

 This work is licensed under a
This work is licensed under a Abstract
Proliferation, differentiation and apoptosis of trophoblast cells are required for normal placental development. Impairment of those processes may lead to pregnancy-related diseases. Disruption of endoplasmic reticulum (ER) homeostasis has been associated with several reproductive pathologies including recurrent pregnancy loss and preeclampsia. In the unfolded protein response (UPR), specific ER-stress signalling pathways are activated to restore ER homeostasis, but if the adaptive response fails, apoptosis is triggered. Protein kinase RNA-like endoplasmic reticulum kinase (PERK), inositol-requiring enzyme 1 (IRE1) and Activating transcription factor 6 (ATF6) are central players in UPR and in ER-stress-induced apoptosis, as well as downstream transcription factors, as C/EBP homologous protein (CHOP). Our previous studies have shown that the endocannabinoid 2-arachidonoylglycerol (2-AG) modulates trophoblast cell turnover. Nevertheless, the role of ER-stress on 2-AG induced apoptosis and cannabinoid signalling in trophoblast has never been addressed. In this work, we used BeWo cells and human primary cytotrophoblasts isolated from term-placenta. The expression of ER-stress markers was analysed by qRT-PCR and Western blotting. ROS generation was assessed by fluorometric methods, while apoptosis was detected by the evaluation of caspase -3/-7 activities and Poly (ADP-ribose) polymerase (PARP) cleavage. Our findings indicate that 2-AG is able to induce ER-stress and apoptosis. Moreover, the eukaryotic initiation factor 2 (eIF2α)/CHOP pathway involved in ER-stress-induced apoptosis is triggered through a mechanism dependent on cannabinoid receptor CB2 activation. The results bring novel insights on the importance of ER-stress and cannabinoid signalling on 2-AG mechanisms of action in placenta.
Introduction
Placental development comprises tightly controlled processes of proliferation, differentiation and apoptosis of the trophoblast cells by the interplay of hormones, growth factors and other signalling mediators. The endocannabinoids (eCBs) anandamide (AEA) and 2-arachidonoylglycerol (2-AG) may play a crucial role in these events and modulate the complex network of cytokines and hormones in reproductive events such as decidualization, implantation and labour (Brents 2016). The cannabinoid receptor type 1 (CB1), the cannabinoid receptor type 2 (CB2), the main eCBs and their metabolic enzymes, that constitute the endocannabinoid system (ECS), have been reported in first trimester and term placenta and in the trophoblastic type BeWo cell line (Kenney et al. 1999, Park et al. 2003, Helliwell et al. 2004, Habayeb et al. 2008, Trabucco et al. 2009, Marczylo et al. 2010). Besides that, the importance of ECS in placental tissues has been demonstrated in several studies with knockout mouse models. In mice CB1−/−, trophoblast cells present reduced proliferation and placenta has a lower weight, when compared with WT mice (Sun et al. 2010).
The tight regulation of eCBs levels throughout the menstrual cycle, decidualization and gestation is well known. Alterations in the ECS homeostasis can lead to abnormal modulation of fundamental cellular processes involved in reproductive pathologies, such as preeclampsia, miscarriage and endometriosis. In women with endometriosis, there is a significant increase in plasmatic AEA and 2-AG levels. High AEA levels have also been shown to be related to failure in the in vitro fertilization embryo transfer and spontaneous miscarriage, while women with preeclampsia exhibit reduced levels of AEA (Maia et al. 2020). Moreover, we previously demonstrated that AEA and 2-AG impair the synthesis of proteins and hormones by the human syncytiotrophoblast (Costa et al. 2015a, 2016). In addition, AEA and 2-AG are able to modulate trophoblast apoptosis by inducing caspase activation and reactive oxygen species (ROS) generation (Costa et al. 2014a,b, 2015b).
Over the last years, placental stress has been linked to the pathophysiology of pregnancy complications such as preeclampsia and intrauterine growth restriction (IUGR), in which oxidative stress and endoplasmic reticulum stress (ER-stress) have gained attention (Burton et al. 2017). In fact, it was reported the association between impaired ER homeostasis and reproductive pathologies including endometriosis, recurrent pregnancy loss, preeclampsia and gestational diabetes (Fu et al. 2015, Yung et al. 2016. Guzel et al. 2017). Moreover, ER-stress has been implicated in cyclic endometrial regeneration and remodelling, folliculogenesis, fertilization, pregnancy and parturition (Schoots et al. 2018).
The ER contributes to the protein production and folding, storage and regulation of calcium and synthesis and storage of lipids. Therefore, ER is inextricably linked to the maintenance of cellular homeostasis and cell fate decisions (Almanza et al. 2019). The ER copes with the burden of unfolded proteins or misfolded proteins in its lumen by activating signalling pathways, collectively known as the unfolded protein response (UPR) (Xu et al. 2005, Szegezdi et al. 2006). Transmembrane protein sensors located in the luminal face of the ER membrane are activated through dissociation of the ER chaperone glucose-regulated protein 78 (GRP78/BiP). Protein kinase RNA-like endoplasmic reticulum kinase (PERK), inositol-requiring enzyme 1 (IRE1) and Activating transcription factor 6 (ATF6) are ER-stress transducers that sense the protein folding status and transmit the information to the cytosol and nucleus (Xu et al. 2005, Szegezdi et al. 2006). These proteins enroll UPR-mediated pathway, an adaptive mechanism that includes up-regulation of the ER-protein folding machinery, ER-associated protein degradation and inhibition of protein synthesis. However, above a certain threshold, unresolved ER-stress elicits apoptosis (Zhang et al. 2019). The players involved in cell death include PERK/ATF4/Eukaryotic Initiation Factor 2 alpha (eIF2α)-dependent induction of the pro-apoptotic C/EBP homologous protein (CHOP) and IRE1-mediated activation of TRAF2 (TNF receptor associated factor 2), which stimulates the ASK1 (apoptosis signal-regulating kinase 1)/JNK (c-Jun N-terminal kinase) cascade and Bax/Bcl2-regulated Ca2+ release from the ER. CHOP has been identified as one of the most important mediators of ER-stress-induced apoptosis (Xu et al. 2005) acting through different mechanisms. CHOP induces the down-regulation of the cell survival BCL-2 family members and the up-regulation of pro-apoptotic Bcl-2 homology 3 (BH-3)-only proteins that play a key role in mitochondrial-dependent apoptosis (Rodriguez et al. 2011). In addition, CHOP activation results in calcium release from ER to the cytosol and ROS production (Zeeshan et al. 2016). Another mechanism of action of CHOP is the modulation of the oxidative state, as overexpression of CHOP leads to an exacerbated increase of ROS at the ER (Malhotra & Kaufman 2007).
