

SUMMARY
Pain thresholds are, in part, set as a function of emotional and internal states by descending modulation of nociceptive transmission in the spinal cord. Neurons of the rostral ventromedial medulla (RVM) are thought to critically contribute to this process; however, the neural circuits and synaptic mechanisms by which distinct populations of RVM neurons facilitate or diminish pain remain elusive. Here we used in vivo opto/chemogenetic manipulations and trans-synaptic tracing of genetically identified dorsal horn and RVM neurons to uncover an RVM-spinal cord-primary afferent circuit controlling pain thresholds. Unexpectedly, we found that RVM GABAergic neurons facilitate mechanical pain by inhibiting dorsal horn enkephalinergic/GABAergic interneurons. We further demonstrate that these interneurons gate sensory inputs and control pain through temporally coordinated enkephalin- and GABA-mediated presynaptic inhibition of somatosensory neurons. Our results uncover a descending disynaptic inhibitory circuit that facilitates mechanical pain, is engaged during stress, and could be targeted to establish higher pain thresholds.
In Brief
François et al. identified a neural circuit that controls mechanical pain thresholds. They demonstrated that GABAergic brainstem neurons regulate the release of the endogenous opioid enkephalin in the spinal cord to modulate inputs from sensory pain fibers.
INTRODUCTION
The brain has long been known to powerfully influence pain thresholds by modulating somatosensory information processing at the level of the spinal cord. This phenomenon, known as the descending control of pain (Basbaum et al., 2009; Porreca et al., 2002), underlies changes in pain thresholds as a function of mood, expectations, and internal states. For example, acute stress and expected pain relief can produce analgesia (i.e., stress-induced and placebo analgesia; Butler and Finn, 2009; Wager and Atlas, 2015), while chronic stress and anxiety can facilitate pain (Jennings et al., 2014), as observed during post-traumatic stress disorder or pain catastrophizing (Palyo and Beck, 2005; Quartana et al., 2009). Previous studies established that descending pain control utilizes neurons of the rostral ventromedial medulla (RVM), an ensemble of functionally related structures, including the raphe magnus and gigantocellular reticular nuclei (Fields et al., 1983a, 1983b; Marinelli et al., 2002; Zhuo and Gebhart, 1990). Classic extracellular recording experiments indicated the existence of several classes of RVM neurons projecting to the spinal cord: on-cells, off-cells, and neutral-cells (Fields et al., 1983a). On-cells are thought to critically contribute to descending pain control by facilitating nociception, presumably via glutamatergic neurotransmission and the excitation of primary afferent terminals and/or excitatory dorsal horn neurons (Heinricher et al., 2009). However, the molecular identity of on-cells is unresolved. Furthermore, the organization of RVM-spinal cord circuits, and mechanisms by which RVM neurons modulate neural activity and nociception at the spinal level, remains poorly understood.
The endogenous opioid system regulates nociception, which includes altering excitability and neurotransmission in the RVM and spinal cord (Basbaum et al., 1976; Heinricher et al., 2009). Exogenous opioid analgesics, such as morphine, act on mu opioid receptors (MORs) on on-cells to reduce pain facilitation and on MORs and delta opioid receptors (DORs) on dorsal root ganglion (DRG) neuron spinal terminals to reduce nociception. By contrast, how endogenous opioids modulate pain remains elusive. Of particular interest are the pentapeptides enkephalins, high-affinity agonists for both DORs and MORs that are particularly abundant in the dorsal horn (Comb et al., 1982; Harlan et al., 1987; Hökfelt et al., 1977; Hughes et al., 1975; Seybold and Elde, 1980). Inhibitors of enzymes degrading enkephalins reduce pain, and intrathecal (i.t.) injection of enkephalins produces analgesia, supporting the critical role of spinal enkephalinergic neuromodulation in pain control (Schreiter et al., 2012; Yaksh et al., 1977). Electrophysiological recordings in spinal cord slices have shown that bath applied exogenous opioids can act on DORs and MORs to presynaptically inhibit neurotransmitter release from DRG axon terminals (Bardoni et al., 2013, 2014; Heinke et al., 2011). Whether enkephalins endogenously released from spinal neurons act in a similar manner and contribute to defining pain thresholds is not known.
Here we report that RVM GABAergic neurons integrate stress information and limit enkephalinergic and GABAergic presynaptic inhibition of DRG neurons in the dorsal horn to facilitate mechanical pain.
RESULTS
Spinal Enkephalinergic Neurons Controlling Nociception Receive Inputs from the RVM
To identify enkephalinergic dorsal horn neurons, manipulate their activity, and define their inputs, we generated knockin mice in which the preproenkephalin gene (Penk) promotor drives Cre recombinase expression. We crossed PenkCre mice with Rosa26-LSL-tdTomato mice (Ai14 line) (Madisen et al., 2010) (Figure S1) and also examined Cre expression patterns by the injection of a Cre-dependent recombinant adeno-associated virus (AAV) expressing yellow fluorescent protein (YFP) (Figure 1A). In situ hybridization experiments confirmed Cre activity in the majority of Penk+ neurons (tdTomato: 78% ± 1.6%; n = 4; YFP: 88% ± 2.5%; n = 6) (Figure S1C; Figure 1B), with very limited Penk expression in DRG neurons (Figure S1B), consistent with previous reports (Harlan et al., 1987; Marvizón et al., 2009; Pohl et al., 1994; Seybold and Elde, 1980). Immunohistochemical and electrophysiological analyses indicated that Penk+ dorsal horn neurons consist of a mixed population of GABAergic and glutamatergic neurons (Figure S1D; Figures 1C and 1D) throughout spinal laminae I to III (37% ± 2.7% of all neurons in LI-III express Penk) (Figure S1E), as shown previously (Chen et al., 2014; Harlan et al., 1987; Huang et al., 2008; Liu et al., 2015; Todd et al., 2003).
Figure 1. Enkephalinergic Neurons in the Dorsal Horn Modulate Mechanical Sensitivity and Receive Inputs from RVM GABAergic Neurons.
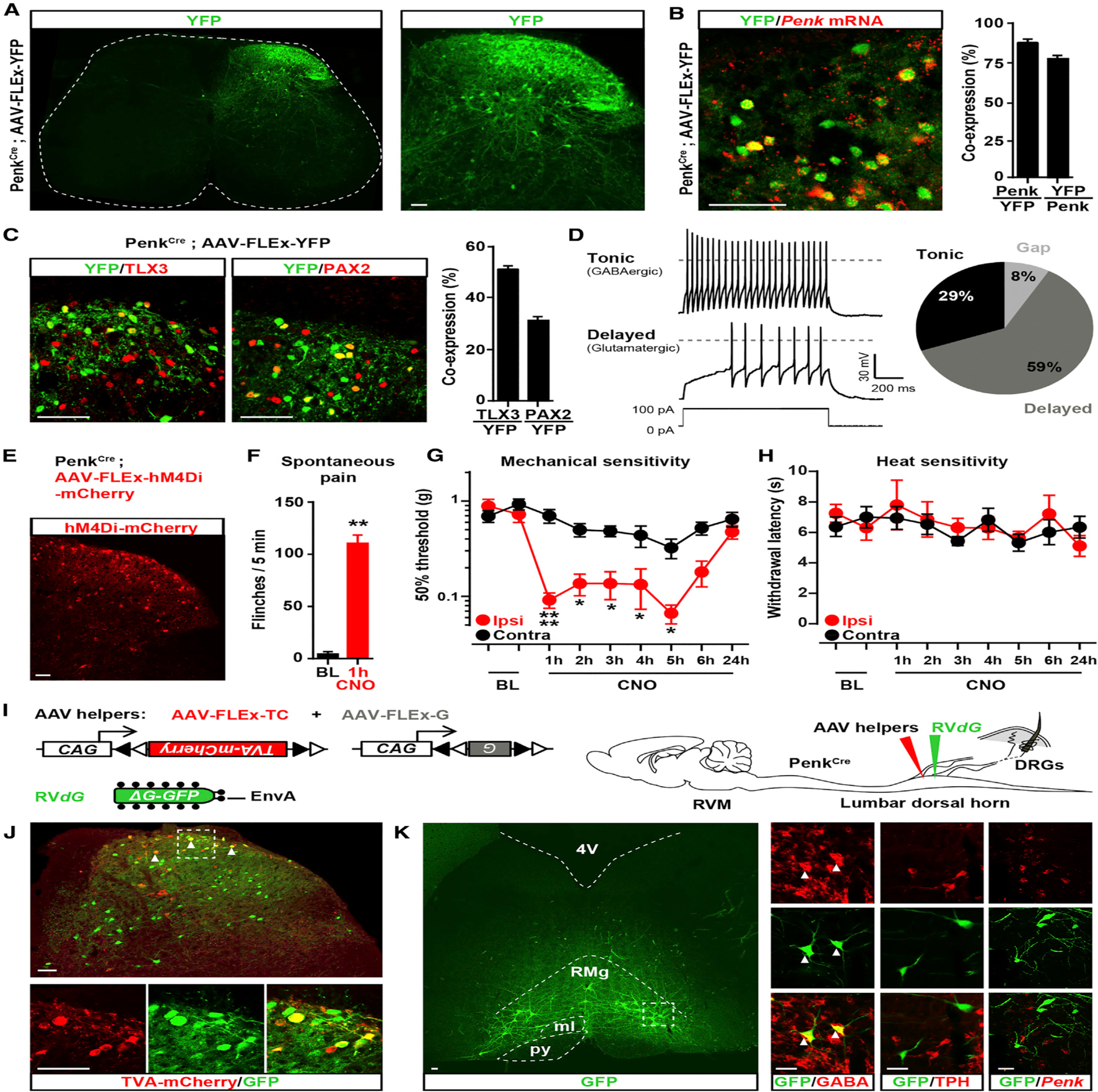
(A) Coronal section of spinal cord from PenkCre mice injected with AAV-FLEx-YFP (green) showing the distribution of Penk+ neurons in laminae I through V (FLEx Cre on).
(B) In situ hybridization shows Penk mRNA (red) in the great majority of YFP+ neurons (green) (88% ± 2.6%; n = 4 mice).
(C) Half of Penk+ neurons (green) coexpress the glutamatergic neuron marker TLX3 (54% ± 4.3%; n = 4) (red, left panel) and a third coexpress the GABAergic/glycinergic neuron marker PAX2 (30% ± 2.4%; n = 4) (red, right panel).
(D) Electrophysiological characterization of Penk+ neurons in PenkCre;Rosa26-LSL-tdTomato mice. Injection of depolarizing currents in tdTomato+ neurons shows that 58% (20/34 neurons) of PenkCre neurons presented a tonic (top panel, pie chart), 29% (10/34) a delayed (bottom panel, pie chart), and 8% (3/34) a gap firing pattern (pie chart).
(E) Injection of AVV-FLEx-hM4Di-mCherry into the right side of the spinal cord dorsal horn of PenkCre mice causes Cre-dependent expression of hM4Di-mCherry 4 weeks after injection.
(F) CNO generated spontaneous nociceptive behaviors 1 hr after administration (Mann-Whitney test, **p < 0.01; n = 5).
(G) CNO induced profound mechanical hypersensitivity in the von Frey test (two-way ANOVA, Bonferroni post hoc test, *p < 0.05, ****p < 0.0001; n = 9).
(H) CNO did not alter heat sensitivity (Hargreaves test).
(I) Strategy for identifying neurons presynaptic to enkephalinergic spinal neurons with rabies virus-mediated trans-synaptic retrograde tracing.
(J) Coronal section of spinal cord dorsal horn from PenkCre mice injected with AAV helpers (red) and RVdG (green) (top panel). Arrows indicate examples of co-infected starter cells (yellow). Bottom panels show a close-up view of the dashed box shown in the top panel.
(K) GFP expression in RVM neurons revealing that enkephalinergic spinal neurons receive input from brainstem descending neurons (left panel). Right panels show a close-up of the dashed box in the left panel. Arrows indicate RVM RVdG GFP+ neurons coexpressing GABA (left column). RVM RVdG GFP+ neurons are TPH-negative (middle column) and rarely Penk positive (right column). RMg, Raphe Magnus nucleus; py, pyramidal tract; ml, medial lemniscus; 4V, fourth ventricle.
All scale bars represent 50 μm. All bar graphs represent mean ± SEM.
See also Figures S1 and S2.
We next used chemogenetics to manipulate the activity of Penk+ spinal neurons and uncover their function in pain processing. We injected an AAV into the right lumbar dorsal horn of PenkCre mice to express the inhibitory G protein-coupled receptor hM4Di in enkephalinergic neurons (Kätzel et al., 2014) (Figure 1E) and administered the hM4Di agonist clozapine-N-oxide (CNO, 10 mg/kg, intraperitoneal [i.p.]) to inhibit Penk+ neurons. Strikingly, mice began to spontaneously flinch, bite, or lick their right paws 1 hr after CNO administration (Figure 1F). Additionally, CNO induced robust mechanical hypersensitivity of the hindpaw ipsilateral to the AAV injection without any change on the contralateral control side (Figure 1G; Figure S2A). Sensitivity to heat (Figure 1H; Figure S2B) and light touch were unaffected (Figures S1F and S1G). To clarify whether glutamatergic or GABAergic Penk+ spinal neurons may be responsible for this phenotype, we inhibited GABAergic neurons with hM4Di in VgatCre mice. Inhibition of GABAergic spinal neurons increased not only mechanical, but also heat sensitivity (Figure S2). As inhibition or deletion of spinal excitatory neurons is conversely anti-nociceptive (Christensen et al., 2016; Duan et al., 2014; Peirs et al., 2015), this finding suggests that the mechanical hypersensitivity resulting from Penk+ interneuron inhibition is due to the GABAergic subpopulation. To determine the contribution of GABA versus enkephalin release to modulation of pain thresholds, we injected i.t. in wild-type mice either the GABAA receptor antagonist bicuculline or the opioid receptor antagonist naloxone. Bicuculline induced strong mechanical and heat hypersensitivity whereas naloxone had no effect on pain thresholds (Figure S2), consistent with previous findings (Grevert and Goldstein, 1978; North, 1978; Yamamoto and Yaksh, 1993). Naloxone blocks the effect of multiple opioid peptides on mu, delta, and kappa opioid receptors, pre- and postsynaptically, at multiple synapses (i.e., between different types of DRG, spinal, and descending neurons). It is thus possible that opioids can have pro- and anti-nociceptive actions at these different spinal loci and that the net effect of blocking all these effects with naloxone is an unchanged sensitivity to mechanical and heat stimuli. These results establish the critical and selective function of GABAergic Penk+ spinal neurons for the inhibition of mechanosensory nociceptive information transmission.
To determine whether brain descending systems engage enkephalinergic spinal neurons, we identified neurons presynaptic to Penk+ neurons using rabies virus-mediated retrograde trans-synaptic tracing (Beier et al., 2015; Wickersham et al., 2007). We injected helper AAVs into the dorsal horn of adult PenkCre mice to express both TVA-mCherry (TC), the receptor for EnvA, and glycoprotein (G) in Penk+ spinal neurons (Figure 1I). Specific infection of TC- and G-expressing Penk+ cells by glycoprotein-deleted and EnvA-pseudotyped rabies virus (RVdG) that expresses GFP resulted in the trans-synaptic spread of RVdG to monosynaptically connected presynaptic neurons (Figure 1J). We examined regions implicated in descending pain control (e.g., locus coeruleus, raphe magnus, gigantocellular reticular [alpha part] nuclei [RVM], and lateral paragigantocellular nucleus) and only found neurons that strongly expressed GFP in the RVM (Figure 1K). We conclude that Penk+ spinal neurons receive monosynaptic inputs from the RVM and may be part of a yet uncharacterized descending pain modulation circuit.
