

Abstract
Inducing protein degradation by proteolysis targeting chimeras (PROTACs) has gained tremendous momentum for its promise to discover and develop new therapies. Based upon our previously reported PROTAC MDM2 degraders, we have designed and synthesized additional analogues. Surprisingly, we found that simple structural modifications of MD-222, a bona fide MDM2 PROTAC degrader, converts it into a “molecular glue”, as exemplified by MG-277. MG-277 induces only moderate MDM2 degradation and fails to activate wild-type p53 but is highly potent in inhibition of tumor cell growth in a p53-independent manner. Our mechanistic investigation established that MG-277 is not a PROTAC MDM2 degrader but instead works as a molecular glue, inducing degradation of a translation termination factor, GSPT1 to achieve its potent anticancer activity. Our study provides the first example that simple structural modifications can convert a bona fide PROTAC degrader into a molecular glue compound, which has a completely different mechanism of action.
INTRODUCTION
The proteolysis targeting chimeric (PROTAC) concept was formally proposed in 2001 with the objective of inducing targeted protein degradation by hijacking the powerful cellular degradation systems.1, 2 The very first PROTAC molecule employed the Skp1-Cullin-F box (SCFβ-TrCP) E3 ligase complex and successfully targeted methionine aminopeptidase-2 (MetAP-2) for degradation.1 In recent years, the PROTAC approach has gained momentum, showing promise for the discovery and development of new therapeutic agents.2–8
A typical PROTAC small-molecule degrader consists of three essential components: a ligand that binds the protein of interest, a second ligand that binds to and recruits an E3 ligase degradation complex, and a linker tethering the two ligands together.2–8 Since 2001, a number of E3 ligases have been tested for the design of PROTAC molecules and have enjoyed different levels of success.1–18 The Cullin4A E3 ligase complex and the Cullin2 E3 ligase complex have emerged as two powerful E3 ligase complexes for the design of highly potent and effective PROTAC molecules, in part due to the availability of potent and druglike small-molecule ligands to recruit these degradation complexes.2, 15 In the case of Cullin4A, a class of well-known small-molecule drugs, represented by thalidomide and lenalidomide and collectively known as immunomodulatory imide drugs (IMiDs), have been discovered as potent small-molecule ligands for cereblon, a receptor protein for the Cullin 4A complex.19, 20
The landmark discovery that cereblon is the molecular target for IMiDs has also revealed a completely new mechanism of drug action.21 By binding to cereblon, an E3 ubiquitin ligase receptor, IMiDs alter the specificity of the Cullin4A E3 ubiquitin ligase and cause it to recruit a set of new substrates, or neo-substrates for ubiquitination by the Cullin4A complex. This is followed by proteasomal degradation.21–24 The term “molecular glue” has been used to describe the unprecedented mechanism of action of IMiDs.25, 26 In addition to IMiDs, indisulam has been recently discovered as another class of molecular glue.27, 28 Indisulam brings together the CUL4-DDB1-DDA1-DCAF15 E3 ubiquitin ligase complex with neo-substrates such as RBM39 for ubiquitination and proteasomal degradation.27, 28 The “molecular glue” mechanism of action has also opened an avenue for the discovery of completely new types of drugs.26
Theoretically, a putative PROTAC molecule can function as either a bona fide PROTAC degrader to bind to and degrade the intended target protein(s) or it can act as a molecular glue by recruiting and degrading neo-substrates. Recently, Ishoey et al.29 demonstrated that a class of phthalimide conjugate putative PROTAC degraders fails to induce degradation of their consensus kinase targets in cells but instead functions as a molecular glue.
We recently reported the discovery of the first-in-class highly potent PROTAC degraders of MDM2, exemplified by MD-222 and MD-224, which induce rapid degradation of the MDM2 protein and activation of wild-type p53 in cells.14 These PROTAC MDM2 degraders potently inhibit cell growth in human cancer cell lines carrying wild-type p53 and demonstrate significant specificity over cancer cell lines with mutated or deleted p53. In our further modifications of MD-222 and its analogues, we identified another type of compounds, exemplified by MG-277. In contrast to MD-222 and MD-224, MG-277 is much less effective than MD-222 in inducing degradation of MDM2 and fails to activate wild-type p53. Furthermore, MG-277 potently inhibits cell growth in cancer cell lines in a p53-independent manner, displaying a completely different mode of action from that of MD-222 or MD-224. Our mechanistic investigation revealed that MG-277 is not a PROTAC MDM2 degrader and instead behaves as a molecular glue. Herein, we report the discovery of MG-277 and our detailed investigation of its mechanism of action.
RESULTS
Design of MG-277 as a new analogue of MD-222, a bona fide PROTAC MDM2 degrader
Previously, we reported the discovery and extensive investigation of MD-222 and its analogues as the first-in-class PROTAC MDM2 degraders.14 MD-222 was designed and synthesized using a potent and selective MDM2 inhibitor (MI-1061, Figure 1A), which was conjugated, through a linker, with a lenalidomide moiety.14
Figure 1. Design of MG-277 as a new, putative PROTAC MDM2 degrader.
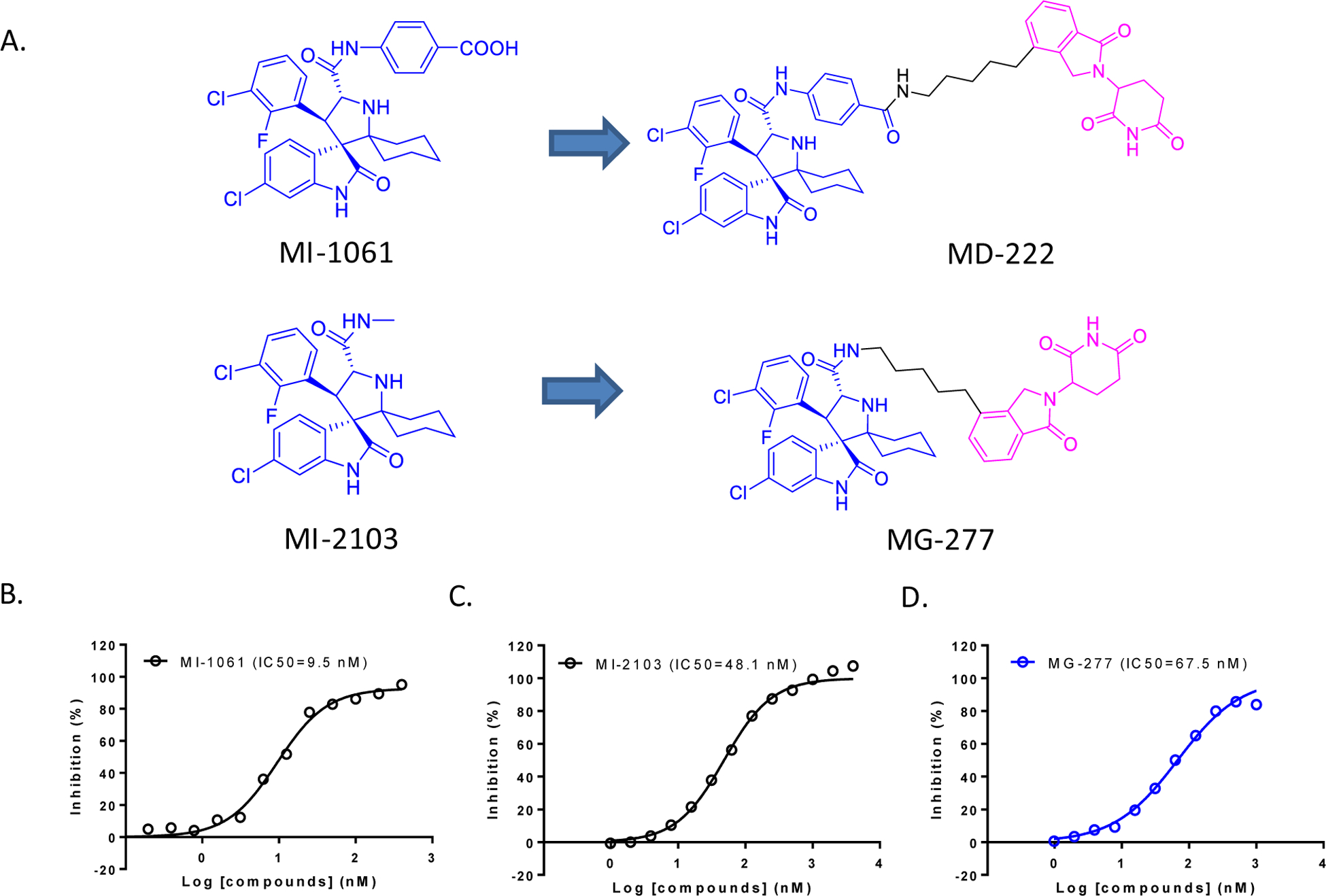
(A). Structures of MDM2 inhibitors MI-1061 and MI-2103, the bona fide MDM2 degrader MD-222 and a putative MDM2 degrader MG-277. (B-D).Binding affinities of MI-1061 (B), MI-2103 (C) and MG-277 (D) to MDM2 by an optimized fluorescence-polarization (FP) competitive binding assay.
We further investigated how the binding affinity of the MDM2 inhibitor component to MDM2 protein in our MDM2 degraders affects their ability to degrade the MDM2 protein. To this end, we replaced the benzoic acid fragment in MI-1061 with a methyl group, obtaining MI-2103 (Figure 1A). In our optimized fluorescence-polarization (FP) based binding assay30, the MDM2 inhibitors MI-1061 and MI-2103, bind to MDM2 with IC50 values of 9.5 nM and 48.1 nM, respectively (Figure 1B–D). While MI-2103 is ~5-times less potent than MI-1061, it is still a high-affinity inhibitor of MDM2. Employing MI-2103 for the MDM2 inhibitor moiety and using the same linker and lenalidomide moiety as those in MD-222, we synthesized MG-277 as a putative PROTAC MDM2 degrader.
MG-277 displays high potencies in inhibition of cell growth in cancer cell lines with different p53 status, in contrast to the bona fide PROTAC MDM2 degrader MD-222
In our previous study14, we showed that acute leukemia cell lines RS4;11, MOLM-13 and MV4;11 carrying wild-type p53, are very responsive to MDM2 inhibitors and degraders. We therefore employed these three leukemia cell lines to evaluate the bona fide MDM2 degrader MD-222, the new putative MDM2 degrader MG-277, and the MDM2 inhibitors MI-1061 and MI-2103.
MD-222 and MG-277 potently and effectively inhibit cell growth in each of these three leukemia cell lines and are much more potent than their corresponding MDM2 inhibitors (Figure S1A–S1C in SI). For example, MG-277 displays IC50 values of 1.3 nM, 24.6 nM and 7.9 nM, respectively, in these three cell lines (Figure S1A–S1C). In comparison, the corresponding MDM2 inhibitor MI-2103 has IC50 values of 669 nM, 1238 nM, 898 nM, respectively, in the same three cell lines (Figure S1A–S1C). These data show that the new, putative MDM2 degrader MG-277 is >50-times more potent than its corresponding MDM2 inhibitor, MI-2103, in inhibition of cell growth in each of these three cell lines.
Our previous study14 showed that similar to MDM2 inhibitors, the PROTAC MDM2 degraders MD-222 and MD-224 also display a high cellular specificity over cancer cell lines carrying mutated or deleted p53. We tested MG-277, MD-222 and MI-2103 for their specificity in the RS4;11/IRMI-2 leukemia cell line31, 32, which contains p53 hot-spot mutations (Y236H and R249G), and in the HL-60 leukemia cell line which has a loss of p53. As expected, the bona fide MDM2 degrader MD-222 and the MDM2 inhibitor MI-2103 are completely ineffective in the RS4;11/IRMI-2 and HL-60 leukemia lines (Figure S1D and S1E). Surprisingly, MG-277 is very potent and effective in inhibition of cell growth in both the RS4;11/IRMI-2 and HL-60 leukemia lines, achieving IC50 values of 3.9 nM and 8.3 nM, respectively (Figure S1D and S1E).
To further investigate their cellular specificity, we evaluated MG-277, MD-222 and MI-2103 for their cell growth inhibitory activity in the MDA-MB-231 and MDA-MB-468 breast cancer cell lines, which harbor a mutated p53 protein.33 While MD-222 and MI-2103 fail to inhibit cell growth in these two breast cancer cell lines, MG-277 potently inhibits cell growth in MDA-MB-231 and MDA-MB-468 breast cancer cell lines, with IC50 values of 39.4 nM and 26.4 nM, respectively (Figures S1F and S1G).
These cell growth inhibition data showed that while the bona fide PROTAC MDM2 degrader, MD-222 displays high cellular specificity for p53 wild-type cancer cell lines over p53 mutated or deleted cell lines, MG-277 is potent and effective against cancer cell lines carrying wild-type, mutated or deleted p53. Therefore, it is very clear that MG-277 and MD-222 have different cellular mechanisms of action.
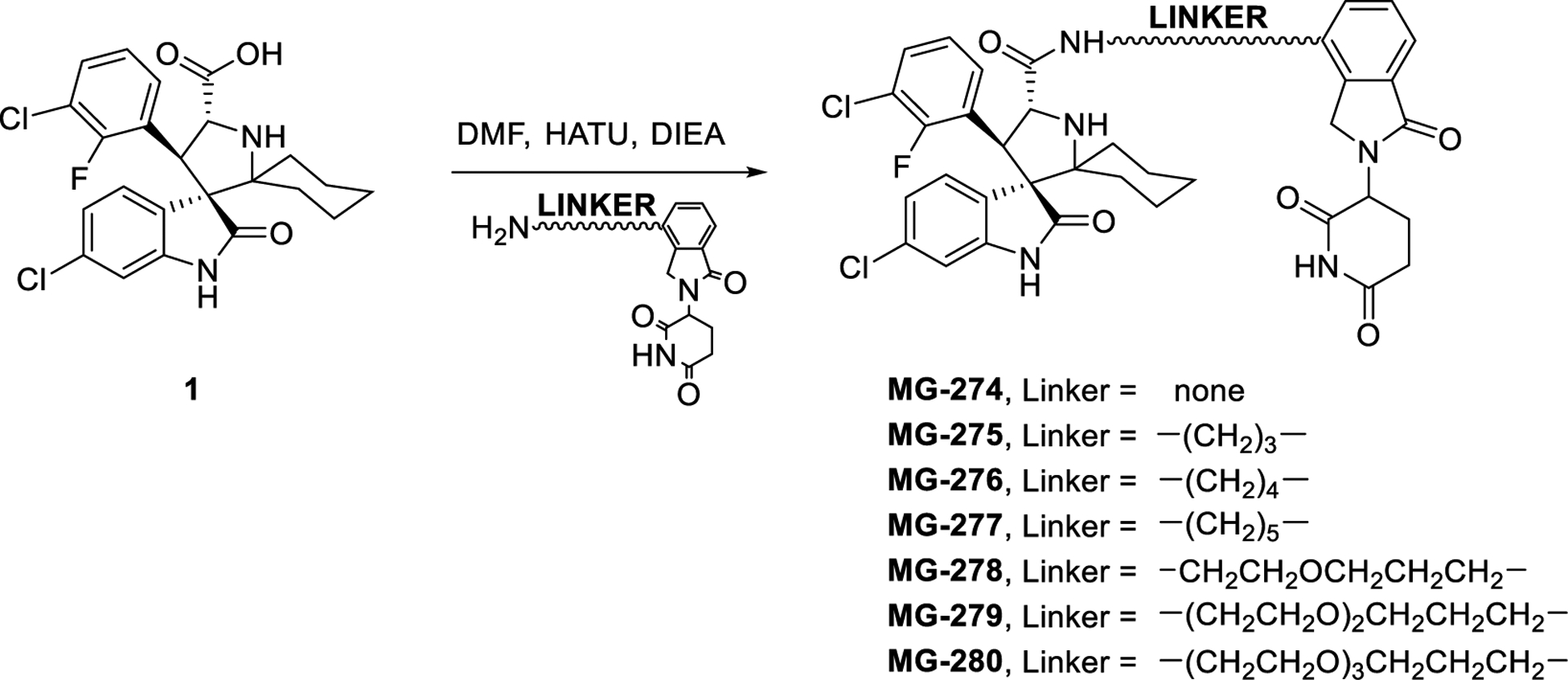
Synthesis and evaluation of MG-277 analogues with different linkers
The linker between the MDM2 inhibitor moiety and the lenalidomide moiety in MG-277 is much shorter than that in MD-222, and this may contribute to their different cellular mechanisms of action. To investigate this, we synthesized a series of analogues of MG-277 with different linker lengths and/or compositions (Table 1). We evaluated these compounds for their cell growth inhibitory activity in the RS4;11 cells with wild-type p53 and RS4;11/IRMI-2 cells carrying a mutated p53, obtaining the data summarized in Table 1 (Table 1, Figure S2A and S2B).
