

Abstract
The global impact of malaria remains staggering despite extensive efforts to eradicate the disease. With increasing drug resistance and the absence of a clinically available vaccine, there is an urgent need for novel, affordable, and safe drugs for prevention and treatment of malaria. Previously we described a novel antimalarial acridone chemotype that is potent against both blood-stage and liver-stage malaria parasites. Here we describe an optimization process that has produced a second-generation acridone series with significant improvements in efficacy, metabolic stability, pharmacokinetics, and safety profiles. These findings highlight the therapeutic potential of dual-stage targeting acridones as novel drug candidates for further preclinical development.
Keywords: Malaria, lead optimization, antimalarials, antiplasmodial activity, second-generation, acridones, Plasmodium falciparum, dual-stage activity, broad-spectrum
Graphical Abstract
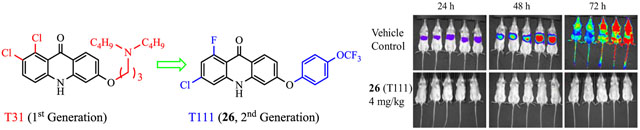
| T31 | T111 (26) | |
|---|---|---|
| In vitro IC50 vs. blood-stage P. falciparum D6/Tm90-C2B: | 0.022/228 | 0.028/5.6 |
| In vivo ED50/ED90 (mg/kg/d) vs. blood-stage P. yoelii: | 1.0/1.6 | 0.045/0.098 |
| In vivo cure (mg/kg×4 days) vs. blood-stage P. yoelii: | 80 | 10 |
| In vivo full protection/cure (mg/kg×3 days) vs. liver-stage P. berghei: | 4.0 | 4.0 |
| In vitro metabolic stability vs. human/mouse (t1/2, min): | 12/21 | >60/50 |
| In vivo PK (mouse, po): plasma/liver half-life (t1/2, h): | 6.00/4.63 | 23.6/14.0 |
| hERG (% inhibition at 10 μM): | 57 | 5.7 |
| hERG (% inhibition at 10 μM): | 57 | 5.7 |
INTRODUCTION
Malaria has been a scourge on humankind down through the centuries of recorded time and even now it accounts for over 200 million clinical cases worldwide each year along with roughly half a million deaths.1, 2 The global eradication effort in the past decade has propelled a surge in antimalarial drug development.3–5 However, many challenges remain for a sustainable elimination campaign, including the failing effectiveness of front-line artemisinin-combination therapy (ACT) due to emerging resistance,6–8 and safety concerns associated with limited radical cure options for relapsing Plasmodium vivax.9, 10 Without any clinically proven vaccine in sight, there is a continuous need to develop new drugs that overcome artemisinin resistance and ideally such agents would be active in both treatment of active bloodstream infections as well as preventing the disease at the liver-stage.3, 4, 11–13
We recently described acridones with dual-stage activity against both liver- and blood-stage malaria parasites.14 However, these acridones only displayed moderate metabolic stability along with an unsatisfactory risk of cardiotoxicity and evidence of cross-resistance to atovaquone (ATV) in P. falciparum. With the aim of improving metabolic stability, safety profile and other desired “drug-like” properties, we created a second-generation of acridones by varying the halogen substitution pattern on ring-A and placement of aryl groups on ring-B (Figure 1). A rigorous lead optimization process produced candidates with enhanced potency against blood- and liver-stage malaria parasites, improved metabolic stability and PK profile, and with a diminished risk of cardiotoxicity and cross-resistance with ATV, while maintaining a high selectivity index toward killing the parasites.
Figure 1.
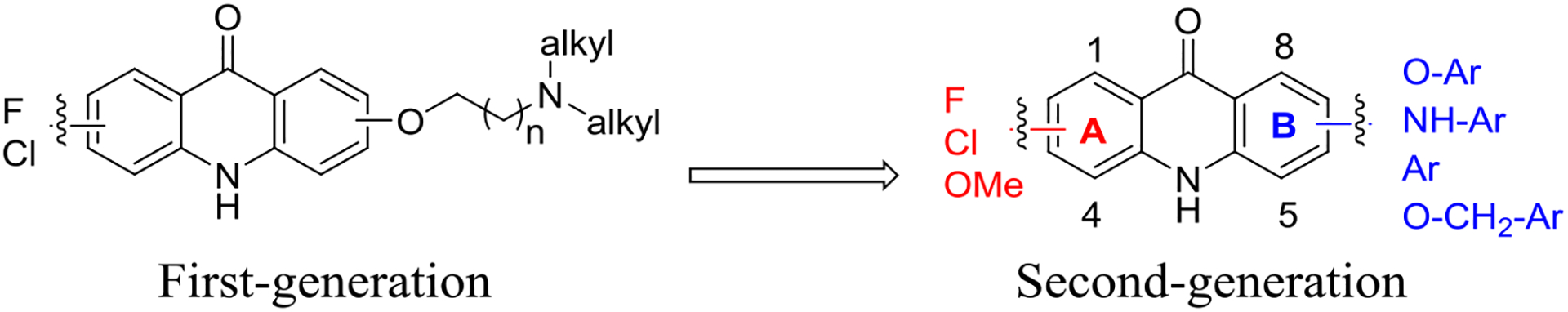
Transition from first-generation acridones to the second-generation acridones.