The cellular adaptive mechanisms, including the ER-stress-induced coping responses, are physiologically important for a normal placental development, since trophoblast cells undergo complex processes of cellular turnover. Bastida-Ruiz et al. (2019) showed that the ER-stress response and UPR play a role in syncytialization both in the human trophoblastic cell line model BeWo and in primary cultures of cytotrophoblasts. ER-stress response inhibition leads to a default in syncytialization, associated with alterations in cell survival. We previously described that 2-AG interferes with cytotrophoblast syncytialization through a CB receptor-dependent mechanism (Costa et al. 2015c). Moreover, in BeWo cells, we found that 2-AG induces trophoblast apoptosis, through mitochondrial membrane potential loss and increase in ROS generation and caspase -3/-7 and -9 activities, suggesting the activation of the mitochondrial pathway (Costa et al. 2014a ). The endocannabinoid 2-AG is therefore implicated in normal trophoblast turnover. However, it is unknown if the ER-stress response and UPR are involved in trophoblast apoptosis induced by cannabinoids and the role of the cannabinoid signalling. Nevertheless, eCBs are able to induce ER-stress in other cell models (Soliman et al. 2016, Almada et al. 2017).
Alterations in proliferation and exacerbation of trophoblast apoptosis have been associated with placental-related complications (Huppertz et al. 2006, Heazell & Crocker 2008). On the other hand, dysregulation of the endocannabinoid levels might lead to adverse pregnancy outcomes, including impairment of implantation, inhibition of decidualization and compromised placentation (Maia et al. 2020).
We have thus hypothesized that the UPR is involved in 2-AG induced apoptosis. Therefore, in this study, we investigated the activation of the ER-stress-induced cell death pathway to address the involvement of ER-stress within the signalling network of 2-AG actions in trophoblast cells.
Materials and methods
Dulbecco’s Modified Eagle Medium F-12 (DMEM/F12), foetal bovine serum (FBS), antibiotic-antimycotic solution (penicillin G sodium, streptomycin sulphate and amphotericin B) and trypsin were from Gibco/Invitrogen Corporation. The endocannabinoid 2-AG and the cannabinoid receptor antagonists AM281 and AM630 were from Tocris Bioscience (Bristol, UK). Percoll was from GE Healthcare and WesternBright™ ECL HRP substrate was from Advansta (Menlo Park, USA). The selective inhibitor of PERK, GSK 2656157 and the ER stress inducers thapsigargin and tunicamycin were from Santa Cruz Biotechnology. All other chemicals were from Sigma-Aldrich Co.
BeWo cell culture
BeWo cell line is a human choriocarcinoma cell line obtained from the American Type Culture Collection, being a well-accepted cytotrophoblast cell model. Cells were cultured in DMEM/F12 medium supplemented with 10% (v/v) FBS and an antibiotic-antimycotic solution (AB-AM), incubated at 37°C and 95% air/5% CO2 humidified atmosphere. For the experiments, cells were seeded in 96- or 6-well plates at densities 1.5 × 104 and 6 × 105 cells/well, respectively. After adherence (12 h), cells were treated with 2-AG (10 µM), in the presence or absence of AM281 or AM630 (1 µM), CB1 and CB2 antagonists, respectively, or in the presence or absence of GSK 2656157 (1 µM), in cell culture medium with 1% (v/v) FBS for 24 h.
Isolation and primary cultures of human cytotrophoblasts
Term placentas of normal pregnancies (38–40 weeks of gestation), from Caucasian women living in the Porto region, were immediately collected after spontaneous delivery or elective caesarean section, from Centro Materno-Infantil do Norte – Centro Hospitalar do Porto after written informed consent. All the procedures were conducted after the approval of the Ethical Committee of Centro Hospitalar do Porto, Porto. Cytotrophoblast cells (hCTs) were isolated as described previously (Kliman et al. 1986, Keating et al. 2007). Briefly, after the removal of decidua, the villous tissue was dissected from at least ten different regions of placenta and the major blood vessels were discarded by fine dissection. Then, the tissue was subjected to a chemical digestion in a solution of trypsin and DNAse I. The resulting cells were separated in a discontinuous Percoll gradient at 1200 g for 10 min. The cytotrophoblasts were seeded and incubated in DMEM/F12 medium supplemented with 10% (v/v) of FBS and antibiotic-antimycotic solution at 37°C in 95% air/5% CO2 humidified atmosphere. For the experiments, cells were seeded in 6-well plates at density of 4.5 × 106 cells/well. After adherence (12 h), cells were treated with 2-AG (10 µM), in the presence or absence of AM281 or AM630 (1 µM), CB1 and CB2 antagonists, respectively, in cell culture medium with 1% (v/v) FBS.
Assessment of intracellular reactive oxygen and nitrogen species
For the evaluation of intracellular reactive oxygen and nitrogen species (ROS/RNS) generation, BeWo cells were seeded in 96-well black plates, treated with 2-AG (10 μM) for 24 h and incubated with the probe 2′-7′-dichlorodihydrofluorescein diacetate (DCDHF-DA) for 1 h at room temperature in the presence or absence of GSK 2656157, a selective PERK inhibitor. Fluorescence, proportional to the cellular levels of ROS/RNS, was measured using the Biotek Synergy HTX Multi-Mode Microplate Reader (Biotek Instruments). As a positive control, phorbol 12-myristate 13-acetate (PMA) (25 ng/ml) was used. The results are expressed in relative fluorescence units (RFU).
Western blotting
BeWo cells were seeded in 6-well plates at a cell density of 6 × 105 and primary cytotrophoblast cells at a cell density of 4.5 × 106 cells/well and treated with 2-AG for 24 h. In some wells, cells were also pre-incubated for 30 min with CB1 and CB2 antagonists or with GSK 2656157, a selective PERK inhibitor. The ER-stress inducer thapsigargin was used as a positive control. Cell extracts were prepared in lysis buffer (20 mM Tris pH 7.5, 150 mM NaCl, 1 mM EDTA, 1% Triton X-100) containing a cocktail of protease and phosphatase inhibitors (1:100 v/v). Samples (30 µg of protein) were separated by 10% SDS–PAGE and transferred onto nitrocellulose membranes. Membranes were subsequently incubated with antibodies against rabbit-CHOP (1:100; Santa Cruz Biotechnology), rabbit p-eIF2α and eIF2α (1:200, Cell Signaling Technology) and anti-PARP (1:200; Cell Signaling Technology), at 4°C overnight. Membranes were then washed and incubated with peroxidase-conjugated secondary antibody anti-rabbit (1:2000; Santa Cruz Biotechnology) and proteins were detected by enhanced chemiluminescence. The membranes were then stripped and reincubated with anti-β-actin or anti-β-tubulin (1:500; Santa Cruz Biotechnology) as loading controls.
qRT-PCR analysis
Cells were collected in TRIzol reagent, and total RNA was extracted according to manufacturer’s instructions and quantified in the NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Inc., Wilmington, DE, USA). RNA quality was assessed using a bioanalyzer (Experion RNA, Bio-Rad Laboratories). One microgram of RNA was reverse-transcribed into cDNA by using the GRS cDNA Synthesis Mastermix (GRiSP Research Solutions, PT). For quantitative PCR, cDNA was amplified with KAPA SYBR® FAST qPCR Master Mix 2× Kit (Kapa Biosystems, Woburn, MA, USA), according to kit instructions within a MiniOpticon Real-Time PCR Detection System (Bio-Rad Laboratories). The PCR conditions and primer sequences are described in Table 1. The specificity of the amplified PCR product was evaluated by the melting curve analysis. The fold change in gene expression was calculated using 2−ΔΔCt method (Livak & Schmittgen 2001) with the housekeeping genes, GAPDH and B2M. As both reference genes revealed stability and similar results, for clarity, we presented data calculated by using GAPDH gene normalized to each control group.
