

Abstract
Small-molecule drugs and toxicants commonly interact with more than a single protein target, each of which may have unique effects on cellular phenotype. Although untargeted metabolomics is often applied to understand the mode of action of these chemicals, simple pairwise comparisons of treated and untreated samples are insufficient to resolve the effects of disrupting two or more independent protein targets. Here, we introduce a workflow for dose-response metabolomics to evaluate chemicals that potentially affect multiple proteins with different potencies. Our approach relies on treating samples with various concentrations of compound prior to analysis with mass spectrometry-based metabolomics. Data are then processed with software we developed called TOXcms, which statistically evaluates dose-response trends for each metabolomic signal according to user-defined tolerances and subsequently groups those that follow the same pattern. Although TOXcms was built upon the XCMS framework, it is compatible with any metabolomic data-processing software. Additionally, to enable correlation of dose responses beyond those that can be measured by metabolomics, TOXcms also accepts data from respirometry, cell death assays, other omic platforms, etc. In this work, we primarily focus on applying dose-response metabolomics to find off-target effects of drugs. Using metformin and etomoxir as examples, we demonstrate that each group of dose-response patterns identified by TOXcms signifies a metabolic response to a different protein target with a unique drug binding affinity. TOXcms is freely available on our laboratory website at http://pattilab.wustl.edu/software/toxcms
Graphical abstract
A dose-response assessment aims to quantify the relationship between the concentration of a chemical and the incidence of its effects in a biological sample.1 The concept dates to the 16th century, when Paracelsus recognized that the same chemical could be both therapeutic and toxic, depending on dose.2 Today, dose-response studies are regularly performed in the fields of pharmacology and toxicology. They are essential to our evaluation of chemical safety, contribute to our understanding of the site of drug action, facilitate selection of drug doses and dosing schedules, help determine drug potency and efficacy, and reveal potential interactions between multiple drugs.3
In principle, any biological readout can be modeled as a function of chemical concentration to assess a potentiai dose-dependent effect. Historically, assays were limited to relatively simple phenotypes like death. More recently, dose-response designs have been extended to microarray experiments where thousands of transcriptional responses can be simultaneously profiled.4 Given that metabolites are downstream of transcripts, serving as substrates for the enzymatic targets of small-molecule drugs and toxicants, it is appealing to consider profiling their response as a function of chemical concentration. To date, however, dose-response experiments performed with untargeted metabolomics have been limited. In part, this is because flexible computational platforms to process the resulting metabolomic data sets have been lacking and researchers have had to rely on ad hoc strategies for processing.
When a small-molecule drug or toxicant is known to elicit a biochemical effect, metabolomics is commonly applied to determine its mode of action.5–7 The idea is that when a small molecule inhibits a protein, the protein’s substrate increases and its product decreases.8 In theory, comprehensive profiling of the metabolome can therefore identify which protein is inhibited de novo. There are two complications with this approach. First, because of the interconnectivity of metabolic networks, changes in metabolite levels are rarely limited to the substrates and products of an impaired protein.9 Second, small molecules are much more promiscuous than previously thought, commonly interacting with more than one protein.10
Given these complexities, a simple pairwise comparison of untreated and treated samples with metabolomics may not be adequate to determine a chemical’s mode of action. A better experimental design is to perform untargeted metabolomics at multiple concentrations of drug or toxicant, so that perturbations of proteins having different binding affinities with the administered chemical can potentially be resolved. Here, we introduce an experimental strategy to perform these dose-response metabolomic experiments. Critical to our workflow is software we developed called TOXcms. Like other programs in the XCMS family that we have introduced, TOXcms is built to run in tandem with XCMS.11–14 Importantly, in addition to accepting other types of omic and phenotypic data, the flexible input format of TOXcms also enables compatibility with any metabolomic software after minimal reformatting (e.g., MZmine, MetaboAnalyst, MS-DIAL, Mass Profiler Professional, Compound Discoverer, etc.).15–17
Dose-response metabolomics is well suited for a broad range of research applications, such as optimization of drug dosing, evaluation of drug synergy, safety assessment of increasing concentrations of environmental exposure chemicals, and identifying off-target effects of small interfering RNAs (siRNAs). Given its platform-independent input, TOXcms can also be applied to multiomics experiments to identify correlations between orthogonal data sets. In this work, we focus on the application of dose-response metabolomics to find off-target effects of small-molecule drugs (Figure 1 and Figure S1). Off-target effects occur when a drug interacts with one or more unintended proteins. It is estimated that the average small-molecule drug interacts with six different proteins.18 Yet, a comprehensive analysis of possible off-targets has not been performed for most drugs currently in research and clinical use, which we recognize as a potential opportunity for metabolomics.
Figure 1. Schematic of our approach to deconvolute protein targets on the basis of dose-response curves.

Drug X binds two protein targets with different affinities. Drug X binds the primary target (target 1) with high affinity. Thus, the accumulation of substrate 1 (S1) and the depletion of product 1 (P1) occurs at low concentrations of drug. In contrast, drug X binds the off-target protein (target 2) with low affinity. The accumulation of substrate 2 (S2) and the depletion of product 2 (P2) occurs only at high concentrations of drug. See Figure S1 for an experimental example of the same schematic
METHODS
BT549 breast cancer cells were treated with etomoxir concentrations that spanned 10-200 μM. HEK-293T cells were treated with metformin concentrations that spanned 0.05-1.5 mM and rotenone concentrations that spanned 10-200 nM. These cell lines were selected for investigation on the basis of previous work.19–21
Metabolites were extracted from cells and analyzed with an Agilent 1260 HPLC system or an Agilent 1290 UPLC system coupled to an Agilent 6540 or an Agilent 6545 quadruple time-of-flight mass spectrometer. Polar metabolites were separated on a Luna aminopropyl column at room temperature or a zic-pHILIC column at 40 °C. Lipids were separated on a Kinetex evo C18 column operated at 35 °C.
After the raw data files were converted from each dose-response experiment to an mzXML format, peak detection and correspondence determination were performed by the XCMS R package with the centWave, obiwarp, and group.density algorithms. The XCMS output files were then analyzed by TOXcms. Statistical comparisons were performed by the calcdosestat function. Monotonically increasing and decreasing dose-response trends were filtered out by the trendfilter function (pval_cutoff = 0.05, pval_thres = 1, anova_cutoff = 0.05, trend = “mono”, relChange_cutoff = 0.10). To estimate ED50 values, each trend was fit with a generalized dose-response logistic model as described previously22. Global dose-response patterns were analyzed and visualized by the plotpca and clusttrend functions (sort.method = c(“clust”, “layer”), sort.thres=20).
Additional method details can be found in Supporting Information.