RESULTS AND DISCUSSION
Chemistry.
Our previous structural optimizations of the first-generation acridones demonstrated that halogen substitutions on ring-A and (dialkylamino)alkoxy moieties at the 6 position on ring-B are required for optimal broad-spectrum antimalarial activity.14 Subsequently, we have designed and synthesized a large library of the second-generation acridones with various mono- and di-substitutions on ring-A and aryloxy, arylamino, aryl or benzyloxy moieties at either the 6 or 7 position of ring-B, via simple and efficient synthetic routes from commercially available and inexpensive starting materials. The synthetic routes for the second-generation acridones are outlined in Schemes 1–3.
Scheme 1.
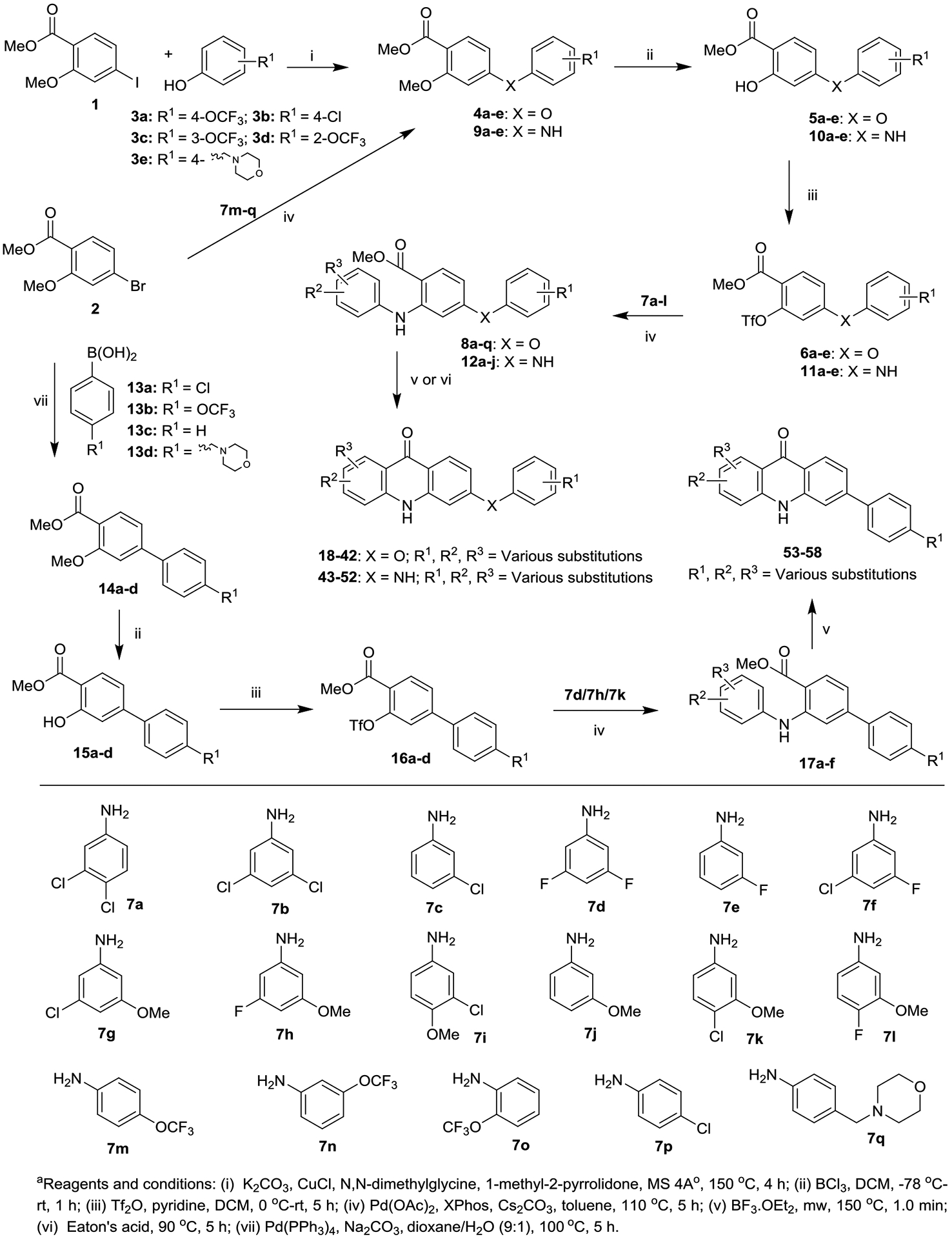
Synthesis of Acridones (18−58) with Aryloxy, Arylamino and Aryl Groups at the 6 Position of Ring-Ba
Scheme 3.