Table 1.
Primer sequences and qPCR conditions used to assess the gene expression of ATF4, HSPA5 and DDIT3, GAPDH and B2M were used as housekeeping controls.
| Gene ID | GenBank | Primer sequence (5′–3′) | Annealing temperature | Amplicon length | Melting temperature |
|---|---|---|---|---|---|
| ATF4 | NM_001675.4 | Sense: ATCCTGCTTGCTGTTGTTGG Anti-sense: GTTCTCCAGCGACAAGGCTA |
61.1°C | 88 | 83.00°C |
| HSPA5 | NM_005347.4 | Sense: TTCTGCTGTATCCTCTTCACCAGT Anti-sense: TGTTCAACCAATTATCAGCAAACTC |
61.1°C | 73 | 78.50°C |
| DDIT3 | NM_001195057.1 | Sense: TCTCCTTCATGCGCTGCTTT Anti-sense: AGAACCAGGAAACGGAAACAGA |
57.0°C | 67 | 80.50°C |
| GAPDH | NM_001289746.1 | Sense: CGGGAAGCTTGTGATCAATGG Anti-sense: GGCAGTGATGGCATGGACTG |
55.0°C | 358 | 83.50°C |
| B2M | NM_004048.2 | Sense: AGCAGCATCATGGAGGTTTG Anti-Sense: AGCCCTCCTAGAGCTACCTG |
59.0°C | 229 | 80.50°C |
The specific primer sequences for ATF4, HSPA5 and DDIT3 were obtained from Oslowski and Urano (2011), whereas GAPDH and B2M primers were in-house designed using BLAST software found on the NCBI website, with the NCBI reported sequences.
Statistical analysis
Statistical analysis was carried out by ANOVA, followed by the Tukey post hoc-test to make pairwise comparisons of individual means when significance was indicated (GraphPad PRISM v. 6.0, GraphPad Software, Inc.). The results are the mean of at least three independent experiments carried out in triplicate. Data are expressed as the mean ± s.e.m., and differences were considered to be statistically significant at P < 0.05.
Results
2-AG effects in ER-stress markers
We have previously reported that 2-AG induces apoptosis in BeWo cells, by a mechanism involving ROS/RNS generation associated with caspase -3/-7 activation (Costa et al. 2014a). In this work, we further explored the pathways of 2-AG mediated cell death by evaluating the involvement of ER-stress and the expression of several ER-stress markers associated with the ER-stress-induced apoptosis. The choriocarcinoma-derived BeWo cell line constitutes a widely accepted in vitro model representative of cytotrophoblasts. These cells are easy to handle and grow in a relatively short period of time surpassing the low availability of fresh tissue samples (Rothbauer et al. 2017). Additionally, they express CB receptors and other members of ECS and respond to cannabinoid stimuli (Habayeb et al. 2008).
We first analysed 2-AG actions on the ER resident chaperone GRP78 (BiP). Besides assisting protein folding, BiP regulates ER-stress-signalling pathways leading to UPR survival/apoptosis responses, as it controls the activation of transmembrane ER-stress sensors (IRE1, PERK, and ATF6) through a direct binding-release mechanism.
BeWo cells were treated with 2-AG (10 μM) for 24 h. We observed an increase in the expression of HSPA5 gene that codifies BiP as a result of the UPR adaptive response. This occurred independently of CB activation (Fig. 1A).
Figure 1.

Evaluation of 2-AG effects in ER-stress markers by qRT-PCR analysis. BeWo cells were treated with 2-AG (10 μM) for 24 h. For the assessment of ER-stress dependency on cannabinoid signalling, cells were pre-incubated with the CB1 and CB2 antagonists AM281 and AM630, respectively. (A) 2-AG increased the transcript levels of BiP encoded by the HSPA5 gene in a CB-independent manner. (B) 2-AG increased the transcript levels of ATF4 through CB2 activation. This effect was reversed by the incubation with the antagonist AM630, indicating a CB2-dependency. Tunicamycin (TN) and thapsigargin (TG) were used as ER-stress inducers. Results show transcript levels normalised against GAPDH. Data are presented as the mean ± s.e.m. (*P < 0.05 vs control; #P < 0.05 vs 2-AG).
When BIP is released, it induces PERK dimerization and subsequent autophosphorylation. In turn, activated PERK phosphorylates eIF2α leading to global translation arrest, though some transcripts such as ATF4 remain preferably translated. If the stress is persistent, ATF4 can stimulate the transcription of the proapoptotic CHOP to induce cell death. Then, we checked whether long-term activation of UPR by 2-AG may evoke paradoxical response with initiation of apoptotic cell death through the PERK-ATF4-CHOP pathway. In fact, ATF4 was also elevated after 2-AG treatment. This increase was dependent on CB2 signalling engagement (Fig. 1B).
Cannabinoid signalling modulates 2-AG-induced CHOP expression
As the prolonged activation of the unfolded protein response may initiate apoptotic cell death via the up-regulation of CHOP, we investigated 2-AG impact on CHOP expression. In BeWo cells, treatment with 2-AG was tested at the mRNA (DDIT3) (Fig. 2A) and protein levels (Fig. 2B). After 24 h, we found that expression levels were significantly enhanced with respect to control. The addition of the CB1 antagonist AM281 did not interfere with the expression, while CB2 antagonist AM630 was able to revert 2-AG induced increase.
Figure 2.
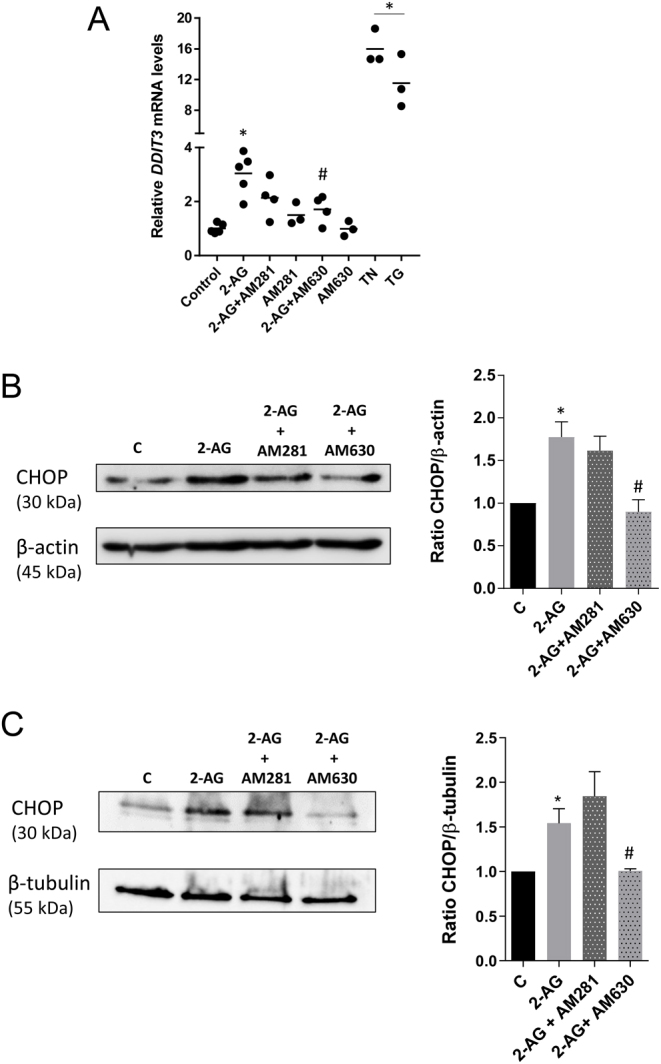
2-AG effects on CHOP expression and modulation by cannabinoid signalling. Cells were treated with 2-AG (10 μM) for 24 h and expression levels of the pro-apoptotic ER-stress factor CHOP were assessed by qRT-PCR and Western blotting. For the assessment of CHOP dependency on cannabinoid signalling, cells were pre-incubated with the CB1 and CB2 antagonists AM281 and AM630, respectively. In BeWo cells, 2-AG treatment increases the transcript levels of CHOP encoded by the DDIT3 gene (A) and CHOP protein expression (B) through a CB2-dependent manner. Results show transcript levels normalised against GAPDH. A representative Western blotting and the densitometry analysis with relative ratios of CHOP/β-actin are shown, as β-actin was used as a loading control. (C) The increase in CHOP protein expression was also observed in cytotrophoblast cells isolated from term placenta and 2-AG effects were also reversed by CB2 receptor blockade with AM631, confirming the involvement of cannabinoid signalling through CB2 activation. A representative Western blotting and the densitometry analysis with relative ratios of CHOP/β-tubulin are shown, as β-tubulin was used as a loading control. Data are presented as the mean ± s.e.m. (*P < 0.05 vs control; #P < 0.05 vs 2-AG).