All data generated or analyzed during this study are included in the publication and supporting files. TOXcms is freely available at pattilab.wustl.edu/software/toxcms.
RESULTS AND DISCUSSION
TOXcms Workflow.
In this work, we will primarily focus on the application of liquid chromatography/mass spectrometry (LC/MS) because of its widespread use in untargeted metabolomics and its relatively broad metabolite coverage compared to other analytical platforms.23 LC/MS analysis of a typical metabolic extract from a biological sample results in the detection of thousands of so-called metabolomic features, which are defined as signals in the LC/MS data having a unique pair of retention time and m/z values. While dose-response curves could theoretically be made for each reported feature without additional software, such an effort would require inspection of tens of thousands of data points and manual statistical assessment. TOXcms is designed to automate this process.
Our approach is to model dose-response curves with TOXcms, which is a program that we designed to process standard files output by the XCMS software. First, data are processed by XCMS for peak detection and correspondence determination. No pairwise comparisons or statistics are needed from XCMS. The XCMS peaks table is then directly imported into TOXcms as a CSV file. We note that, even though TOXcms was designed to work in tandem with XCMS, it is compatible with any data having dose and intensity coordinates (Figure 2). This includes peaks tables from other commonly used software in metabolomics, both freely available and commercial. An advantage of a platform-independent input is that TOXcms can be used to analyze data from a variety of mass spectrometry methods, ranging from gas chromatography/mass spectrometry to flow injection analysis. Moreover, dose-response curves of metabolites derived from mass spectrometry can easily be correlated with dose-response curves of other physical parameters such as oxygen consumption and proliferation rate by using TOXcms.
Figure 2. Informatic workflow of TOXcms.
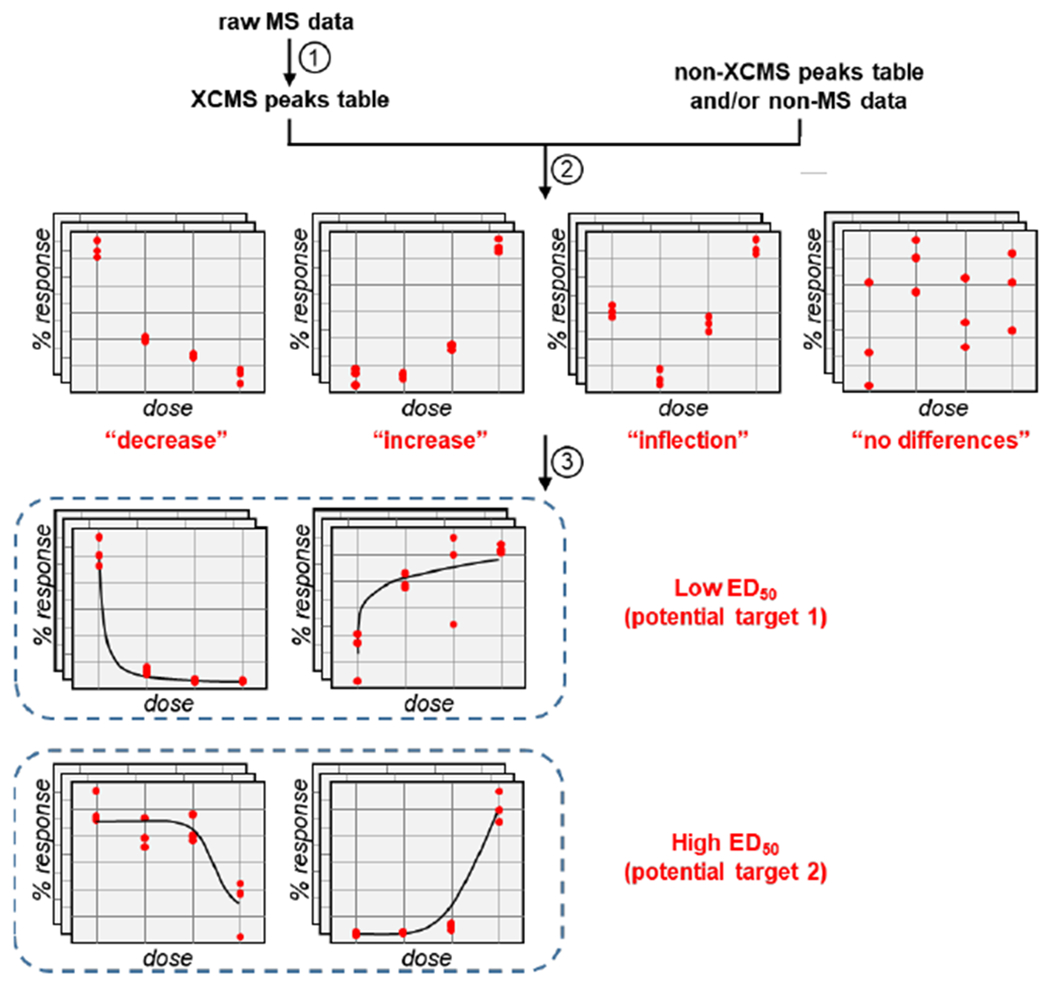
There are three general steps to the TOXcms workflow. First, raw mass spectrometry (MS) data are converted into a peaks table by XCMS (left) or any other appropriate peak detection and alignment algorithm (right). Second, from the peaks table, the intensities of each feature are plotted as a function of dose. Statistical comparisons between each dose are then performed to determine whether there are any significant changes to a feature’s intensity. Features showing no statistically significant changes are placed into the “no differences” group. Features showing only decreases or increases in intensity are placed into the “decrease” or “increase” group, respectively. Features showing significant increases and decreases are placed into the “inflection” group. Last, the third data-processing step of TOXcms is to cluster dose-response curves that have similar shapes by combining ED50 values with hierarchical clustering or principal component analysis (see Figure 3 and Figure S3)
A key function of TOXcms is organizing the dose-response plots on the basis of statistical trends, which is essential to correlating changes between features. Features that monotonically decrease with dose (i.e., features whose intensity only decreases with increasing drug concentration) are assigned to the “decrease” group. Features that monotonically increase with dose (i.e., features whose intensity only increases with increasing drug concentration) are assigned to the “increase” group. When a feature shows both an increase and a decrease with dose, it is assigned to the “inflection” group. Although an inflection trend may not be insightful when searching for drug targets, we anticipate that this group will be most relevant to other applications of TOXcms such as rescue experiments that are discussed later. Finally, features that do not show any statistically significant changes between doses are assigned to the “no differences” group. Each detected feature is assigned to one of these four groups (Figure S2). To start the assignment process, a simple t-test is performed between each pair of adjacent doses. If none of the p-values calculated from adjacent doses of a particular feature are above a user-defined threshold, then the feature is automatically placed into the “no differences” group. On the other hand, if at least one t-test produces a p-value that passes the user’s filter, then the data are evaluated by an ANOVA test. Provided the difference between the means of all doses is not above a user-specified significance threshold, the feature is moved to the “no differences” group. If the feature does pass the ANOVA test, then it is assessed to be monotonically increasing or decreasing by applying eqs 1–3:
| (1) |
| (2) |
| (3) |
where Ci,i−1 = 1 if the intensity measurements of dose i and dose i−1 are significantly different and Ci,i−1 = 0 if they are not. When the mean intensity at dose i is greater than that at dose i−1, then reli,i−1 = 1. If it is lower at dose i than at dose i−1 then reli,i−1 = −1, and if the mean intensities do not change, reli,i−1 = 0. By superimposing C and rel using eqs 1–3, a trend is classified into monotonically increasing, monotonically decreasing, or inflection groups. Next, for the monotonic-increase group and the monotonic-decrease group, the effective dose at 50% maximal value (ED50) is calculated by fitting a logistic dose-response model to the measured intensities as has been previously described.22 We note that the calculated ED50 values may not equal the actual ED50 values when insufficient data have been input into TOXcms to create a well-defined dose-response curve (e.g., insufficient replicates and/or doses). In this work, we observed meaningful changes with only three biological replicates but additional replicates may be required in other studies. Finally, to aid in the interpretation of TOXcms results at the global level, principal component analysis (PCA) is performed on the normalized intensities of all input features. The first two components are plotted and colored according to the predicted ED50 values. Additionally, hierarchical clustering of feature intensities is performed to provide an orthogonal visualization (Figure S3). For detailed analysis, dose-response curves are plotted for all detected features and the full TOXcms results are provided in an automatically generated CSV file.