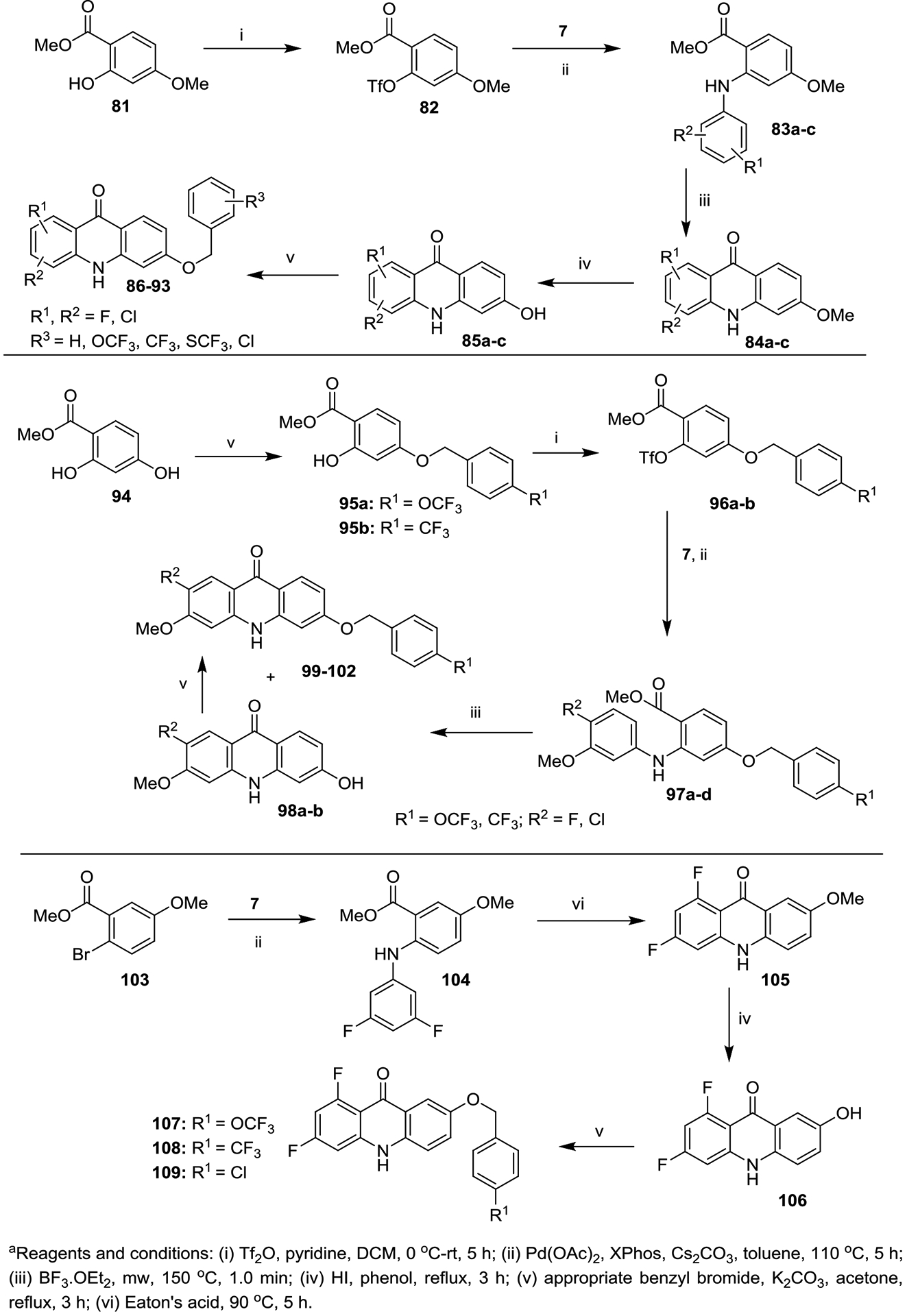
Synthesis of Acridones (86−93, 99−102 and 107−109) with Benzyloxy Groups at the 6 Position (Top and Middle Panel) and the 7 Position (Bottom Panel) of the Ring-Ba
Synthesis of 6-aryloxy-, 6-arylamino- and 6-aryl-acridones (18–58).