To further confirm CHOP activation, primary cultures of term cytotrophoblasts were used. Quantification and normalization of Western blotting results (Fig. 2C) showed a similar protein expression profile to the one observed in BeWo cells. Together, these results suggest that the pro-apoptotic UPR response is not only activated by 2-AG treatment but is also dependent on cannabinoid signalling through CB2 activation.
PERK/eIF2α/ATF4/CHOP pathway involvement in 2-AG-induced apoptosis
In response to ER-stress, phosphorylation of the α subunit of eukaryotic initiation factor 2 (eIF2α) reduces general translation initiation, though selectively it enhances the translation of ATF4, involved in the regulation of redox status of cells and apoptosis. Therefore, increased eIF2α phosphorylation induces the ATF4/CHOP pathway. The compound GSK 2656157 selectively inhibits PERK activity in cells by inhibiting stress-induced PERK autophosphorylation, eIF2α substrate phosphorylation, together with corresponding decreases in ATF4 and CHOP. We observed that cells treated with 2-AG present enhanced eIF2α phosphorylation that is reversed by pre-incubation with the selective PERK inhibitor, GSK 2656157 (Fig. 3A). Moreover, GSK alleviates 2-AG induced ROS/RNS generation (Fig. 3B), pointing to the interdependence between oxidative stress and ER-stress. A crosstalk among different cellular components appears essential to recruit pathways leading to cell death. Results demonstrate an increase in poly(ADP-ribose) polymerase (PARP) cleavage, a characteristic of the apoptotic process, that was attenuated by pretreatment with GSK 2656157, indicating the involvement of the PERK arm of the ER-stress in the apoptotic process (Fig. 3C).
Figure 3.

2-AG effects on eIF2α/ATF4/CHOP apoptotic pathway. BeWo cells were treated with 2-AG (10 μM) for 24 h. For the assessment of PERK/eIF2α/ATF4/CHOP apoptotic pathway activation, cells were incubated with the selective PERK inhibitor GSK 2656157 (1 µM). For the evaluation of the dependency on cannabinoid signalling, cells were pre-incubated with the CB1 and CB2 antagonists AM281 and AM630, respectively. (A) Through Western blotting analysis, it was observed an increased phosphorylation of eIF2α, mediated by CB2. The selective PERK inhibitor, GSK 2656157, prevented eIF2α activation. Thapsigargin (TG) was used as a stress inducer. Representative Western blotting, densitometry analysis and relative ratios of phosphorylated eIF2α to total eIF2α are shown. (B) 2-AG induced ER-stress leads to accumulation of ROS. Pretreatment with GSK attenuates ROS generation. Phorbol 12-myristate 13-acetate (PMA) was used as a positive control. (C) Cleaved PARP was determined by Western blotting analysis. Effects of 2-AG treatment were significantly attenuated by pretreatment with GSK. Ratios of cleaved PARP/β-actin are shown. Data are presented as mean ± s.e.m. Significant differences between control and treated cells are denoted by *P < 0.05 vs control and #P < 0.05 vs 2-AG.
Discussion
Placental development involves well-coordinated processes of trophoblast proliferation, differentiation and apoptosis, in which eCBs play an important role. Moreover, a dysregulation on this balance may be associated with pregnancy-related disorders and infertility (Cuffe et al. 2017). High metabolism and oxidative stress are essential to regulate gene transcription associated to trophoblast turnover, invasion and angiogenesis. However, excessive ROS may harm placental development and lead to miscarriage, preeclampsia and intrauterine growth restriction (Schoots et al. 2018). ER-stress/UPR signalling-mediated pathways are involved in a broad range of physiologic events including cell differentiation and survival/apoptosis, migration, invasion and angiogenesis (Guzel et al. 2017). In a mouse model of constitutive ER-stress, due to a dysfunctional mutation in eIF2α, a premature differentiation of cytotrophoblast cells was observed (Yung et al. 2012b), whereas genetic knock-out of IRE-1 pathway led to aberrant placental development and decreased trophoblast proliferation (Iwawaki et al. 2009). In addition, it has also been suggested that ER-stress is involved in foetal growth restriction, preeclampsia, low birth weight and recurrent pregnancy loss (Yung et al. 2008, 2014, Burton et al. 2009, Burton & Yung 2011, Kawakami et al. 2014, Guzel et al. 2017, Lorenzon-Ojea et al. 2019). Interestingly, the levels of p-eIF2α, ER chaperone BiP and CHOP proteins were elevated in pregnancies where the growth restriction was complicated by preeclampsia when compared to normal placentas (Yung et al. 2008). Moreover, inhibition of eIF2α phosphorylation is associated with reduced cell proliferation and placental villous trees (Yung et al. 2012a). Increased levels of ER-stress response genes were also observed in preeclamptic and in early pregnancy loss decidualised endometrium (Loset et al. 2011, Lian et al. 2011).
The role of the eCBs and the cannabinoid signalling in placental ER-stress is totally unknown, though eCBs are involved in ER-stress-induced apoptosis in other cells. In fact, in human pancreatic tumor cells, CB2 dependent accumulation of ceramide upregulates ER-stress related genes (Carracedo et al. 2006). In non-melanoma skin cancer, AEA induces ER stress-apoptosis mediated by CHOP expression and oxidative stress (Soliman & Van Dross 2016). Moreover, AEA-oxidative metabolites activate ER-stress apoptosis in tumorigenic keratinocytes through engagement of CHOP and PERK, IRE1 and ATF6 arms of UPR (Soliman et al. 2016). Recently, it was reported that the phytocannabinoid Δ9-tetrahydrocannabinol, the main psychoactive compound in Cannabis sativa, also induces ER-stress in BeWo cells, involving an up-regulation of CHOP levels through the eIF2α/ATF4/CHOP pathway (Lojpur et al. 2019).