Table 1.
Linker exploration between MI-2103 and lenalidomide moiety and the corresponding IC50 in the RS4;11 cell line and the p53 mutant cell line RS4;11/IRMI-2.
 | |||
|---|---|---|---|
| Compound No. | Linker | IC50(nM) in cell growth inhibition | |
| RS4;11 | RS4;ll/IRMI-2 | ||
| MG-274 | No linker | 760±96 | 25801450 |
| MG-275 | 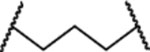 |
130±39 | 180145 |
| MG-276 | 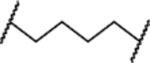 |
2.5±0.9 | 2.6+0.5 |
| MG-277 | 3.5±1.3 | 3.4±0.7 | |
| MG-278 | 3.6±0.9 | 3.5±0.9 | |
| MG-279 | 4.9±1.2 | 4.9±0.8 | |
| MG-280 | 14±3.7 | 1412.6 | |
MG-274, in which MI-2103 and lenalidomide are connected directly without a linker, display similar potencies in both cell lines, and MG-274 is slightly more potent than MI-2103 (Figure S2A and S2B). MG-275 has a (CH2)3 linker, two methylenes less than the linker in MG-277, and is approximately 10-times more potent than MG-274 in both cell lines and displays essentially no selectivity between the RS4;11 cell line and the RS4;11/IRMI-2 cell line (Figure S2A and S2B). MG-276, which has a linker one methylene shorter than that in MG-277, is equipotent with MG-277 in both cell lines and shows no selectivity (Figure S2A and S2B). Insertion of one oxygen atom into the linker in MG-277 yielded MG-278, which is also equipotent with MG-277 in both cell lines and displays no cellular selectivity (Figure S2A and S2B). Replacement of the (CH2)5 linker in MG-277 with the longer and more hydrophilic linker – (CH2)2O-CH2O-(CH2)3-, afforded MG-279, which is similarly potent and non-selective as MG-277 (Figure S2A and S2B). Changing the (CH2)5 linker in MG-277 to a (CH2O)3(CH2)3 linker yielded MG-280, which displays IC50 values of 10 nM and 11 nM in the RS4;11 cell line carrying wild-type p53 and the RS4;11/IRMI-2 cell line (Figure S2A and S2B), respectively. MG-280 is therefore 5-times less potent than MG-277, and displays no selectivity between these two cell lines (Figure S2A and S2B). Taken together, these data show that while the linker length and composition have clear influence on the cellular potencies, the lack of cellular selectivity for MG-277 between cell lines carrying wild-type p53 and cell lines containing a mutated or deleted p53 is not simply due to its shorter linker than that in MD-222.
MG-277 is much less effective and less potent than MD-222 in inducing MDM2 degradation and fails to activate wild-type p53
A bona fide PROTAC MDM2 degrader such as MD-222 is potent and effective in inducing MDM2 degradation and in activating wild-type p53 in cancer cells.14 We evaluated MG-277 for its ability to induce MDM2 degradation and activation of p53 in the RS4;11 cell line, with MD-222 and MI-2103 used as controls.
Western blotting analysis showed that MD-222 is very effective in reducing the levels of MDM2 protein and increasing the levels of p53 after treatment for just 1 h in RS4;11 cells (Figure 2A). This is consistent with our previous data14 and its mechanism as an MDM2 degrader. In comparison, with a treatment time of 1 h MI-2103 induces upregulation of both MDM2 and p53 proteins (Figure 2A), consistent with its mechanism as an MDM2 inhibitor. In contrast, MG-277 fails to reduce the levels of MDM2 protein and increase the levels of p53 with 1 h treatment time in RS4;11 cells (Figure 2A). Treatment of the RS4;11 cells with MD-222 for 2 h shows that it reduces the level of MDM2 and increases the level of p53 more than was observed with a treatment time of 1 h (Figure 2B). Interestingly, with a 2 h treatment time, MG-277 reduces the MDM2 level moderately but fails to increase the level of p53 in the RS4;11 cells (Figure 2B).
Figure 2. MG-277 is much less effective and less potent than MD-222 in inducing MDM2 degradation and fails to activate wild-type p53.
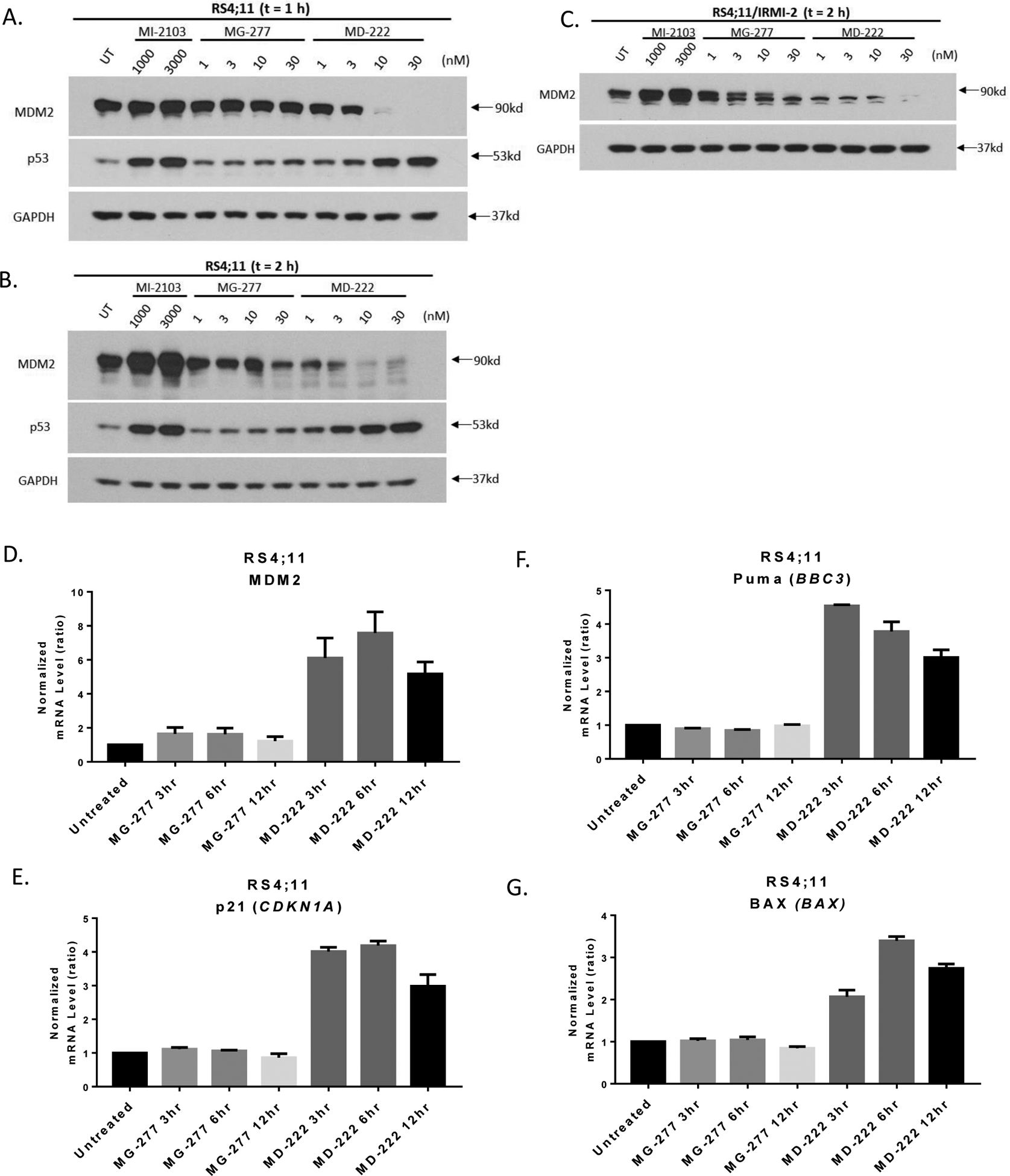
(A-B) Western blotting analysis of MDM2 and p53 protein levels in RS4;11 cells after treatment with MI-2103, MG-277 and MD-222 for 1 h (A) or 2 h (B), and in p53 mutant RS4;11/IRMI-2 cell line after 2 h treatment (C). The cells were treated for 1 h or 2 h with the compounds at the indicated concentrations, and proteins were probed by specific antibodies. (D-G) Changes of mRNA levels of p53 downstream target genes, including MDM2, CDKN1A, PUMA and BAX were analyzed after treatment with 3 nM MG-277 or 10 nM MD-222 in the RS4;11 cell line by qRT-PCR.
In the RS4;11/IRMI-2 cell line carrying a mutated p53, both MD-222 and MG-277 can reduce the levels of MDM2 with 2 h treatment time but again MD-222 is much more potent and effective than MG-277 (Figure 2C).
Quantitative reverse transcription polymerase chain reaction (qRT-PCR) experiments show that MD-222 is effective in increasing the expression of several p53-targeted genes, such as MDM2, CDKN1A, PUMA and BAX (Figure 2D–2G) in RS4;11 cells, indicating strong activation of p53. In contrast, MG-277 fails entirely to increase the expression of any of these p53-targeted genes (Figure 2D–2G), showing lack of p53 activation.
Hence, while MD-222 is very effective in reducing the levels of MDM2 and activating wild-type p53 in RS4;11 cells, MG-277 is much less potent and effective than MD-222 in reducing the levels of MDM2 and fails to activate wild-type p53.
The cell growth inhibition activity of MG-277 is MDM2-independent
MG-277 is much less potent and effective than MD-222 in reducing the levels of MDM2 in the RS4;11 cells but retains a moderate effect (Figure 2A and 2B). We examined if the cell growth inhibition activity of MG-277 is MDM2-dependent. Because it is difficult to knock-down MDM2 efficiently in leukemia cells, we knocked down MDM2 by siRNA in the MDA-MB-231 and MDA-MB-468 breast cancer cell lines and assessed the cell growth inhibitory activity of MG-277 in these MDM2 knocked-down cell lines.
Our data show that efficient knock-down of MDM2 by siRNA in these two cell lines has no effect on the cell growth inhibitory activity of MG-277 (Figure 3C and 3D), when compared to the siRNA controls (Figure 3A and 3B). These data also show that MG-277 achieves its cell growth inhibitory activity in both the MDA-MB-231 and MDA-MB-468 cell lines through an MDM2-independent mechanism.
Figure 3. The cell growth inhibition activity of MG-277 is MDM2-independent.
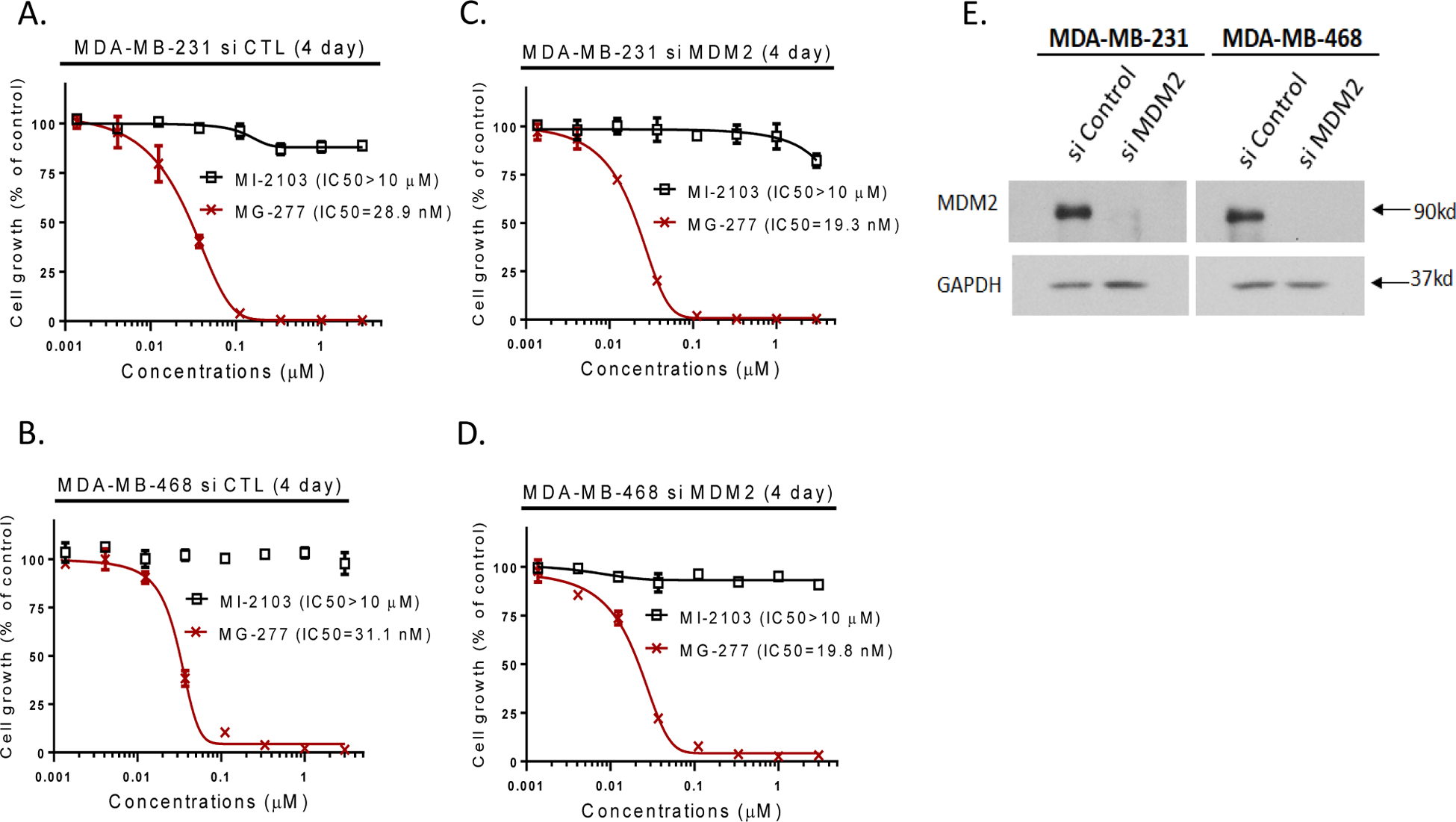
(A-D) Effect of knockdown of MDM2 on the cell growth inhibitory activity of MI-2103 and MG-277 in MDA-MB-231 and MDA-MB-468 cells after transfection with MDM2 siRNAs (C-D) or non-targeting siRNA control vector (A-B) for 2 days. (E) MDM2 knockdown efficiency was determined by Western blotting analysis.
The cell growth inhibition activity of MG-277 does not require binding to MDM2
MG-277 consists of an MDM2 binding moiety, and a cereblon binding moiety, tethered together through a linker. We investigated if the binding of MG-277 to MDM2 is required for its cellular growth inhibition activity by synthesizing and evaluating a number of analogues using an MDM2 inhibitor which is much weaker than MG-277.