RVM Neurons Projecting onto Spinal Enkephalinergic Neurons Are GABAergic but Facilitate Nociception
The RVM contains several classes of spinally projecting neurons previously classified based on their firing pattern, expression of MOR, and pro- versus anti-nociceptive actions (Barbaro et al., 1986; Basbaum et al., 1976; Budai and Fields, 1998; Fields et al., 1983b). We thus characterized GFP+ RVM neurons projecting onto enkephalinergic neurons and found that the great majority display GABA immunoreactivity (i.r.) (88% ± 1.1%; n = 5), but few express Penk (6.54% ± 1.62%; n = 3) (Figure 1K; Figure S1H). Consistent with the idea that the RVM contains GABAergic neurons projecting to the spinal cord, injection of the retrograde tracer fluorogold (FG) in the lumbar dorsal horn of VgatCre;Rosa26-LSL-tdTomato mice resulted in accumulation of FG in a population of Vgat+ RVM neurons (45% ± 4.4%) (Figures 2A and 2B). Furthermore, injection of an AAV expressing the trans-synaptic anterograde tracer wheat germ agglutinin (WGA) in the RVM of VgatCre;Rosa26-LSL-tdTomato mice caused transport of WGA predominantly in tdTomato+ TLX3-negative (a glutamatergic neuron marker) dorsal horn neurons (Figures 2C–2E). We conclude that the RVM contains a population of GABAergic neurons projecting onto GABAergic/enkephalinergic spinal neurons.
Figure 2. Inhibition of RVM GABAergic Neurons Projecting onto GABAergic Dorsal Horn Neurons Causes Mechanical Hyposensitivity.
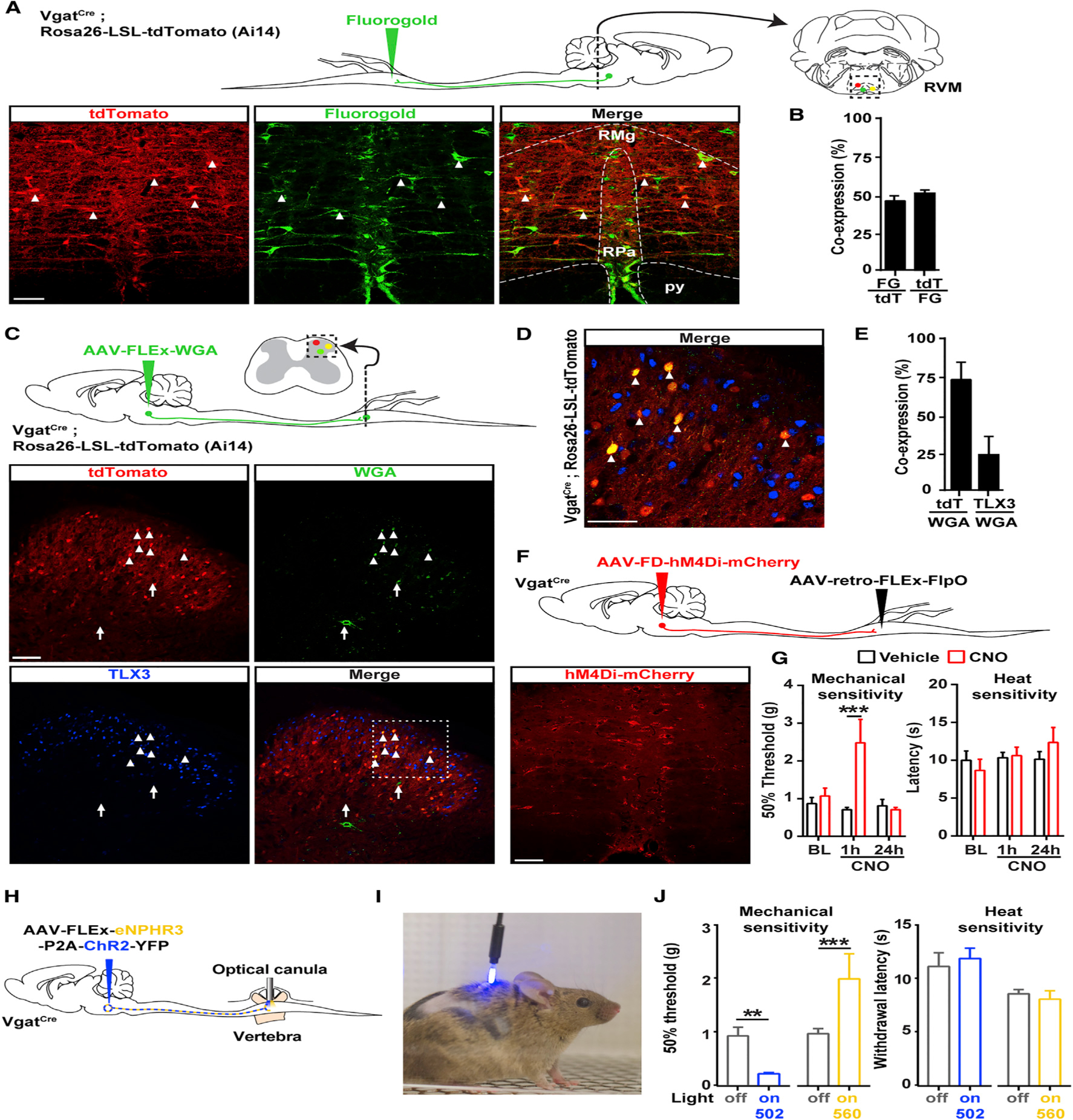
(A) Top: strategy to identify GABAergic RVM neurons projecting to the spinal cord using the retrograde tracer Fluorogold and VgatCre; Rosa26-LSL-tdTomato reporter mice. Bottom: representative image of Fluorogold in the RVM of VgatCre;Rosa26-LSL-tdTomato mice.
(B) Approximately half of the RVM neurons projecting to the dorsal horn (Fluorogold+, green) are GABAergic (VgatCre+, red) (n = 3 mice).
(C) Top: experimental approach to identify the output of GABAergic RVM neurons projecting to the spinal cord using an AAV to express the anterograde tracer WGA in a Cre-dependent manner. Bottom: representative image of WGA in the dorsal horn of VgatCre;Rosa26-LSL-tdTomato mice.
(D) Close-up view of the dashed box shown in (C). The majority of WGA-containing dorsal horn neurons are tdTomato+ (arrowheads) and TLX3-negative, indicating that GABAergic RVM neurons predominantly project onto GABAergic spinal neurons. Arrows indicate neurons containing WGA that express neither tdTomato nor TLX3.
(E) Quantification of (D) (n = 3 mice).
(F) Spinal injection of the retrograde AAV-retro-FLEx-FlpO in VgatCre mice allows expression of hM4Di-mCherry in a Cre and FlpO-dependent manner to target only RVM GABAergic spinal projections (see also Figure S4).
(G) CNO caused strong mechanical hyposensitivity in the von Frey test, but not in the Hargreaves test (two-way ANOVA, Bonferroni post hoc test, ***p < 0.001; n = 5).
(H) Experimental approach to stimulate spinal cord terminals of RVM Vgat neurons expressing ChR2 and eNPHR3 in freely moving animals.
(I) Photograph of a VgatCre mouse injected with AAV-FLEx-ChR2-P2A-eNPHAR3-YFP in the RVM and with an optical fiber implanted in the lumbar vertebra.
(J) Stimulation of RVM Vgat spinal terminals caused a strong mechanical hypersensitivity while inhibition causes mechanical hyposensitivity (von Frey test; Mann-Whitney test, **p < 0.01, ***p < 0.001; n = 5), but no change in heat thresholds.
Scale bars represent 100 μm. All bar graphs represent mean ± SEM.
See also Figures S3, S4, and S5.
To determine whether RVM GABAergic neurons facilitate or reduce nociception, we virally expressed hM4Di in Cre+ RVM neurons in VgatCre mice and inhibited these cells with i.p. CNO (Figure S3A). Remarkably, we found that CNO-treated mice developed significant mechanical hyposensitivity compared to vehicle-treated mice (Figure S3B), while behavioral responses in the Hargreaves’ heat pain, light touch, motor coordination, and anxiety tests were unaffected (Figures S3C–S3G). As manipulation of all RVM GABAergic neurons in the RVM may lead to non-specific modulation of non-nociceptive pathways, we next used an intersectional approach. We injected the retrograde virus AAV-retro (Tervo et al., 2016) expressing FlpO in a Cre-dependent manner (AAV2-retro-FLEx-FlpO) in the dorsal horn of VgatCre mice and AAV-FD(FlpO-dependent)-hM4Di-mCherry in the RVM to restrict hM4Di expression to Vgat+ RVM neurons projecting to the spinal cord (Figure 2F; Figure S4A). Inhibition of these RVM GABAergic projection neurons increased mechanical threshold without altering heat sensitivity, as previously observed (Figure 2G; Figures S4B and S4C). Unexpectedly, these results suggest that despite their inhibitory nature, RVM Vgat+ GABAergic neurons normally facilitate nociception. To further test this possibility, we employed in vivo spinal optogenetics (Figure 2I). We injected an AAV in the RVM of VgatCre mice to express the excitatory Channelrhodopsin 2 (ChR2) and the inhibitory Halorhodopsin (eNPHR) in a Cre-dependent manner (Rashid et al., 2016) and implanted an optical fiber in the lumbar spine for light stimulation of GABAergic descending axons during behavioral testing (Figures 2H and 2I; Figure S4D) (Christensen et al., 2016). Consistent with chemogenetic inhibition experiments, yellow light activation of eNPHR (continuous pulse, laser 561 nm, 10 mW) in RVM GABAergic axons in the spinal cord increased mechanical threshold but had no effect on heat sensitivity (Figure 2J). In contrast, blue light activation of ChR2 in these axons (15 Hz pulse, 473 nm LED light 5–8 mW) caused robust mechanical hypersensitivity, without altering heat pain thresholds (Figure 2J). Altogether, these experiments indicate that RVM Vgat+ GABAergic neurons projecting to the dorsal horn facilitate mechanical pain.
Previous studies indicated that pain facilitating on-cells express MOR (Barbaro et al., 1986; Heinricher et al., 1992; Marinelli et al., 2002). We found that in MOR-mCherry reporter mice (Erbs et al., 2015), approximately 67% of MOR+ RVM neurons are Vgat+ (Figure S5A), and more than half of MOR+ RVM neurons project to the lumbar spinal cord, consistent with previous findings (Pedersen et al., 2011) (Figure S5B). In contrast, MOR or Vgat are very rarely expressed by serotonergic RVM neurons (13.8% ± 3.9% and 4.7% ± 0.4%, respectively) (Figures S5C and S5D). Characterization of Cre+ RVM neurons in VgatCre mice by in situ hybridization indicated that while most neurons expressing Vgat coexpress Gad1 and Gad2, large populations of Vgat+ neurons express only one or the other (Gad1 only: 21.3% ± 7.9%; Gad2 only: 23.9% ± 0.5%; n = 3) (Figures S5E and S5F). Collectively, our experiments suggest that pro-nociceptive and MOR+ RVM neurons include Vgat+ GABAergic neurons projecting onto GABAergic/enkephalinergic neurons.
GABAergic RVM Cells Promote Nociception by Inhibiting Enkephalinergic Spinal Neurons
Our tracing analysis suggested the existence of a previously uncharacterized descending pain control system exerting inhibition over inhibitory spinal neurons. To functionally test this model, we used electrophysiology and optogenetics to interrogate neurotransmission between descending RVM neurons and Penk+ spinal neurons.
We injected an AAV to express ChR2 in RVM neurons and then recorded from Penk+ neurons in spinal cord slices from PenkCre;Rosa26-LSL-tdTomato mice (Figures 3A and 3B). We observed that application of blue light caused robust inward anion currents when holding Penk+ neuron membrane potential at −40 mV (Veq Cl− = −64.4 mV). These light-evoked currents were blocked by bath application of the GABAA and glycine receptor antagonists bicuculline and strychnine, indicating that they are inhibitory postsynaptic currents (IPSCs) (Figure 3C). Interestingly, IPSCs were only evoked in Penk+ neurons presenting a tonic firing pattern, a hallmark of spinal GABAergic interneurons, in contrast to spinal glutamatergic interneurons that show delayed or gap firing patterns (Todd, 2010) (Figures 3C–3E). Furthermore, light-evoked inhibitory inputs strongly reduced action potential firing and excitability of GABAergic Penk+ neurons (Figures 3F and 3G). No IPSCs were observed in Penk-negative neurons.
Figure 3. GABAergic RVM Neurons Control the Excitability of Spinal Enkephalinergic Neurons.
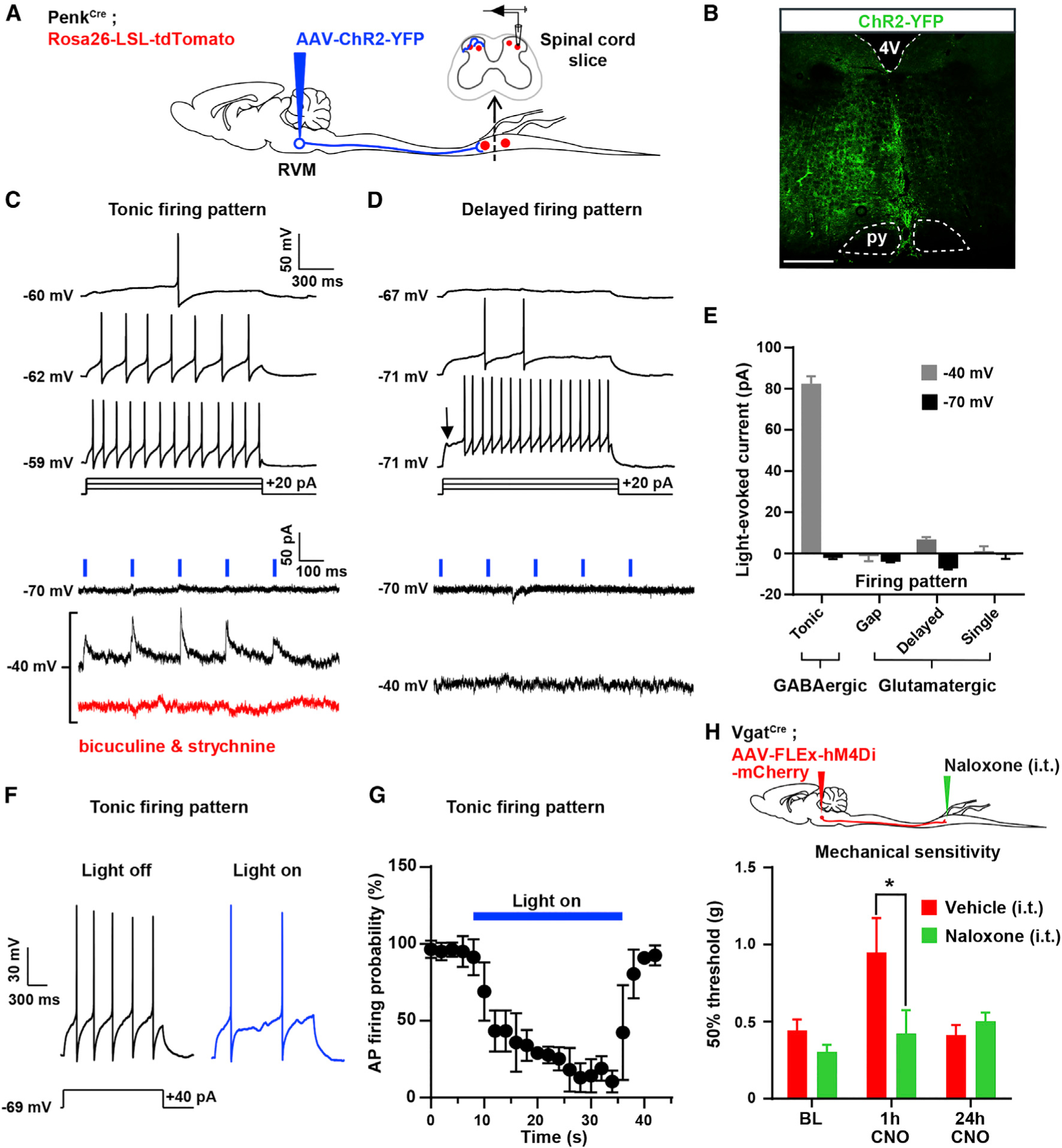
(A) Experimental approach used to test the functional connectivity between RVM neurons and Penk+ spinal neurons. An AAV was injected into the RVM of PenkCre;Rosa26-LSL-tdTomato mice to express ChR2-YFP in RVM neurons. Recordings from tdTomato+ neurons in spinal cord slices were performed during optogenetic stimulation of ChR2-YFP+ axons of RVM descending neurons.
(B) Expression of ChR2-YFP in the RVM.