In this work, we investigated if the endocannabinoid 2-AG was implicated in placental ER-stress and apoptosis, as we have previously reported that, in BeWo cells, 2-AG was able to induce the production of ROS and caspase -3/-7 activation (Costa et al. 2014a). Here, we demonstrate that 2-AG, via CB2 signalling, leads to the ER-stress apoptotic cell death through the PERK-ATF4-CHOP pathway activation.
We observed that the transcript levels of HSPA5 gene that codifies BiP were increased in response to 2-AG treatment, independently of CB activation. Nevertheless, the observed alterations in HSPA5 mRNA expression may also indicate an activation of ATF6 arm of UPR (Mizuuchi et al. 2016) besides the PERK-ATF4-CHOP pathway. The release of BiP from stress sensors initiates the transduction of the UPR signals. Although the regulation of ER-stress response at both mRNA and protein level contributes to the overall change in the system (Cheng et al. 2016), we decided to study BiP expression only at the mRNA level. In fact, elevated mRNA levels encoding BiP are considered a sensitive and early indicator of ER-stress and have been observed in diseases linked to ER-stress and apoptosis (Ni & Lee 2007, Kroeger et al. 2012). In human SH-SY5Y neuroblastoma cells, the ER-stress inducer tunicamycin increased protein and mRNA levels of BiP, as a protective response, while prolonged treatment resulted in apoptotic cell death and up-regulation of CHOP (Reimertz et al. 2003). If UPR-induced mechanisms fail to alleviate ER-stress, both the intrinsic and extrinsic pathways for apoptosis can be activated (Sano & Reed 2013).
In addition, we observed that 2-AG increased mRNA levels of ATF4 and mRNA and protein levels of CHOP involving CB2 activation. PERK phosphorylates eIF2α, which blocks cell proliferation and reduces mRNA translation, but selectively ensues ATF4 translation, that induces CHOP expression (Xu et al. 2005). This eIF2α/ATF4/CHOP pathway has been widely explored and plays a crucial role in cell death, particularly in mitochondrial apoptotic pathway (Szegezdi et al. 2006). In B-cell chronic lymphocytic leukemia (B-CLL) cells, ER-stress-induced apoptosis is accompanied by increased BiP and CHOP expression (Rosati et al. 2010). In endometrial cells and in endometrial cancer cells, eCB-induced apoptosis was also associated to CHOP up-regulation (Almada et al. 2017, Fonseca et al. 2018). This is also observed for trophoblast cells, as we demonstrate 2-AG sustained increase in the levels of CHOP and apoptosis activation through CB2 receptor both in BeWo cells and term primary cytotrophoblasts. Although placental samples were obtained from a mixture of cesarean and vaginal births, there was no significant variation between controls, though it was previously described that the labour process strongly activates the UPR/ER stress pathways (Cindrova-Davies et al. 2007). The PERK pathway is predominant in CHOP activation. However, the observed increase in CHOP expression may result from the activation of different arms of UPR response that we did not explore. For example, IRE1α upon dimerization and autophosphorylation splices XBP1 mRNA and allows translation of an active transcriptional factor XBP1 (Szegezdi et al. 2006). IRE1α can also recruit TNF-associated factor-2 (TRAF-2) and apoptosis signal-regulating kinase-1 (ASK1), which causes phosphorylation of p38 MAPK that is associated with CHOP and c-Jun N-terminal kinase 1 (JNK) activation (Sano & Reed 2013, Redza-Dutordoir & Averill-Bates 2016).
Our findings indicate that the PERK/ATF4/CHOP pathway may be associated with 2-AG-induced apoptosis. Interestingly, this UPR arm has also been linked with early-onset of preeclampsia and uterine growth restriction, through the down-regulation of placental growth factor (PlGF), an important regulator of angiogenesis (Mizuuchi et al. 2016). In addition, vascular endothelial growth factor A expression, which is necessary for a correct placenta vessel formation is regulated by the UPR-related proteins IRE1α, PERK and ATF6 (Ghosh et al. 2010). Importantly, Bastida-Ruiz et al. demonstrated that ER-stress and the UPR are involved in trophoblast cell fusion and syncytialization in early-first trimester, late first-trimester and at term (Bastida-Ruiz et al. 2019). Moreover, the induction of ER-stress and the UPR processes accompanies the decidualization (Soczewski et al. 2020). Therefore, these are all processes that involve the endocannabinoid modulation and the possible crosstalk between eCBs and ER-stress.
The observed attenuation of ROS levels with the pretreatment with the specific inhibitor of the PERK arm of ER-stress, GSK 2656157, suggests the occurrence of an ER-stress-associated ROS production. Many studies have indicated a crosstalk between the generation of ROS and the ER-stress response (Cao & Kaufman 2014). ROS formation in the ER occurs as a regular by-product of disulfide bond formation oxidative protein folding. Protein misfolding may contribute to oxidative stress and ROS generation (Haynes et al. 2004, Eletto et al. 2014). When ER-stress-associated ROS production is sustained, apoptosis may be triggered (Tabas & Ron 2011). 2-AG induces an increase in ROS, p-eIF2 and CHOP levels that leads to apoptosis as verified by the increase in PARP cleavage, an effect reversed by the addition of GSK. ER-stress and ROS balance are crucial points for placental development, and exacerbation of these processes may lead to pregnancy-related complications.
To the best of our knowledge, this is the first time that it is shown the association of the endocannabinoid 2-AG with ER-stress in placenta. In this work, we propose 2-AG as an ER-stress and apoptotic inducer through the cannabinoid receptor CB2 activation (Fig. 4). These findings along with the antiproliferative effects of 2-AG and AEA on trophoblasts (Costa et al. 2014a,b) further support a crosstalk between cannabinoid signalling, cytotrophoblast turnover and ER-stress that may be implicated in the pathophysiology of some pregnancy complications, such as pregnancy loss, preeclampsia and intrauterine growth restriction.
Figure 4.

Alterations in 2-AG levels may condition placentation through ER-stress and apoptosis activation: a proposed model. The endocannabinoid 2-AG through cannabinoid receptor 2 (CB2) signalling induces ER-stress and UPR via PERK arm, activating the PERK/eIF2α/ATF4/CHOP pathway leading to ROS generation and apoptosis. Moreover, as we previously reported, 2-AG is also involved in caspase -3/-7 activation and plays a role in trophoblast syncytialization. The signalling pathways involved in ER-stress and UPR play key roles in the normal trophoblast apoptosis and syncytialization. Changes in 2-AG levels/cannabinoid signalling may disturb those processes and therefore the normal trophoblast turnover and promote altered placentation and consequently pregnancy disorders, such as preeclampsia and IUGR.
Declaration of interest
The authors declare that there is no conflict of interest that could be perceived as prejudicing the impartiality of the research reported.