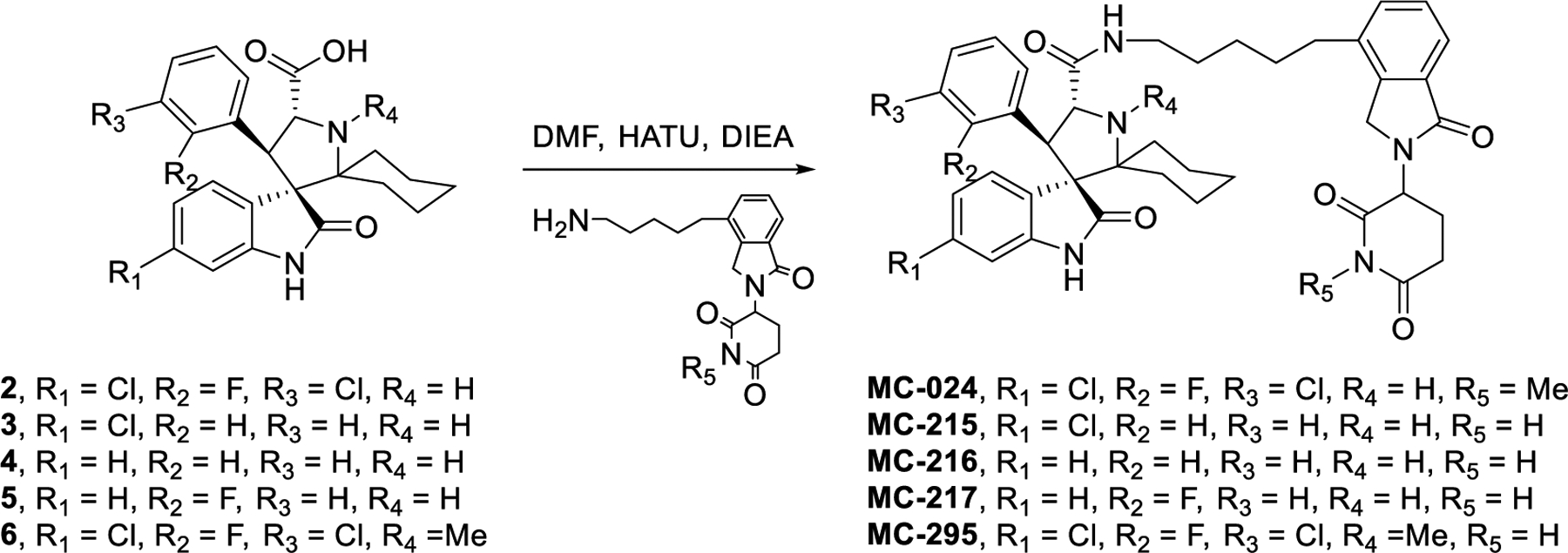
In our previous studies,34, 35 we showed that the halogen atoms in our MDM2 inhibitors play an important role in enhancing the binding affinity to MDM2. Therefore, we made three MDM2 inhibitors by removal of one or more of the Cl or F atoms. Using these three much weaker MDM2 inhibitors, we have synthesized three analogues of MG-277 using the same linker and the lenalidomide moiety as those in MG-277 (Figure 4A–4C). Our binding data showed that the conjugated compounds based on these weaker MDM2 inhibitors show minimal binding to MDM2 (IC50 > 20 μM) (Figure 4F).
Figure 4. The cell growth inhibition activity of MG-277 does not require binding to MDM2.
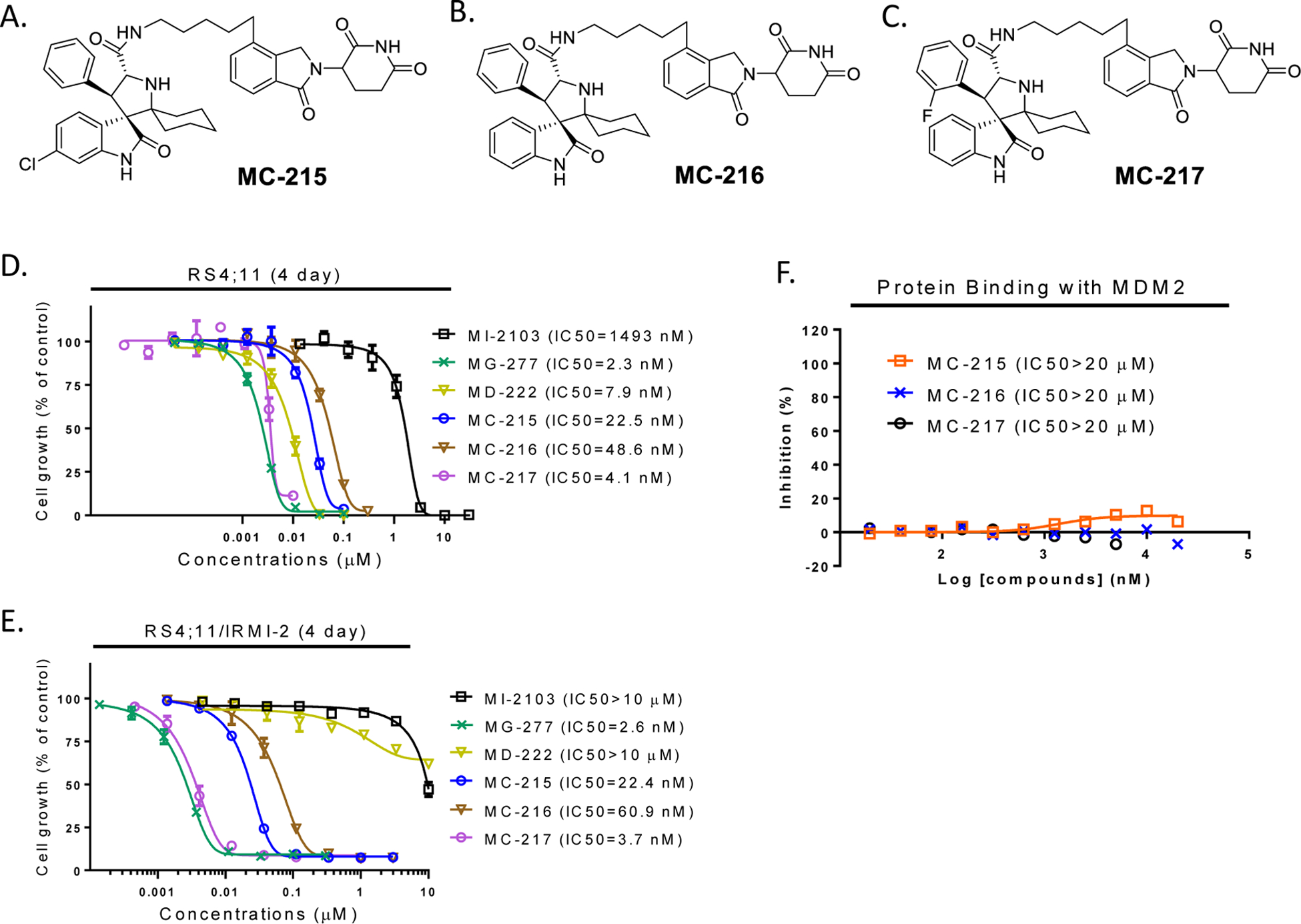
(A-C) Structures of MC-215, MC-216 and MC-217, three control analogues of MG-277 formed by removal of fluorine and/or one or two chlorine atoms from the MI-2103 portion of MG-277. (D-E) Cell growth inhibitory activity of MG-277, MC-215, MC-216 and MC-217 in RS4;11 (D) and RS4;11/IRMI-2 (E) cell lines. (F) Binding affinity of the three analogues of MG-277 with MDM2 protein was determined by an FP assay.
These three new analogues, MC-215, MC-216 and MC-217, however, all display potent cell growth inhibitory activity in both the RS4;11 and RS4;11/IRMI-2 cell lines and show no selectivity over the RS4;11/IRMI-2 cell line (Figure 4D and 4E). These data indicate that the cell growth inhibition activity by MC-215, MC-216, MC-217, and MG-277 in different cell lines is independent of their binding to MDM2.
The cell growth inhibitory activity of MG-277 is dependent on cereblon binding
Because MG-277 contains a cereblon-binding moiety, we investigated if its cell growth inhibitory activity is dependent upon its binding to cereblon using several complementary methods.
The co-crystal structure of lenalidomide in a complex with cereblon shows that the amino group of the glutarimide in lenalidomide forms a hydrogen bond with cereblon and methylation of this amino group should completely block the binding to cereblon.36, 37 We therefore synthesized MC-024 in which the amino group of the glutarimide in MG-277 is methylated (Figure 5I). MC-024 is >1000-times less potent than MG-277 in inhibition of cell growth in RS4;11, RS4;11/IRMI-2, MDA-MB-231 and MDA-MB-468 cell lines (Figure 5E–5H), indicating that binding of MG-277 to cereblon is required for its cell growth inhibitory activity in cell lines with wild-type and mutated p53.
Figure 5. The cellular activity of MG-277 is dependent on cereblon binding.
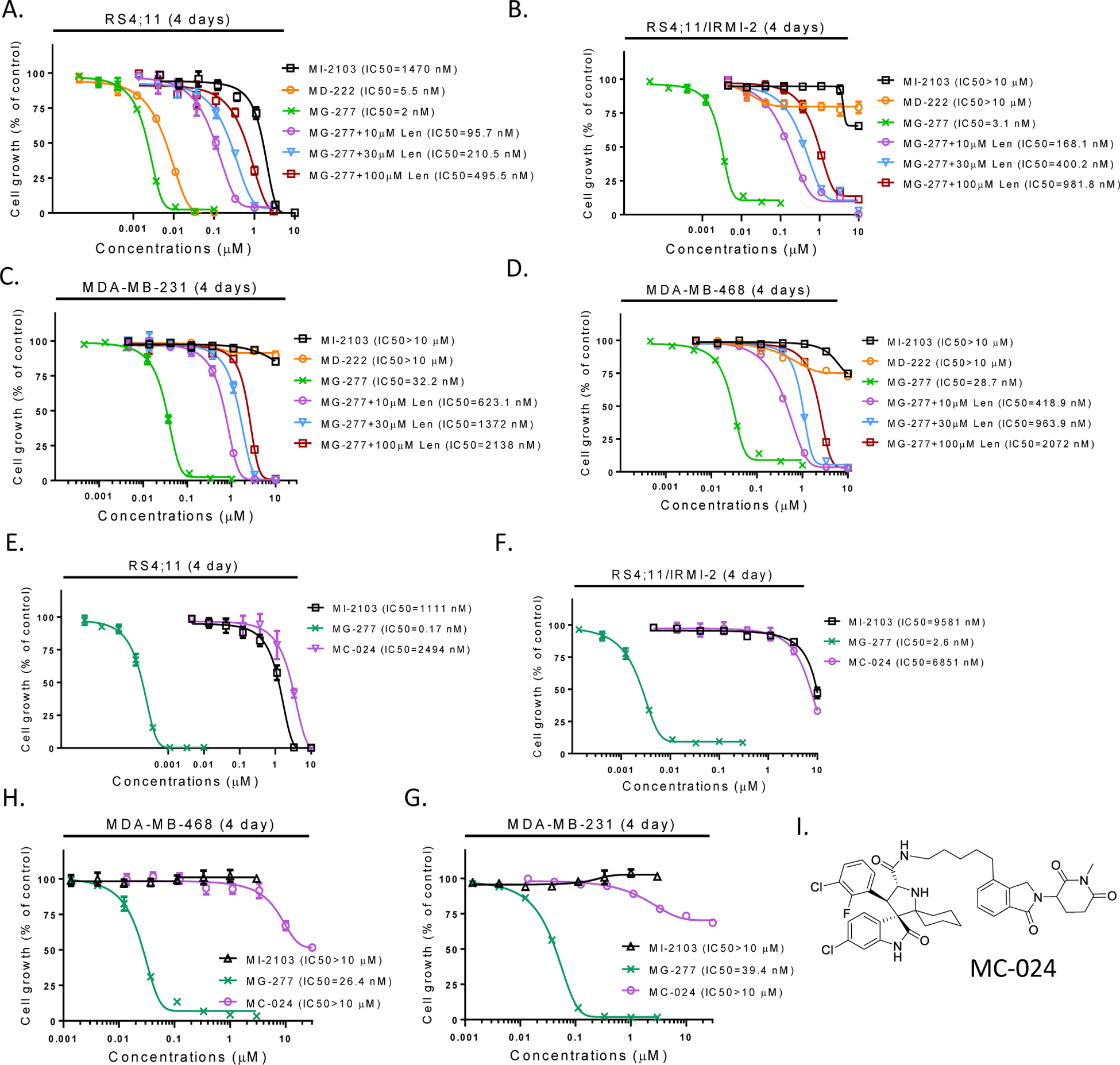
(A-D) Cell growth inhibitory activity of MI-2103, MD-222 and MG-277 in RS4;11, RS4;11/IRMI-2, MDA-MB-231 and MDA-MB-468 cells in the presence or absence of excess lenalidomide to occupy the binding site on cereblon. (E-H) Cell growth inhibitory activity of MI-2103, MG-277 and MC-024 with no cereblon binding in RS4;11, RS4;11/IRMI-2, MDA-MB-231 and MDA-MB-468 cell lines by a 4-day cell viability assay. (I) Structure of MC-024 with the amino group of the glutarimide in MG-277 methylated.
To further examine the effect of cereblon binding on cell growth inhibition by MG-277, we used an excess amount of lenalidomide to compete with MG-277 for binding with cereblon. Our data show that lenalidomide is very effective in reducing the cell growth inhibitory activity of MG-277 in a dose-dependent manner in p53 wild-type RS4;11 cells and p53 mutant RS4;11/IRMI-2, MDA-MB-231 and MDA-MB-468 cells (Figure 5A–5D).
We knocked down CRBN in the MDA-MB-231 and MDA-MB-468 cell lines to examine the effect on cell growth inhibition by MG-277, and found that efficient knock-down of CRBN dramatically reduces the cell growth inhibition by MG-277 in both cell lines (Figure 6).
Figure 6. The cell growth inhibition activity of MG-277 requires binding to cereblon.
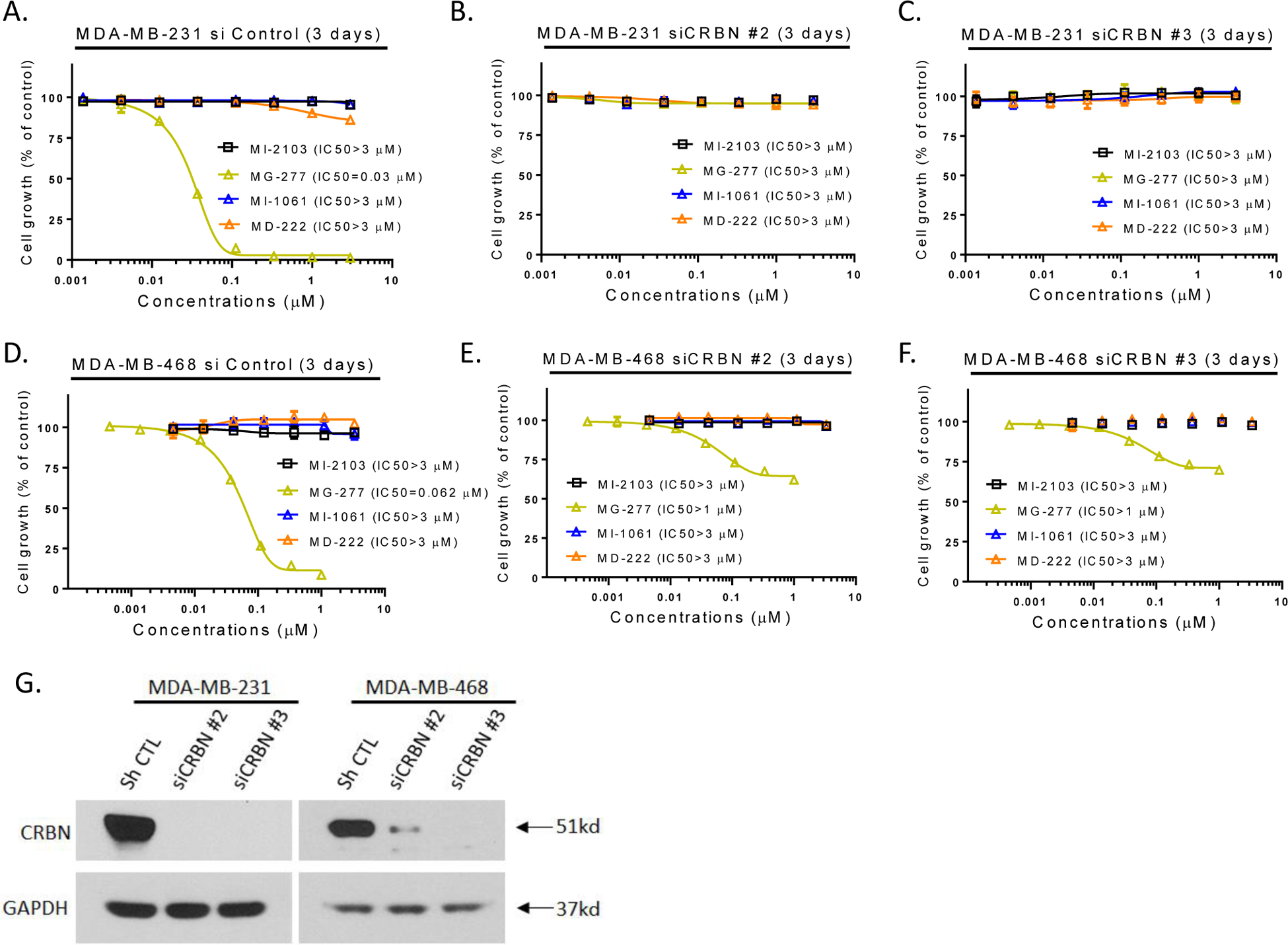
Effect of siRNA CRBN knockdown on the cell growth inhibitory activity of MG-277. (A-F) MDA-MB-231 (A-C) and MDA-MB-468 (D-F) cells were transfected with two different CRBN siRNAs targeting two different sequences (B-C, E-F) or non-targeting siRNA control vector (A, D) before treatment with MI-2103 or MG-277 for 3 days. Cell growth inhibition was determined by cell viability assay. (G) Western blotting for cereblon protein level was used to determine the knockdown efficiency.