(C) 5 Hz blue light pulses induced robust positive inward (inhibitory) currents at −40 mV without failure. These currents were blocked by bath application of 10 μM bicuculline and 2 μM strychnine. All neurons presenting blue light-evoked IPSCs showed a tonic action potential firing pattern, a hallmark of GABAergic spinal neurons.
(D) No EPSCs or IPSCs were evoked by light stimulation at either −70 mV or −40 mV in Penk+ neurons presenting a delayed firing pattern (glutamatergic spinal neurons).
(E) Summary graph showing the amplitude of light-evoked currents recorded in spinal Penk+ neurons presenting a tonic, gap, delayed, or single firing pattern. IPSCs were only observed in neurons with a tonic firing pattern.
(F) Light stimulation reduced action potential firing triggered by current injection in Penk+ neurons showing a tonic firing pattern.
(G) Light stimulation reduced the probability of action potential firing in Penk+ GABAergic neurons (n = 16 neurons).
(H) I.t. naloxone reversed the mechanical hyposensitivity induced by CNO in VgatCre mice injected with AAV-FLEx-hM4Di-mCherry in the RVM (two-way ANOVA, Bonferroni post hoc test, *p < 0.05, n = 12).
16 neurons were recorded for these electrophysiological experiments. Scale bar represents 500 μm. All graphs represent mean ± SEM.
Taken together with our tracing experiments, these results indicate that RVM Vgat+ neurons project to and inhibit Penk+ neurons in the spinal cord, uncovering a disynaptic inhibitory circuit controlling nociception. It follows that the anti-nociceptive effect we observed with hM4Di-mediated inhibition of RVM GABAergic neurons (Figure S3) might have resulted, at least in part, from disinhibition of Penk+ neurons and subsequent increase in dorsal horn enkephalinergic tone. To test this hypothesis, we injected naloxone (i.t.) and repeated the experiment described in Figure S3. Naloxone abolished the anti-nociceptive effect of CNO on mechanical sensitivity (Figure 3H). We conclude that the RVM may modulate pain thresholds via a population of GABAergic RVM neurons that project to the dorsal horn and tonically regulate mechanical sensitivity by inhibiting enkephalinergic spinal neurons.
GABA and Enkephalins from Penk+ Neurons Presynaptically Inhibit Primary Afferents in a Temporally Coordinated Manner
We next investigated the synaptic mechanisms by which spinal enkephalinergic neuron activity regulates nociceptive processing. We and others have previously shown that exogenous opioid agonists can act presynaptically on DOR and MOR to control neurotransmission at the synapse between primary sensory neurons and spinal interneurons (Bardoni et al., 2014; Heinke et al., 2011; Jessell and Iversen, 1977; Yaksh et al., 1980). We thus hypothesized that enkephalins from Penk+ spinal neurons might also presynaptically inhibit primary afferents.
To test this, we expressed ChR2 in Penk+ neurons to trigger enkephalin release and assayed for potential enkephalinergic presynaptic inhibition (Figure 4; Figure S6). We recorded excitatory postsynaptic currents (EPSCs) monosynaptically evoked by primary afferent stimulation in randomly selected Penk-negative dorsal horn neurons (Figure 4A). We observed that light stimulation of Penk+/ChR2+ neurons caused a strong reduction in synaptic transmission between primary afferents and spinal interneurons lasting up to 2,000 ms (Figure 4B; Figure S6D). Remarkably, we found that this inhibition consists of two phases: a first bicuculline/strychnine-sensitive inhibition lasting up to 300 ms after stimulation and a second, delayed CTOP/Tipp-psi-sensitive (MOR and DOR antagonists, respectively) (Hawkins et al., 1989; Schiller et al., 1993) and thus opioidergic inhibition lasting up to 2,000 ms (Figures 4B–4H). Analysis of the paired-pulse ratio (PPR), which is inversely related to neurotransmitter release probability, suggests that both GABA- and enkephalin-mediated reductions in neurotransmission occur through presynaptic inhibition (Figures 4E and 4H).
Figure 4. Temporally Coordinated Presynaptic Inhibition of Primary Afferents by GABA/Glycine and Enkephalins from Penk+ Neurons.
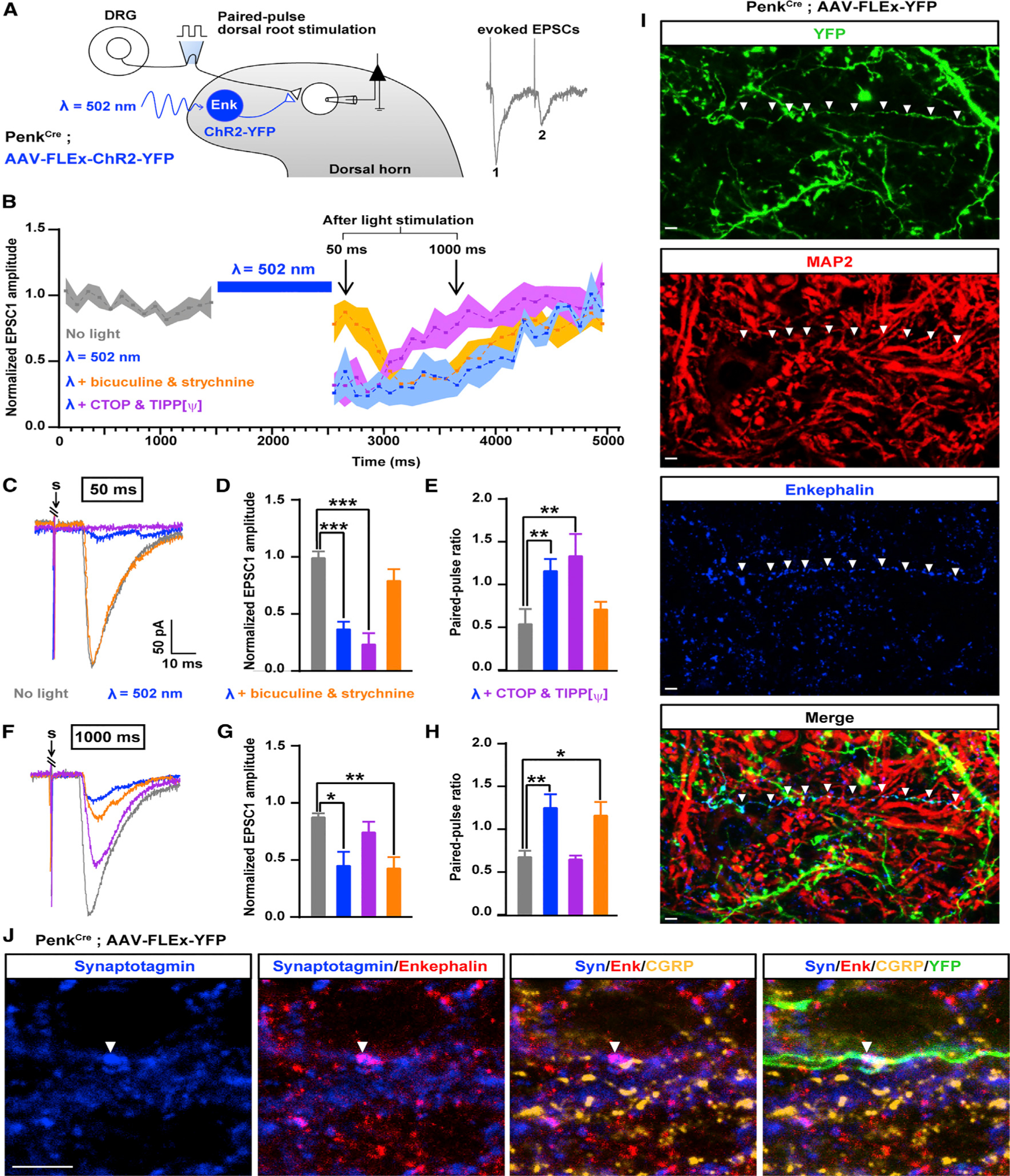
(A) Experimental design used to assess the effect of ChR2-mediated activation of Penk+ neurons on synaptic transmission between primary afferent and spinal neurons based on the amplitude of EPSCs evoked by dorsal root stimulation (Penk-negative neurons were recorded).
(B) Activation of Penk+ neurons reduced synaptic transmission between primary afferent and spinal neurons for up to 2 s after light stimulation. Results are expressed as mean ± SEM.
(C) Example traces of EPSCs evoked by dorsal root stimulation and modulated by light during the early phase of inhibition of synaptic transmission (50 ms after light stimulation). Bicuculline and strychnine, but not the DOR and MOR antagonists Tipp-psi and CTOP, blocked the reduction in EPSC amplitude during the early phase of synaptic transmission inhibition. “S” indicates dorsal root stimulation artifacts.
(D) Quantification of (C).
(E) Light-evoked increase in the paired-pulse ratio (PPR), which indicates presynaptic inhibition, was also blocked by bicuculline and strychnine during the early phase of synaptic transmission inhibition.
(F) Example traces of EPSCs evoked by dorsal root stimulation and regulated by light during the late phase of presynaptic inhibition (1,000 ms after light). DOR and MOR antagonists Tipp-psi and CTOP, but not bicuculline and strychnine, prevented the reduction in EPSC amplitude during the early phase of presynaptic inhibition. “S” indicates dorsal root stimulation artifacts.
(G) Quantification of (H).
(H) The increase in the PPR during the late phase of light-induced presynaptic inhibition was also blocked by Tipp-psi and CTOP.
(I) Immunostaining in spinal cord sections from PenkCre mice injected with AAV-FLEx-YFP (green) showed that enkephalins detected in the processes of Penk+ neurons do not co-localize with the somato-dendritic marker MAP2 (red), suggesting enkephalin presence in axons.
(J) Enkephalins (red) co-localized with the presynaptic marker synaptotagmin (blue) and were present in close proximity to primary afferent axon terminals containing CGRP (gold) and synaptotagmin. Arrow heads indicate a process from YFP+ Penk+ neuron (green) forming an enkephalinergic en passant synapse with a CGRP+ primary afferent axon terminal.
Kruskal-Wallis test, *p < 0.05, **p < 0.01, ***p < 0.001. Scale bars represent 10 μm. All bar graphs represent mean ± SEM.
See also Figure S6.
We next immunostained spinal cord sections from PenkCre mice injected with an AAV to sparsely express YFP in Cre+ cells with antibodies against calcitonin gene-related peptide (CGRP), the somatic and dendritic marker microtubule-associated protein 2 (MAP2), the axonal and presynaptic marker synaptotagmin, and enkephalin. Enkephalin-i.r. was concentrated in varicosities of YFP+ MAP2-negative neural processes of Penk+ neurons (Figure 4I). Furthermore, enkephalin-i.r. co-localized with synaptotagmin and opposed CGRP+ primary afferents, providing evidence that Penk+ neurons might form enkephalin-containing axo-axonic synapses with primary afferents (Figure 4J).
These results uncover a combined GABAergic and enkephalinergic presynaptic inhibition mechanism in which fast and slow neurotransmitters acting on ion channels and G protein-coupled receptors cooperate to regulate neurotransmitter release in a temporally coordinated manner.
We also examined whether enkephalinergic neurons could influence the activity of dorsal horn neurons. We injected an AAV to express the anterograde tracer WGA in Penk+ dorsal horn neurons and found that WGA accumulated in Penk-negative neurons in laminae IIinner(i)/III. Interestingly, these WGA+ neurons mostly consisted of glutamatergic (TLX3+) neurons (Figures 5A–5F). Consistent with the idea that dorsal horn neuron function might also be regulated by Penk+ neurons, activation of Penk+ neurons with ChR2 induced both excitatory and inhibitory polysynaptic currents (Figures 5G and 5K) in recorded cells, presumably due to the mixed excitatory and inhibitory nature of the Penk+ population. Most of these recorded neurons presented a delayed firing pattern (36/51), suggesting that they are glutamatergic, in agreement with TLX3-i.r (Figures 5H and 5L). Additionally, we occasionally observed slow positive outward currents after light stimulation in lamina II interneurons presenting a delayed firing pattern (3/14) (Figures 5H and 5J). The kinetics of these currents are similar to those of GIRK channels, suggesting postsynaptic expression of opioid receptors in this lamina, consistent with previous studies (Eckert and Light, 2002; Grudt and Williams, 1994; Yoshimura and North, 1983). Finally, these neurons located downstream of Penk+ interneurons receive monosynaptic Aβ/δ inputs (Figures 5I, 5M, and 5N), suggesting, along with their localization in laminae IIi/III, a function in mechanosensation (Bourane et al., 2015; Duan et al., 2014; Peirs et al., 2015; Petitjean et al., 2015).
Figure 5. Dorsal Horn Postsynaptic Targets of Penk+ Neurons.
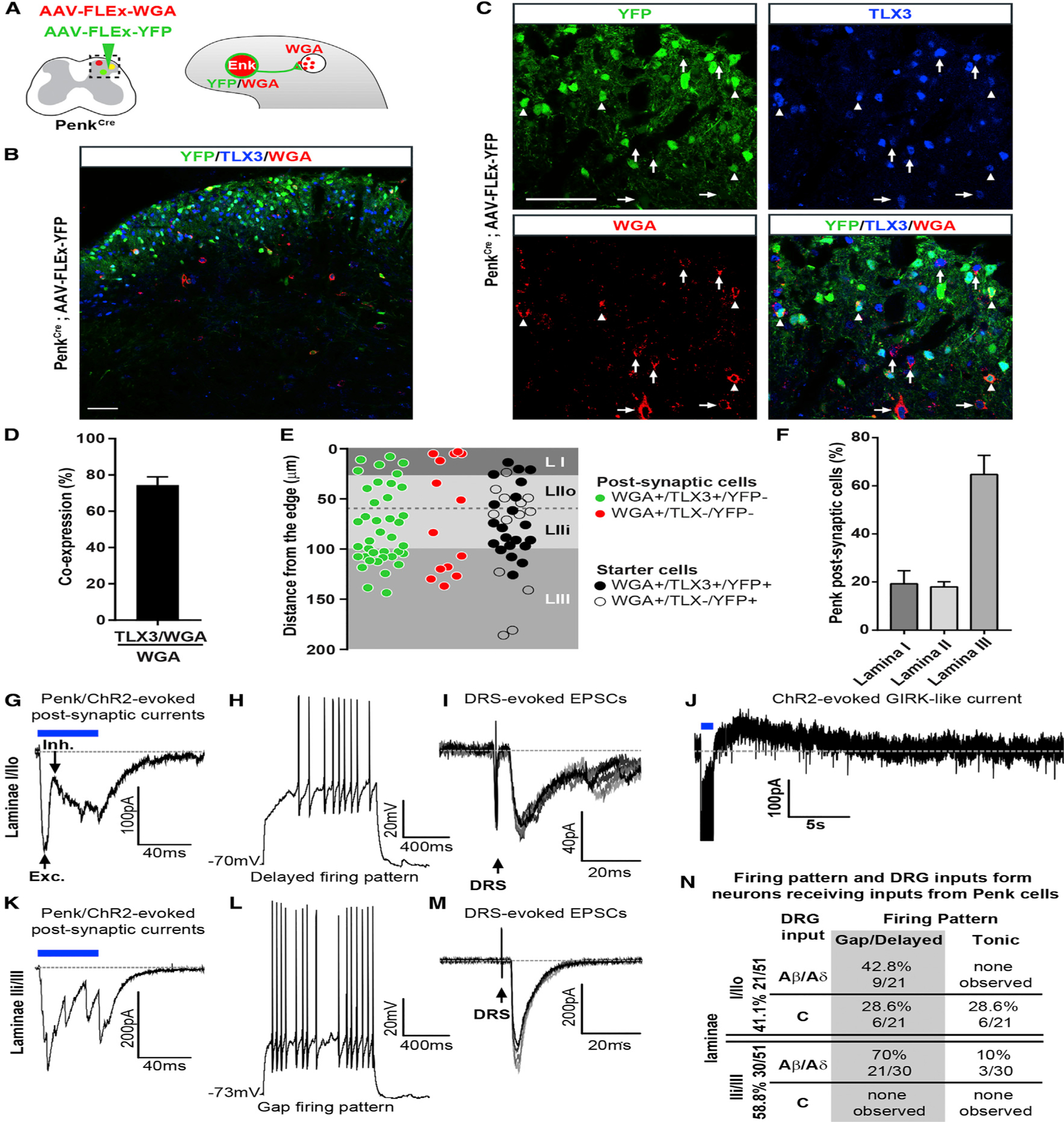
(A) Spinal injection of AAV-FLEx-WGA and AAV-FLEx-YFP in PenkCre mice allows expression of WGA and YFP in Penk+ neurons and transport of WGA to postsynaptic cells.
