2005). [DOI] [PubMed] [Google Scholar]
- 78.Dorhoi A, Kaufmann SHE, Versatile myeloid cell subsets contribute to tuberculosis-associated inflammation. European journal of immunology 45, 2191–2202 (2015). [DOI] [PubMed] [Google Scholar]
- 79.Kanehisa M, Furumichi M, Tanabe M, Sato Y, Morishima K, KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Research 45, D353–d361 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Szklarczyk D, Gable AL, Lyon D, Junge A, Wyder S, Huerta-Cepas J, Simonovic M, Doncheva NT, Morris JH, Bork P, Jensen LJ, Mering CV, STRING v11: protein-protein association networks with increased coverage, supporting functional discovery in genome-wide experimental datasets. Nucleic Acids Research 47, D607–d613 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Walzl G, Ronacher K, Hanekom W, Scriba TJ, Zumla A, Immunological biomarkers of tuberculosis. Nature reviews. Immunology 11, 343–354 (2011). [DOI] [PubMed] [Google Scholar]
- 82.Xu W, Roos A, Schlagwein N, Woltman AM, Daha MR, van Kooten C, IL-10-producing macrophages preferentially clear early apoptotic cells. Blood 107, 4930–4937 (2006). [DOI] [PubMed] [Google Scholar]
- 83.Turner J, Gonzalez-Juarrero M, Ellis DL, Basaraba RJ, Kipnis A, Orme IM, Cooper AM, In vivo IL-10 production reactivates chronic pulmonary tuberculosis in C57BL/6 mice. Journal of immunology (Baltimore, Md. : 1950) 169, 6343–6351 (2002). [DOI] [PubMed] [Google Scholar]
- 84.Anders S, Pyl PT, Huber W, HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.McCarthy DJ, Chen Y, Smyth GK, Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res 40, 4288–4297 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Svenson KL, Gatti DM, Valdar W, Welsh CE, Cheng R, Chesler EJ, Palmer AA, McMillan L, Churchill GA, High-resolution genetic mapping using the Mouse Diversity outbred population. Genetics 190, 437–447 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schraml BU, Hildner K, Ise W, Lee WL, Smith WA, Solomon B, Sahota G, Sim J, Mukasa R, Cemerski S, Hatton RD, Stormo GD, Weaver CT, Russell JH, Murphy TL, Murphy KM, The AP-1 transcription factor Batf controls T(H)17 differentiation. Nature 460, 405–409 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Barnes N, Gavin AL, Tan PS, Mottram P, Koentgen F, Hogarth PM, FcgammaRI-deficient mice show multiple alterations to inflammatory and immune responses. Immunity 16, 379–389 (2002). [DOI] [PubMed] [Google Scholar]
- 89.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, Calderon B, Schraml BU, Unanue ER, Diamond MS, Schreiber RD, Murphy TL, Murphy KM, Batf3 deficiency reveals a critical role for CD8alpha+ dendritic cells in cytotoxic T cell immunity. Science (New York, N.Y.) 322, 1097–1100 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Tussiwand R, Lee W-L, Murphy TL, Mashayekhi M, Kc W, Albring JC, Satpathy AT, Rotondo JA, Edelson BT, Kretzer NM, Wu X, Weiss LA, Glasmacher E, Li P, Liao W, Behnke M, Lam SSK, Aurthur CT, Leonard WJ, Singh H, Stallings CL, Sibley LD, Schreiber RD, Murphy KM, Compensatory dendritic cell development mediated by BATF-IRF interactions. Nature 490, 502 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Khader SA, Bell GK, Pearl JE, Fountain JJ, Rangel-Moreno J, Cilley GE, Shen F, Eaton SM, Gaffen SL, Swain SL, Locksley RM, Haynes L, Randall TD, Cooper AM, IL-23 and IL-17 in the establishment of protective pulmonary CD4+ T cell responses after vaccination and during Mycobacterium tuberculosis challenge. Nature immunology 8, 369–377 (2007). [DOI] [PubMed] [Google Scholar]