These three sets of complementary experiments clearly demonstrate that binding of MG-277 to cereblon is required for the cell growth inhibitory activity of MG-277 in cancer cell lines with wild-type or mutated p53.
MG-277 effectively and specifically induces degradation of GSPT1
Because the cell growth inhibitory activity of MG-277 is MDM2-independent and cereblon-dependent, we hypothesized that MG-277 may in fact function as a molecular glue and promote recruitment of certain cell growth-related protein(s) to the CUL4-DDB1-CRBN E3 ligase complex (CRL4CRBN) for ubiquitination and subsequent proteasomal degradation.
To identify the actual cellular target(s) of MG-277, we performed an unbiased proteomic analysis using the RS4;11 cells with wild-type p53 and the RS4;11/IRMI-2 cells carrying mutated p53. This analysis showed that upon treatment with 0.1 μM of MG-277 for 3 h, the G1 to S phase transition 1 (GSPT1) protein, a translation termination factor, is the only protein whose level is greatly reduced in both RS4;11 (5.3-fold, P<0.001) and RS4;11/IRMI-2 (3.7-fold, P<0.001) cell lines (Figure 7A and 7B).
Figure 7. MG-277 effectively and specifically induces the degradation of GSPT1.
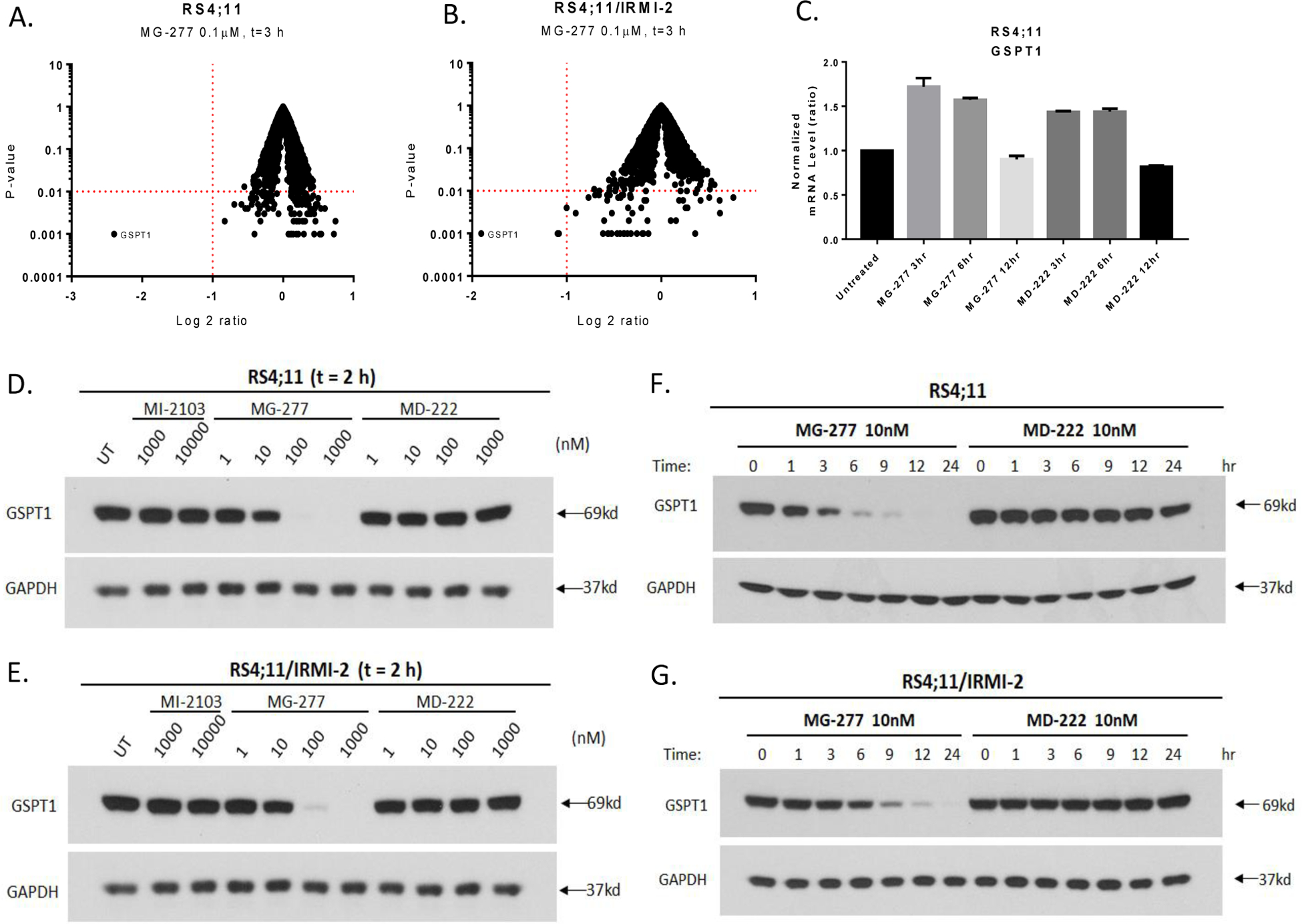
(A-B) Proteomic analysis/protein profiling after a 3 h treatment with 0.1 μM of MG-277 in p53 wild-type RS4;11 (A) and p53 mutated RS4;11/IRMI-2 (B) cells. The Y-axis represents P-value and X-axis represents the log 2 ratio of the level of a certain protein after the treatment to its protein level before the treatment. (C) GSPT1 mRNA level was measured by qRT-PCR after treatment with MG-277 and MD-222 at indicated time points. (D-G) Western blotting analysis of GSPT1 protein level after treatment with MI-2103, MG-277 and MD-222 for 2 h (D-E) or treatment with MG-277 or MD-222 for different time lengths as indicated (F-G) in RS4;11 and RS4;11/IRMI-2 cells.
GSPT1, also known as eRF3a, mediates stop codon recognition and nascent protein release from the ribosome through interaction with a release factor, eRF1.38, 39 A recent study reported the similar finding that two series of phthalimide conjugate degraders originally designed to target multiple kinases for degradation actually induce significant GSPT1 degradation and fail to degrade their consensus targets.29
GSPT1 protein level is greatly reduced by treatment with MG-277 and its analogues but not by treatment with MD-222, a bona fide PROTAC MDM2 degrader
The data in Figure 7A–B suggest that GSPT1 may the true molecular target for MG-277. Accordingly, we investigated if degradation of GSPT1 protein is required for the cell growth inhibitory activity of MG-277.
Western blot analysis showed that MG-277, but not MD-222, effectively reduces the level of the GSPT1 protein in a dose- and time-dependent manner in both p53 wild-type and mutant cell lines (Figure 7D–7G). The kinetics data showed that GSPT1 protein level is markedly decreased starting from 1–3 h with MG-277 at a concentration of as low as 10 nM (Figure 7F and 7G), indicating rapid degradation of GSPT1 induced by MG-277. No reduction of GSPT1 mRNA level is observed after treatment with MG-277, indicating that MG-277 has no effect on GSPT1 transcription (Figure 7C).
We further evaluated additional analogues of MG-277 containing linkers of different lengths and composition (Table 1). All of these analogues, with the exception of MG-274 and MG-275, effectively reduce the GSPT1 protein level after 2 h of treatment (Figure S2C and S2D). MG-274 with no linker and MG-275 with a (CH2)3 linker are also both capable of reducing the level of GSPT1 but require higher concentrations (1–3 μM) and 24 h treatment time (Figure S2E and S2F), consistent with their weaker potencies in cell growth inhibition. MG-276, MG-277, MG-278 and MG-279 achieve DC50 (concentration required to degrade 50% of protein) values of 1.1 nM, 1.3 nM, 1.8 nM and 1.2 nM, respectively, in the RS4;11 cell line with 24 h treatment. MG-276, MG-277, MG-278 and MG-279 are also highly potent in inducing degradation of GSPT1 (Figures S3B–S3D and S3F, Table S1). MG-274 (DC50= 2.3 μM) with no linker and MG-275 (DC50= 0.12 μM) with the shortest linker are much less potent than MG-277 in inducing GSPT1 degradation (Figure S3A and S3F, Table S1). The MDM2 inhibitor MI-2103 has no effect on the level of GSPT1 protein (Figure S2C and S2D).
These data establish an excellent correlation between the induced degradation of GSPT1 and the cell growth inhibition by MG-277 and its analogues in both p53 wild-type and p53-mutated cell lines.
Decrease of GSPT1 protein level induced by MG-277 is dependent on its binding with cereblon, proteasome and cullin-RING E3 ligases (CRLs) function but independent of MDM2
Our data show that cell growth inhibition by MG-277 depends upon cereblon but not on MDM2. We next investigated the dependence of GSPT1 degradation induced by MG-277 on cereblon, proteasome, CUL E3 ligase and MDM2.
Co-treatment with an excess amount of lenalidomide (30 μM) effectively blocks the degradation of GSPT1 protein induced by MG-277 in both MDA-MB-231 and MDA-MB-468 cell lines (Figure 8A and 8B). Efficient siRNA knock-down of CRBN in these two breast cancer cell lines also blocks the degradation of GSPT1 induced by MG-277 (Figure 8C and 8D). These data establish that degradation of GSPT1 protein induced by MG-277 depends upon cereblon.
Figure 8. Decrease of GSPT1 protein level induced by MG-277 is dependent on its binding with cereblon, proteasome and cullin-RING E3 ligases (CRLs) but independent of MDM2.
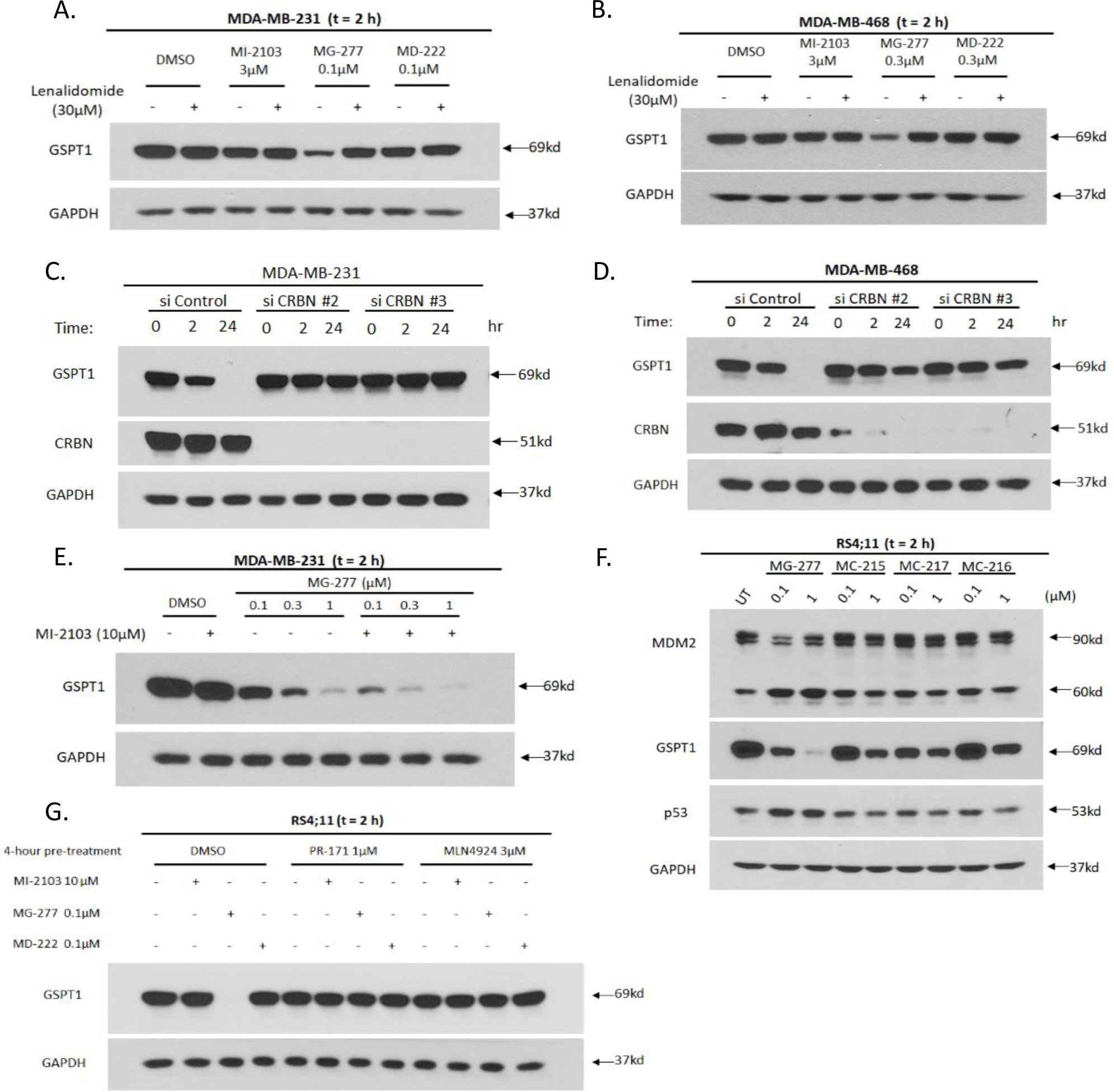
(A-B) Western blotting for GSPT1 protein level after 2 h treatment with MI-2103, MG-277 or MD-222 in MDA-MB-231 (A) and MDA-MB-468 (B) cells with or without lenalidomide (30 μM) to block binding with cereblon. (C-D) MDA-MB-231 (C) and MDA-MB-468 (D) cells were transfected with CRBN siRNAs targeting two different sequences or non-targeting siRNA control. Protein levels of GSPT1 and cereblon were determined by Western blot analysis after treatment with 0.1 μM MG-277 for 2 h or 24 h in the transfected cells. (E) Western blot analysis for GSPT1 protein level after 2 h treatment with MG-277 in the presence or absence of 10 μM MI-2103 (or DMSO as control) to block binding with MDM2 in MDA-MB-231 cells. (F) Western blotting for MDM2 and GSPT1 protein levels after 2 h treatment with MG-277 and three analogues (MC-215, MC-216 and MC-217) with weaker MDM2 binding affinity in RS4;11 cells. (G) RS4;11 cells were pre-incubated with a proteasome inhibitor, PR-171 or a Nedd8 inhibitor, MLN4924, or DMSO as control for 4 h to block proteasome or CRL function. GSPT1 protein level was measured by Western blotting after 2 h treatment with MI-2103, MG-277 or MD-222.
We investigated if degradation of GSPT1 protein induced by MG-277 requires binding to MDM2. An excess of MI-2103 was found to fail to negate degradation of GSPT1 protein by MG-277 in the MDA-MB-231 cell line (Figure 8E). We evaluated the ability of several analogues of MG-277 (MC-215, MC-216 and MC-217) (Figure 4A–4C), each of which contains a very weak MDM2 inhibitor, to reduce the level of GSPT1 protein in the RS4;11 cells. These compounds are all very potent and effective in reducing the GSPT1 protein level but have no obvious effect on the level of MDM2 protein (Figure S3D–S3F and 8F). Hence, the binding to MDM2 of MG-277 and its analogues is not required for their effective degradation of GSPT1 protein.