(B and C) Co-staining for WGA and TLX3 in PenkCre mice 3 weeks after injections of AAV-FLEx-WGA and AAV-FLEx-YFP (B). (C) is a close-up of (B). Arrowheads indicate cell initially infected (starter cells; YFP+ and WGA+), arrows indicate WGA+ and TLX3+ postsynaptic neurons.
(D) More than 75% of spinal neurons receiving input from Penk+ neurons are TLX3+.
(E and F) The majority of postsynaptic cells receiving inputs from Penk+ neurons are located in lamina III. Distribution of WGA cells among spinal laminae (E). Summary of the laminar distribution of WGA+ neurons (F) (n = 3).
(G–M) Electrophysiological recordings from Penk-negative neurons in PenkCre mice injected with AAV-FLEx-ChR2-YFP; laminae I/IIouter neurons (G–J) and laminae IIinner/IIIi neurons (K–M). Blue light stimulation evoked polysynaptic excitatory and inhibitory postsynaptic currents in laminae I/IIouter neurons (G) presenting a delayed/gap firing pattern (H) and receiving Aδ/β inputs (I). Blue light stimulation also evoked outward potassium current in some laminae II neurons (3/14) (J). ChR2 stimulation in laminae IIi/III (K) triggered polysynaptic excitatory postsynaptic currents in neurons also presenting a gap/delayed firing pattern (L) and receiving large Aβ/δ inputs (M).
(N) The majority of interneurons receiving inputs from Penk interneurons are observed in laminae Iii/III, present a gap/delayed firing pattern, and receive Aβ/δ inputs.
Scale bars represent 50 μm. All bar graphs represent mean ± SEM.
Organizational Logic of Sensory and Descending Input Processing by Penk+ Neurons
We next determined the specific contribution of MOR and DOR to enkephalin-mediated, long-lasting presynaptic inhibition. We found that in laminae I/IIo, the majority of neurons presenting an increase in PPR following blue light stimulation receive C-fiber inputs, in which case the increase in PPR was blocked exclusively by CTOP and Tipp-psi applied together, but not by either alone. In contrast, in a smaller proportion of neurons in laminae I/IIo, and in deeper laminae IIi/III, neurons that showed an increase in PPR received Aβ- and Aδ-fiber inputs, and the PPR increase was blocked by Tipp-psi, but not by CTOP (Figure 6). These data uncover a topographically organized gating mechanism of primary afferent inputs by the endogenous opioid system for the control of sensory information transmitted from mostly DOR-expressing DRG neurons in distinct laminae.
Figure 6. Laminar Organization and DOR/MOR Contribution to Enkephalinergic Presynaptic Inhibition of C- and A-fibers.
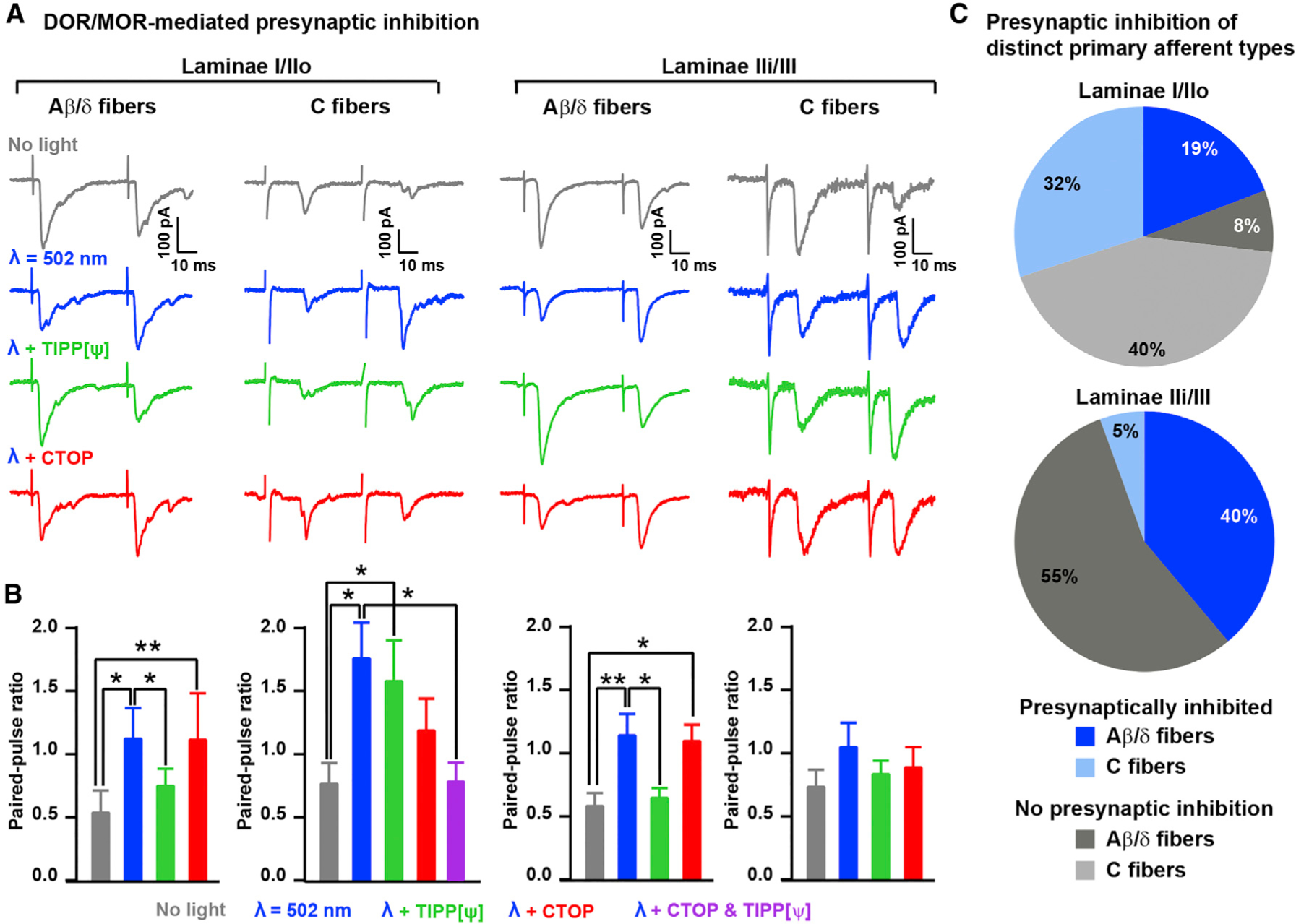
(A) Example traces showing that the MOR antagonist CTOP (1 μM) reversed the light-induced enkephalinergic presynaptic inhibition of C fibers in lamina I/IIo, whereas the DOR antagonist Tipp-psi (1 μM) reversed presynaptic inhibition of A-fibers in lamina IIi/III.
(B) Quantification of (A) indicating the effect of CTOP and Tipp-psi on the light-induced increase in PPR 1,000 ms after light stimulation (Kruskal-Wallis test, *p < 0.05; **p < 0.01).
(C) Pie charts indicating the proportions of neurons in which light induced a significant increase in PPR (i.e., presynaptically inhibited) in laminae I/IIo and IIi/III and for C- and A-fibers (n = 26 neurons for lamina I/IIo and 18 for lamina IIi/III).
All bar graphs represent mean ± SEM.
To determine whether Penk+ spinal neurons receive primary afferent input, we used RVdG-based tracing strategies in PenkCre mice as in Figure 1 and observed GFP+ DRG neurons (Figures 7A–7D; Figure S7). These included unmyelinated CGRP+ nociceptors and Ret+ myelinated mechanoreceptors innervating hair follicles, which express MOR and DOR, respectively (Bardoni et al., 2014; Scherrer et al., 2009; Usoskin et al., 2015). We thus analyzed light-induced enkephalin release and presynaptic inhibition in Penk+ neurons (Figure 7E). We found that only Penk+ neurons presenting a gap or delayed firing pattern, presumably glutamatergic, showed a reduction in amplitude of dorsal root stimulation-evoked EPSCs and an increase in PPR following light application. By contrast, Penk+ neurons presenting a tonic firing pattern, presumably GABAergic, also received inputs from DRG neurons, but evoked EPSCs were insensitive to light-induced stimulation of Penk+ neurons (Figures 7F and 7G). Therefore, Penk+ spinal interneurons integrate inputs from both the periphery and the brain, with Penk+ glutamatergic neurons receiving inputs only from opioid receptor-containing DRG neurons, whereas Penk+ GABAergic neurons receive inputs from DRG neurons lacking opioid receptors as well as from the RVM.
Figure 7. Differential Primary Sensory and Descending Neuron Inputs onto Glutamatergic and GABAergic Penk+ Neurons.
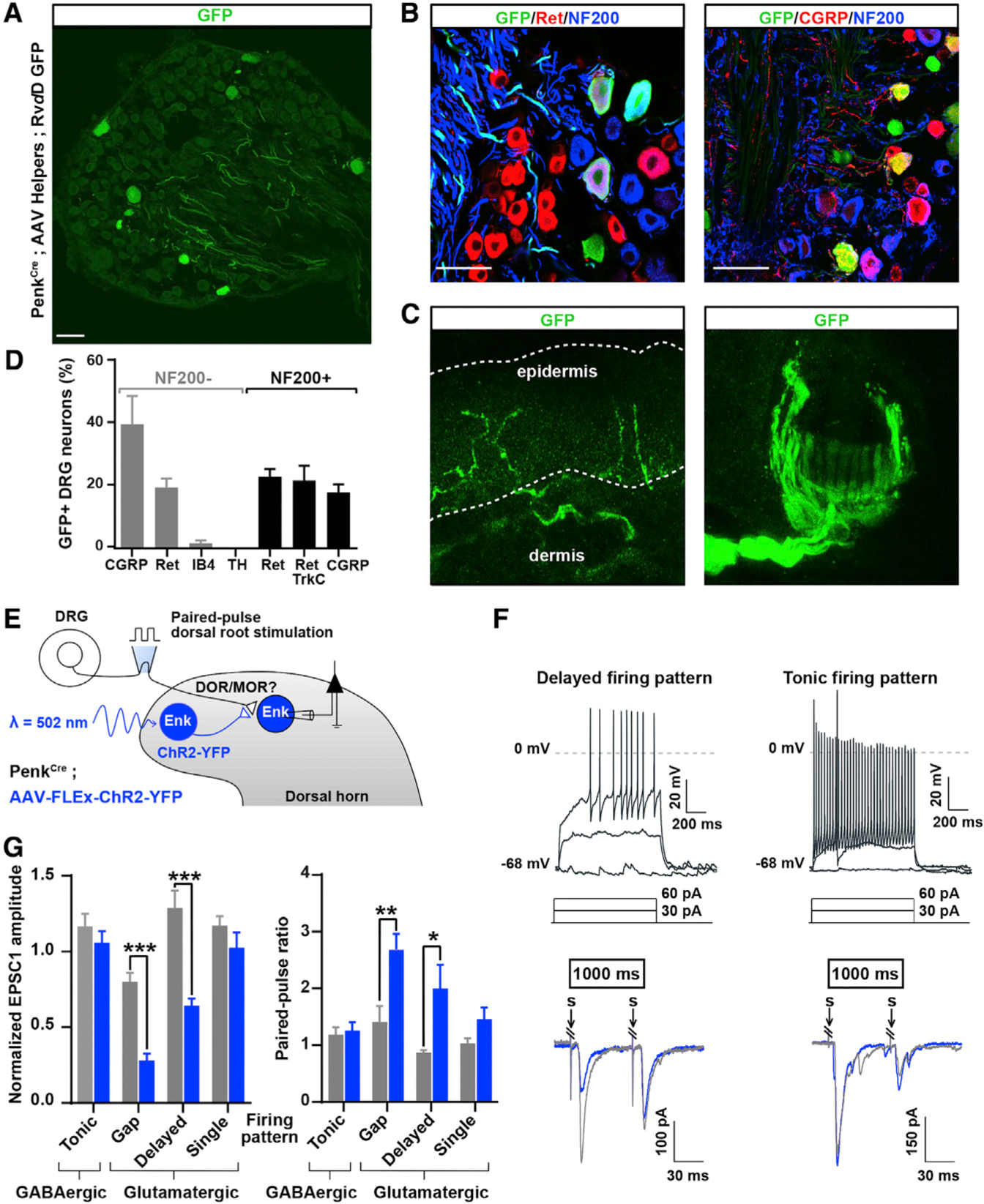
(A) DRG sections from PenkCre mice in which AAV helpers and GFP-expressing RVdG were injected in the dorsal horn, as in Figure 1J, showing that DRG neurons with both small- and large-diameter cell bodies express GFP and thus project onto Penk+ spinal neurons.
(B) Immunostaining indicating that GFP+ DRG neurons shown in (A) include Ret+ myelinated (NF200+) mechanoreceptors and CGRP+ unmyelinated (NF200−) nociceptors.
(C) Skin analysis confirmed that GFP+ DRG neurons were cutaneous afferents, including myelinated mechanoreceptors forming circumferential and longitudinal lanceolate endings around hair follicles, and nociceptors forming epidermal free nerve endings.
(D) Molecular identity of GFP+ DRG neurons.
(E) Experimental approach used to determine whether Penk+ neurons receive inputs from primary afferent neurons expressing DOR or MOR.
(F) Penk+ neurons showing a delayed firing pattern (top left) present a decrease in EPSC amplitude and increase in PPR (bottom left) following light stimulation. In contrast, Penk+ neurons presenting a tonic firing pattern (top right) do not show any change in EPSC amplitude or PPR (bottom right). Gray and blue traces represent paired EPSCs before and 1,000 ms after light stimulation, respectively.
(G) Quantification of (F) (two-way ANOVA, Bonferroni post hoc test, *p < 0.05, **p < 0.01, ***p < 0.001).
“S” indicates dorsal root stimulation artifacts. Scale bars represent 50 μm. All bar graphs represent mean ± SEM.
See also Figure S7.
GABAergic RVM Neurons Are at the Crossroads of Ascending and Descending Pain Pathways
To elucidate what conditions might recruit these RVM-Penk+ spinal neuron-primary afferent mechanisms for pain modulation, we identified cells presynaptic to GABAergic RVM neurons projecting to the dorsal horn using cTRIO-based tracing in VGATCre mice (Schwarz et al., 2015). In addition to other brainstem nuclei (e.g., periaqueductal gray, lateral cerebellar nucleus, and paralemniscal nucleus; Figure S8), we notably found GFP+ neurons in the posterior hypothalamus (PH) and lateral parabrachial nucleus (LPB), both of which are implicated in stress responses (Figures 8A and 8B).
Figure 8. RVM GABAergic Neurons Receive Inputs from Brain Structures Critical for Stress Responses and Differentially Engage Penk+ GABAergic Neurons in the Spinal Cord.