Dose Design.
The experiments described herein necessitate that samples be treated with multiple doses of a compound. A critical question is which dose levels should be applied (Figure S4). The potency of drugs and toxicants can span a large range of concentrations. In theory, a wide distribution of doses could be tested with fine spacing. Practically, however, sample sizes and dose ranges are limited by resources, sample type, and ethical considerations (e.g., when studies involve human subjects and animals). Additionally, the number of dose levels evaluated may be constrained by the availability of a compound, its solubility, manufactured dose sizes, etc. Strategies to identify optimal doses most efficiently when performing a dose-response study have therefore been the focus of decades of research, with many statistical theories and mathematical models having been applied. A discussion of these efforts is beyond the scope of the current work, but details can be found elsewhere.24–27
Although our TOXcms-based approach can be readily applied to animal and patient samples, the data presented in this study were generated from cells in culture. We selected doses on the basis of ED50 values available in the literature, doses used in previous studies to elicit various end points, and clinical relevance.21,28, 29 It is interesting to consider other examples, however, when no prior knowledge is available. In these instances, a trial experiment may be performed first to determine the approximate range of sensitivity to the compound. It has been suggested that a 10-fold spacing in compound concentration is adequate for such preliminary cell-culture experiments.30 Rather than doing LC/MS-based metabolomics on samples from each dose, we recommend monitoring other end points that are easier to measure (e.g., viability, respiration, etc.). Upon identifying concentration ranges where phenotypes are altered, more narrowly spaced doses around the responsive range can then be tested in a subsequent experiment by metabolomics. In our experiences, it is not essential that these doses be evenly spaced. Rather, doses should be selected to maximize fitting of the dose-response curve. Ideally, at least one dose should elicit a minimal metabolic response, at least one dose should elicit a maximal metabolic response, and several doses should elicit a metabolic response on the transition part of the ED50 curve (Figure S4).
Evaluating TOXcms Performance.
We next sought to assess the ability of TOXcms to construct dose-response curves and accurately group them on the basis of statistical trends. To create well-defined data sets for evaluation, we used in silico methods to simulate metabolomic data sets from which false positive rates (FPR) and true positive rates (TPR) were determined. We started by performing a metabolomic analysis on three biological replicates of BT549 breast cancer cells. These three experimental data files were used to simulate data sets for four different drug doses. First, we randomly selected 1000 features from the 26 853 in the experimental data and calculated their mean intensity. Next, we fit a normal distribution centered around the calculated mean intensity with a standard deviation equal to the mean intensity multiplied by a standard error that was randomly selected from the experimental measurements. We simulated three replicates for each dose by using the distribution that was fit for each of the 1000 randomly selected features. Given that the intensity of the randomly selected 1000 features exhibited no statistically significant dose-response trend between the data sets but shared the same statistical characteristics of real metabolomic data, these 1000 features allowed us to assess the FPR of TOXcms. To assess the TPR, we repeated a similar procedure for a unique set of 100 randomly selected features from the experimental BT549 data. We simulated a dose-dependent trend for these 100 features by using the generalized logistic model of dose responses with a bimodal distribution of ED50 values (Figure S5). Of the 100 features, one-half were simulated to increase as a function of dose and one-half were simulated to decrease as a function of dose. For each of the 50 increasing and decreasing features, 25 had ED50 values normally distributed around 40 arbitrary units and 25 had ED50 values normally distributed around 150 arbitrary units. For each simulated dose, we added variance equal to the mean standard error of a representative drug (etomoxir, as measured by nine experimental LC/MS runs).
These simulations resulted in three replicate data sets for each of four doses. The data contained 1000 features that showed no statistically significant trends and 100 features that showed dose-dependent trends. Each of the data files were run through the TOXcms workflow with t-test and ANOVA cutoffs of p = 0.05 and a relative change cutoff of 0.1. The TOXcms output was analyzed by recording the number of false positives and true positives. False positives occurred when any of the 1000 features not showing a dose-dependent trend were reported to have a trend by TOXcms. True positives occurred when any of the 100 features showing a dose-dependent trend were indeed reported to have a trend by TOXcms. We repeated the process 100 times, randomly selecting 1000 features to show no statistical trend and randomly selecting 100 features to show a dose-dependent trend in each simulation. We estimated 95% confidence intervals for the FPR, TPR, and false discovery rate (FDR) by using a bootstrapping method where we resampled the true positive/false positive counts of each TOXcms output file 1000 times. The FPR, TPR, and FDR were calculated for each resampled data set. The 25th and 975th lowest and highest values were used as the upper and lower bounds of the confidence interval for each metric, respectively. We determined the FPR to be <0.4%, the TPR to be ~85%, and the FDR to be <4% (Figure 3, Figure S6, and Table S1). The accuracy of the ED50 values returned by TOXcms was also evaluated by PCA. As shown in Figure 3 and Figure S6, a PCA plot of the ED50 values output by TOXcms was able to recover the underlying bimodal distribution of the true ED50 values. These results suggest that PCA plots produced by TOXcms can be used to visualize clusters of metabolomic features that change with correlated dose-response trends and unimodal ED50 value distributions.