We investigated whether proteasomal activity and cullin-RING E3 ligases (CRLs) activity are essential for MG-277 induced GSPT1 degradation. Both PR-171, a proteasome inhibitor, and MLN4924, a Nedd8 conjugating enzyme (NAE) inhibitor, completely block the degradation of GSPT1 induced by MG-277 (Figure 8G). Hence the GSPT1 degradation induced by MG-277 requires the activity of both the CUL4 E3 ubiquitin ligase and the proteasome.
Structural requirements within the PROTAC molecules for the GSPT1 recruitment to cereblon
To shed light on the structural requirements for MG-277 to induce GSPT1 degradation, we modeled the binding of MG-277 with the cereblon:GSPT1 complex based upon the co-crystal structure of CC-885 in a complex with cereblon and GSPT1 (Figure 9)40. Our modeling showed that MG-277 captures some of the key interactions with residues uniquely critical for recruiting GSPT1 to cereblon that had been shown in the previous study.40 Specifically, Phe150 in cereblon is hydrophobically packed with GPST1 and the cyclohexyl group of MG-277. Glu377 of cereblon enjoys hydrogen bonding and electrostatic interactions with residues in GSPT1 and the positively charged nitrogen atom in the pyrrolidine group of MG-277. Additional interactions among the ligand and the two proteins are also observed (see Figure S4 for more details).
Figure 9.
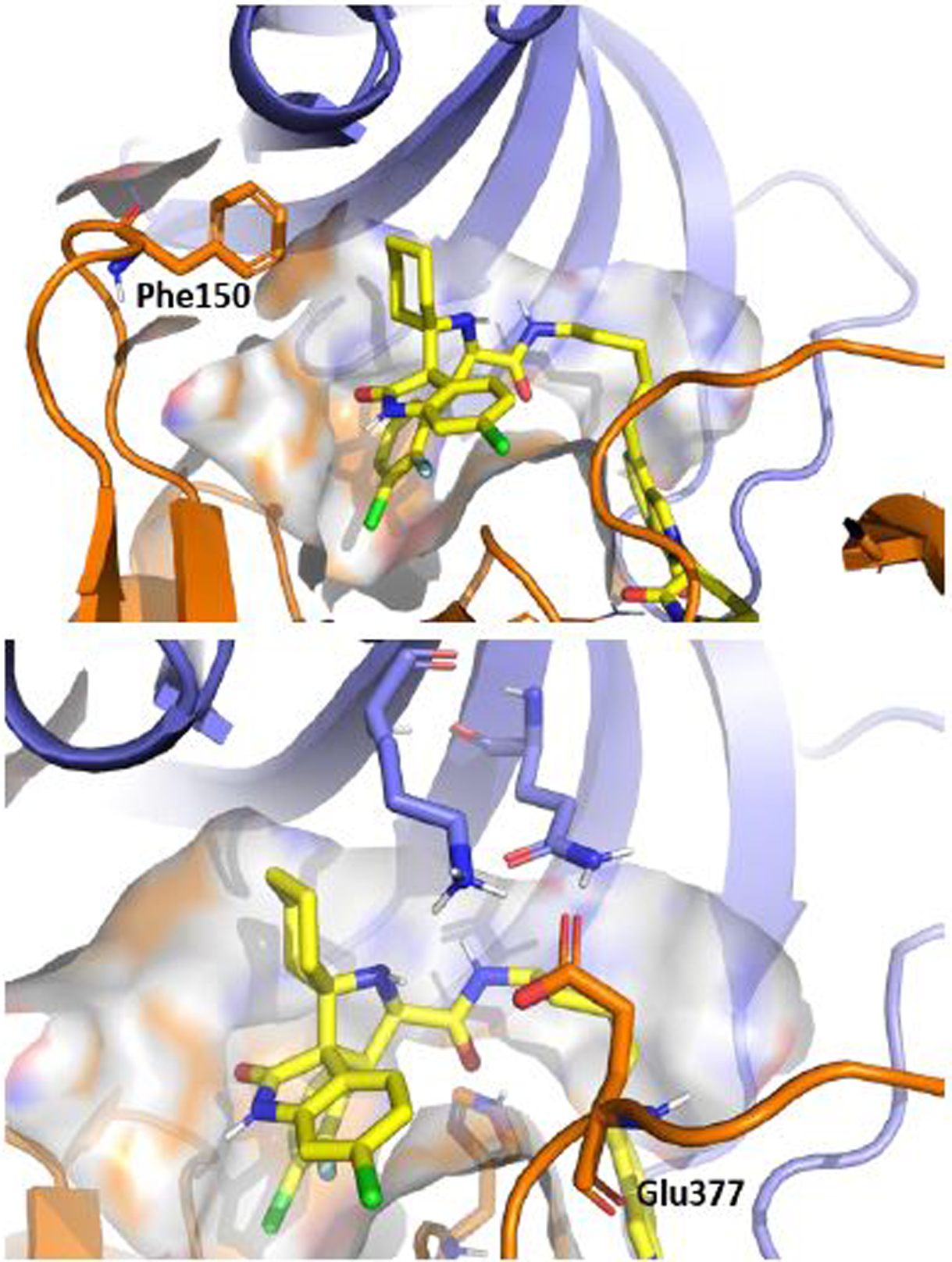
Molecular modeling of MG-277 (yellow) binding with GSPT1 (blue) and cereblon (orange) with highlighted residues, Phe150 and Glu377, which are uniquely critical for recruitment of GSPT1. The available structure of GSPT1-cereblon complex co-crystalized with small ligand CC-88540 (PDB code: 5HXB) was used.
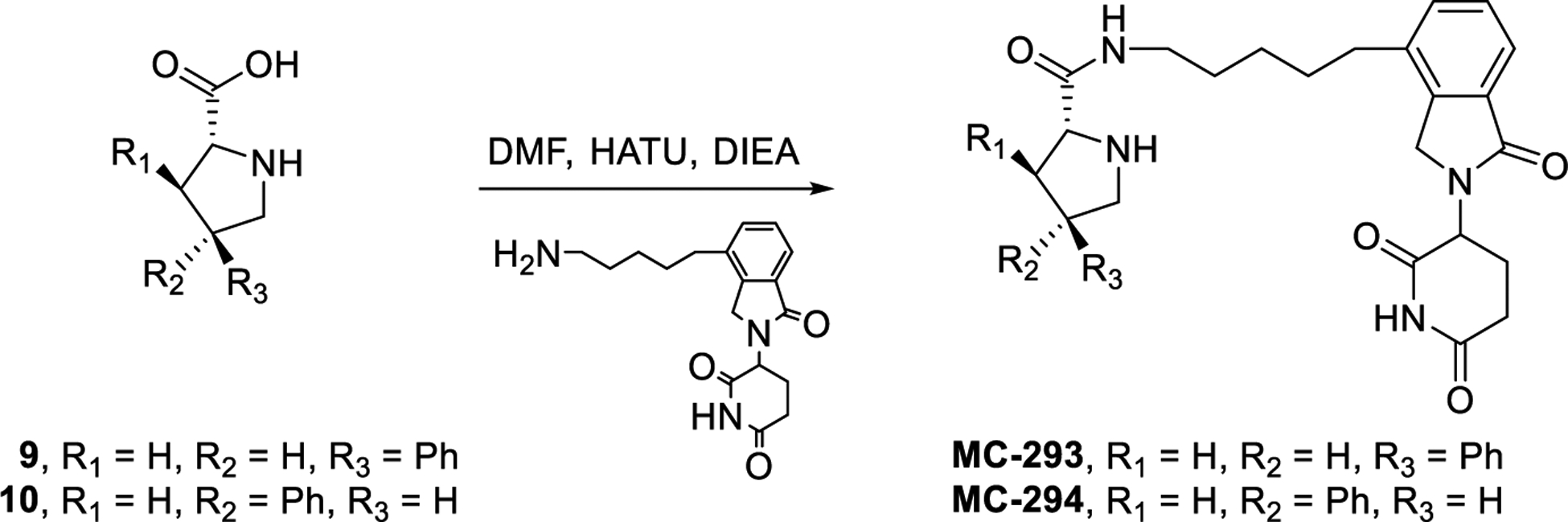
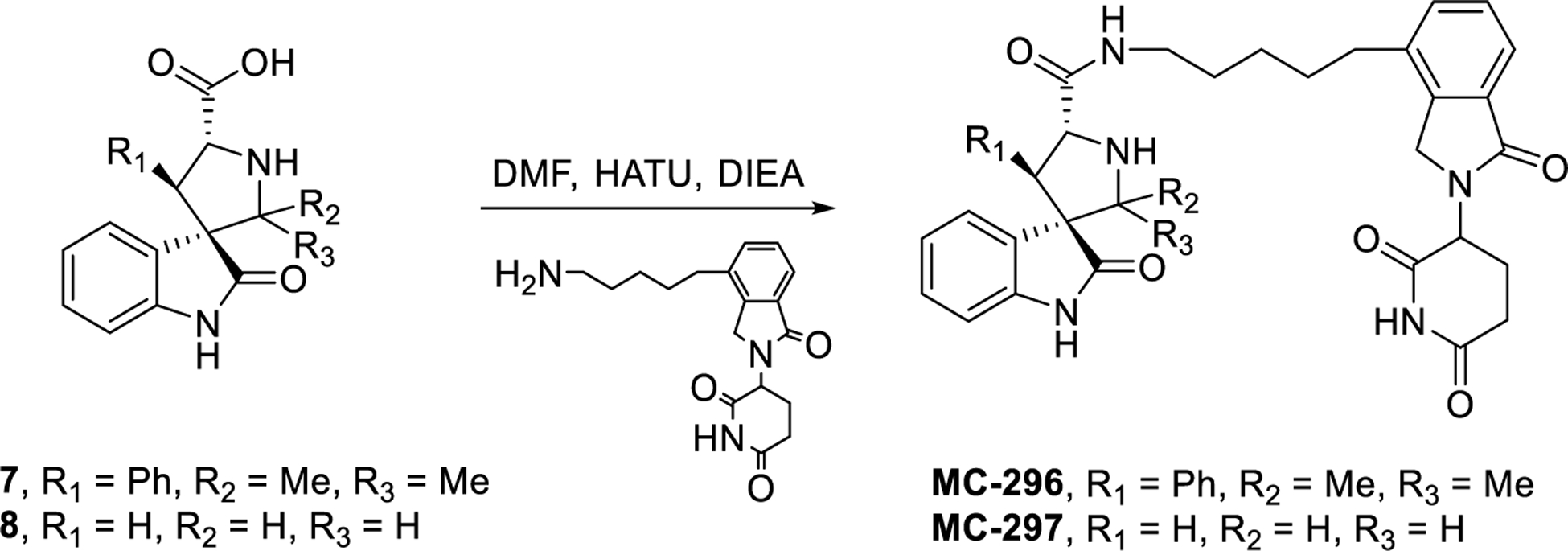
We further evaluated the structural requirements for the potent cell activity and GSPT-1 degradation by MG-277. The reported DDB1-GSPT1-CC-885 complex shows hydrogen bonding interaction between the two urea nitrogens of CC-885 and cereblon.40 We postulated that it may be possible that the adjacent carboxamide and pyrrolidine nitrogens in MG-277 mimic this interaction. If this hypothesis is correct, blocking one of the nitrogens would abolish the activity. Blocking the nitrogen of the pyrrolidine produced MC-295 which retains the cellular activity, suggesting that this position plays a minor role in the GSPT-1 degradation (Figure 10). Next, the substituents around the pyrrolidine core were systematically removed. Removal of the cyclohexyl ring on carbon-2, resulted in MC-296, which the same activity as MG-277 (Figure 10). However, modification of the 4-aryl substituent produced compounds MC-297, MC-293 and MC-294, all having weak cell potency suggesting both aryl groups are necessary for GSPT-1 degradation and consequently potent cell growth-inhibitory activity (Figure 10). Further studies will be performed to precisely define the minimal structural requirements for induced GSPT1 protein degradation.
Figure 10. Chemical structural requirements for the GSPT1 recruitment to cereblon.
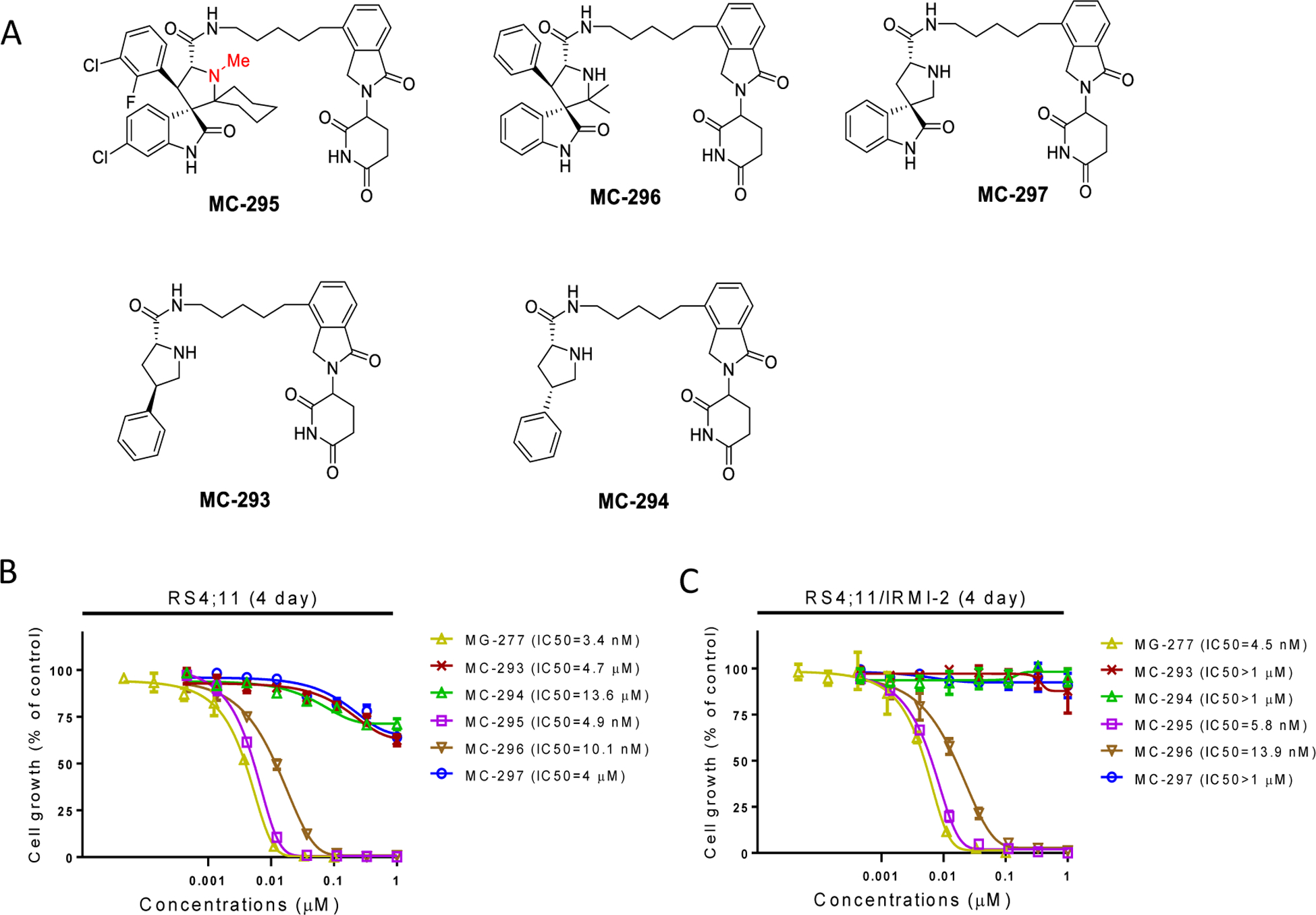
(A) Chemical structures of the new compounds after modifications of the inhibitor portion to investigate the structural requirement of GSPT1 recruitment. (B-C) Potencies of the compounds in cell growth inhibition were evaluated in both p53 wild-type RS4;11 cell line (B) and the p53 mutant RS4;11/IRMI-2 cell line (C) by a 4-day cell viability assay.