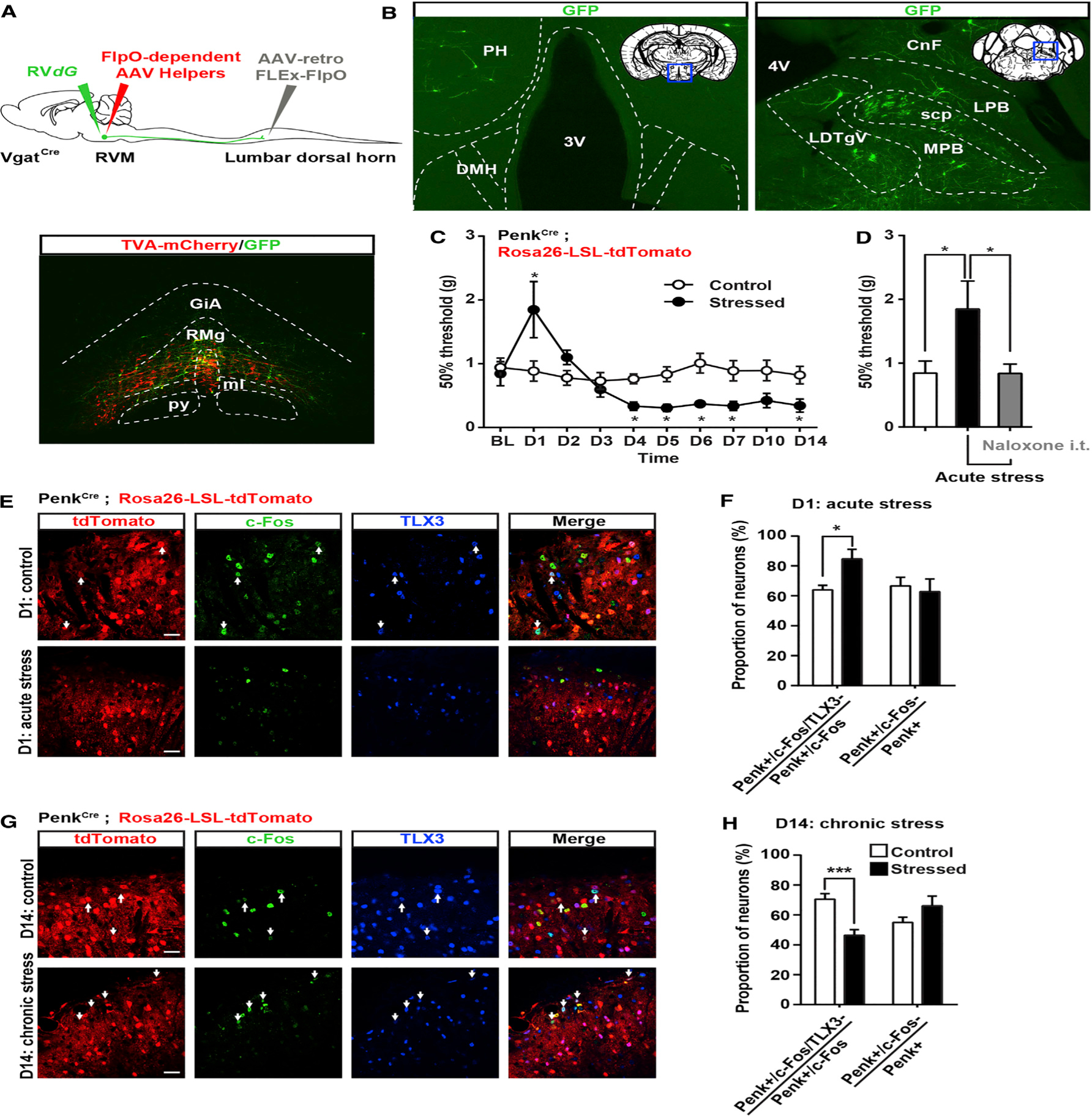
(A) Similar strategy as in Figures 2 and S4 to infect VgatCre neurons projecting to the spinal cord with AAV-retro-FLEx-FlpO in the spinal cord, flpO-dependent AAV helpers (red), and RVdG (green) in the RVM.
(B) Expression of GFP in the lateral hypothalamus (left) or lateral parabrachial nucleus (right) reveals that RVM GABAergic neurons receive inputs from brain structures critical for stress.
(C) Mechanical sensitivity in mice restrained 2 hr daily for 2 weeks and in unstressed littermate controls. Acute stress induced analgesia, whereas chronic stress increased mechanical sensitivity (two-way ANOVA (F(9, 144) = 4.525; * = p < 0.05).
(D) Stress-induced analgesia can be reversed by intrathecal injection of 5 μg naloxone (one-way ANOVA *p < 0.05).
(E and G) Coimmunostaining of c-Fos (green) and TLX3 (blue) PenkCre;Rosa26-LSL-tdTomato mice (red) before and after acute (E) or chronic (G) stress.
(F) Acute stress-induced analgesia was accompanied by an increase in the number of c-Fos+ Penk+ neurons that do not express TLX3 (presumably GABAergic). The total number of c-Fos+ Penk+ neurons is similar in both conditions.
(H) Chronic stress-induced hyperalgesia was accompanied by a decrease in the number of c-Fos+ Penk+ neurons not expressing TLX3 (blue) without affecting the overall population of c-Fos+ Penk+ neurons (Mann-Whitney test * = p < 0.05; *** = p < 0.0001).
PH, posterior hypothalamus; DMH, dorsomedial hypothalamus; LPB, lateral parabrachial nu; MPB, medial parabrachial nu; scp, superior cerebellar peduncle; LDTg, laterodorsal tegmental nu ventral part; CnF, cuneiform nu.
Scale bars represent 20 μm. All bar graphs represent mean ± SEM.
See also Figure S8.
Stress influences pain thresholds: acutely, stress can induce analgesia (Butler and Finn, 2009), while chronic stress can cause hypersensitivity (Jennings et al., 2014). We hypothesized that enkephalins from Penk+ spinal neurons might contribute to such changes. We used c-Fos-i.r. to determine the extent to which acute and chronic stress influence the activity of GABAergic Penk+ neurons mediating presynaptic inhibition under RVM control. We restrained PenkCre;Rosa26-LSL-tdTomato mice daily for 2 hr for 14 days. After an initial period during which stress-induced antinociception occurred, chronic restraint caused significant mechanical hypersensitivity (Figure 8C). Because reliable antibodies against c-Fos and GABA or PAX2 were generated in the same species, we identified GABAergic Penk+ neurons as TLX3-negative Penk+ neurons (Figures 8E–8G). Chronic stress decreased the number of GABAergic Penk+ neurons showing c-Fos-i.r. (i.e., tdTomato+,TLX3-negative) (Figures 8G and 8H). In contrast, we found that acute stress increased the number of c-Fos-immunoreactive GABAergic Penk+ neurons (Figures 8E and 8F), suggesting that recruitment of GABAergic Penk+ neurons mediating presynaptic inhibition might contribute to stress-induced antinociception. To test this, we injected naloxone (i.t.) and observed a significant reduction in the hyposensitivity induced by acute stress (Figure 8D). Collectively, these data suggest that the circuit described in this study is implicated in changes in pain thresholds following stress. Finally, given that several other brain structures contain GFP+ cells (i.e., RVdG-infected and presynaptic to GABAergic RVM neurons) (Figure S8), a variety of stimuli, internal states, and other experiences might activate or inhibit this descending circuit for pain modulation.
DISCUSSION
Pain thresholds are set as a function of emotional and internal states by descending modulation of nociceptive transmission in the spinal cord. In this study, we identified the components of a circuit and synaptic mechanisms for the descending modulation of mechanical sensitivity. We propose that dorsal horn GABAergic/enkephalinergic neurons integrate both sensory input and internal state information from RVM GABAergic neurons and act as gatekeepers for mechanical pain (Figure S9). The endogenous opioids enkephalins function as a molecular hinge of the gate along with GABA, by inhibiting neurotransmitter release from primary afferent neurons.
Organization and Function of Enkephalinergic Neuromodulation via DOR and MOR
Enkephalins are high-affinity agonists for both DOR and MOR (Kieffer and Gavériaux-Ruff, 2002). The precise function and necessity of each receptor for enkephalinergic modulation of synaptic transmission is less well understood, given that DOR and MOR reportedly regulate similar effectors, including presynaptic voltage-gated calcium channels. In DRG, MOR and DOR are predominantly expressed by peptidergic C nociceptors and myelinated mechanoreceptors, respectively, and are transported to their central terminals in the dorsal horn (Bardoni et al., 2014; Scherrer et al., 2009; Usoskin et al., 2015). Here we demonstrate that endogenous enkephalins act on DOR expressed in primary afferents to control neurotransmission within functionally distinct dorsal horn microcircuits.
Surprisingly, inhibition of enkephalinergic neurons exacerbated only mechanical sensitivity but had no effect on heat sensitivity. MOR is predominantly expressed by TRPV1+ peptidergic C nociceptors, which are essential to cutaneous heat sensitivity (Cavanaugh et al., 2009). Among the limited number of C fibers synapsing in lamina I and in which we saw enkephalinergic presynaptic inhibition (8/24), only half were exclusively sensitive to the MOR agonist CTOP (4/8). Thus, this population of MOR+ C fibers might be too restricted for enkephalins to significantly modulate heat sensitivity. Furthermore, peptidergic C fibers carrying heat information may not receive inhibitory axo-axonic input from GABAergic or enkephalinergic spinal interneurons (Ralston and Ralston, 1979; Ribeiro-da-Silva et al., 1989; Todd and Spike, 1993). Finally, among spinal cord interneurons receiving inputs from Penk+ interneurons, the vast majority were in laminae IIi/III, were likely glutamatergic, received A-fiber input, and occasionally presented GIRK-like currents following ChR2 activation of Penk+ neurons. This dorsal horn circuit might regulate mechanical sensitivity and contribute to the phenotype observed when inhibiting Penk+ neurons. Previous studies have established that subpopulations of dorsal horn neurons respond to opioid agonists and, in particular, enkephalin (Eckert and Light, 2002; Grudt and Williams, 1994; Yoshimura and North, 1983); however, the type of opioid receptors involved and their precise distribution in spinal circuits remain to be established.
Cooperative Enkephalinergic and GABAergic Presynaptic Inhibition for Gating Cutaneous Mechanosensory Inputs
GABA-mediated presynaptic inhibition of sensory inputs is well established for several types of primary afferents (Bardoni et al., 2013; Zeilhofer et al., 2012); however, the identity of the spinal interneurons contributing to this process and the consequences of presynaptic inhibition on pain information processing remain unclear. To our knowledge, this study provides the first demonstration that enkephalin release from spinal neurons causes presynaptic inhibition of mechanosensory neurons and reduces mechanical pain, confirming the gating mechanism proposed by Jessell and Iversen (1977).
The specific function of presynaptic versus postsynaptic inhibition has been described for spinal circuits underlying motor coordination (Betley et al., 2009; Fink et al., 2014). Presynaptic inhibition of proprioceptors may tune the gain of or scale sensory inputs to motor neurons for fine motor control, whereas postsynaptic inhibition contributes to gross motor control (Brenner et al., 2000; Fink et al., 2014). Similarly, GABAergic presynaptic inhibition of mechanonociceptors may contribute to fine-tuning of mechanical sensory inputs to shape the cutaneous mechanosensory experience. Mechanosensation results from activity in a variety of primary mechanosensory neurons and receptors with overlapping activation properties (Abraira and Ginty, 2013; Delmas et al., 2011), and the coordination and integration of mechanoreceptor inputs is likely necessary for the emergence of selected aspects that ultimately dominate mechanosensory experience (e.g., mechanical pain versus touch perception).
GABAergic neurons making axo-axonic synapses with proprioceptors rarely express neuropeptides, unlike their dorsal horn counterparts, raising the question of the specific function of enkephalins in gating cutaneous sensory information. While GABA is stored in readily available small synaptic vesicles, enkephalins are contained in dense core vesicles and only released following strong/sustained stimulation (McMahon et al., 1992; Yaksh et al., 1983). Consequently, enkephalins are expected to be released only in specific circumstances, such as when enkephalinergic neurons receive convergent excitatory inputs from different circuits, or following disinhibition. We found that spinal inhibitory interneurons not only receive cutaneous A-fiber inputs, consistent with recent findings (Duan et al., 2014; Foster et al., 2015), but that GABAergic/enkephalinergic spinal neurons also receive inputs from GABAergic RVM neurons. Given that inhibition of RVM GABAergic neurons diminishes mechanical sensitivity, our results suggest that RVM inputs tonically inhibit enkephalinergic neurons in basal conditions and that disinhibition at the brain or spinal level, together with increased activity in primary afferents, might generate enkephalinergic presynaptic inhibition. We propose that GABA can finely tune cutaneous mechanosensory information, whereas endogenous opioids, by their prolonged action on MOR and/or DOR, will shut down transmission of sensory information in instances of abnormal activity in descending and ascending pathways, resulting in analgesia: GABA may close the gate, and enkephalins, controlled by the RVM, may persistently lock it.
Descending RVM-Spinal Cord Circuits for Pain Facilitation and Inhibition
Several previous studies on brainstem descending systems for pain modulation focused on RVM serotonergic and noradrenergic inputs to spinal and primary afferent neurons (Dogrul et al., 2009; Kato et al., 2006; Lu and Perl, 2007; Zhao et al., 2014). These studies suggested that the dorsal horn receives descending excitatory and pro-nociceptive projections, including direct input from RVM serotonergic cells onto TRPV1+ nociceptors (Zhao et al., 2014). Others indicated that the dorsal horn receives substantial GABAergic RVM input (Belin et al., 1983; Pedersen et al., 2011; Potrebic et al., 1994; Reichling and Basbaum, 1990; Skagerberg and Björklund, 1985). Interestingly, a recent tracing analysis identified a population of GABAergic RVM neurons that synapse onto primary afferents in the dorsal horn (Zhang et al., 2015) and decrease pain, possibly counteracting serotonergic nociception facilitation through presynaptic inhibition of nociceptors.
We found that Penk+ spinal inhibitory neurons receive inputs from RVM GABAergic neurons revealing the existence of a disynaptic inhibitory circuit for pain modulation. As these RVM neurons often display MOR-i.r. and facilitate pain, our results suggest that they may functionally correspond to a class of on-cells that is primarily involved in the regulation of mechanical pain thresholds. GAD1 and GAD2 are thought to be expressed by subsets of GABAergic neurons that make axo-somatic and axo-axonic inhibitory boutons, respectively (Fink et al., 2014; Mende et al., 2016), while Vgat is expressed by both neuronal populations. Our results suggest that the RVM might contain several GABAergic descending pathways; for example, Penk+/GAD2+ neurons directly synapsing onto primary afferent DRG neurons and inhibiting pain (Zhang et al., 2015) and Penk-negative/presumably GAD1+ neurons synapsing onto Penk+ dorsal horn interneurons and facilitating mechanical pain. Consistent with our disynaptic inhibitory circuit model, RVM GABAergic neurons reportedly project onto parvalbumin+ spinal interneurons (Antal et al., 1996; Petitjean et al., 2015), suggesting that this class of neuron might also contribute to descending pain modulation, as demonstrated in this study for enkephalinergic neurons. The observations that the circuit described here modulates only mechanical pain—and that both Penk+ and parvalbumin+ neurons receive RVM GABAergic inputs, but are located predominantly in distinct laminae and likely receive distinct primary afferent inputs—suggest that multiple parallel GABAergic systems may exist for independent descending control of distinct somatosensory modalities.
We also identified neurons projecting specifically onto RVM GABAergic descending neurons, including in the lateral hypothalamus (LH) and lateral parabrachial nucleus (LPB). The position of the LPB at the intersection of ascending and descending pain pathways suggests a critical role for tuning sensory experiences as a function of internal states, including through a pain-stress loop that might facilitate pain in patients with psychiatric disorders (Ohayon and Schatzberg, 2003; Zhuo, 2016) or underlie the emergence of pain catastrophizing (Quartana et al., 2009).
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for reagents may be directed to, and will be fulfilled by, the corresponding author and Lead Contact, Dr. Grégory Scherrer (gs25@stanford.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All procedures followed animal care guidelines approved by Stanford University’s Administrative Panel on Laboratory Animal Care (APLAC) and the recommendations of the International Association for the Study of Pain. Mice were housed up to 5 per cage and maintained on a 12 hr light/dark cycle with ad lib access to food and water. All animals were healthy naive males unless indicated, randomly assigned to experimental groups. Group sizes were estimated based on literature, availability, and power analysis with an expected change of 20 to 30%.