Figure 3. PCA plot produced by TOXcms can be used to visualize clusters of metabolomic features that change with similar dose-response classifications.

Each dot on the plot represents a distinct feature. Features in the in silico data showing a monotonic increase are on the left, and features showing a monotonic decrease are on the right. Data are color-coded based on their ED50 values. Features in the in silico data that do not show a statistically significant trend are colored gray. The plot shows that the ED50 values calculated by TOXcms are in general agreement with the actual ED50 values (see Figure S6) and that TOXcms is able to capture the bimodal distribution of dose-response curves in the data set, reflected by the separation of low ED50 values (circled in purple) and high ED50 values (circled in green)
TOXcms Applied to Metformin.
We aimed to apply TOXcms to assess the possibility that metformin has multiple protein targets with different drug binding affinities. We first treated HEK-293T cells with 0, 0.05, 0.5, 1, or 1.5 mM concentrations of metformin for 48 h. Cellular metabolites were then extracted, analyzed by LC/MS, and the data were processed with the TOXcms workflow outlined above. Out of the 19 506 metabolomic features detected, 371 showed monotonic increases or monotonic decreases in intensity as a function of metformin dose (p-value and ANOVA ≤ 0.05). Metabolites identified to have a monotonic increase are consistent with the expected mechanism of metformin action (Figure S7a–d).31 Most interestingly, the PCA plot produced by TOXcms showed only two clusters corresponding to features that increased or decreased as a function of metformin with similar ED50 values (Figure S7e). The metformin plot is distinct from that of the in silico data above (Figure 3), where increasing or decreasing features were observed for two distinct clusters of ED50 values. Our results suggest that only a single metabolic network effect occurs over the drug concentrations of metformin evaluated.
TOXcms Applied to Etomoxir.
Etomoxir is known to inhibit the enzyme carnitine palmitoyltransferase I (CPT1), which transforms acyl-CoAs to acyl-carnitines (ACs). Given that neither long-chain fatty acids nor acyl-CoAs can directly cross the inner mitochondrial membrane, CPT1 activity is required to transport long acyl chains into the mitochondrial matrix for oxidation. Beyond its applications in metabolic research, etomoxir has also drawn interest as a potential therapeutic for conditions ranging from multiple sclerosis to cancer.32–34 Notably, however, concerns of hepatotoxicity from a clinical trial of etomoxir in congestive heart failure patients have limited applications of etomoxir to treat human disease.35
A benefit of evaluating our approach with etomoxir is that CPT1 has many substrates and products having a variable acyl chain length. Each chemical species should yield similar dose-response trends that are grouped by TOXcms. As an experimental test, we treated BT549 breast cancer cells with 0, 10, 50, or 200 μM of etomoxir for 48 h. Complementary methods were applied to analyze polar and nonpolar metabolites in separate LC/MS experiments. Processing of the resulting data with XCMS led to the detection of 26 920 total features. We point out that, although XCMS must process data from reversed-phase and hydrophilic interaction liquid chromatography experiments separately, TOXcms can model dose-response curves for any set of input features (irrespective of the m/z values and retention times assigned by XCMS). This allowed us to process all XCMS files simultaneously, enabling TOXcms to make correlations between the different experiments. From the XCMS input files, TOXcms identified 908 features (<4% of the total features) having a statistically significant monotonic increase or monotonic decrease as a function of etomoxir dose. Visualization of the features on the PCA plot produced by TOXcms showed a bimodal distribution of ED50 values that was similar to the pattern observed for our in silico data but distinct from the pattern of our metformin data, indicating that etomoxir has at least two biochemical targets at the concentrations tested (Figure 4a).
Figure 4.
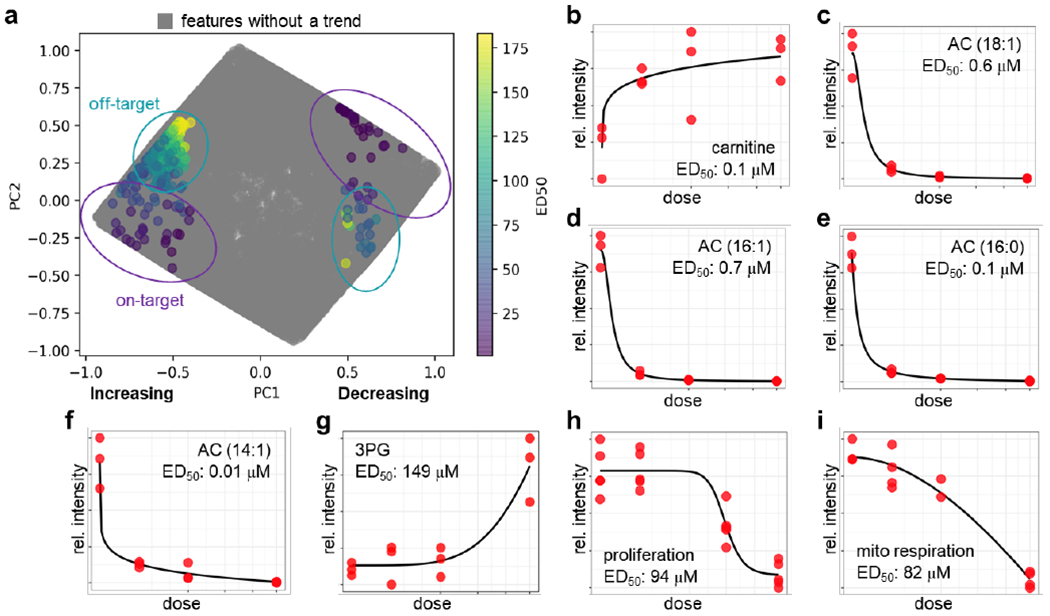
TOXcms reveals that etomoxir has a second protein target at 200 μM in BT549 breast cancer cells. (a) PCA plot shows distribution of ED50 values calculated by TOXcms. Each dot on the plot represents a distinct feature. Features with increasing dose-response trends are shown on the left of the plot, and features with decreasing dose-response trends are shown on the right. ED50 values are color-coded. Features in the data that do not show a statistically significant trend are colored gray. Similar to the in silico data plotted in Figure 3, data from etomoxir-treated samples show a bimodal distribution of ED50 values (purple and green circles). (b-f) Features in the purple circles were identified as substrates and products of the primary target of etomoxir (i.e., CPT1). (g) Features in the green circles were identified to be in pathways distinct from CPT1, and hence were annotated as off-target. (h) At hundreds of micromolar, etomoxir decreases proliferation of BT549 cells. The dose-response curve of this phenotype does not correlate with the dose-response curve of the substrates and products of CPT1. Instead, the dose-response curve of proliferation correlates with the off-target effect of etomoxir. (i) Similarly, the dose-response curve of mitochondrial respiration correlates with the dose-response curve of etomoxir’s off-target effect. Taken together, these data demonstrate that etomoxir inhibits CPT1 at low concentrations but inhibits mitochondrial respiration via an off-target effect at high concentrations
We first focused on features from the PCA plot having ED50 values less than 2 μM (colored purple on Figure 4a), which we expected to be associated with CPT1. For these features, we found a subgroup exhibiting a monotonic decrease in intensity as a function of etomoxir dose (right of Figure 4a) and a subgroup of features exhibiting a monotonic increase in intensity as a function of etomoxir dose (left of Figure 4a). As anticipated, among the subgroup of features showing a monotonic increase in intensity was carnitine (Figure 4b). In contrast, among the subgroup of features showing a monotonic decrease in intensity was many ACs, including those with acyl chains of 18:0, 18:1, 16:0, 16:1, 14:0, 14:1, 12:0, 10:0, and 8:0 (Figure 4c–f and Figure S8). We note that the methods we applied here are not optimized for analysis of CoA species, which are substrates for CPT1. Our ability to detect accumulated acylCoAs was therefore limited. Notwithstanding, it is clear that the cluster of features from the PCA plot having low ED50 values (<2 μM) is a result of altered CPT1 activity. Our observations indicate that low micromolar concentrations of etomoxir are sufficient to inhibit its primary target, which is consistent with previous reports.36, 37