DISCUSSION
We previously reported the discovery of a class of bona fide PROTAC MDM2 degraders, exemplified by MD-222 and MD-224, that are highly potent and effective in inducing degradation of MDM2 and in activating wild-type p53 in cells.14 In the present study, we report the discovery of another type of compounds, exemplified by MG-277 which was obtained by removal of the benzoic acid moiety of the MDM2 inhibitor segment of MD-222. In contrast to MD-222 and MD-224, MG-277 has a significantly decreased potency in reducing the level of MDM2 protein in cells and fails to activate wild-type p53 (Figure 2A–2C). However, MG-277 is highly potent and effective in inhibition of cell growth in cancer cell lines with wild-type p53, mutated p53, or deleted p53, indicating a p53-independent mechanism. We found that that MG-277 and a number of its analogues achieve cell growth inhibition in a cereblon-dependent and MDM2-independent manner. Our unbiased proteomic analysis revealed that GSPT1 is the only protein whose level is dramatically and significantly reduced by MG-277. Furthermore, MG-277 induces rapid GSPT1 degradation in cancer cells in a p53- and MDM2-independent manner but in a manner dependent upon cereblon, CUL4 E3 ubiquitin ligase and proteasomes. Moreover, the potencies of different analogues of MG-277 in induction of GSPT1 degradation by correlate well with their cell growth inhibitory activity (Figure S3, Table S1). Our data therefore strongly support our proposal that induction of GSPT1 degradation by MG-277 and its analogues is responsible for their cell growth inhibitory activity.
A recent study by Ishoey et al.29 has demonstrated that while phthalimide conjugate degraders fail to induce degradation of their consensus kinase targets in cells, these compounds effectively induce degradation of GSPT1 protein. Their study also provided convincing evidence that degradation of GSPT1 is responsible for the cell growth inhibitory activity of these phthalimide conjugate degraders in leukemia cell lines.29 Our present study provides an example that while some of our designed putative MDM2 degraders such as MD-222 and MD-224 indeed function as bona fide PROTAC MDM2 degraders14, structural modifications of the MDM2 inhibitor portion of MD-222 yielded compounds such MG-277 which has a cellular mode of action completely different from that of MD-222. Despite the fact that MG-277 contains an MDM2 inhibitor portion, which binds to MDM2 with a high affinity (IC50 = 48.1 nM) (Figure 1C), it is much less potent and effective in inducing degradation of MDM2 than MD-222 and fails to activate p53. Instead, MG-277 achieves its remarkable cell growth inhibitory activity by inducing degradation of the GSPT1 protein.
Our data suggest that for any designed phthalimide conjugate degrader, there are two potential mechanisms of action. On the one hand, the phthalimide conjugate can function as a bona fide degrader to degrade the intended protein(s) of interest. On the other hand, the conjugate can function as a molecular glue to recruit neo-substrate protein(s) to cereblon and Cullin 4A for ubiquitination and subsequent degradation. Although further studies are needed to determine the precise structural requirements for a conjugate to operate either as a bona fide PROTAC degrader or as a molecular glue, both cellular mechanisms of action must be considered in the design of phthalimide conjugate degraders.
CONCLUSIONS
By modification of our previously discovered bona fide PROTAC MDM2 degraders, we discovered a new type of compounds with a completely different mechanism of action. This new type of compounds, exemplified by MG-277, exerts potent anticancer activity by functioning as molecular glues. In contrast to our previously reported bona fide PROTAC MDM2 degraders, MG-277 shows very weak activity in inducing degradation of MDM2 and fails to activate p53 but is potent and effective in inducing degradation of GSPT1. Our study shows that for a designed phthalimide conjugate degrader, it can function either as a bona fide degrader for the intended protein(s) of interest, or as a molecular glue which recruits neo-substrate protein(s) to CRL4CRBN E3 ligase for ubiquitination and subsequent degradation. Both mechanisms must be considered in the design of a PROTAC degrader. Further investigation is ongoing to define the precise structural requirements for a phthalimide conjugate to operate either as a bona fide PROTAC degrader or as a molecular glue.
CHEMISTRY
Spiroxindoles 1–741, 842 and all amino-lenalidomide linkers14 were obtained as previously described. Amide coupling of the respective cores and amino-lenalidomide linkers produced the target molecules as illustrated in Schemes 1 – 4.
Scheme 1. Linker modifications.
Scheme 4. Removal of pyrrolidine substituents.
EXPERIMENTAL SECTION
Compound Synthesis
Unless otherwise stated, all commercial reagents were used as supplied without further purification. NMR spectra were obtained on a Bruker 400 Ascend™ spectrometer at a 1H frequency of 400 MHz. Chemical shifts (δ) are reported in parts per million (ppm) relative to an internal standard. The final products were purified on a preparative HPLC column (Waters 2545, Quaternary Gradient Module) with a SunFire Prep C18 OBD 5 μm 50 × 100 mm reverse phase column. The mobile phase was a gradient of solvent A (H2O with 0.1% TFA) and solvent B (MeCN with 0.1% TFA) at a flow rate of 60 mL/min and 1%/min increase of solvent B. All final compounds have purity ≥ 95% as determined by Waters ACQUITY UPLC using a reverse phase column (SunFire, C18–5 μm, 4.6 × 150 mm) and a solvent gradients of A (H2O with 0.1% of TFA) and B (MeCN with 0.1% of TFA). Mass spectral analyses were carried out with a Waters ACQUITY UPLC/QDa mass detector.
All the compounds were synthesized according to the procedure described for MG-277.
(3’R,4’S,5’R)-6”-Chloro-4’-(3-chloro-2-fluorophenyl)-N-(5-(2-(1-methyl-2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)pentyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (MC-024).
LC-MS (ESI) m/z (MH)+ calcd. for C42H45Cl2FN5O5+ = 788.28; found 788.31; >95% purity. 1H NMR (400 MHz, Methanol-d4) δ 7.69 − 7.61 (m, 2H), 7.50 − 7.35 (m, 4H), 7.17 (t, J = 8.0 Hz, 1H), 7.12 (dt, J = 8.2, 2.0 Hz, 1H), 6.77 (d, J = 1.9 Hz, 1H), 5.21 (dt, J = 13.5, 4.8 Hz, 1H), 5.08 (d, J = 10.9 Hz, 1H), 4.75 (dd, J = 11.1, 2.5 Hz, 1H), 4.53 − 4.36 (m, 2H), 3.47 − 3.35 (m, 1H), 3.15 (s, 3H), 3.05 − 2.86 (m, 3H), 2.84 − 2.68 (m, 1H), 2.60 (t, J = 7.7 Hz, 2H), 2.50 (qd, J = 12.9, 5.5 Hz, 1H), 2.24 − 2.10 (m, 2H), 2.01 − 1.82 (m, 3H), 1.76 (d, J = 13.3 Hz, 2H), 1.61 − 1.14 (m, 8H), 1.14 − 1.00 (m, 2H).
(3’R,4’R,5’R)-6”-Chloro-N-(5-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)pentyl)-2”-oxo-4’-phenyldispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (MC-215).
LC-MS (ESI) m/z (MH)+ calcd. for C41H45ClN5O5+ = 722.30; found 722.41; >95% purity. 1H NMR (400 MHz, Methanol-d4) δ 7.66 (t, J = 7.2 Hz, 1H), 7.55 (dd, J = 8.2, 5.6 Hz, 1H), 7.52 − 7.42 (m, 1H), 7.40 (ddd, J = 7.6, 2.7, 1.1 Hz, 1H), 7.26 − 7.08 (m, 6H), 6.71 (dd, J = 2.0, 0.9 Hz, 1H), 5.27 − 5.15 (m, 2H), 4.54 − 4.36 (m, 2H), 4.25 (dd, J = 23.8, 11.2 Hz, 1H), 3.04 − 2.86 (m, 2H), 2.86 − 2.71 (m, 2H), 2.61 − 2.45 (m, 3H), 2.27 − 2.10 (m, 2H), 2.02 − 1.80 (m, 3H), 1.82 − 1.70 (m, 2H), 1.58 − 1.14 (m, 8H), 1.11 − 0.97 (m, 2H).
(3’R,4’R,5’R)-N-(5-(2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)pentyl)-2”-oxo-4’-phenyldispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (MC-216).
LC-MS (ESI) m/z (MH)+ calcd. For C41H46N5O5+ = 688.35; found 688.45; >95% purity. 1H NMR (400 MHz, Methanol-d4) δ 7.66 (t, J = 7.6 Hz, 1H), 7.60 − 7.52 (m, 1H), 7.52 − 7.42 (m, 1H), 7.40 (ddd, J = 7.6, 2.4, 1.1 Hz, 1H), 7.28 − 7.05 (m, 7H), 6.69 (d, J = 7.8 Hz, 1H), 5.28 − 5.14 (m, 2H), 4.53 − 4.37 (m, 2H), 4.25 (dd, J = 24.0, 11.1 Hz, 1H), 3.05 − 2.86 (m, 3H), 2.85 − 2.73 (m, 2H), 2.60 − 2.46 (m, 2H), 2.26 − 2.13 (m, 2H), 2.00 − 1.85 (m, 3H), 1.81 − 1.69 (m, 2H), 1.58 − 1.13 (m, 8H), 1.11 − 0.98 (m, 2H).
(3’R,4’S,5’R)-N-(5-(2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)pentyl)-4’-(2-fluorophenyl)2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (MC-217).
LC-MS (ESI) m/z (MH)+ calcd. for C41H45FN5O5+ = 706.34; found 706.50; >95% purity. 1H NMR (400 MHz, Methanol-d4) δ 7.72 (td, J = 7.6, 1.8 Hz, 1H), 7.66 (dd, J = 7.4, 1.3 Hz, 1H), 7.51 − 7.39 (m, 3H), 7.26 − 7.17 (m, 2H), 7.14 (t, J = 7.7 Hz, 1H), 7.07 (t, J = 7.7 Hz, 1H), 6.90 − 6.79 (m, 1H), 6.71 (d, J = 7.8 Hz, 1H), 5.24 − 5.14 (m, 1H), 4.77 (d, J = 10.8 Hz, 1H), 4.55 − 4.37 (m, 2H), 3.08 − 2.86 (m, 2H), 2.86 − 2.66 (m, 2H), 2.60 (t, J = 7.8 Hz, 2H), 2.58 − 2.43 (m, 1H), 2.25 − 2.08 (m, 2H), 1.99 − 1.80 (m, 3H), 1.80 − 1.65 (m, 2H), 1.64 − 1.24 (m, 7H), 1.24 − 1.00 (m, 4H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-chloro-2-fluorophenyl)-N-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (MG-274).
LC-MS (ESI) m/z (MH)+ calcd. for C36H33Cl2FN5O5+ = 704.19; found 704.34; >95% purity. 1H NMR (400 MHz, Methanol-d4) δ 7.78 − 7.63 (m, 3H), 7.60 − 7.51 (m, 2H), 7.38 (t, J = 7.5 Hz, 1H), 7.18 (t, J = 8.1 Hz, 1H), 7.10 (dd, J = 8.2, 2.0 Hz, 1H), 6.79 (t, J = 1.6 Hz, 1H), 5.34 − 5.21 (m, 1H), 5.21 − 5.10 (m, 1H), 4.29 − 3.95 (m, 3H), 3.02 − 2.86 (m, 2H), 2.86 − 2.74 (m, 1H), 2.76 − 2.63 (m, 1H), 2.43 − 2.28 (m, 1H), 2.23 − 2.08 (m, 2H), 1.97 − 1.69 (m, 5H), 1.65 − 1.50 (m, 2H), 1.25 − 1.12 (m, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-chloro-2-fluorophenyl)-N-(3-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)propyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (MG-275).
LC-MS (ESI) m/z (MH)+ calcd. for C39H39Cl2FN5O5+ = 746.23; found 746.45; >95% purity. 1H NMR (400 MHz, Methanol-d4) δ 7.73 − 7.58 (m, 2H), 7.50 (dd, J = 8.3, 2.5 Hz, 1H), 7.43 (td, J = 7.6, 2.1 Hz, 1H), 7.34 (dddd, J = 8.6, 7.3, 5.7, 1.6 Hz, 1H), 7.24 (ddd, J = 7.6, 5.4, 1.0 Hz, 1H), 7.18 (tt, J = 8.1, 1.2 Hz, 1H), 7.13 − 7.06 (m, 1H), 6.78 (d, J = 1.9 Hz, 1H), 5.26 − 5.11 (m, 2H), 4.82 (d, J = 11.2 Hz, 1H), 4.51 − 4.29 (m, 2H), 3.50 − 3.34 (m, 1H), 3.12 − 2.99 (m, 1H), 2.99 − 2.74 (m, 3H), 2.57 − 2.36 (m, 3H), 2.27 − 2.09 (m, 2H), 2.01 − 1.83 (m, 3H), 1.82 − 1.64 (m, 4H), 1.50 (dd, J = 15.3, 11.9 Hz, 1H), 1.21 (td, J = 13.8, 4.0 Hz, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-chloro-2-fluorophenyl)-N-(4-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)butyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (MG-276).
LC-MS (ESI) m/z (MH)+ calcd. for C40H41Cl2FN5O5+ = 760.25; found 760.51; >95% purity. 1H NMR (400 MHz, Methanol-d4) δ 7.69 − 7.58 (m, 2H), 7.54 − 7.41 (m, 2H), 7.41 − 7.31 (m, 1H), 7.31 − 7.06 (m, 3H), 6.77 (d, J = 2.0 Hz, 1H), 5.22 − 5.06 (m, 2H), 4.77 (d, J = 11.1 Hz, 1H), 4.52 − 4.34 (m, 2H), 3.47 − 3.33 (m, 1H), 3.27 − 3.11 (m, 1H), 3.11 − 2.99 (m, 1H), 3.00 − 2.84 (m, 1H), 2.85 − 2.73 (m, 2H), 2.63 − 2.45 (m, 3H), 2.25 − 2.11 (m, 2H), 2.01 − 1.82 (m, 3H), 1.76 (d, J = 13.4 Hz, 2H), 1.58 − 1.30 (m, 5H), 1.29 − 1.11 (m, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-chloro-2-fluorophenyl)-N-(5-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)pentyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (MG-277).
HATU (9 mg, 0.0237 mmol) was added to a solution of 141 (10 mg, 0.0216 mmol), 214 (10 mg, 0.0226 mmol) and DIEA (16 μL, 0.094 mmol) in DMF (0.5 mL). After 10 min at rt the reaction was complete as determined by a UPLC/QDa mass detector. The reaction was diluted with MeOH (0.5 mL), acidified with TFA, then H2O (1 mL) was added and this solution was injected into a preparative HPLC for purification. The HPLC fractions containing the product were partially concentrated and the remaining solution was frozen and lyophilized to produce 6.4 mg of MG-277 as a white powder. LC-MS (ESI) m/z (MH)+ calcd. for C41H43Cl2FN5O5+ = 774.26; Found 774.42; >95% purity. 1H NMR (400 MHz, Methanol-d4) δ 7.65 (d, J = 7.3 Hz, 2H), 7.53 − 7.38 (m, 3H), 7.40 − 7.31 (m, 1H), 7.16 (t, J = 8.0 Hz, 1H), 7.10 (dt, J = 8.2, 2.4 Hz, 1H), 6.77 (d, J = 2.0 Hz, 1H), 5.19 (dt, J = 13.3, 5.5 Hz, 1H), 5.08 (d, J = 11.0 Hz, 1H), 4.75 (d, J = 11.0 Hz, 1H), 4.55 − 4.38 (m, 2H), 3.45 − 3.33 (m, 1H), 3.08 − 2.86 (m, 2H), 2.85 − 2.65 (m, 2H), 2.65 − 2.45 (m, 3H), 2.40 − 2.30 (m, 1H), 2.25 − 2.07 (m, 2H), 1.99 − 1.80 (m, 3H), 1.79 − 1.67 (m, 2H), 1.60 − 1.26 (m, 5H), 1.23 − 1.04 (m, 3H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-chloro-2-fluorophenyl)-N-(2-(3-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)propoxy)ethyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (MG-278).