Funding
This work was supported by the European Regional Development Fund (ERDF) through the Operational Competitiveness Factors Program – COMPETE and by National Funds through FCT – Foundation for Science and Technology within the scope of the project ‘PTDC/DTP-FTO/5651/2014 – POCI-01-0145-FEDER-016562’. Marta Almada and Bruno Fonseca thank for the grants from national funds and PORTUGAL 2020 Partnership Agreement, HEALTH_RL2_PHD_BIOK_01, NORTE-01-0145-FEDER-000024.
Author contribution statement
A M, F B M, T N and C G S designed and directed the study. A M, C L and A P performed experiments and analysed data. B J and G D were responsible for sample collection. A M, F B M, T N and C S G wrote and prepared the manuscript. All authors provided critical feedback and helped to shape the manuscript.
References
- Almada M, Fonseca BM, Amaral C, Diniz-da-Costa M, Correia-da-Silva G, Teixeira N. 2017. Anandamide oxidative metabolism-induced endoplasmic reticulum stress and apoptosis. Apoptosis 22 816–826. ( 10.1007/s10495-017-1356-4) [DOI] [PubMed] [Google Scholar]
- Almanza A, Carlesso A, Chintha C, Creedican S, Doultsinos D, Leuzzi B, Luís A, McCarthy N, Montibeller L, More S. et al. 2019. Endoplasmic reticulum stress signalling – from basic mechanisms to clinical applications. FEBS Journal 286 241–278. ( 10.1111/febs.14608) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bastida-Ruiz D, Yart L, Wuillemin C, Ribaux P, Morris N, Epiney M, de Tejada BM, Cohen M. 2019. The fine-tuning of endoplasmic reticulum stress response and autophagy activation during trophoblast syncytialization. Cell Death and Disease 10 651. ( 10.1038/s41419-019-1905-6) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brents LK. 2016. Marijuana, the endocannabinoid system and the female reproductive system. Yale Journal of Biology and Medicine 89 175–191. [PMC free article] [PubMed] [Google Scholar]
- Burton GJ, Yung HW. 2011. Endoplasmic reticulum stress in the pathogenesis of early-onset pre-eclampsia. Pregnancy Hypertension 1 72–78. ( 10.1016/j.preghy.2010.12.002) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton GJ, Yung HW, Cindrova-Davies T, Charnock-Jones DS. 2009. Placental endoplasmic reticulum stress and oxidative stress in the pathophysiology of unexplained intrauterine growth restriction and early onset preeclampsia. Placenta 30 (Supplement A) S43–S48. ( 10.1016/j.placenta.2008.11.003) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burton GJ, Yung HW, Murray AJ. 2017. Mitochondrial – endoplasmic reticulum interactions in the trophoblast: stress and senescence. Placenta 52 146–155. ( 10.1016/j.placenta.2016.04.001) [DOI] [PubMed] [Google Scholar]
- Cao SS, Kaufman RJ. 2014. Endoplasmic reticulum stress and oxidative stress in cell fate decision and human disease. Antioxidants and Redox Signaling 21 396–413. ( 10.1089/ars.2014.5851) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carracedo A, Gironella M, Lorente M, Garcia S, Guzmán M, Velasco G, Iovanna JL. 2006. Cannabinoids induce apoptosis of pancreatic tumor cells via endoplasmic reticulum stress-related genes. Cancer Research 66 6748–6755. ( 10.1158/0008-5472.CAN-06-0169) [DOI] [PubMed] [Google Scholar]
- Cheng Z, Teo G, Krueger S, Rock TM, Koh HWL, Choi H, Vogel C. 2016. Differential dynamics of the mammalian mRNA and protein expression response to misfolding stress. Molecular Systems Biology 12 855. ( 10.15252/msb.20156423) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cindrova-Davies T, Yung HW, Johns J, Spasic-Boskovic O, Korolchuk S, Jauniaux E, Burton GJ, Charnock-Jones DS. 2007. Oxidative stress, gene expression, and protein changes induced in the human placenta during labor. American Journal of Pathology 171 1168–1179. ( 10.2353/ajpath.2007.070528) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa MA, Fonseca BM, Keating E, Teixeira NA, Correia-da-Silva G. 2014a. 2-arachidonoylglycerol effects in cytotrophoblasts: metabolic enzymes expression and apoptosis in BeWo cells. Reproduction 147 301–311. ( 10.1530/REP-13-0563) [DOI] [PubMed] [Google Scholar]
- Costa MA, Fonseca BM, Keating E, Teixeira NA, Correia-da-Silva G. 2014b. Transient receptor potential vanilloid 1 is expressed in human cytotrophoblasts: induction of cell apoptosis and impairment of syncytialization. International Journal of Biochemistry and Cell Biology 57 177–185. ( 10.1016/j.biocel.2014.10.008) [DOI] [PubMed] [Google Scholar]
- Costa MA, Fonseca BM, Mendes A, Braga J, Teixeira NA, Correia da Silva G. 2015a. The endocannabinoid anandamide affects the synthesis of human syncytiotrophoblast-related proteins. Cell and Tissue Research 362 441–446. ( 10.1007/s00441-015-2236-2) [DOI] [PubMed] [Google Scholar]
- Costa MA, Fonseca BM, Teixeira NA, Correia-da-Silva G. 2015b. The endocannabinoid anandamide induces apoptosis in cytotrophoblast cells: involvement of both mitochondrial and death receptor pathways. Placenta 36 69–76. ( 10.1016/j.placenta.2014.10.011) [DOI] [PubMed] [Google Scholar]
- Costa MA, Keating E, Fonseca BM, Teixeira NA, Correia-da-Silva G. 2015c. 2-Arachidonoylglycerol impairs human cytotrophoblast cells syncytialization: influence of endocannabinoid signalling in placental development. Molecular and Cellular Endocrinology 399 386–394. ( 10.1016/j.mce.2014.09.005) [DOI] [PubMed] [Google Scholar]
- Costa MA, Fonseca BM, Mendes A, Braga J, Teixeira NA, Correia-da-Silva G. 2016. The endocannabinoid 2-arachidonoylglycerol dysregulates the synthesis of proteins by the human syncytiotrophoblast. Biochimica et Biophysica Acta 1861 205–212. ( 10.1016/j.bbalip.2015.12.008) [DOI] [PubMed] [Google Scholar]
- Cuffe JSM, Holland O, Salomon C, Rice GE, Perkins AV. 2017. Review: Placental derived biomarkers of pregnancy disorders. Placenta 54 104–110. ( 10.1016/j.placenta.2017.01.119) [DOI] [PubMed] [Google Scholar]
- Eletto D, Chevet E, Argon Y, Appenzeller-Herzog C. 2014. Redox controls UPR to control redox. Journal of Cell Science 127 3649–3658. ( 10.1242/jcs.153643) [DOI] [PubMed] [Google Scholar]
- Fonseca BM, Correia-da-Silva G, Teixeira NA. 2018. Cannabinoid-induced cell death in endometrial cancer cells: involvement of TRPV1 receptors in apoptosis. Journal of Physiology and Biochemistry 74 261–272. ( 10.1007/s13105-018-0611-7) [DOI] [PubMed] [Google Scholar]
- Fu J, Zhao L, Wang L, Zhu X. 2015. Expression of markers of endoplasmic reticulum stress-induced apoptosis in the placenta of women with early and late onset severe pre-eclampsia. Taiwanese Journal of Obstetrics and Gynecology 54 19–23. ( 10.1016/j.tjog.2014.11.002) [DOI] [PubMed] [Google Scholar]