METHOD DETAILS
PenkCre Mice Generation
Targeting vector construction: The targeting vector was constructed using recombineering techniques described by Liu (Liu et al., 2003). An 8,528 bp genomic DNA fragment containing the last exon of the Penk gene was retrieved from BAC clone RP23-RP23–365K8 to a vector containing the DT gene, a negative selection marker. A cassette of IRES-Cre frt-PGKNeo-frt was inserted between stop codon TGA and 3′ UTR. The length of the 5′ homologous arm is 3,064 bp and that for the 3′ arm is 5,462 kb. ES cell targeting and screening: The targeting vector was electroporated into F1 hybrid of 129S6 × C57BL/6J ES cells derived by Janelia Transgenic Facility. The G418 resistant ES clones were screened by nested PCR using primers outside the construct paired with primers inside the neo cassette. The primers sequences were as following: 5′ arm forward primers: Penk scr F1 (5′-ACTTGGCCAGGAAAGCACTA-3′) and Penk scr F2 (5′-CCTATAGTCAGGAGCTTGCA-3′). Reverse primers: IRES R1 (5′–AGGAACTGCTTCCTTCACGA-3′) and IRES R2 (5′-CCTAGGAATGCTCGTCAAGA-3′). 3′ arm forward primers: Neo F3 (5′-CTTCCTCGTGCTTTACGGTA-3′) and Neo F4 (5′-ACGAGTTCTTCTGAGGGGAT-3′); Reverse primers: Penk scr R3 (5′-GAGACACGGCTATCTTGTAC-3′) and Penk scr R4 (5′-TGTAGGTCCTCAGAAGAGCA-3′). Generation of chimera and genotyping: The PCR positive ES clones and were expanded for generation of chimeric mice. The ES cells were aggregated with 8-cell embryos of CD-1 strain. The neo cassette was removed by breeding germline chimeras with Rosa26FLP1 (Jax stock#: 003946) homozygous females. The F1 pups were genotyped by PCR using primers flanking the insertion site and a primer in IRES for 5′ arm. The primer Penk gt P1 (5′-CTGGCAGTGACGAAAGTGAA-3′); Penk gt P2 (5′-GGACTTGCATCTTAAGCCTG-3′) and IRES P3 (5′-GCTTCGGCCAGTAACGTTAG-3′). The PCR products are 292 bp for the wild-type allele and 203 bp for the mutant allele. The primer set for the 3′ arm is Cre P4 (5′-ATCCGTAACCTGGATAGTGAA-3′) and Penk gt P5 (5′-ATCACAGCTTTCAGGCAGTG-3′). The generated PCR products are 347 bp for the mutant allele. The correct targeting was further confirmed by obtaining homozygotes from chimera × F1 heterozygous females mating. The mouse line passed homozygosity testing was bred for experiments. Genotyping PCR: The template DNA was obtained by digesting an ear piece in 50 ul proteinase K buffer (50 mM Tris pH8.8, 1mM EDTA pH 8.0, 0.5% Tween-20 and proteinase K 0.6 mg/ml). The reaction was incubated at 55°C overnight and heat inactivated at 100°C for 10 min. 0.5 ul of the template was used in 12 ul PCR reaction. The reaction was carried out with an initial denature cycle of 94°C for 3 min, followed by 35 cycles of 94°C 30 s, 55°C 30 s and 72°C 30 s and ended with one cycle of 72°C for 5 min.
Viral Trans-Synaptic Tracing
AAV helper viruses (AAV-FLEx-TVA-mCherry and AAV-FLEx-G) were stereotaxically injected into the lumbar spinal cord (as described in detail below) of PenkCre animals. 2 to 3 weeks later, a recombinant rabies virus (RVdG) was injected using the same coordinates. Brain, spinal cord, and DRG were then collected 5 to 7 days after injection of rabies virus.
Stereotaxic Injections
6 to 8 week old mice were anesthetized by inhalation of a 2% isoflurane/1.5% oxygen mixture. For spinal injections, thoracic vertebra T12 was exposed by carefully removing the paraspinal muscles. The animal was then placed into a stereotaxic frame (Kopf Instruments) and vertebrae were immobilized using a pair of spinal adaptors. The T12 dorsal spinous process was removed to expose the dura mater and lumbar spinal cord. Viral vectors were injected 300 μm to the right of the posterior median spinal vein at a depth of 300 μm. Pulled glass micropipettes were used to inject 200nL of vector solution at a speed of 40nl/min. For medullary injections, the animal was placed into a stereotaxic frame (Kopf Instruments) to immobilize the cranium. An incision was made to expose the calvarium and 30% hydrogen peroxide was applied to visualize the sutures. A burr hole was made in the skull over the desired injection site. Pulled glass micropipettes were used to inject 250nL of viral vectors or 100nL 2% Fluorogold in normal saline into the rostral ventromedial medulla (bregma −6.0, lateral 0.0, ventral −5.5) at a speed of 40nL/min. The micropipette was kept in the injection site for 10 min after infusion was complete and then retracted slowly over 15 min. Wounds were sutured and surgical sites infiltrated with 2% lidocaine in saline. Animals were placed on a heating pad and monitored until fully recovered.
Implantation of Fiber Optic Cannulae
Mice were anesthetized under 2 – 3% isoflurane. Once a stable plane of anesthesia was reached, the area surrounding the dorsal hump was shaved, and then cleansed with alternating applications of betadine and alcohol. A 1 – 2 cm incision was made slightly caudal to the peak of the dorsal hump in order to expose the lumbar spinal region. The vertebra of interest was identified, and then a small incision was made between the tendons and the vertebral column on either side. The vertebra was then secured using spinal adaptor clamps, and all the tissue was removed from the surface of the bone. Using a micro drill, we removed the spinous process, and roughed the surface of the vertebra. Next a small hole was drilled about 2 mm from midline, centrally on the rostral caudal axis.
We first cleaved the end of the fiber optic to a length less than 1 mm, and then positioned it above the drilled hole. We used a small amount of super glue around the drill hole and over the surface of the bone, to reduce the possibility of bone bleeds, and to secure the cannula in place. Next we dental cemented the cannula in place, and then after the dental cement dried, sutured the skin surrounding the dental cement. Mice were allowed to recover under a heat lamp before being returned to their cage. Mice continued to be group housed after this procedure was performed. Experiments were performed 8 to 12 weeks after injection.
Histology
Tissue Collection and Processing
Animals were transcardially perfused with phosphate-buffered saline (PBS) followed by 10% formaldehyde in PBS. Brain, spinal cord, or skin were dissected, post-fixed in 10% formaldehyde, and cryoprotected in 30% sucrose in PBS. DRGs were cryoprotected directly after formaldehyde perfusion. Tissues were then frozen in Optimum Cutting Temperature (OCT, Tissue Tek) and sectioned using a cryostat (Leica). Spinal cord and brain were sectioned at 40 μm and stored in PBS at 4°C if used immediately. Skin was sectioned at 20 μm. For longer storage, tissue sections were placed in glycerol-based cryoprotectant solution and kept at −20°C. For DRGs and in situ hybridization, tissues were sectioned at 12 μm, collected on Superfrost Plus slides (Fisher Scientific), and stored at −80°C.
Immunofluorescence
Tissues were incubated for 1 hr and blocked in a solution consisting of 0.1 M PBS with 0.3% Triton X-100 (Sigma) plus 5% normal donkey serum. Primary and secondary antibodies were diluted in 0.1 M PBS with 0.3% Triton X-100 plus 1% normal donkey serum. Sections were then incubated overnight at 4°C in primary antibody solution, washed in 0.1 M PBS with 0.3% Triton X-100 for 40 min, incubated for 2 hr in secondary antibody at room temperature (RT), and washed again in 0.1 M PB for 40 min. Sections were then mounted using Fluoromount-G. Images were acquired with a Leica TCS SPE confocal microscope. Primary antibodies: anti-CGRP: Abcam (sheep; 1:2000); anti-GFP: Molecular Probes (rabbit; 1:1000), Abcam (chicken; 1:1000) or Biosystems (sheep; 1:500); anti-NF200: Sigma (mouse; 1:1000) or Aves Labs (chicken; 1:1000); anti-TH: Millipore (Rabbit; 1:1000); anti-TPH: Millipore (rabbit; 1:800); anti-Ret: R&D Systems (goat; 1:60); anti-TrkA: Millipore (rabbit; 1:500); anti-TrkC: R&D Systems (goat; 1:500); anti-GABA: Sigma (rabbit; 1:1000); anti-PAX2: Life Technologies (rabbit; 1:50); anti-TLX3: gift from Dr. Carmen Birchmeier (guinea pig; 1:10000); anti-met-enkephalin: Immunostar (rabbit; 1:1000); anti-MAP2: Millipore (mouse; 1:1000); anti-synaptotagmin: R&D Systems (mouse; 1:500); anti-NeuN: Millipore (mouse; 1:1000); anti-PKCγ: Gift from Dr. Allan Basbaum (guinea pig; 1:10000); anti-calbindin: Sigma (mouse; 1:1000); anti-calretinin: Swant (goat; 1:2000); anti-fluorogold: Millipore (rabbit; 1:1000); anti-RFP: Abcam (rabbit; 1:1000), anti-Cfos: Abcam (Rabbit 1:500), anti-WGA: Sigma (rabbit; 1:50000). To identify IB4-binding cells, biotinylated IB4 (Sigma, 1:500) and fluorophore-conjugated streptavidin (Molecular Probes, 1:1000) were used in place of primary and secondary antibodies. Secondary antibodies: Alexa Fluor®-conjugated secondary antibodies were acquired from Invitrogen and Jackson Immunoresearch Labs.
In Situ Hybridization
The Cre expression pattern in PenkCre; Rosa26-LSL-tdTomato mice or PenkCre; AAV-FLEx-YFP was validated using the RNAscope Multiplex Fluorescent Assay (Advanced Cell Diagnostics, Hayward, CA). Slides were washed in PBS for 5 min and pretreated per manufacturer’s instructions. Protease treatment was applied for 15 min at 40°C. Target probes for Penk, Gad1, Gad2, eGFP, and/or tdTomato were combined and hybridized for 2 hr at 40°C using the HybEZ Hybridization System. Following amplification and label application, slides were imaged with a Leica TCS SPE confocal microscope. Only cells with visible DAPI+ nuclei were examined for the presence of labeled mRNA.
Electrophysiology
Spinal cord slice preparation and electrophysiology were performed as previously described in Bardoni et al., 2014. Briefly, two weeks after virus injections, mice were anesthetized with isoflurane, decapitated, and the vertebral column was rapidly removed and placed in oxygenated ice-cold dissection solution (in mM: 95 NaCl, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 50 sucrose, 25 glucose, 6 MgCl2, 1.5 CaCl2, and 1 kynurenic acid, pH 7.4, 320 mOsm). The lumbar spinal cord was isolated, embedded in a 3% agarose block, and transverse slices (400 μm thick) were made using a vibrating microtome (Leica VT1200). Special care was used to avoid any damage to the dorsal roots during this process. Slices were incubated in oxygenated recovery solution (in mM: 125 NaCl, 2.5 KCl, 1.25 NaH2PO4, 26 NaHCO3, 25 glucose, 6 MgCl2, and 1.5 CaCl2, pH 7.4, 320 mOsm) at 35°C for 1 hr. Neurons were visualized with an Olympus BX51WI with Nomarski optics connected to a camera (Q-imaging) and patch-clamp recording in whole-cell configuration was performed at RT. Recordings were performed in current-clamp or voltage-clamp mode at a holding potential of −70 mV, unless otherwise indicated. Thick-walled borosilicate pipettes, with a resistance of 3–5 MOhm, were filled with internal solution (in mM: 120 K-methyl-sulfonate, 10 NaCl, 10 EGTA, 1 CaCl2, 10 HEPES, 0.5 NaGTP, 5 MgATP, pH adjusted to 7.2 with KOH, osmolarity adjusted to 305 with sucrose). Light stimulations were evoked via Lambda TLED controller at a wavelength of 530nm (Sutter Instrument). Dorsal root simulation was performed using a suction electrode connected to a DS4 Digitimer. Paired-pulse recordings were obtained by briefly stimulating the dorsal root two times with an interval of 50ms. Stimulation intensity and conduction velocity (CV) were used to selectively stimulate Aβ/δ and C fibers and distinguish corresponding evoked EPSCs. CVs were determined by calculating the ratio between the stimulation artifact - EPSC latency, and the length between the suction electrode and the dorsal entry zone. A stimulation intensity of 5 to 50 μA with a conduction velocity less than or equal to 0.1 m/s were considered to correspond to Aβ/δ-fiber activity, whereas a stimulation intensity of 50 μA to 500 μA with a conduction velocity equal or greater to 0.5 m/s were considered to correspond to C-fiber activity. EPSCs with constant latency and no failures during a train stimulus of 10 Hz were considered monosynaptic responses mediated by Aβ/δ fibers, and EPSCs with constant latency and no failures during a train stimulus of 2 Hz were considered monosynaptic responses mediated by C fibers. For paired-pulse root stimulation combined with blue light stimulation, the ratio between the amplitude of the second and first EPSCs was calculated to determine the PPR. The blue light stimulation protocol (1 s at 15Hz) was followed by a paired-pulse stimulation of the dorsal root. The interval between the light stimulation and paired-pulse was then gradually increased by 50ms between every sweep. Enkephalinergic neurons with a significant increase in the PPR after blue light application were classified as “presynaptically inhibited” (Mann Whitney test; 5 to 7 traces for each condition per neuron). Data were acquired using a Multiclamp 700A amplifier and pClamp10 software (Molecular Devices, USA). Sampling rate was 10 kHz and data were filtered at 2 kHz. Analysis was done with Clampfit software (pClamp9, Molecular Devices, USA). Graphs and statistical analyses were generated using Graphpad and Microsoft Excel. EPSC peaks were manually detected for each trace.
Healthy cells were randomly selected by the experimenter. No data were excluded.
Behavior
All mice were habituated to each test apparatus for 60 min on two consecutive days prior to testing. All experiments were conducted blind to genotype and treatment group. Randomization was done by the experimenter and no subjet or data were excluded for any reason. For optogenetic experiments, light was turned on 5 s before the beginning of the test and remained on for the duration of the von Frey test. For Hargreaves, the light was turned off after each temperature ramp. The experimenter was blind to the light stimulation.
Spontaneous Nocifensive Behaviors
Mice were placed in a red plastic cylinder and acclimatized for 60 min. The number of flitching, biting, or licking bouts over a 5 min interval were scored, before and after the administration of CNO.
Von Frey Test
Mice were placed in a red plastic cylinder on an elevated wire grid and the plantar surface of the hindpaw was stimulated with a set of calibrated von Frey filaments (0.008 – 6 g). The 50% paw withdrawal threshold was determined using the Dixon’s up-down method (Chaplan et al., 1994).
Hargreaves’ Test
Mice were placed in a red plastic cylinder on a glass floor maintained at 30°C. A radiant heat beam was focused onto the hindpaw (Radiant heat apparatus purchased from the Department of Anesthesiology, University of California San Diego, La Jolla, CA). The latency to hindpaw withdrawal was recorded with at least 3 trials per animal repeated ≥ 5 min apart. Beam intensity was adjusted so that control mice displayed a latency of 8 – 12 s. A cut-off latency of 30 s was set to avoid tissue damage.
Sticky Tape Test
Animals were placed in a red plastic cylinder and acclimatized for 10 min. A one-quarter square inch piece of scotch tape was placed on the plantar surface of the hindpaw and latency to hindpaw shaking was recorded. 3 trials per animal were repeated ≥ 5 min apart.
Hot Plate Test
Mice were placed on a metal plate preheated to 52°C. Latency to hindpaw licking, shaking, or jumping was recorded. 3 trials per animal were repeated ≥ 5 min apart.
Rotarod Test
Mice were trained on a rotarod for 2 consecutive days before testing. For training, mice were placed on the rotarod at 5rpm for 5 min. Mice that fell were placed back on the rotarod and the 5 min trial restarted. The day after training was completed, mice were placed on a still rotarod, which was then ramped up to 20rpm. The time to fall was recorded. 3 trials were repeated per animal.
Open-Field Test
Mice were placed in a 45×60×15cm open field arena and allowed to explore freely for 15 min. Their activity was recorded with a video camera. The experimenter was not present in the room during the experiment. Videos were analyzed with EthoVision ET to determine the distance and velocity of travel during the 15 min trial.
Chronic Restraint Stress
Mice were place in 50ml tube pierced with multiple holes to allow appropriate ventilation for 2h under normal light between 10 a.m. and 12 p.m. Control littermates were placed individually in new cages without water or food. 2h after mechanical stimulation and assessment, mice were perfused and tissues processed as described above for immunostaining.