We next turned our attention to features from the PCA plot having high ED50 values, which we associated with an off-target effect of etomoxir. These features also partitioned into those showing a monotonic increase and a monotonic decrease in intensity as a function of etomoxir dose. Although there were many features in the cluster that we could not identify, which is typical in untargeted metabolomics 38, we did find a change in central carbon metabolism. The change was found to have a statistically significant difference at 200 μM etomoxir but showed no significant differences at lower doses of etomoxir (Figure 4g). The dose-dependent trends were distinct from that of carnitine and ACs, which showed 90% of their change at 10 μM etomoxir. The disparity in ED50 values suggests that alterations in central carbon metabolism are independent of CPT1 inhibition and instead an off-target effect of etomoxir interacting with a second protein having a lower drug binding affinity. Follow-up experiments revealed that high concentrations of etomoxir inhibit complex I of the electron transport chain (Figure S9). The data are consistent with recent reports that concentrations of etomoxir ~100 μM and higher impair mitochondrial respiration through direct inhibition of complex I of the electron transport chain and adenine nucleotide translocase, which subsequently causes alterations in central carbon metabolism.37, 39, 40
TOXcms Applied to Rotenone.
The doses we used to study etomoxir and metformin are higher than those that are typical when evaluating chemical toxicity.30 To ensure that our approach is applicable to toxicological studies involving lower ranges of chemical concentrations, we performed a dose-response analysis of rotenone. Rotenone is a potent inhibitor of complex I of the electron transport chain and is known to be toxic to HEK-293 cells at nanomolar concentrations.21 We treated HEK-293T cells with 0, 10, 50, and 200 nM concentrations of rotenone for 48 h prior to performing metabolomic analysis. Processing of the resulting data with XCMS led to the detection of 18 963 total features, of which 1240 were identified by TOXcms to have a statistically significant monotonic increase or decrease as a function of rotenone dose. Visualization of the features on the PCA plot produced by TOXcms showed a single cluster of ED50 values (Figure S10), consistent with rotenone having one metabolic network effect over the concentrations tested. Among the metabolites identified to have dose-dependent changes were those in central carbon metabolism that would be expected to change upon complex I inhibition (Figure S11).
Correlating Phenotypic Data with Metabolomic Data.
We sought to develop a method that could better correlate alterations in metabolomic features with alterations in other measurable phenotypes. Our approach is to acquire dose-response data on the phenotype of interest, which we then interpret in conjunction with metabolomic dose-response experiments. By “phenotype of interest”, we mean any physical measurement that is distinct from the LC/MS-based metabolomic analyses. Examples of phenotypic measurements might include cell viability, size, growth rate, respiration, etc. Although we suspect that phenotypic measurements will typically not be made by using a mass spectrometer, our approach does not prevent it (e.g., using LC/MS to measure protein levels). Data for one or more phenotypes can be input into TOXcms in the format of dose-intensity coordinates, along with a metabolomic peaks table (Figure 2). Dose-dependent trends in phenotypic data can then be statistically compared to dose-dependent trends in metabolomic features.
To illustrate the value of our approach, we build upon the etomoxir example detailed above. We and others have shown that proliferation of BT549 cancer cells is sensitive to etomoxir at 200 μM.19, 37 An interesting mechanistic question is why. When we compared BT549 cells treated with 200 μM etomoxir to untreated BT549 control cells by using a conventional pairwise analysis of untargeted metabolomic data, we found 1731 features that were statistically different. Not only is this large number of metabolomic features difficult to interpret, it can also be misleading because included in the list of altered features is acylcarnitines. Importantly, by integrating additional dose data into our analysis with TOXcms, we found that the dose-response trend of cell proliferation (ED50 = 94 μM) was poorly correlated with the dose-response trend of acylcarnitine levels (ED50 < 1 μM). In contrast, the dose-response trend of a subset of the 1731 altered features was highly correlated with cell proliferation (Figure 4h). Some of these features corresponded to metabolites in central carbon metabolism whose alteration was a result of etomoxir interacting with off-target proteins, which have recently been identified as complex I of the electron transport chain and adenine nucleotide translocase (Figure S9).37, 39, 40 As expected, TOXcms also reported a strong correlation between this subset of features and mitochondrial respiration as measured with a Seahorse Analyzer (ED50 = 82 μM, Figure 4i). Taken together, this example shows that by using TOXcms to analyze metabolomic dose-response curves, we can correlate a specific phenotype (i.e., decreased proliferation) with a specific metabolic response (i.e., impaired respiration).
Data Normalization for PCA.
Given that normalization strategies are known to affect the results of PCA, we evaluated six different methods to normalize our etomoxir data: range scaling, autoscaling, Pareto scaling, vast scaling, level scaling, and square-root scaling. The advantages and disadvantages of each approach for metabolomic data are context dependent and have been considered in detail previously.41 Here, we found that range scaling was best suited for our dose-response metabolomic approach, and therefore we chose to implement it within TOXcms to normalize feature intensities. In range scaling, intensity values are transformed such that the minimum intensity of a feature is set to zero and the maximum intensity is set to one. The approach removes the large dynamic range between different features, which spans 4–5 orders of magnitude in the raw data. In comparison to other approaches, range scaling and autoscaling were the only methods that led to effective PCA separation of dose-response patterns from our etomoxir data set (Figure S12). A benefit of range scaling over autoscaling is that it has been shown in the past to allow accurate estimation of ED50 values in dose-response modeling.42 In addition, range-scaled data can be input directly into the four-parameter logistic model that we apply within TOXcms.