- Ghosh R, Lipson KL, Sargent KE, Mercurio AM, Hunt JS, Ron D, Urano F. 2010. Transcriptional regulation of VEGF-A by the unfolded protein response pathway. PLoS ONE 5 e9575. ( 10.1371/journal.pone.0009575) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzel E, Arlier S, Guzeloglu-Kayisli O, Tabak MS, Ekiz T, Semerci N, Larsen K, Schatz F, Lockwood CJ, Kayisli UA. 2017. Endoplasmic reticulum stress and homeostasis in reproductive physiology and pathology. International Journal of Molecular Sciences 18 792. ( 10.3390/ijms18040792) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habayeb OM, Taylor AH, Bell SC, Taylor DJ, Konje JC. 2008. Expression of the endocannabinoid system in human first trimester placenta and its role in trophoblast proliferation. Endocrinology 149 5052–5060. ( 10.1210/en.2007-1799) [DOI] [PubMed] [Google Scholar]
- Haynes CM, Titus EA, Cooper AA. 2004. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Molecular Cell 15 767–776. ( 10.1016/j.molcel.2004.08.025) [DOI] [PubMed] [Google Scholar]
- Heazell AEP, Crocker IP. 2008. Live and let die – regulation of villous trophoblast apoptosis in normal and abnormal pregnancies. Placenta 29 772–783. ( 10.1016/j.placenta.2008.07.003) [DOI] [PubMed] [Google Scholar]
- Helliwell RJ, Chamley LW, Blake-Palmer K, Mitchell MD, Wu J, Kearn CS, Glass M. 2004. Characterization of the endocannabinoid system in early human pregnancy. Journal of Clinical Endocrinology and Metabolism 89 5168–5174. ( 10.1210/jc.2004-0388) [DOI] [PubMed] [Google Scholar]
- Huppertz B, Kadyrov M, Kingdom JCP. 2006. Apoptosis and its role in the trophoblast. American Journal of Obstetrics and Gynecology 195 29–39. ( 10.1016/j.ajog.2005.07.039) [DOI] [PubMed] [Google Scholar]
- Iwawaki T, Akai R, Yamanaka S, Kohno K. 2009. Function of IRE1 alpha in the placenta is essential for placental development and embryonic viability. PNAS 106 16657–16662. ( 10.1073/pnas.0903775106) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawakami T, Yoshimi M, Kadota Y, Inoue M, Sato M, Suzuki S. 2014. Prolonged endoplasmic reticulum stress alters placental morphology and causes low birth weight. Toxicology and Applied Pharmacology 275 134–144. ( 10.1016/j.taap.2013.12.008) [DOI] [PubMed] [Google Scholar]
- Keating E, Gonçalves P, Lemos C, Costa F, Campos I, Smith SB, Bridges CC, Martel F. 2007. Progesterone inhibits folic acid transport in human trophoblasts. Journal of Membrane Biology 216 143–152. ( 10.1007/s00232-007-9057-5) [DOI] [PubMed] [Google Scholar]
- Kenney SP, Kekuda R, Prasad PD, Leibach FH, Devoe LD, Ganapathy V. 1999. Cannabinoid receptors and their role in the regulation of the serotonin transporter in human placenta. American Journal of Obstetrics and Gynecology 181 491–497. ( 10.1016/s0002-9378(99)70583-1) [DOI] [PubMed] [Google Scholar]
- Kliman HJ, Nestler JE, Sermasi E, Sanger JM, Strauss JF. 1986. Purification, characterization, and in vitro differentiation of cytotrophoblasts from human term placentae. Endocrinology 118 1567–1582. ( 10.1210/endo-118-4-1567) [DOI] [PubMed] [Google Scholar]
- Kroeger H, Messah C, Ahern K, Gee J, Joseph V, Matthes MT, Yasumura D, Gorbatyuk MS, Chiang WC, Lavail MM. et al. 2012. Induction of endoplasmic reticulum stress genes, BiP and chop, in genetic and environmental models of retinal degeneration. Investigative Ophthalmology and Visual Science 53 7590–7599. ( 10.1167/iovs.12-10221) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lian IA, Løset M, Mundal SB, Fenstad MH, Johnson MP, Eide IP, Bjørge L, Freed KA, Moses EK, Austgulen R. 2011. Increased endoplasmic reticulum stress in decidual tissue from pregnancies complicated by fetal growth restriction with and without pre-eclampsia. Placenta 32 823–829. ( 10.1016/j.placenta.2011.08.005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak KJ, Schmittgen TD. 2001. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25 402–408. ( 10.1006/meth.2001.1262) [DOI] [PubMed] [Google Scholar]
- Lojpur T, Easton Z, Raez-Villanueva S, Laviolette S, Holloway AC, Hardy DB. 2019. Delta9-tetrahydrocannabinol leads to endoplasmic reticulum stress and mitochondrial dysfunction in human BeWo trophoblasts. Reproductive Toxicology 87 21–31. ( 10.1016/j.reprotox.2019.04.008) [DOI] [PubMed] [Google Scholar]
- Lorenzon-Ojea AR, Yung HW, Burton GJ, Bevilacqua E. 2019. The potential contribution of stromal cell-derived factor 2 (SDF2) in endoplasmic reticulum stress response in severe preeclampsia and labor-onset. Biochimica et Biophysica Acta (BBA): Molecular Basis of Disease 1866 165386. ( 10.1016/j.bbadis.2019.01.012) [DOI] [PubMed] [Google Scholar]
- Loset M, Mundal SB, Johnson MP, Fenstad MH, Freed KA, Lian IA, Eide IP, Bjorge L, Blangero J, Moses EK. et al. 2011. A transcriptional profile of the decidua in preeclampsia. American Journal of Obstetrics and Gynecology 204 84.e1–84.e27. ( 10.1016/j.ajog.2010.08.043) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maia J, Fonseca BM, Teixeira N, Correia-da-Silva G. 2020. The fundamental role of the endocannabinoid system in endometrium and placenta: implications in pathophysiological aspects of uterine and pregnancy disorders. Human Reproduction Update dmaa005. ( 10.1093/humupd/dmaa005) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malhotra JD, Kaufman RJ. 2007. Endoplasmic reticulum stress and oxidative stress: a vicious cycle or a double-edged sword? Antioxidants and Redox Signaling 9 2277–2293. ( 10.1089/ars.2007.1782) [DOI] [PubMed] [Google Scholar]
- Marczylo TH, Lam PM, Amoako AA, Konje JC. 2010. Anandamide levels in human female reproductive tissues: solid-phase extraction and measurement by ultraperformance liquid chromatography tandem mass spectrometry. Analytical Biochemistry 400 155–162. ( 10.1016/j.ab.2009.12.025) [DOI] [PubMed] [Google Scholar]
- Mizuuchi M, Cindrova-Davies T, Olovsson M, Charnock-Jones DS, Burton GJ, Yung HW. 2016. Placental endoplasmic reticulum stress negatively regulates transcription of placental growth factor via ATF4 and ATF6β: implications for the pathophysiology of human pregnancy complications. Journal of Pathology 238 550–561. ( 10.1002/path.4678) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni M, Lee AS. 2007. ER chaperones in mammalian development and human diseases. FEBS Letters 581 3641–3651. ( 10.1016/j.febslet.2007.04.045) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oslowski CM, Urano F. 2011. Measuring ER stress and the unfolded protein response using mammalian tissue culture system. Methods in Enzymology 490 71–92. ( 10.1016/B978-0-12-385114-7.00004-0) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park B, Gibbons HM, Mitchell MD, Glass M. 2003. Identification of the CB1 cannabinoid receptor and fatty acid amide hydrolase (FAAH) in the human placenta. Placenta 24 990–995. ( 10.1016/s0143-4004(03)00165-6) [DOI] [PubMed] [Google Scholar]