QUANTIFICATION AND STATISTICAL ANALYSIS
All Statistical analyses were done using Graphpad prism 6. No assumption on the distribution of the data was done, and only non-parametric tests were used. Values are presented as mean ± SEM. Statistical significance was assessed using a Mann-Whitney t test, a one-way ANOVA with Kruskal-Wallis post hoc test, or a two-way ANOVA with Bonferroni post hoc test for repeated-measures at a significance level of 0.05. No statistical methods were used to predetermine sample sizes, but our sample sizes were similar to those generally employed in the field. Numbers of replications (n) are equal to the number of animals used in behavior and immunohistochemistry experiments and to the number of neurons recorded from for patch clamp experiments.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Anti-CGRP Sheep | Abcam | Cat# ab22560; RRID: AB_725809 |
| Anti-GFP Rabbit | Molecular Probes | Cat# A-11122; RRID: AB_221569 |
| Anti-GFP Chicken | Aves Labs | Cat# GFP-1020; RRID: AB_10000240 |
| Anti-GFP goat | Abcam | Cat# ab6673; RRID: AB_305643 |
| Anti-NF200 Mouse | Sigma-Aldrich | Cat# N0142; RRID: AB_477257 |
| Anti-NF200 Chicken | Aves Labs | Cat# NFH0211 |
| Anti-TH Rabbit | Millipore | Cat# AB152; RRID: AB_390204 |
| Anti-Ret Goat | R&D Systems | Cat# AF482; RRID: AB_2301030 |
| Anti-TrkA Rabbit | Millipore | Cat# 06–574; RRID: AB_310180 |
| Anti-TrkC Goat | R&D Systems | Cat# AF1404; RRID: AB_2155412 |
| Anti-GABA Rabbit | Sigma-Aldrich | Cat# A2052; RRID: AB_477652 |
| Anti-Pax2 Rabbit | Thermo Fisher Scientific | Cat# 716000; RRID: AB_2533990 |
| Anti-TLX3 Guinea Pig | Dr. Carmen Birchmeier | N/A |
| Anti-Met-Enkephalin Rabbit | ImmunoStar | Cat# 20065 RRID: AB_572250 |
| Anti-MAP2 Mouse | Millipore | Cat# MAB3418; RRID: AB_11212326 |
| Anti-Synaptotagmin Mouse | R&D Systems | Cat# ASV48; RRID: AB_1643138 |
| Anti-NeuN Mouse | Millipore | Cat# MAB377; RRID: AB_2298772 |
| Anti-PKCg Guinea Pig | Dr. Allan Basbaum | N/A |
| Anti-Calbindin Mouse | Sigma | Cat# C9848; RRID: AB_476894 |
| Anti-Calretinin Goat | Swant | Cat# CG1; RRID: AB_10000342 |
| Anti-Fluorogold Rabbit | Millipore | Cat# AB153; RRID: AB_90738 |
| Anti-RFP Rabbit | Abcam | Cat# ab62341; RRID: AB_945213 |
| Anti-Cfos Rabbit | Abcam | Cat# ab7963–1; RRID: AB_306177 |
| Anti-WGA Rabbit | Sigma-Aldrich | Cat# T4144; RRID: AB_261669 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| IB4 biotinylated | Sigma-Aldrich | Cat# L2140 |
| Fluorogold | Fluorochrome | Fluoro-Gold |
| Clozapine N-oxide (CNO) | Tocris Biosciences | Cat# 4936 |
| Naloxone | Tocris Biosciences | Cat# 0599 |
| Tipp-psi | Dr. Peter Schiller | Schiller et al., 1993 |
| CTOP | Tocris Biosciences | Cat# 1578 |
| Bicuculline | Tocris Biosciences | Cat# 2503 |
| Strychnine | Sigma-Aldrich | Cat# s0532 |
| Critical Commercial Assays | ||
| RNAscope Multiplex Fluorescent Assay | Advanced Cell Diagnostics | Cat# 320850 |
| RNAscope Probe - Mm-Gad1 | Advanced Cell Diagnostics | 400951 |
| RNAscope Probe - Mm-Gad2-C2 | Advanced Cell Diagnostics | 415071-C2 |
| RNAscope Probe - EGFP-C3 | Advanced Cell Diagnostics | 400281-C3 |
| RNAscope Probe- Mm-Penk | Advanced Cell Diagnostics | 318761 |
| RNAscope Probe- tdTomato-C2 | Advanced Cell Diagnostics | 317041-C2 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Penk-Cre | Dr. Adam Hantman | This paper |
| Mouse: Vgat-Cre | The Jackson Laboratory | Stock# 016962; RRID: IMSR_JAX:016962 |
| Mouse: Rosa26-LSL-tdTomato | The Jackson Laboratory | Stock# 007914; RRID: IMSR_JAX:007914 |
| Mouse: R26-LSL-ZsGreen | The Jackson Laboratory | Stock# 007906; RRID: IMSR_JAX:007906 |
| Recombinant DNA | ||
| Mutated rabies: RVdG | Dr. Kevin Beier, Dr. Liqun Luo, and Dr. Robert C. Malenka | Beier et al., 2015; Schwarz et al., 2015 |
| AAV8-CAG-FLEx-G | Dr. Kevin Beier, Dr. Liqun Luo, and Dr. Robert C. Malenka | Beier et al., 2015; Schwarz et al., 2015 |
| AAV5-CAG-FLEx-TVA-mCherry | Dr. Kevin Beier, Dr. Liqun Luo, and Dr. Robert C. Malenka | Beier et al., 2015; Schwarz et al., 2015 |
| AAV8-CAG-FD-G | Dr. Kevin Beier, Dr. Liqun Luo, and Dr. Robert C. Malenka | Beier et al., 2015; Schwarz et al., 2015 |
| AAV5-CAG-FD-TVA-mCherry | Dr. Kevin Beier, Dr. Liqun Luo, and Dr. Robert C. Malenka | Beier et al., 2015; Schwarz et al., 2015 |
| AAV-DJ-ef1a-DIO-eYFP | Stanford University Gene Vector and Virus Core | N/A |
| AAV-DJ-ef1a-DIO-mCherry | Stanford University Gene Vector and Virus Core | N/A |
| AAV-DJ-ef1a-DIO-hChR2 (ET/TC) p2a-eYFP-WPRE | Stanford University Gene Vector and Virus Core | N/A |
| AAV8 CMV hChR2 (ET/TC) p2a-eYFP-WPRE | Stanford University Gene Vector and Virus Core | N/A |
| AAV5-hsyn-DIO-hM4Di-mcherry | University of North Carolina Vector Core | N/A |
| AAV2-retro-Syn-FLEx-FlpO | Dr. Adam Hantman | Tervo et al., 2016 |
| AAV2-CBA-FLEx-WGA | Dr. Reza Sharif-Naeini | Petitjean et al., 2015 |
| AAV-DJ-hSyn-FD-hM4Di-mCherry | Dr. Karl Deisseroth and Dr. Charu Ramakrishnan | This paper |
| AAV-DJ-nEF-DIO-eNphR3-P2A-hChR2-eYFP | Dr. Karl Deisseroth and Dr. Charu Ramakrishnan | Rashid et al., 2016 |
| Sequence-Based Reagents | ||
| Penk scr F1 (5′-ACTTGGCCAGGAAAGCACTA-3′) | Dr. Adam Hantman | N/A |
| Penk scr F2 (5′-CCTATAGTCAGGAGCTTGCA-3′) | Dr. Adam Hantman | N/A |
| Penk scr R3 (5′-GAGACACGGCTATCTTGTAC-3′) | Dr. Adam Hantman | N/A |
| Penk scr R4 (5′-TGTAGGTCCTCAGAAGAGCA-3′) | Dr. Adam Hantman | N/A |
| Penk gt P1 (5′-CTGGCAGTGACGAAAGTGAA-3′) | Dr. Adam Hantman | N/A |
| Penk gt P2 (5′-GGACTTGCATCTTAAGCCTG-3′) | Dr. Adam Hantman | N/A |
| Cre P4 (5′-ATCCGTAACCTGGATAGTGAA-3′) | Dr. Adam Hantman | N/A |
| Penk gt P5 (5′-ATCACAGCTTTCAGGCAGTG-3′) | Dr. Adam Hantman | N/A |
| IRES R1 (5′-AGGAACTGCTTCCTTCACGA-3′) | Dr. Adam Hantman | N/A |
| IRES R2 (5′-CCTAGGAATGCTCGTCAAGA-3′) | Dr. Adam Hantman | N/A |
| Software and Algorithms | ||
| Ethovision ET | Noldus | N/A |
| pClamp10 | Molecular Devices | N/A |
| Clampfit | Molecular Devices | N/A |
| Prism6 | GraphPad | N/A |
| Photoshop CS | Adobe | N/A |
| Illustrator CS | Adobe | N/A |
| Excel | Microsoft | N/A |
Highlights.
Primary afferents and descending pain pathways project onto spinal Penk+ neurons
A population of GABA+ RVM neurons control spinal Penk+ neurons and mechanical pain
Together, spinal enkephalins and GABA presynaptically modulate mechanonociception
Brain regions processing stress recruit this RVM/spinal/primary afferent circuit
ACKNOWLEDGMENTS
This work was funded by NIH/NIDA grant DA031777 (G.S.), a Rita Allen Foundation and American Pain Society award (G.S.), a Stanford Dean’s fellowship (A.F.), an HHMI medical research fellowship (S.A.L.), a NDSEG fellowship (E.I.S), and HHMI (K.D.R. and A.W.H.). We thank Dr. Carmen Birchmeier for the TLX3 and PAX2 antibodies, Dr. Brigitte Kieffer for providing MOR-mCherry mice, and Dr. Peter Schiller for providing Tipp-psi.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes nine figures and can be found with this article online at http://dx.doi.org/10.1016/j.neuron.2017.01.008.
A video abstract is available at http://dx.doi.org/10.1016/j.neuron.2017.01.008#mmc3.
SUPPORTING CITATIONS
The following references appear in the Supplemental Information: Van Dort et al. (2015); Uusisaari and Knöpfel (2011); Zemlan and Behbehani (1988); Miczek and Winslow (1987); Goetz et al. (2016); Bellchambers et al. (1998); Baker et al. (2010).
REFERENCES
- Abraira VE, and Ginty DD (2013). The sensory neurons of touch. Neuron 79, 618–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antal M, Petkó M, Polgár E, Heizmann CW, and Storm-Mathisen J (1996). Direct evidence of an extensive GABAergic innervation of the spinal dorsal horn by fibres descending from the rostral ventromedial medulla. Neuroscience 73, 509–518. [DOI] [PubMed] [Google Scholar]
- Baker KB, Schuster D, Cooperrider J, and Machado AG (2010). Deep brain stimulation of the lateral cerebellar nucleus produces frequency-specific alterations in motor evoked potentials in the rat in vivo. Exp. Neurol 226, 259–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbaro NM, Heinricher MM, and Fields HL (1986). Putative pain modulating neurons in the rostral ventral medulla: reflex-related activity predicts effects of morphine. Brain Res 366, 203–210. [DOI] [PubMed] [Google Scholar]
- Bardoni R, Takazawa T, Tong CK, Choudhury P, Scherrer G, and Macdermott AB (2013). Pre- and postsynaptic inhibitory control in the spinal cord dorsal horn. Ann. N Y Acad. Sci 1279, 90–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardoni R, Tawfik VL, Wang D, François A, Solorzano C, Shuster SA, Choudhury P, Betelli C, Cassidy C, Smith K, et al. (2014). Delta opioid receptors presynaptically regulate cutaneous mechanosensory neuron input to the spinal cord dorsal horn. Neuron 81, 1312–1327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum AI, Clanton CH, and Fields HL (1976). Opiate and stimulus-produced analgesia: functional anatomy of a medullospinal pathway. Proc. Natl. Acad. Sci. USA 73, 4685–4688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basbaum AI, Bautista DM, Scherrer G, and Julius D (2009). Cellular and molecular mechanisms of pain. Cell 139, 267–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier KT, Steinberg EE, DeLoach KE, Xie S, Miyamichi K, Schwarz L, Gao XJ, Kremer EJ, Malenka RC, and Luo L (2015). Circuit architecture of VTA dopamine neurons revealed by systematic input-output mapping. Cell 162, 622–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belin MF, Nanopoulos D, Didier M, Aguera M, Steinbusch H, Verhofstad A, Maitre M, and Pujol JF (1983). Immunohistochemical evidence for the presence of gamma-aminobutyric acid and serotonin in one nerve cell. A study on the raphe nuclei of the rat using antibodies to glutamate decarboxylase and serotonin. Brain Res 275, 329–339. [DOI] [PubMed] [Google Scholar]
- Bellchambers CE, Chieng B, Keay KA, and Christie MJ (1998). Swim-stress but not opioid withdrawal increases expression of c-fos immunoreactivity in rat periaqueductal gray neurons which project to the rostral ventromedial medulla. Neuroscience 83, 517–524. [DOI] [PubMed] [Google Scholar]
- Betley JN, Wright CV, Kawaguchi Y, Erdélyi F, Szabó G, Jessell TM, and Kaltschmidt JA (2009). Stringent specificity in the construction of a GABAergic presynaptic inhibitory circuit. Cell 139, 161–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourane S, Grossmann KS, Britz O, Dalet A, Del Barrio MG, Stam FJ, Garcia-Campmany L, Koch S, and Goulding M (2015). Identification of a spinal circuit for light touch and fine motor control. Cell 160, 503–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner N, Bialek W, and de Ruyter van Steveninck R (2000). Adaptive rescaling maximizes information transmission. Neuron 26, 695–702. [DOI] [PubMed] [Google Scholar]
- Budai D, and Fields HL (1998). Endogenous opioid peptides acting at muopioid receptors in the dorsal horn contribute to midbrain modulation of spinal nociceptive neurons. J. Neurophysiol 79, 677–687. [DOI] [PubMed] [Google Scholar]
- Butler RK, and Finn DP (2009). Stress-induced analgesia. Prog. Neurobiol 88, 184–202. [DOI] [PubMed] [Google Scholar]
- Cavanaugh DJ, Lee H, Lo L, Shields SD, Zylka MJ, Basbaum AI, and Anderson DJ (2009). Distinct subsets of unmyelinated primary sensory fibers mediate behavioral responses to noxious thermal and mechanical stimuli. Proc. Natl. Acad. Sci. USA 106, 9075–9080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, and Yaksh TL (1994). Quantitative assessment of tactile allodynia in the rat paw. J. Neurosci. Methods 53, 55–63. [DOI] [PubMed] [Google Scholar]
- Chen J, Huang J, Wei YY, Sun XX, Wang W, Bai L, Wang YY, Kaneko T, Li YQ, and Wu SX (2014). Birth-date dependent arrangement of spinal enkephalinergic neurons: evidence from the preproenkephalin-green fluorescent protein transgenic mice. Neuroscience 260, 47–58. [DOI] [PubMed] [Google Scholar]
- Christensen AJ, Iyer SM, François A, Vyas S, Ramakrishnan C, Vesuna S, Deisseroth K, Scherrer G, and Delp SL (2016). In vivo interrogation of spinal mechanosensory circuits. Cell Rep 17, 1699–1710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comb M, Seeburg PH, Adelman J, Eiden L, and Herbert E (1982). Primary structure of the human Met- and Leu-enkephalin precursor and its mRNA. Nature 295, 663–666. [DOI] [PubMed] [Google Scholar]
- Delmas P, Hao J, and Rodat-Despoix L (2011). Molecular mechanisms of mechanotransduction in mammalian sensory neurons. Nat. Rev. Neurosci 12, 139–153. [DOI] [PubMed] [Google Scholar]
- Dogrul A, Ossipov MH, and Porreca F (2009). Differential mediation of descending pain facilitation and inhibition by spinal 5HT-3 and 5HT-7 receptors. Brain Res 1280, 52–59. [DOI] [PubMed] [Google Scholar]
- Duan B, Cheng L, Bourane S, Britz O, Padilla C, Garcia-Campmany L, Krashes M, Knowlton W, Velasquez T, Ren X, et al. (2014). Identification of spinal circuits transmitting and gating mechanical pain. Cell 159, 1417–1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eckert WA 3rd, and Light AR (2002). Hyperpolarization of substantia gelatinosa neurons evoked by mu-, kappa-, delta 1-, and delta 2-selective opioids. J. Pain 3, 115–125. [DOI] [PubMed] [Google Scholar]
- Erbs E, Faget L, Scherrer G, Matifas A, Filliol D, Vonesch JL, Koch M, Kessler P, Hentsch D, Birling MC, et al. (2015). A mu-delta opioid receptor brain atlas reveals neuronal co-occurrence in subcortical networks. Brain Struct. Funct 220, 677–702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields HL, Bry J, Hentall I, and Zorman G (1983a). The activity of neurons in the rostral medulla of the rat during withdrawal from noxious heat. J. Neurosci 3, 2545–2552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fields HL, Vanegas H, Hentall ID, and Zorman G (1983b). Evidence that disinhibition of brain stem neurones contributes to morphine analgesia. Nature 306, 684–686. [DOI] [PubMed] [Google Scholar]
- Fink AJ, Croce KR, Huang ZJ, Abbott LF, Jessell TM, and Azim E (2014). Presynaptic inhibition of spinal sensory feedback ensures smooth movement. Nature 509, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster E, Wildner H, Tudeau L, Haueter S, Ralvenius WT, Jegen M, Johannssen H, Hösli L, Haenraets K, Ghanem A, et al. (2015). Targeted ablation, silencing, and activation establish glycinergic dorsal horn neurons as key components of a spinal gate for pain and itch. Neuron 85, 1289–1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goetz L, Piallat B, Bhattacharjee M, Mathieu H, David O, and Chabardès S (2016). On the role of the pedunculopontine nucleus and mesencephalic reticular formation in locomotion in nonhuman primates. J. Neurosci 36, 4917–4929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grevert P, and Goldstein A (1978). Endorphins: naloxone fails to alter experimental pain or mood in humans. Science 199, 1093–1095. [DOI] [PubMed] [Google Scholar]