TOXcms Identifies Off-target Effects of Genetic Knockdown.
In addition to finding metabolomic features that show monotonic increases or monotonic decreases in intensity, TOXcms also identifies features whose intensities are statistically altered in different directions between sample classes. When the intensity of a feature changes direction between sample classes, we refer to it as having an “inflection” pattern (Figure 2). Although inflection points within a trend may not be applicable in the assessment of drug targets, it is compelling to consider the insight they might offer in other types of studies such as rescue experiments. A challenge when knocking down a gene with siRNAs, for instance, is that siRNAs can nonspecifically target unintended genes with only partial sequence complementarity.43, 44 The gold-standard control to confirm that the observed phenotype is a result of siRNA-induced silencing of a specific gene is rescue of the observed phenotype by expressing a functional version of the target gene that is resistant to the siRNA.45 As a simple example, when the gene being knocked down reduces expression of a metabolic enzyme, the metabolite produced by the target protein should decrease in concentration with siRNA (Figure 5). Rescuing the cells with a functional version of the target gene resistant to the siRNA should then restore the concentration of the metabolite produced by the target protein. Thus, when a wild-type, a knockdown, and a rescue sample class are considered in that order, a “V” trend should be observed for the metabolite produced by the target protein. Conversely, the concentration of the target protein’s substrate would be expected to increase upon knockdown and display a “Λ” trend in the wild-type, knockdown, and rescue sample classes, respectively.
Figure 5.

Application of TOXcms to identify off-target effects of siRNA. (a) Theoretical schematic showing expected results for a biochemical reaction in which substrate S is converted to product P by enzyme E. P is detected in wild-type (WT) samples, but not after knockdown (KD) of E. To ensure that P decreases due to enzyme knockdown, siRNA-resistant E is overexpressed in the KD samples to restore activity. Overexpression of E is expected to “rescue” levels of P. (b) In WT samples, acylcarntines are readily detected by LC/MS. After KD of CPT1, however, AC levels drop. AC levels are restored by expressing CPT1 that is resistant to siRNA. Data shown were output by TOXcms. (c) Example of a metabolic alteration identified by TOXcms to be a result of an off-target effect of siRNA, rather than an effect of CPT1 KD.
TOXcms automates the processing of metabolomic data from these kind of rescue experiments. Using TOXcms to analyze rescue data at the comprehensive level, rather than merely evaluating the substrates and products of the target protein manually, has the major benefit of potentially discovering unexpected off-target effects. Metabolites that decrease with knockdown but that do not increase after rescue may not be associated with the target gene. To extend our etomoxir study to rescue experiments, we treated BT549 breast cancer cells with scrambled siRNA or with siRNA targeting CPT1A to serve as a control group and a CPT1 knockdown group, respectively. We then rescued knockdown cells by overexpressing siRNA-resistant CPT1A to restore CPT1 protein levels (Figure S13). As expected, acylcarnitines were successfully identified by TOXcms as having a “V” trend (Figure 5b, Figure S14a–c). Interestingly, we confirmed that a number of features showing “V” or “Λ” trends were not substrates or products of CPT1. It is likely that these signals correspond to metabolic adaptations of impaired fatty acid oxidation. We point out that we did find features that decreased with siRNA but whose levels were not restored by overexpression of siRNA-resistant CPT1, indicating that our siRNA had off-target metabolic effects (Figure 5c and Figure S14d). Additionally, we discovered features that did not change between control and knockdown conditions, but whose levels did change upon overexpression of CPT1 (Figure S14e–f). These results suggest that overexpression of the knocked down protein can itself have metabolic consequences that potentially complicate interpretation of rescue experiments in metabolism research.
Limitations.
The output of TOXcms should be viewed as a source of leads from which potential protein targets can be inferred on the basis of alterations in substrate and product levels. The approach provides an indirect readout of drug-protein interactions and is complicated by the possibility that the levels of multiple metabolites can change in response to a single impaired protein.9 Despite the challenge of determining the source of metabolic dysregulation, however, many successful applications have demonstrated that metabolomics can effectively identify alterations in specific protein activities.46, 47 Here, we build upon this prior work to facilitate deconvolution of metabolic changes associated with different protein targets. We wish to point out three possible limitations of the deconvolution approach we have introduced. First, because impairing enzymes in the same metabolic pathway can lead to similar metabolomic profiles, deconvoluting these protein targets is likely to be challenging with our approach. Second, even when they are in different metabolic pathways, enzymes with similar binding affinities for a test compound will produce overlapping dose-response profiles that cannot be deconvolved with the strategies that we presented here. Third, given that it uses metabolites as a readout, dose-response metabolomics alone may not be effective at deconvoluting nonmetabolic targets (e.g., ion channels, receptors, etc.). In these cases, integrating metabolomics with other “omic” technologies and biochemical assays may prove helpful.
CONCLUSION
Here, we introduce the TOXcms software to support dose-response metabolomics. The basis of the experimental paradigm is to trace each of the individual features detected in an untargeted metabolomic experiment through a series of related perturbations or exposures, such as an increasing dose of drug or toxicant. After each feature intensity is plotted as a function of perturbation or exposure, TOXcms then performs a statistical assessment to classify each trend as increasing, decreasing, increasing and decreasing, or not changing over the sample groups. The strategy is distinct from other metabolomic workflows that have been applied previously to multiple sample groups, which are not designed to assess stimulus-response curves.48, 49
In this work, we focused on applying TOXcms to study off-target drug effects. We examined two drugs with contrasting safety profiles, metformin and etomoxir. Metformin has been used in the clinic for over 60 years without any notable side effects or safety issues.50 In contrast, clinical development of etomoxir has been limited due to hepatotoxicity.35 A metabolomic dose-response analysis of cells treated with metformin showed evidence that metformin impairs mitochondrial respiration, which is known to be its primary target. A metabolomic dose-response analysis of cells treated with etomoxir, on the other hand, provided clear evidence that etomoxir has multiple protein targets at high concentrations. Our results revealed that, at 200 μM, etomoxir inhibits both CPT1 activity and mitochondrial respiration. The former is the well-established primary target of etomoxir, whereas the latter is an off-target effect.
Supplementary Material
Footnotes
Supporting Information
Refer to Web version on PubMed Central for supplementary material.