LC-MS (ESI) m/z (MH)+ calcd. for C41H43Cl2FN5O6+ = 790.26; found 790.23; >95% purity. 1H NMR (400 MHz, Methanol-d4) δ 7.69 − 7.60 (m, 2H), 7.53 − 7.44 (m, 2H), 7.41 (dd, J = 7.6, 1.2 Hz, 1H), 7.27 (dddd, J = 23.7, 8.5, 7.1, 1.5 Hz, 1H), 7.17 − 7.06 (m, 2H), 6.76 (dd, J = 1.9, 0.8 Hz, 1H), 5.25 − 5.12 (m, 1H), 4.79 (d, J = 11.0 Hz, 1H), 4.55 − 4.35 (m, 2H), 3.58 (dtd, J = 13.9, 6.2, 4.1 Hz, 1H), 3.38 − 3.09 (m, 5H), 3.02 − 2.85 (m, 2H), 2.84 − 2.76 (m, 2H), 2.63 (td, J = 7.5, 2.3 Hz, 2H), 2.53 (qd, J = 13.2, 4.6 Hz, 1H), 2.24 − 2.12 (m, 2H), 2.00 − 1.87 (m, 3H), 1.81 − 1.68 (m, 4H), 1.51 (q, J = 13.8 Hz, 1H), 1.25 − 1.15 (m, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-chloro-2-fluorophenyl)-N-(2-(2-(3-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)propoxy)ethoxy)ethyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (MG-279).
LC-MS(ESI) m/z (MH)+ calcd. for C43H47Cl2FN5O7+ = 834.28; found 834.35; >95% purity. 1H NMR (400 MHz, Methanol-d4) δ 7.67 (dd, J = 6.4, 2.3 Hz, 1H), 7.65 − 7.58 (m, 1H), 7.57 − 7.41 (m, 3H), 7.29 (dd, J = 8.0 Hz, 1H), 7.19 − 7.01 (m, 2H), 6.75 (d, J = 2.0 Hz, 1H), 5.19 (dd, J = 13.3, 5.2 Hz, 1H), 4.75 (d, J = 10.4 Hz, 1H), 4.56 − 4.37 (m, 2H), 3.66 − 3.31 (m, 12H), 3.00 − 2.86 (m, 1H), 2.86 − 2.72 (m, 3H), 2.56 − 2.45 (m, 1H), 2.22 − 2.13 (m, 1H), 2.08 − 1.61 (m, 8H), 1.58 − 1.45 (m, 1H), 1.22 − 1.01 (m, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-chloro-2-fluorophenyl)-N-(2-(2-(2-(3-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)propoxy)ethoxy)ethoxy)ethyl)-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (MG-280).
LC-MS(ESI) m/z (MH)+ calcd. for C45H51Cl2FN5O8+ = 878.31; found 878.65; >95% purity. 1H NMR (400 MHz, Methanol-d4) δ 7.73 − 7.61 (m, 2H), 7.55 − 7.45 (m, 3H), 7.35 (t, J = 7.5 Hz, 1H), 7.20 −7.04 (m, 2H), 6.78 (d, J = 1.9 Hz, 1H), 5.21 (dt, J = 13.3, 4.8 Hz, 1H), 4.79 (d, J = 10.7 Hz, 1H), 4.60 − 4.40 (m, 2H), 3.74 − 3.38 (m, 13H), 3.33 − 3.18 (m, 2H), 2.95 (ddd, J = 17.6, 13.5, 5.4 Hz, 1H), 2.88 − 2.77 (m, 3H), 2.75 − 2.61 (m, 1H), 2.61 − 2.45 (m, 1H), 2.25 − 2.16 (m, 1H), 2.09 (d, J = 13.8 Hz, 1H), 2.02 − 1.67 (m, 7H), 1.55 (q, J = 13.9 Hz, 1H), 1.27 − 1.05 (m, 2H).
(2R,4R)-N-(5-(2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)pentyl)-4-phenylpyrrolidine-2-carboxamide (MC-293).
LC-MS (ESI) m/z (MH)+ calcd. for C29H35N4O4+ = 503.26; found 503.17; >95% purity. 1H NMR (400 MHz, Methanol-d4) δ 7.60 (ddd, J = 7.4, 3.1, 1.3 Hz, 1H), 7.48 − 7.34 (m, 4H), 7.34 − 7.25 (m, 3H), 5.17 (td, J = 13.0, 5.2 Hz, 1H), 4.57 − 4.39 (m, 3H), 3.87 − 3.77 (m, 1H), 3.53 − 3.40 (m, 1H), 3.29 − 3.16 (m, 2H), 2.98 − 2.84 (m, 1H), 2.84 − 2.68 (m, 3H), 2.60 − 2.38 (m, 2H), 2.38 − 2.25 (m, 1H), 2.25 − 2.14 (m, 1H), 1.83 − 1.51 (m, 4H), 1.47 − 1.32 (m, 2H).
(2R,4S)-N-(5-(2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)pentyl)-4-phenylpyrrolidine-2-carboxamide (MC-294).
LC-MS (ESI) m/z (MH)+ calcd. for C29H35N4O4+ = 503.26; found 503.17; >95% purity. 1H NMR (400 MHz, Methanol-d4) δ 7.57 (ddd, J = 7.5, 3.0, 1.2 Hz, 1H), 7.44 (dd, J = 7.6, 1.2 Hz, 1H), 7.42 − 7.34 (m, 3H), 7.36 − 7.24 (m, 3H), 5.16 (ddd, J = 13.5, 8.6, 5.2 Hz, 1H), 4.57 − 4.28 (m, 3H), 3.82 − 3.71 (m, 1H), 3.69 − 3.53 (m, 1H), 3.40 − 3.33 (m, 1H), 3.28 − 3.17 (m, 1H), 2.97 − 2.69 (m, 5H), 2.53 (qd, J = 13.2, 4.7 Hz, 1H), 2.24 − 2.13 (m, 1H), 2.01 − 1.87 (m, 1H), 1.80 − 1.52 (m, 5H), 1.49 − 1.32 (m, 2H).
(3’R,4’S,5’R)-6”-Chloro-4’-(3-chloro-2-fluorophenyl)-N-(5-(2-(2,6-dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)pentyl)-1’-methyl-2”-oxodispiro[cyclohexane-1,2’-pyrrolidine-3’,3”-indoline]-5’-carboxamide (MC-295).
LC-MS (ESI) m/z (MH)+ calcd. for C42H45Cl2FN5O5+ = 788.27; found 788.19; >95% purity. 1H NMR (400 MHz, Methanol-d4) δ 7.66 (dd, J = 7.4, 1.2 Hz, 1H), 7.60 − 7.50 (m, 2H), 7.50 − 7.40 (m, 2H), 7.36 (q, J = 6.7 Hz, 1H), 7.16 − 7.09 (m, 2H), 6.77 (dd, J = 2.0, 0.8 Hz, 1H), 5.19 (dt, J = 13.3, 5.0 Hz, 1H), 5.02 − 4.93 (m, 3H), 4.66 (dd, J = 11.9, 4.4 Hz, 1H), 4.54 − 4.37 (m, 2H), 3.44 − 3.33 (m, 2H), 3.25 (d, J = 4.6 Hz, 3H), 3.12 − 2.97 (m, 1H), 2.99 − 2.87 (m, 1H), 2.85 − 2.75 (m, 1H), 2.65 − 2.43 (m, 4H), 2.31 − 2.10 (m, 3H), 1.81 − 1.45 (m, 7H), 1.45 − 1.27 (m, 2H), 1.27 − 1.15 (m, 1H), 1.15 − 0.95 (m, 2H).
(3R,4’R,5’R)-N-(5-(2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)pentyl)-2’,2’-dimethyl-2-oxo-4’-phenylspiro[indoline-3,3’-pyrrolidine]-5’-carboxamide (MC-296).
LC-MS (ESI) m/z (MH)+ calcd. for C38H42N5O5+ = 648.31; found 648.24; >95% purity. 1H NMR (400 MHz, Methanol-d4) δ 7.68 − 7.63 (m, 1H), 7.50 − 7.43 (m, 1H), 7.44 − 7.35 (m, 2H), 7.26 − 7.02 (m, 7H), 6.73 (dd, J = 18.1, 7.8 Hz, 1H), 5.25 − 5.14 (m, 1H), 5.02 − 4.94 (m, 1H), 4.53 − 4.25 (m, 3H), 3.41 − 3.34 (m, 1H), 3.08 − 2.96 (m, 1H), 2.96 − 2.87 (m, 1H), 2.85 − 2.75 (m, 1H), 2.64 − 2.45 (m, 3H), 2.26 − 2.13 (m, 1H), 1.94 − 1.80 (m, 3H), 1.59 − 1.44 (m, 2H), 1.44 − 1.19 (m, 4H), 1.14 − 0.97 (m, 2H).
(3R,5’R)-N-(5-(2-(2,6-Dioxopiperidin-3-yl)-1-oxoisoindolin-4-yl)pentyl)-2-oxospiro[indoline-3,3’-pyrrolidine]-5’-carboxamide (MC-297).
LC-MS (ESI) m/z (MH)+ calcd. for C30H34N5O5+ = 544.25; found 544.15; >95% purity. 1H NMR (400 MHz, Methanol-d4) δ 7.63 − 7.54 (m, 1H), 7.45 − 7.35 (m, 3H), 7.36 − 7.26 (m, 1H), 7.11 (qd, J = 7.5, 1.1 Hz, 1H), 7.00 − 6.91 (m, 1H), 5.18 (td, J = 13.7, 5.2 Hz, 1H), 4.56 − 4.39 (m, 2H), 3.76 − 3.63 (m, 2H), 3.40 − 3.33 (m, 1H), 3.28 − 3.17 (m, 1H), 3.00 − 2.85 (m, 1H), 2.86 − 2.74 (m, 2H), 2.76 − 2.44 (m, 4H), 2.34 − 2.22 (m, 1H), 2.25 − 2.13 (m, 1H), 1.78 − 1.49 (m, 4H), 1.43 − 1.32 (m, 2H).
Fluorescence Polarization (FP)-Based Protein Binding Assay
The binding affinity of the compounds to MDM2 protein was determined by an optimized, sensitive and quantitative fluorescence polarization (FP)-based binding assay using a recombinant human His-tagged MDM2 protein (residues 1–118) and a FAM-tagged p53-based peptide as the fluorescent probe, as described previously.32, 43
Cell lines
The human leukemia RS4;11 cell lines (ATCC® CRL-1873™), MV-4–11 (ATCC® CRL-9591™), HL-60 (ATCC® CCL-240™) and breast cancer cell lines MDA-MB-468 (ATCC® HTB-132™) and MDA-MB-231 (ATCC® HTB-26™) were purchased from the American Type Culture Collection (ATCC). MOLM-13 (ACC 554, DSMZ) cell line was purchased from DSMZ. RS4;11/IRMI-2 cell line is an induced MDM2 inhibitor-resistant cell line generated from p53 wild-type RS4;11 cells by repeated treatment with a MDM2 inhibitor, SAR405838 (MI-73301) as described in our earlier study.31, 32 The p53 mutations (Y236 Y/H, R249 R/G) in RS4;11/IRMI-2 cells were confirmed by DNA sequencing.31, 32 In all experiments, RS4;11, MOLM-13, MDA-MB-468 and MDA-MB-231 cells were cultured in RPMI 1640 media (Cat#11875119, ThermoFisher), MV-4–11 and HL-60 cells were cultured in IMDM media (Cat# 12440061, ThermoFisher), supplemented with 10% HyClone™ fetal bovine serum (Cat#SH30910.03, GE Healthcare Life Sciences) and 1% penicillin–streptomycin (Cat#15140122, Life Technologies) at 37 °C in a humidified atmosphere containing 5% CO2 and were used within three months of thawing fresh vials.
Cell Growth Inhibition Assay
In cell growth inhibition experiments, cells were seeded in 96-well cell culture plates at densities of 10000–20000 cells/well for leukemia cell lines and 5000–10000 cells/well for breast cancer cell lines in 100 μL of culture medium. Each compound that was tested was serially diluted in the culture medium, and 100 μL of the diluted solution containing the tested compound was added to the appropriate wells of the cell plate. After addition of the tested compound, the cells were incubated for 4 days at 37 °C in an atmosphere of 5% CO2. Cell growth inhibition was then evaluated by a lactate dehydrogenase-based colorimetric assay (Dojindo Molecular Technologies) using a water soluble tetrazolium salt, WST-8.43 After the WST-8 reagent was added (20 μL/well), the plates were incubated for at least 1 h, and read at 450 nm using Tecan Infinite M1000 multimode microplate reader (Tecan, Morrisville, NC).
Western Blotting Assay and Quantitative Proteomics Profiling
For Western blot analysis, 2 × 106 cells/well were added into 6-well plates and treated with compounds at the indicated concentrations for various lengths of time. Cells were then collected and lysed in ice-cold Cell Lysis Buffer (Cell Signaling Technology, #9803) containing a protease inhibitor. 25 μg of lysate protein was run in each lane of a PAGE-SDS and blotted into polyvinylidene fluoride (PVDF) membranes. Expression levels of the indicated proteins were probed by the following primary antibodies: MDM2 (SMP14) (sc-965, Santa Cruz), p53 (DO-1) (sc-126, Santa Cruz), HRP-conjugated GAPDH (sc-25778 HRP, sc-47724 HRP, Santa Cruz) from Santa Cruz and eRF3/GSPT1 (ab126090, ab49878) from Abcam.
Total protein samples after treatment with indicated compounds in RS4;11 and RS4;11/IRMI-2 cell lines were prepared with ice-cold RIPA buffer containing protease inhibitor. The abundance of 5264 cellular proteins in each sample was analyzed by high resolution Orbitrap liquid chromatography mass spectrometry (LC-MS/MS) using TMT10plex™ Isobaric Label Reagent Set (Cat#90111, ThermoFisher) for protein labeling.
Quantitative Real-Time PCR (qRT-PCR)
Total RNA was prepared from RS4;11 cells after the indicated treatments using RNeasy Mini Kit (74106; QIAGEN) according to the manufacturer’s protocol. RNA concentration in each sample was determined with a Tecan Infinite M1000 multimode microplate reader (Tecan, Morrisville, NC). cDNA was generated using High-Capacity RNA-to-cDNA™ Kit (4387406; ThermoFisher Scientific). qRT-PCR was performed in triplicate with MDM2, p21, Puma, Bax and GAPDHspecific primer pairs using the Applied Biosystems QuantStudio 7 Flex Real-Time PCR System with TaqMan™ Fast Advanced Master Mix (Cat#4444557; Applied Biosystems). Target gene expression levels were normalized to GAPDH levels, made relative to untreated control samples and presented as 2−ΔΔCt. All primers used for qRT-PCR were purchase from ThermoFisher and as follows: MDM2 (Hs01066930_m1; ThermoFisher); p21 (Hs00355782_m1; ThermoFisher); Puma (Hs00248075_m1; ThermoFisher); Bax (Hs00180269_m1; ThermoFisher), GSPT1 (Hs01093019_m1; ThermoFisher) and GAPDH (Hs02758991_g1; ThermoFisher).
Small interfering RNA (siRNA) Knockdown
To down-regulate MDM2 and cereblon, ON-TARGETplus siRNAs (SMARTpool) for human MDM2 (LU-003279-00-0005; Dharmacon), ON-TARGETplus siRNAs (individual) for cereblon (LQ-021086-10-0002, LQ-021086-11-0002 ; Dharmacon) and non-targeting negative control siRNAs (D-001810-10-20; Dharmacon) were used. Transfection was performed using Lipofectamine RNAiMAX (Cat#13778150, ThermoFisher). MDA-MB-231 and MDA-MB-468 cells were transfected with MDM2 or cereblon siRNAs or non-targeting negative control siRNAs for 2 days, followed by indicated drug treatment.
Quantification and Statistical Analysis
All the cell data of protein binding assay, cell growth inhibition assay and qRT-PCR were presented as mean ± standard error from at least three independent experiments. Statistical analyses presented in all figures were performed using GraphPad Prism software (version 7.00).
Supplementary Material
Scheme 2. Removal of halogens and methylation of pyrrolidine and glutarimide nitrogens.
Scheme 3. Systematic removal of pyrrolidine substituents.
ACKNOWLEDGMENTS
We are grateful for the financial support from the National Cancer Institute, NIH (1R01CA208267 to S.W.), the University of Michigan Rogel Cancer Center support grant from the National Cancer Institute, NIH (P30 CA046592) and the Endowment for the Basic Sciences (EBS) EDGE Student Fellowship from the University of Michigan Medical School (to J.Y.).