- Grudt TJ, and Williams JT (1994). mu-Opioid agonists inhibit spinal trigeminal substantia gelatinosa neurons in guinea pig and rat. J. Neurosci 14, 1646–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harlan RE, Shivers BD, Romano GJ, Howells RD, and Pfaff DW (1987). Localization of preproenkephalin mRNA in the rat brain and spinal cord by in situ hybridization. J. Comp. Neurol 258, 159–184. [DOI] [PubMed] [Google Scholar]
- Hawkins KN, Knapp RJ, Lui GK, Gulya K, Kazmierski W, Wan YP, Pelton JT, Hruby VJ, and Yamamura HI (1989). [3H]-[H-D-Phe-Cys-Tyr-D-Trp-Orn-Thr-Pen-Thr-NH2] ([3H]CTOP), a potent and highly selective peptide for mu opioid receptors in rat brain. J. Pharmacol. Exp. Ther 248, 73–80. [PubMed] [Google Scholar]
- Heinke B, Gingl E, and Sandkühler J (2011). Multiple targets of μ-opioid receptor-mediated presynaptic inhibition at primary afferent Aδ- and C-fibers. J. Neurosci 31, 1313–1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinricher MM, Morgan MM, and Fields HL (1992). Direct and indirect actions of morphine on medullary neurons that modulate nociception. Neuroscience 48, 533–543. [DOI] [PubMed] [Google Scholar]
- Heinricher MM, Tavares I, Leith JL, and Lumb BM (2009). Descending control of nociception: Specificity, recruitment and plasticity. Brain Res. Brain Res. Rev 60, 214–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hökfelt T, Ljungdahl A, Terenius L, Elde R, and Nilsson G (1977). Immunohistochemical analysis of peptide pathways possibly related to pain and analgesia: enkephalin and substance P. Proc. Natl. Acad. Sci. USA 74, 3081–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J, Wang Y, Wang W, Wei Y, Li Y, and Wu S (2008). Preproenkephalin mRNA is expressed in a subpopulation of GABAergic neurons in the spinal dorsal horn of the GAD67-GFP knock-in mouse. Anat. Rec. (Hoboken) 291, 1334–1341. [DOI] [PubMed] [Google Scholar]
- Hughes J, Smith TW, Kosterlitz HW, Fothergill LA, Morgan BA, and Morris HR (1975). Identification of two related pentapeptides from the brain with potent opiate agonist activity. Nature 258, 577–580. [DOI] [PubMed] [Google Scholar]
- Jennings EM, Okine BN, Roche M, and Finn DP (2014). Stress-induced hyperalgesia. Prog. Neurobiol 121, 1–18. [DOI] [PubMed] [Google Scholar]
- Jessell TM, and Iversen LL (1977). Opiate analgesics inhibit substance P release from rat trigeminal nucleus. Nature 268, 549–551. [DOI] [PubMed] [Google Scholar]
- Kato G, Yasaka T, Katafuchi T, Furue H, Mizuno M, Iwamoto Y, and Yoshimura M (2006). Direct GABAergic and glycinergic inhibition of the substantia gelatinosa from the rostral ventromedial medulla revealed by in vivo patch-clamp analysis in rats. J. Neurosci 26, 1787–1794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kätzel D, Nicholson E, Schorge S, Walker MC, and Kullmann DM (2014). Chemical-genetic attenuation of focal neocortical seizures. Nat. Commun 5, 3847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kieffer BL, and Gavériaux-Ruff C (2002). Exploring the opioid system by gene knockout. Prog. Neurobiol 66, 285–306. [DOI] [PubMed] [Google Scholar]
- Liu P, Jenkins NA, and Copeland NG (2003). A highly efficient recombineering-based method for generating conditional knockout mutations. Genome Res 13, 476–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T, Li J, Liu H, Wang X, Fan F, Zhang P, Tu Y, and Zhang Y (2015). The coexistence of VGluT2 and neurotensin or leu-enkephalin in the medullary dorsal horn: a confocal and electron microscopic immunohistochemical study in the rat. Neurosci. Lett 584, 390–394. [DOI] [PubMed] [Google Scholar]
- Lu Y, and Perl ER (2007). Selective action of noradrenaline and serotonin on neurones of the spinal superficial dorsal horn in the rat. J. Physiol 582, 127–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marinelli S, Vaughan CW, Schnell SA, Wessendorf MW, and Christie MJ (2002). Rostral ventromedial medulla neurons that project to the spinal cord express multiple opioid receptor phenotypes. J. Neurosci 22, 10847–10855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marvizón JC, Chen W, and Murphy N (2009). Enkephalins, dynorphins, and beta-endorphin in the rat dorsal horn: an immunofluorescence colocalization study. J. Comp. Neurol 517, 51–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon HT, Foran P, Dolly JO, Verhage M, Wiegant VM, and Nicholls DG (1992). Tetanus toxin and botulinum toxins type A and B inhibit glutamate, gamma-aminobutyric acid, aspartate, and met-enkephalin release from synaptosomes. Clues to the locus of action. J. Biol. Chem 267, 21338–21343. [PubMed] [Google Scholar]
- Mende M, Fletcher EV, Belluardo JL, Pierce JP, Bommareddy PK, Weinrich JA, Kabir ZD, Schierberl KC, Pagiazitis JG, Mendelsohn AI, et al. (2016). Sensory-derived glutamate regulates presynaptic inhibitory terminals in mouse spinal cord. Neuron 90, 1189–1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miczek KA, and Winslow JT (1987). Analgesia and decrement in operant performance in socially defeated mice: selective cross-tolerance to morphine and antagonism by naltrexone. Psychopharmacology (Berl.) 92, 444–451. [DOI] [PubMed] [Google Scholar]
- North MA (1978). Naloxone reversal of morphine analgesia but failure to alter reactivity to pain in the formalin test. Life Sci 22, 295–302. [DOI] [PubMed] [Google Scholar]
- Ohayon MM, and Schatzberg AF (2003). Using chronic pain to predict depressive morbidity in the general population. Arch. Gen. Psychiatry 60, 39–47. [DOI] [PubMed] [Google Scholar]
- Palyo SA, and Beck JG (2005). Post-traumatic stress disorder symptoms, pain, and perceived life control: associations with psychosocial and physical functioning. Pain 117, 121–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen NP, Vaughan CW, and Christie MJ (2011). Opioid receptor modulation of GABAergic and serotonergic spinally projecting neurons of the rostral ventromedial medulla in mice. J. Neurophysiol 106, 731–740. [DOI] [PubMed] [Google Scholar]
- Peirs C, Williams SP, Zhao X, Walsh CE, Gedeon JY, Cagle NE, Goldring AC, Hioki H, Liu Z, Marell PS, and Seal RP (2015). Dorsal horn circuits for persistent mechanical pain. Neuron 87, 797–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petitjean H, Pawlowski SA, Fraine SL, Sharif B, Hamad D, Fatima T, Berg J, Brown CM, Jan LY, Ribeiro-da-Silva A, et al. (2015). Dorsal horn parvalbumin neurons are gate-keepers of touch-evoked pain after nerve injury. Cell Rep 13, 1246–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pohl M, Collin E, Bourgoin S, Conrath M, Benoliel JJ, Nevo I, Hamon M, Giraud P, and Cesselin F (1994). Expression of preproenkephalin A gene and presence of Met-enkephalin in dorsal root ganglia of the adult rat. J. Neurochem 63, 1226–1234. [DOI] [PubMed] [Google Scholar]
- Porreca F, Ossipov MH, and Gebhart GF (2002). Chronic pain and medullary descending facilitation. Trends Neurosci 25, 319–325. [DOI] [PubMed] [Google Scholar]
- Potrebic SB, Fields HL, and Mason P (1994). Serotonin immunoreactivity is contained in one physiological cell class in the rat rostral ventromedial medulla. J. Neurosci 14, 1655–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quartana PJ, Campbell CM, and Edwards RR (2009). Pain catastrophizing: a critical review. Expert Rev. Neurother 9, 745–758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ralston HJ, and Ralston DD (1979). Identification of dorsal root synaptic terminals on monkey ventral horn cells by electron microscopic autoradiography. J. Neurocytol 8, 151–166. [DOI] [PubMed] [Google Scholar]
- Rashid AJ, Yan C, Mercaldo V, Hsiang HL, Park S, Cole CJ, De Cristofaro A, Yu J, Ramakrishnan C, Lee SY, et al. (2016). Competition between engrams influences fear memory formation and recall. Science 353, 383–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reichling DB, and Basbaum AI (1990). Contribution of brainstem GABAergic circuitry to descending antinociceptive controls: I. GABA-immunoreactive projection neurons in the periaqueductal gray and nucleus raphe magnus. J. Comp. Neurol 302, 370–377. [DOI] [PubMed] [Google Scholar]
- Ribeiro-da-Silva A, Tagari P, and Cuello AC (1989). Morphological characterization of substance P-like immunoreactive glomeruli in the superficial dorsal horn of the rat spinal cord and trigeminal subnucleus caudalis: a quantitative study. J. Comp. Neurol 281, 497–515. [DOI] [PubMed] [Google Scholar]
- Scherrer G, Imamachi N, Cao YQ, Contet C, Mennicken F, O’Donnell D, Kieffer BL, and Basbaum AI (2009). Dissociation of the opioid receptor mechanisms that control mechanical and heat pain. Cell 137, 1148–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiller PW, Weltrowska G, Nguyen TM, Wilkes BC, Chung NN, and Lemieux C (1993). TIPP[psi]: a highly potent and stable pseudopeptide delta opioid receptor antagonist with extraordinary delta selectivity. J. Med. Chem 36, 3182–3187. [DOI] [PubMed] [Google Scholar]
- Schreiter A, Gore C, Labuz D, Fournie-Zaluski MC, Roques BP, Stein C, and Machelska H (2012). Pain inhibition by blocking leukocytic and neuronal opioid peptidases in peripheral inflamed tissue. FASEB J 26, 5161–5171. [DOI] [PubMed] [Google Scholar]
- Schwarz LA, Miyamichi K, Gao XJ, Beier KT, Weissbourd B, DeLoach KE, Ren J, Ibanes S, Malenka RC, Kremer EJ, and Luo L (2015). Viral-genetic tracing of the input-output organization of a central noradrenaline circuit. Nature 524, 88–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seybold V, and Elde R (1980). Immunohistochemical studies of peptidergic neurons in the dorsal horn of the spinal cord. J. Histochem. Cytochem 28, 367–370. [DOI] [PubMed] [Google Scholar]
- Skagerberg G, and Björklund A (1985). Topographic principles in the spinal projections of serotonergic and non-serotonergic brainstem neurons in the rat. Neuroscience 15, 445–480. [DOI] [PubMed] [Google Scholar]
- Tervo DG, Hwang BY, Viswanathan S, Gaj T, Lavzin M, Ritola KD, Lindo S, Michael S, Kuleshova E, Ojala D, et al. (2016). A designer AAV variant permits efficient retrograde access to projection neurons. Neuron 92, 372–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd AJ (2010). Neuronal circuitry for pain processing in the dorsal horn. Nat. Rev. Neurosci 11, 823–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd AJ, and Spike RC (1993). The localization of classical transmitters and neuropeptides within neurons in laminae I-III of the mammalian spinal dorsal horn. Prog. Neurobiol 41, 609–645. [DOI] [PubMed] [Google Scholar]
- Todd AJ, Hughes DI, Polgár E, Nagy GG, Mackie M, Ottersen OP, and Maxwell DJ (2003). The expression of vesicular glutamate transporters VGLUT1 and VGLUT2 in neurochemically defined axonal populations in the rat spinal cord with emphasis on the dorsal horn. Eur. J. Neurosci 17, 13–27. [DOI] [PubMed] [Google Scholar]
- Usoskin D, Furlan A, Islam S, Abdo H, Lönnerberg P, Lou D, Hjerling-Leffler J, Haeggström J, Kharchenko O, Kharchenko PV, et al. (2015). Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci 18, 145–153. [DOI] [PubMed] [Google Scholar]
- Uusisaari M, and Knöpfel T (2011). Functional classification of neurons in the mouse lateral cerebellar nuclei. Cerebellum 10, 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dort CJ, Zachs DP, Kenny JD, Zheng S, Goldblum RR, Gelwan NA, Ramos DM, Nolan MA, Wang K, Weng FJ, et al. (2015). Optogenetic activation of cholinergic neurons in the PPT or LDT induces REM sleep. Proc. Natl. Acad. Sci. USA 112, 584–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wager TD, and Atlas LY (2015). The neuroscience of placebo effects: connecting context, learning and health. Nat. Rev. Neurosci 16, 403–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickersham IR, Finke S, Conzelmann KK, and Callaway EM (2007). Retrograde neuronal tracing with a deletion-mutant rabies virus. Nat. Methods 4, 47–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaksh TL, Huang SP, and Rudy TA (1977). The direct and specific opiate-like effect of met5-enkephalin and analogues on the spinal cord. Neuroscience 2, 593–596. [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Jessell TM, Gamse R, Mudge AW, and Leeman SE (1980). Intrathecal morphine inhibits substance P release from mammalian spinal cord in vivo. Nature 286, 155–157. [DOI] [PubMed] [Google Scholar]
- Yaksh TL, Terenius L, Nyberg F, Jhamandas K, and Wang JY (1983). Studies on the release by somatic stimulation from rat and cat spinal cord of active materials which displace dihydromorphine in an opiate-binding assay. Brain Res 268, 119–128. [DOI] [PubMed] [Google Scholar]
- Yamamoto T, and Yaksh TL (1993). Effects of intrathecal strychnine and bicuculline on nerve compression-induced thermal hyperalgesia and selective antagonism by MK-801. Pain 54, 79–84. [DOI] [PubMed] [Google Scholar]
- Yoshimura M, and North RA (1983). Substantia gelatinosa neurones hyper-polarized in vitro by enkephalin. Nature 305, 529–530. [DOI] [PubMed] [Google Scholar]
- Zeilhofer HU, Wildner H, and Yévenes GE (2012). Fast synaptic inhibition in spinal sensory processing and pain control. Physiol. Rev 92, 193–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemlan FP, and Behbehani MM (1988). Nucleus cuneiformis and pain modulation: anatomy and behavioral pharmacology. Brain Res 453, 89–102. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Zhao S, Rodriguez E, Takatoh J, Han BX, Zhou X, and Wang F (2015). Identifying local and descending inputs for primary sensory neurons. J. Clin. Invest 125, 3782–3794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao ZQ, Liu XY, Jeffry J, Karunarathne WK, Li JL, Munanairi A, Zhou XY, Li H, Sun YG, Wan L, et al. (2014). Descending control of itch transmission by the serotonergic system via 5-HT1A-facilitated GRP-GRPR signaling. Neuron 84, 821–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhuo M (2016). Neural mechanisms underlying anxiety-chronic pain interactions. Trends Neurosci 39, 136–145. [DOI] [PubMed] [Google Scholar]
- Zhuo M, and Gebhart GF (1990). Characterization of descending inhibition and facilitation from the nuclei reticularis gigantocellularis and gigantocellularis pars alpha in the rat. Pain 42, 337–350. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.