- Redza-Dutordoir M, Averill-Bates DA. 2016. Activation of apoptosis signalling pathways by reactive oxygen species. Biochimica et Biophysica Acta 1863 2977–2992. ( 10.1016/j.bbamcr.2016.09.012) [DOI] [PubMed] [Google Scholar]
- Reimertz C, Kögel D, Rami A, Chittenden T, Prehn JHM. 2003. Gene expression during ER stress-induced apoptosis in neurons: induction of the BH3-only protein BBC3/PUMA and activation of the mitochondrial apoptosis pathway. Journal of Cell Biology 162 587–597. ( 10.1083/jcb.200305149) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez D, Rojas-Rivera D, Hetz C. 2011. Integrating stress signals at the endoplasmic reticulum: the BCL-2 protein family rheostat. Biochimica et Biophysica Acta 1813 564–574. ( 10.1016/j.bbamcr.2010.11.012) [DOI] [PubMed] [Google Scholar]
- Rosati E, Sabatini R, Rampino G, De Falco F, Di Ianni M, Falzetti F, Fettucciari K, Bartoli A, Screpanti I, Marconi P. 2010. Novel targets for endoplasmic reticulum stress-induced apoptosis in B-CLL. Blood 116 2713–2723. ( 10.1182/blood-2010-03-275628) [DOI] [PubMed] [Google Scholar]
- Rothbauer M, Patel N, Gondola H, Siwetz M, Huppertz B, Ertl P. 2017. A comparative study of five physiological key parameters between four different human trophoblast-derived cell lines. Scientific Reports 7 5892. ( 10.1038/s41598-017-06364-z) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano R, Reed JC. 2013. ER stress-induced cell death mechanisms. Biochimica et Biophysica Acta 1833 3460–3470. ( 10.1016/j.bbamcr.2013.06.028) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schoots MH, Gordijn SJ, Scherjon SA, Van Goor H, Hillebrands JL. 2018. Oxidative stress in placental pathology. Placenta 69 153–161. ( 10.1016/j.placenta.2018.03.003) [DOI] [PubMed] [Google Scholar]
- Soczewski E, Grasso E, Gallino L, Hauk V, Fernández L, Gori S, Paparini D, Leirós CP, Ramhorst R. 2020. Immunoregulation of the decidualization program: focus on the endoplasmic reticulum stress. Reproduction 159 203–211. ( 10.1530/REP-19-0391) [DOI] [PubMed] [Google Scholar]
- Soliman E, Henderson KL, Danell AS, Van Dross R. 2016. Arachidonoyl-ethanolamide activates endoplasmic reticulum stress-apoptosis in tumorigenic keratinocytes: role of cyclooxygenase-2 and novel J-series prostamides. Molecular Carcinogenesis 55 117–130. ( 10.1002/mc.22257) [DOI] [PubMed] [Google Scholar]
- Soliman E, Van Dross R. 2016. Anandamide-induced endoplasmic reticulum stress and apoptosis are mediated by oxidative stress in non-melanoma skin cancer: receptor-independent endocannabinoid signaling. Molecular Carcinogenesis 55 1807–1821. ( 10.1002/mc.22429) [DOI] [PubMed] [Google Scholar]
- Sun X, Xie H, Yang J, Wang H, Bradshaw HB, Dey SK. 2010. Endocannabinoid signaling directs differentiation of trophoblast cell lineages and placentation. PNAS 107 16887–16892. ( 10.1073/pnas.1010892107) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szegezdi E, Logue SE, Gorman AM, Samali A. 2006. Mediators of endoplasmic reticulum stress-induced apoptosis. EMBO Reports 7 880–885. ( 10.1038/sj.embor.7400779) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Ron D. 2011. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nature Cell Biology 13 184–190. ( 10.1038/ncb0311-184) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trabucco E, Acone G, Marenna A, Pierantoni R, Cacciola G, Chioccarelli T, Mackie K, Fasano S, Colacurci N, Meccariello R. et al. 2009. Endocannabinoid system in first trimester placenta: low FAAH and high CB1 expression characterize spontaneous miscarriage. Placenta 30 516–522. ( 10.1016/j.placenta.2009.03.015) [DOI] [PubMed] [Google Scholar]
- Xu C, Bailly-Maitre B, Reed JC. 2005. Endoplasmic reticulum stress: cell life and death decisions. Journal of Clinical Investigation 115 2656–2664. ( 10.1172/JCI26373) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung HW, Calabrese S, Hynx D, Hemmings BA, Cetin I, Charnock-Jones DS, Burton GJ. 2008. Evidence of placental translation inhibition and endoplasmic reticulum stress in the etiology of human intrauterine growth restriction. American Journal of Pathology 173 451–462. ( 10.2353/ajpath.2008.071193) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung HW, Cox M, Tissot Van Patot M, Burton GJ. 2012a. Evidence of endoplasmic reticulum stress and protein synthesis inhibition in the placenta of non-native women at high altitude. FASEB Journal 26 1970–1981. ( 10.1096/fj.11-190082) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung HW, Hemberger M, Watson ED, Senner CE, Jones CP, Kaufman RJ, Charnock-Jones DS, Burton GJ. 2012b. Endoplasmic reticulum stress disrupts placental morphogenesis: implications for human intrauterine growth restriction. Journal of Pathology 228 554–564. ( 10.1002/path.4068) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung HW, Atkinson D, Campion-Smith T, Olovsson M, Charnock-Jones DS, Burton GJ. 2014. Differential activation of placental unfolded protein response pathways implies heterogeneity in causation of early- and late-onset pre-eclampsia. Journal of Pathology 234 262–276. ( 10.1002/path.4394) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung HW, Alnaes-Katjavivi P, Jones CJ, El-Bacha T, Golic M, Staff AC, Burton GJ. 2016. Placental endoplasmic reticulum stress in gestational diabetes: the potential for therapeutic intervention with chemical chaperones and antioxidants. Diabetologia 59 2240–2250. ( 10.1007/s00125-016-4040-2) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeeshan HMA, Lee GH, Kim HR, Chae HJ. 2016. Endoplasmic reticulum stress and associated ROS. International Journal of Molecular Sciences 17 327. ( 10.3390/ijms17030327) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Zhang L, Zhou L, Lei Y, Zhang Y, Huang C. 2019. Redox signaling and unfolded protein response coordinate cell fate decisions under ER stress. Redox Biology 25 101047. (doi:10.1016/j.redox.2018.11.005) [DOI] [PMC free article] [PubMed] [Google Scholar]