All data generated or analyzed during this study are included in the publication and supporting files. TOXcms is freely available at pattilab.wustl.edu/software/toxcms.
The authors declare no conflict of interest.
REFERENCES
- 1.Snyder R, Basic Concepts of the Dose-Response Relationship. In Assessment and Management of Chemical Risks, American Chemical Society: 1984; Vol. 239, pp 37–55. [Google Scholar]
- 2.Waddell WJ, History of dose response. J Toxicol Sci 2010, 35 (1), 1–8. [DOI] [PubMed] [Google Scholar]
- 3.Lowe ES; Balis FM, CHAPTER 18 - Dose-Effect and Concentration-Effect Analysis In Principles of Clinical Pharmacology (Second Edition), Atkinson AJ; Abernethy DR; Daniels CE; Dedrick RL; Markey SP, Eds. Academic Press: Burlington, 2007; pp 289–300. [Google Scholar]
- 4.Ji RR; de Silva H; Jin Y; Bruccoleri RE; Cao J; He A; Huang W; Kayne PS; Neuhaus IM; Ott KH; Penhallow B; Cockett MI; Neubauer MG; Siemers NO; Ross-Macdonald P, Transcriptional profiling of the dose response: a more powerful approach for characterizing drug activities. PLoS Comput Biol 2009, 5 (9), e1000512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Kaddurah-Daouk R; Kristal BS; Weinshilboum RM, Metabolomics: a global biochemical approach to drug response and disease. Annu Rev Pharmacol Toxicol 2008, 48, 653–83. [DOI] [PubMed] [Google Scholar]
- 6.Southam AD; Lange A; Hines A; Hill EM; Katsu Y; Iguchi T; Tyler CR; Viant MR, Metabolomics reveals target and off-target toxicities of a model organophosphate pesticide to roach (Rutilus rutilus): implications for biomonitoring. Environ Sci Technol 2011, 45 (8), 3759–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Warth B; Spangler S; Fang ML; Johnson CH; Forsberg EM; Granados A; Martin RL; Domingo-Almenara X; Huan T; Rinehart D; Montenegro-Burke JR; Hilmers B; Aisporna A; Hoang LT; Uritboonthai W; Benton HP; Richardson SD; Williams AJ; Siuzdak G, Exposome-Scale Investigations Guided by Global Metabolomics, Pathway Analysis, and Cognitive Computing. Analytical Chemistry 2017, 89 (21), 11505–11513. [DOI] [PubMed] [Google Scholar]
- 8.Saghatelian A; Trauger SA; Want EJ; Hawkins EG; Siuzdak G; Cravatt BF, Assignment of endogenous substrates to enzymes by global metabolite profiling. Biochemistry 2004, 43 (45), 14332–9. [DOI] [PubMed] [Google Scholar]
- 9.Kell DB; Goodacre R, Metabolomics and systems pharmacology: why and how to model the human metabolic network for drug discovery. Drug Discovery Today 2014, 19 (2), 171–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jalencas X; Mestres J, On the origins of drug polypharmacology. MedChemComm 2013, 4 (1), 80–87. [Google Scholar]
- 11.Smith CA; Want EJ; O’Maille G; Abagyan R; Siuzdak G, XCMS: processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal Chem 2006, 78 (3), 779–87. [DOI] [PubMed] [Google Scholar]
- 12.Huang X; Chen YJ; Cho K; Nikolskiy I; Crawford PA; Patti GJ, X13CMS: global tracking of isotopic labels in untargeted metabolomics. Anal Chem 2014, 86 (3), 1632–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mahieu NG; Genenbacher JL; Patti GJ, A roadmap for the XCMS family of software solutions in metabolomics. Curr Opin Chem Biol 2016, 30, 87–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Patti GJ; Tautenhahn R; Siuzdak G, Meta-analysis of untargeted metabolomic data from multiple profiling experiments. Nat Protoc 2012, 7 (3), 508–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Olivon F; Grelier G; Roussi F; Litaudon M; Touboul D, MZmine 2 Data-Preprocessing To Enhance Molecular Networking Reliability. Anal Chem 2017, 89 (15), 7836–7840. [DOI] [PubMed] [Google Scholar]
- 16.Xia J; Sinelnikov IV; Han B; Wishart DS, MetaboAnalyst 3.0-making metabolomics more meaningful. Nucleic Acids Res 2015, 43 (W1), W251–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsugawa H; Cajka T; Kind T; Ma Y; Higgins B; Ikeda K; Kanazawa M; VanderGheynst J; Fiehn O; Arita M, MS-DIAL: data-independent MS/MS deconvolution for comprehensive metabolome analysis. Nat Methods 2015, 12 (6), 523–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mestres J; Gregori-Puigjane E; Valverde S; Sole RV, Data completeness--the Achilles heel of drug-target networks. Nat Biotechnol 2008, 26 (9), 983–4. [DOI] [PubMed] [Google Scholar]
- 19.Camarda R; Zhou AY; Kohnz RA; Balakrishnan S; Mahieu C; Anderton B; Eyob H; Kajimura S; Tward A; Krings G; Nomura DK; Goga A, Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat Med 2016, 22 (4), 427–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liu X; Romero IL; Litchfield LM; Lengyel E; Locasale JW, Metformin Targets Central Carbon Metabolism and Reveals Mitochondrial Requirements in Human Cancers. Cell Metab 2016, 24 (5), 728–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hsieh JH; Huang R; Lin JA; Sedykh A; Zhao J; Tice RR; Paules RS; Xia M; Auerbach SS, Real-time cell toxicity profiling of Tox21 10K compounds reveals cytotoxicity dependent toxicity pathway linkage. PLoS One 2017, 12 (5), e0177902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ritz C; Baty F; Streibig JC; Gerhard D, Dose-Response Analysis Using R PLoS One 2015, 10 (12), e0146021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Milne SB; Mathews TP; Myers DS; Ivanova PT; Brown HA, Sum of the parts: mass spectrometry-based metabolomics. Biochemistry 2013, 52 (22), 3829–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fedorov VV; Leonov SL, Optimal Design of Dose Response Experiments: A Model-Oriented Approach. Drug Information Journal 2001, 35 (4), 1373–1383. [Google Scholar]
- 25.Holland-Letz T; Kopp-Schneider A, Optimal experimental designs for dose-response studies with continuous endpoints. Arch Toxicol 2015, 89 (11), 2059–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hamlett A; Ting N; Hanumara RC; Finman JS, Dose Spacing in Early Dose Response Clinical Trial Designs. Drug Information Journal 2002, 36 (4), 855–864. [Google Scholar]