- 92.Mehra S, Golden NA, Dutta NK, Midkiff CC, Alvarez X, Doyle LA, Asher M, Russell-Lodrigue K, Monjure C, Roy CJ, Blanchard JL, Didier PJ, Veazey RS, Lackner AA, Kaushal D, Reactivation of latent tuberculosis in rhesus macaques by coinfection with simian immunodeficiency virus. Journal of Medical Primatology 40, 233–243 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gautam US, Foreman TW, Bucsan AN, Veatch AV, Alvarez X, Adekambi T, Golden NA, Gentry KM, Doyle-Meyers LA, Russell-Lodrigue KE, Didier PJ, Blanchard JL, Kousoulas KG, Lackner AA, Kalman D, Rengarajan J, Khader SA, Kaushal D, Mehra S, In vivo inhibition of tryptophan catabolism reorganizes the tuberculoma and augments immune-mediated control of Mycobacterium tuberculosis. Proceedings of the National Academy of Sciences 115, E62–E71 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Kaushal D, Foreman TW, Gautam US, Alvarez X, Adekambi T, Rangel-Moreno J, Golden NA, Johnson AM, Phillips BL, Ahsan MH, Russell-Lodrigue KE, Doyle LA, Roy CJ, Didier PJ, Blanchard JL, Rengarajan J, Lackner AA, Khader SA, Mehra S, Mucosal vaccination with attenuated Mycobacterium tuberculosis induces strong central memory responses and protects against tuberculosis. Nature communications 6, 8533 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Foreman TW, Mehra S, LoBato DN, Malek A, Alvarez X, Golden NA, Bucşan AN, Didier PJ, Doyle-Meyers LA, Russell-Lodrigue KE, Roy CJ, Blanchard J, Kuroda MJ, Lackner AA, Chan J, Khader SA, Jacobs WR, Kaushal D, CD4+ T-cell-independent mechanisms suppress reactivation of latent tuberculosis in a macaque model of HIV coinfection. Proceedings of the National Academy of Sciences 113, E5636–E5644 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Bolger AM, Lohse M, Usadel B, Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kim D, Langmead B, Salzberg SL, HISAT: a fast spliced aligner with low memory requirements. Nature Methods 12, 357–360 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Leinonen R, Sugawara H, Shumway M, on C behalf of the International Nucleotide Sequence Database, The Sequence Read Archive. Nucleic Acids Research 39, D19–D21 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Liao Y, Smyth GK, Shi W, featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 100.Anders S, Huber W, Differential expression analysis for sequence count data. Genome biology 11, R106 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Benjamini Y, Hochberg Y, Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society. Series B (Methodological) 57, 289–300 (1995). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. RNA-Seq sample metadata and accessions.
Table S3. Significance testing for overlaps of differentially expressed genes between species.
Table S4. Correlation analysis between ACS signature genes and tuberculosis metadata (lung inflammation and bacterial bacteria) among mouse controller and progressor samples.
Table S5. Flow cytometric quantification of lung cellular subsets in control and gene-deficient models infected with Mtb.
Table S6. Significantly enriched REACTOME pathways among gene sets of interest.
Table S2. FPKM gene expression values and differential expression statistics for the top 1000 mouse genes, including information about ortholog matches in human and macaque.
Fig.S1. Differential gene expression (DESeq2) across species.
Fig.S2. Serping1 regulates inflammation but not Mtb control in mice.
Fig.S3. T cell cytokine profile and myeloid cell recruitment in Mtb-infected Tap1−/− mice.
Fig.S4. Protection from Mtb HN878 infection is mediated by Batf2.
Fig.S5. IL-17-producing CD4+ T cell cytokines are increased in Mtb-infected Batf2/3−/− mice.
Fig.S6. T cell cytokine profile in Mtb-infected Fcgr1−/− mice.
Fig.S7. SCARF1 expression drives inflammation within TB granulomas.
Fig. S8. Hierarchical gating strategy used to identify myeloid populations in mice lungs.
Data file S1. Primary Data