ABBREVIATIONS USED
- MDM2
Human murine double minute 2
- PROTAC
proteolysis targeting chimera
- GSPT1
G1 to S Phase Transition 1
- CRBN
cereblon
- PUMA
p53 upregulated modulator of apoptosis
- MetAP-2
methionine aminopeptidase-2
- IMiDs
immunomodulatory imide drugs
- CRL4CRBN
CUL4-DDB1-RBX1-CRBN E3 ligase complex
- FP
fluorescence-polarization
- qRT-PCR
quantitative reverse transcription polymerase chain reaction
- CRLs
Cullin-RING E3 ligases
- NAE
Nedd8 E1 conjugating enzyme
Footnotes
Supporting Information Availability
Cell growth inhibitory activity of the MDM2 inhibitors, bona fide MDM2 degraders, MD-222 and the new putative MDM2 degrader MG-277 in p53 wild-type and p53 mutated/deleted cell lines; The potency of different lenalidomide conjugated analogues of MG-277 in cell growth inhibition and GSPT1 degradation; Determination of the half maximal degradation concentration (DC50) of GSPT1 degradation by different analogues of MG-277 in RS4;11 cell line after a 24 h treatment; Correlation between GSPT1 degradation DC50 of the compounds in RS4;11 cells and IC50 in growth inhibition in RS4;11 and IRMI-2 cell lines; Detailed illustration of the predicted binding mode of MG-277 to the cereblon-GSPT1 complex; NMR spectra of MG-277; Mass spectrum and purity spectrum of MG-277; CSV file containing SMILES, binding and cellular data for final target compounds.
The authors declare no competing interests.
REFERENCES
- 1.Sakamoto KM; Kim KB; Kumagai A; Mercurio F; Crews CM; Deshaies RJ, Protacs: Chimeric Molecules That Target Proteins to the Skp1–Cullin–F Box Complex for Ubiquitination and Degradation. Proceedings of the National Academy of Sciences 2001, 98 (15), 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gu S; Cui D; Chen X; Xiong X; Zhao Y, PROTACs: An Emerging Targeting Technique for Protein Degradation in Drug Discovery. 2018, 40 (4), 1700247. [DOI] [PubMed] [Google Scholar]
- 3.Salami J; Crews CM, Waste Disposal-An Attractive Strategy for Cancer Therapy. Science (New York, N.Y.) 2017, 355 (6330), 1163–1167. [DOI] [PubMed] [Google Scholar]
- 4.Ottis P; Crews CM, Proteolysis-Targeting Chimeras: Induced Protein Degradation as a Therapeutic Strategy. ACS chemical biology 2017, 12 (4), 892–898. [DOI] [PubMed] [Google Scholar]
- 5.Neklesa TK; Winkler JD; Crews CM, Targeted Protein Degradation by PROTACs. Pharmacology & Therapeutics 2017, 174, 138–144. [DOI] [PubMed] [Google Scholar]
- 6.Lai AC; Crews CM, Induced Protein Degradation: An Emerging Drug Discovery Paradigm. Nature reviews. Drug discovery 2017, 16 (2), 101–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cromm PM; Crews CM, Targeted Protein Degradation: from Chemical Biology to Drug Discovery. Cell chemical biology 2017, 24 (9), 1181–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Toure M; Crews CM, Small-Molecule PROTACS: New Approaches to Protein Degradation. 2016, 55 (6), 1966–1973. [DOI] [PubMed] [Google Scholar]
- 9.Neklesa TK; Jin M; Crew AP; Rossi AK; Willard RR; Dong H; Siu K; Wang J; Gordon DA; Chen X; Ferraro C; Crews CM; Coleman K; Winkler JD, ARV-330: Androgen Receptor PROTAC Degrader for Prostate Cancer. 2016, 34 (2_suppl), 267–267. [Google Scholar]
- 10.Lu J; Qian Y; Altieri M; Dong H; Wang J; Raina K; Hines J; Winkler James D.; Crew Andrew P.; Coleman K; Crews Craig M., Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chemistry & Biology 2015, 22 (6), 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zengerle M; Chan K-H; Ciulli A, Selective Small Molecule Induced Degradation of the BET Bromodomain Protein BRD4. ACS chemical biology 2015, 10 (8), 1770–1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crew AP; Raina K; Dong H; Qian Y; Wang J; Vigil D; Serebrenik YV; Hamman BD; Morgan A; Ferraro C; Siu K; Neklesa TK; Winkler JD; Coleman KG; Crews CM, Identification and Characterization of Von Hippel-Lindau-Recruiting Proteolysis Targeting Chimeras (PROTACs) of TANK-Binding Kinase 1. Journal of medicinal chemistry 2018, 61 (2), 583–598. [DOI] [PubMed] [Google Scholar]
- 13.Lai AC; Toure M; Hellerschmied D; Salami J; Jaime-Figueroa S; Ko E; Hines J; Crews CM, Modular PROTAC Design for the Degradation of Oncogenic BCR-ABL. 2016, 55 (2), 807–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y; Yang J; Aguilar A; McEachern D; Przybranowski S; Liu L; Yang C-Y; Wang M; Han X; Wang S, Discovery of MD-224 as a First-in-Class, Highly Potent, and Efficacious Proteolysis Targeting Chimera Murine Double Minute 2 Degrader Capable of Achieving Complete and Durable Tumor Regression. Journal of medicinal chemistry 2019, 62 (2), 448–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bondeson DP; Mares A; Smith IED; Ko E; Campos S; Miah AH; Mulholland KE; Routly N; Buckley DL; Gustafson JL; Zinn N; Grandi P; Shimamura S; Bergamini G; Faelth-Savitski M; Bantscheff M; Cox C; Gordon DA; Willard RR; Flanagan JJ; Casillas LN; Votta BJ; den Besten W; Famm K; Kruidenier L; Carter PS; Harling JD; Churcher I; Crews CM, Catalytic in vivo Protein Knockdown by Small-Molecule PROTACs. Nature chemical biology 2015, 11 (8), 611–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bai L; Zhou B; Yang C-Y; Ji J; McEachern D; Przybranowski S; Jiang H; Hu J; Xu F; Zhao Y; Liu L; Fernandez-Salas E; Xu J; Dou Y; Wen B; Sun D; Meagher J; Stuckey J; Hayes DF; Li S; Ellis MJ; Wang S, Targeted Degradation of BET Proteins in Triple-Negative Breast Cancer. Cancer research 2017, 77 (9), 2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sakamoto KM; Kim KB; Verma R; Ransick A; Stein B; Crews CM; Deshaies RJ, Development of Protacs to Target Cancer-promoting Proteins for Ubiquitination and Degradation. Molecular & Cellular Proteomics 2003, 2 (12), 1350–1358. [DOI] [PubMed] [Google Scholar]
- 18.Winter GE; Buckley DL; Paulk J; Roberts JM; Souza A; Dhe-Paganon S; Bradner JE, Phthalimide Conjugation as A Strategy for in vivo Target Protein Degradation. Science (New York, N.Y.) 2015, 348 (6241), 1376–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ito T; Ando H; Suzuki T; Ogura T; Hotta K; Imamura Y; Yamaguchi Y; Handa H, Identification of A Primary Target of Thalidomide Teratogenicity. Science (New York, N.Y.) 2010, 327 (5971), 1345–1350. [DOI] [PubMed] [Google Scholar]
- 20.Lopez-Girona A; Mendy D; Ito T; Miller K; Gandhi AK; Kang J; Karasawa S; Carmel G; Jackson P; Abbasian M; Mahmoudi A; Cathers B; Rychak E; Gaidarova S; Chen R; Schafer PH; Handa H; Daniel TO; Evans JF; Chopra R, Cereblon is A Direct Protein Target for Immunomodulatory and Antiproliferative Activities of Lenalidomide and Pomalidomide. Leukemia 2012, 26 (11), 2326–2335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Petzold G; Fischer ES; Thomä NH, Structural Basis of Lenalidomide-Induced CK1α Degradation by the CRL4CRBN Ubiquitin Ligase. Nature 2016, 532, 127–130. [DOI] [PubMed] [Google Scholar]
- 22.Lu G; Middleton RE; Sun H; Naniong M; Ott CJ; Mitsiades CS; Wong KK; Bradner JE; Kaelin WG Jr., The Myeloma Drug Lenalidomide Promotes the Cereblon-Dependent Destruction of Ikaros Proteins. Science (New York, N.Y.) 2014, 343 (6168), 305–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kronke J; Udeshi ND; Narla A; Grauman P; Hurst SN; McConkey M; Svinkina T; Heckl D; Comer E; Li X; Ciarlo C; Hartman E; Munshi N; Schenone M; Schreiber SL; Carr SA; Ebert BL, Lenalidomide Causes Selective Degradation of IKZF1 and IKZF3 in Multiple Myeloma Cells. Science (New York, N.Y.) 2014, 343 (6168), 301–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Krönke J; Fink EC; Hollenbach PW; MacBeth KJ; Hurst SN; Udeshi ND; Chamberlain PP; Mani DR; Man HW; Gandhi AK; Svinkina T; Schneider RK; McConkey M; Järås M; Griffiths E; Wetzler M; Bullinger L; Cathers BE; Carr SA; Chopra R; Ebert BL, Lenalidomide Induces Ubiquitination and Degradation of CK1α in del(5q) MDS. Nature 2015, 523 (7559), 183–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fischer ES; Park E; Eck MJ; Thomä NH, SPLINTS: Small-molecule Protein Ligand Interface Stabilizers. Curr Opin Struct Biol 2016, 37, 115–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hughes SJ; Ciulli A, Molecular Recognition of Ternary Complexes: A New Dimension in the Structure-guided Design of Chemical Degraders. Essays Biochem 2017, 61 (5), 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Han T; Goralski M; Gaskill N; Capota E; Kim J; Ting TC; Xie Y; Williams NS; Nijhawan D, Anticancer Sulfonamides Target Splicing by Inducing RBM39 Degradation via Recruitment to DCAF15. Science (New York, N.Y.) 2017, 356 (6336), eaal3755. [DOI] [PubMed] [Google Scholar]
- 28.Uehara T; Minoshima Y; Sagane K; Sugi NH; Mitsuhashi KO; Yamamoto N; Kamiyama H; Takahashi K; Kotake Y; Uesugi M; Yokoi A; Inoue A; Yoshida T; Mabuchi M; Tanaka A; Owa T, Selective Degradation of Splicing Factor CAPERα by Anticancer Sulfonamides. Nature chemical biology 2017, 13, 675–680. [DOI] [PubMed] [Google Scholar]
- 29.Ishoey M; Chorn S; Singh N; Jaeger MG; Brand M; Paulk J; Bauer S; Erb MA; Parapatics K; Müller AC; Bennett KL; Ecker GF; Bradner JE; Winter GE, Translation Termination Factor GSPT1 Is a Phenotypically Relevant Off-Target of Heterobifunctional Phthalimide Degraders. ACS Chemical Biology 2018, 13 (3), 553–560. [DOI] [PubMed] [Google Scholar]
- 30.Zhao Y; Liu L; Sun W; Lu J; McEachern D; Li X; Yu S; Bernard D; Ochsenbein P; Ferey V; Carry J-C; Deschamps JR; Sun D; Wang S, Diastereomeric Spirooxindoles as Highly Potent and Efficacious MDM2 Inhibitors. Journal of the American Chemical Society 2013, 135 (19), 7223–7234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hoffman-Luca CG; Ziazadeh D; McEachern D; Zhao Y; Sun W; Debussche L; Wang S, Elucidation of Acquired Resistance to Bcl-2 and MDM2 Inhibitors in Acute Leukemia in Vitro and in Vivo. Clinical cancer research : an official journal of the American Association for Cancer Research 2015, 21 (11), 2558–2568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang S; Sun W; Zhao Y; McEachern D; Meaux I; Barrière C; Stuckey JA; Meagher JL; Bai L; Liu L; Hoffman-Luca CG; Lu J; Shangary S; Yu S; Bernard D; Aguilar A; Dos-Santos O; Besret L; Guerif S; Pannier P; Gorge-Bernat D; Debussche L, SAR405838: An Optimized Inhibitor of MDM2-p53 Interaction That Induces Complete and Durable Tumor Regression. Cancer research 2014, 74 (20), 5855–5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Olivier M; Eeles R; Hollstein M; Khan MA; Harris CC; Hainaut P, The IARC TP53 Database: New Online Mutation Analysis and Recommendations to Users. Human Mutation 2002, 19 (6), 607–614. [DOI] [PubMed] [Google Scholar]
- 34.Ding K; Lu Y; Nikolovska-Coleska Z; Qiu S; Ding Y; Gao W; Stuckey J; Krajewski K; Roller PP; Tomita Y; Parrish DA; Deschamps JR; Wang S, Structure-Based Design of Potent Non-Peptide MDM2 Inhibitors. Journal of the American Chemical Society 2005, 127 (29), 10130–10131. [DOI] [PubMed] [Google Scholar]
- 35.García-Echeverría C; Chène P; Blommers MJJ; Furet P, Discovery of Potent Antagonists of the Interaction between Human Double Minute 2 and Tumor Suppressor p53. Journal of Medicinal Chemistry 2000, 43 (17), 3205–3208. [DOI] [PubMed] [Google Scholar]
- 36.Lu J; Qian Y; Altieri M; Dong H; Wang J; Raina K; Hines J; Winkler JD; Crew AP; Coleman K; Crews CM, Hijacking the E3 Ubiquitin Ligase Cereblon to Efficiently Target BRD4. Chemistry & biology 2015, 22 (6), 755–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Fischer ES; Böhm K; Lydeard JR; Yang H; Stadler MB; Cavadini S; Nagel J; Serluca F; Acker V; Lingaraju GM; Tichkule RB; Schebesta M; Forrester WC; Schirle M; Hassiepen U; Ottl J; Hild M; Beckwith REJ; Harper JW; Jenkins JL; Thomä NH, Structure of the DDB1–CRBN E3 Ubiquitin Ligase in Complex with Thalidomide. Nature 2014, 512, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cheng Z; Saito K; Pisarev AV; Wada M; Pisareva VP; Pestova TV; Gajda M; Round A; Kong C; Lim M; Nakamura Y; Svergun DI; Ito K; Song H, Structural Insights Into eRF3 and Stop Codon Recognition by eRF1. Genes & development 2009, 23 (9), 1106–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Preis A; Heuer A; Barrio-Garcia C; Hauser A; Eyler, Daniel E.; Berninghausen, O.; Green, R.; Becker, T.; Beckmann, R., Cryoelectron Microscopic Structures of Eukaryotic Translation Termination Complexes Containing eRF1-eRF3 or eRF1-ABCE1. Cell Reports 2014, 8 (1), 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Matyskiela ME; Lu G; Ito T; Pagarigan B; Lu C-C; Miller K; Fang W; Wang N-Y; Nguyen D; Houston J; Carmel G; Tran T; Riley M; Nosaka LA; Lander GC; Gaidarova S; Xu S; Ruchelman AL; Handa H; Carmichael J; Daniel TO; Cathers BE; Lopez-Girona A; Chamberlain PP, A Novel Cereblon Modulator Recruits GSPT1 to the CRL4CRBN Ubiquitin Ligase. Nature 2016, 535, 252–257. [DOI] [PubMed] [Google Scholar]
- 41.Aguilar A; Sun W; Liu L; Lu J; McEachern D; Bernard D; Deschamps JR; Wang S, Design of Chemically Stable, Potent, and Efficacious MDM2 Inhibitors That Exploit the Retro-Mannich Ring-Opening-Cyclization Reaction Mechanism in Spiro-oxindoles. Journal of medicinal chemistry 2014, 57 (24), 10486–10498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Efremov IV; Vajdos FF; Borzilleri KA; Capetta S; Chen H; Dorff PH; Dutra JK; Goldstein SW; Mansour M; McColl A; Noell S; Oborski CE; O’Connell TN; O’Sullivan TJ; Pandit J; Wang H; Wei B; Withka JM, Discovery and Optimization of a Novel Spiropyrrolidine Inhibitor of β-Secretase (BACE1) through Fragment-Based Drug Design. Journal of medicinal chemistry 2012, 55 (21), 9069–9088. [DOI] [PubMed] [Google Scholar]
- 43.Zhao Y; Yu S; Sun W; Liu L; Lu J; McEachern D; Shargary S; Bernard D; Li X; Zhao T; Zou P; Sun D; Wang S, A Potent Small-Molecule Inhibitor of the MDM2-p53 Interaction (MI-888) Achieved Complete and Durable Tumor Regression in Mice. Journal of medicinal chemistry 2013, 56 (13), 5553–5561. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.