- 27.Müller H-G; Schmitt T, Choice of Number of Doses for Maximum Likelihood Estimation of the ED50 for Quantal Dose-Response Data. Biometrics 1990, 46 (1), 117–129. [PubMed] [Google Scholar]
- 28.Ceccarelli SM; Chomienne O; Gubler M; Arduini A, Carnitine palmitoyltransferase (CPT) modulators: a medicinal chemistry perspective on 35 years of research. J Med Chem 2011, 54 (9), 3109–52. [DOI] [PubMed] [Google Scholar]
- 29.He L; Wondisford FE, Metformin action: concentrations matter. Cell Metab 2015, 21 (2), 159–162. [DOI] [PubMed] [Google Scholar]
- 30.Niepel M; Hafner M; Chung M; Sorger PK, Measuring Cancer Drug Sensitivity and Resistance in Cultured Cells. Curr Protoc Chem Biol 2017, 9 (2), 55–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Andrzejewski S; Siegel PM; St-Pierre J, Metabolic Profiles Associated With Metformin Efficacy in Cancer. Front Endocrinol (Lausanne) 2018, 9, 372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Caspary F; Elliott G; Nave BT; Verzaal P; Rohrbach M; Das PK; Nagelkerken L; Nieland JD, A new therapeutic approach to treat psoriasis by inhibition of fatty acid oxidation by Etomoxir. Br J Dermatol 2005, 153 (5), 937–44. [DOI] [PubMed] [Google Scholar]
- 33.Shriver LP; Manchester M, Inhibition of fatty acid metabolism ameliorates disease activity in an animal model of multiple sclerosis. Sci Rep 2011, 1, 79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Carracedo A; Cantley LC; Pandolfi PP, Cancer metabolism: fatty acid oxidation in the limelight. Nat Rev Cancer 2013, 13 (4), 227–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Holubarsch CJ; Rohrbach M; Karrasch M; Boehm E; Polonski L; Ponikowski P; Rhein S, A double-blind randomized multicentre clinical trial to evaluate the efficacy and safety of two doses of etomoxir in comparison with placebo in patients with moderate congestive heart failure: the ERGO (etomoxir for the recovery of glucose oxidation) study. Clin Sci (Lond) 2007, 113 (4), 205–12. [DOI] [PubMed] [Google Scholar]
- 36.Lopaschuk GD; Wall SR; Olley PM; Davies NJ, Etomoxir, a carnitine palmitoyltransferase I inhibitor, protects hearts from fatty acid-induced ischemic injury independent of changes in long chain acylcarnitine. Circ Res 1988, 63 (6), 1036–43. [DOI] [PubMed] [Google Scholar]
- 37.Yao CH; Liu GY; Wang R; Moon SH; Gross RW; Patti GJ, Identifying off-target effects of etomoxir reveals that carnitine palmitoyltransferase I is essential for cancer cell proliferation independent of beta-oxidation. PLoS Biol 2018, 16 (3), e2003782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Blazenovic I; Kind T; Ji J; Fiehn O, Software Tools and Approaches for Compound Identification of LC-MS/MS Data in Metabolomics. Metabolites 2018, 8 (2). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Raud B; Roy DG; Divakaruni AS; Tarasenko TN; Franke R; Ma EH; Samborska B; Hsieh WY; Wong AH; Stuve P; Arnold-Schrauf C; Guderian M; Lochner M; Rampertaap S; Romito K; Monsale J; Bronstrup M; Bensinger SJ; Murphy AN; McGuire PJ; Jones RG; Sparwasser T; Berod L, Etomoxir Actions on Regulatory and Memory T Cells Are Independent of Cpt1a-Mediated Fatty Acid Oxidation. Cell Metab 2018, 28 (3), 504–515 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Divakaruni AS; Hsieh WY; Minarrieta L; Duong TN; Kim KKO; Desousa BR; Andreyev AY; Bowman CE; Caradonna K; Dranka BP; Ferrick DA; Liesa M; Stiles L; Rogers GW; Braas D; Ciaraldi TP; Wolfgang MJ; Sparwasser T; Berod L; Bensinger SJ; Murphy AN, Etomoxir Inhibits Macrophage Polarization by Disrupting CoA Homeostasis. Cell Metab 2018, 28 (3), 490–503 e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.van den Berg RA; Hoefsloot HCJ; Westerhuis JA; Smilde AK; van der Werf MJ, Centering, scaling, and transformations: improving the biological information content of metabolomics data. Bmc Genomics 2006, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Weimer M; Jiang XQ; Ponta O; Stanzel S; Freyberger A; Kopp-Schneider A, The impact of data transformations on concentration-response modeling. Toxicol Lett 2012, 213 (2), 292–298. [DOI] [PubMed] [Google Scholar]
- 43.Pei Y; Tuschl T, On the art of identifying effective and specific siRNAs. Nat Methods 2006, 3 (9), 670–6. [DOI] [PubMed] [Google Scholar]
- 44.Jackson AL; Bartz SR; Schelter J; Kobayashi SV; Burchard J; Mao M; Li B; Cavet G; Linsley PS, Expression profiling reveals off-target gene regulation by RNAi. Nat Biotechnol 2003, 21 (6), 635–7. [DOI] [PubMed] [Google Scholar]
- 45.Cullen BR, Enhancing and confirming the specificity of RNAi experiments. Nat Methods 2006, 3 (9), 677–81. [DOI] [PubMed] [Google Scholar]
- 46.Ismail IT; Showalter MR; Fiehn O, Inborn Errors of Metabolism in the Era of Untargeted Metabolomics and Lipidomics. Metabolites 2019, 9 (10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vincent IM; Creek DJ; Burgess K; Woods DJ; Burchmore RJ; Barrett MP, Untargeted metabolomics reveals a lack of synergy between nifurtimox and eflornithine against Trypanosoma brucei. PLoS Negl Trop Dis 2012, 6 (5), e1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Tautenhahn R; Patti GJ; Kalisiak E; Miyamoto T; Schmidt M; Lo FY; McBee J; Baliga NS; Siuzdak G, metaXCMS: second-order analysis of untargeted metabolomics data. Anal Chem 2011, 83 (3), 696–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Gowda H; Ivanisevic J; Johnson CH; Kurczy ME; Benton HP; Rinehart D; Nguyen T; Ray J; Kuehl J; Arevalo B; Westenskow PD; Wang J; Arkin AP; Deutschbauer AM; Patti GJ; Siuzdak G, Interactive XCMS Online: simplifying advanced metabolomic data processing and subsequent statistical analyses. Anal Chem 2014, 86 (14), 6931–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Long-term safety, tolerability, and weight loss associated with metformin in the Diabetes Prevention Program Outcomes Study. Diabetes Care 2012, 35 (4), 731–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.