Synthesis of the first series of acridones 18–58 with various mono and di-substitutions on ring-A and aryloxy, arylamino and aryl moieties at the 6 position of ring-B started from commercially available methyl 4-iodo-2-methoxybenzoate (1) and methyl 4-bromo-2-methoxybenzoate (2). Initially the copper-catalyzed arylation15 of 1 with phenols 3a−e delivered the corresponding diaryl ether intermediates 4a−e, which were then converted into 2-hydroxy-diaryl ethers 5a−e, by selective deprotection of the methoxy (OMe) group with boron trichloride (BCl3).16 Intermediates 5a−e were treated with triflic anhydride (Tf2O) in the presence of pyridine to give the triflates 6a−e, which were further subjected to Buchwald-Hartwig cross coupling with various substituted anilines 7a–l to give the key anthranilate intermediates 8a−q (Scheme 1).14, 17 Conversely, synthesis of anthranilate intermediates 12a−j and 17a−f, key synthons in the synthesis of acridones 43−52 and 53−58, commenced with the coupling of 2 with corresponding anilines 7m−q and boronic acids 13a−d under standard Buchwald-Hartwig and Suzuki cross-coupling reaction conditions to derive intermediates 9a−e and 14a−d, respectively. Intermediates 9a−e and 14a−d then delivered 12a−j and 17a−f in good yields, via the appropriate intermediates 10a−e, 11a−e and 15a−d, 16a−d, respectively. Finally, our recently developed method (boron trifluoride etherate (BF3·Et2O)/microwave irradiation)18 efficiently delivered the desired acridones 18−42 and 53−58 in good to excellent yields in short reaction time (1.0 min), via an intramolecular cyclization of the anthranilate intermediates 8a−q and 17a−f, respectively (Scheme 1). Similarly, Eaton’s acid-mediated cyclization of 12a−j provided the corresponding acridones 43−52 in good yields.14 Positional isomers were separated either by recrystallization or flash column chromatography.
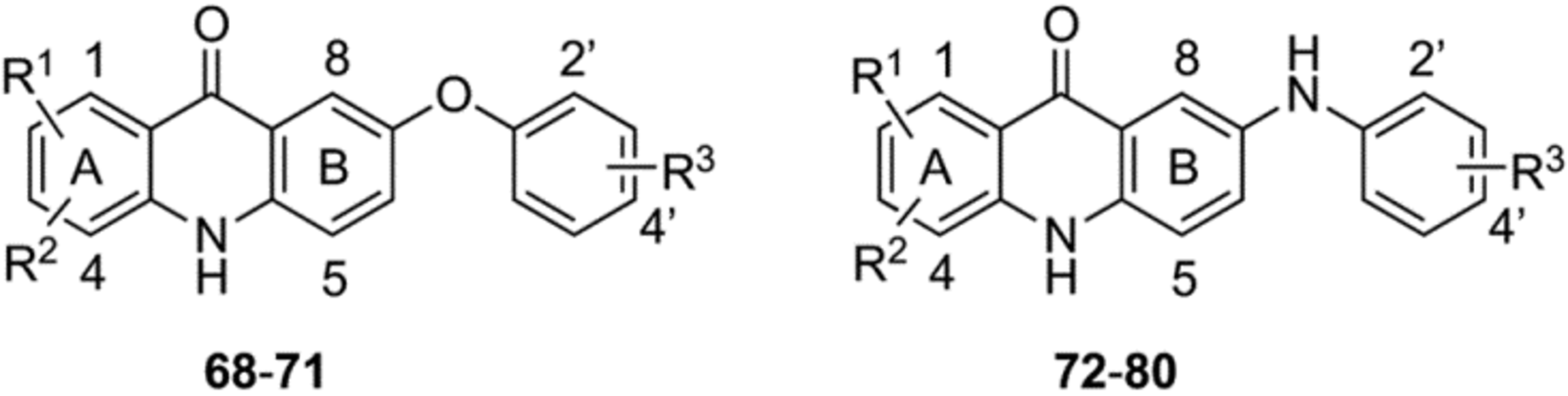
Synthesis of 7-aryloxy- and 7-arylamino-acridones (68–80).
The second series of acridones 68−80 with di-substitutions on ring-A, and aryloxy and arylamino moieties at the 7 position of ring-B were synthesized from commercially available methyl 5-iodo-2-methoxybenzoate (59) accompanying 3a and methyl 5-bromo-2-hydroxybenzoate (60) with appropriate anilines 7, via the same synthetic methods that were employed in the Scheme 1. Eaton’s acid or polyphosphoric acid (PPA)-mediated cyclization of the key anthranilate intermediates 66a−c and 67a−i provided the desired acridones 68−71 and 72−80, respectively, in good yields (Scheme 2).
Scheme 2.
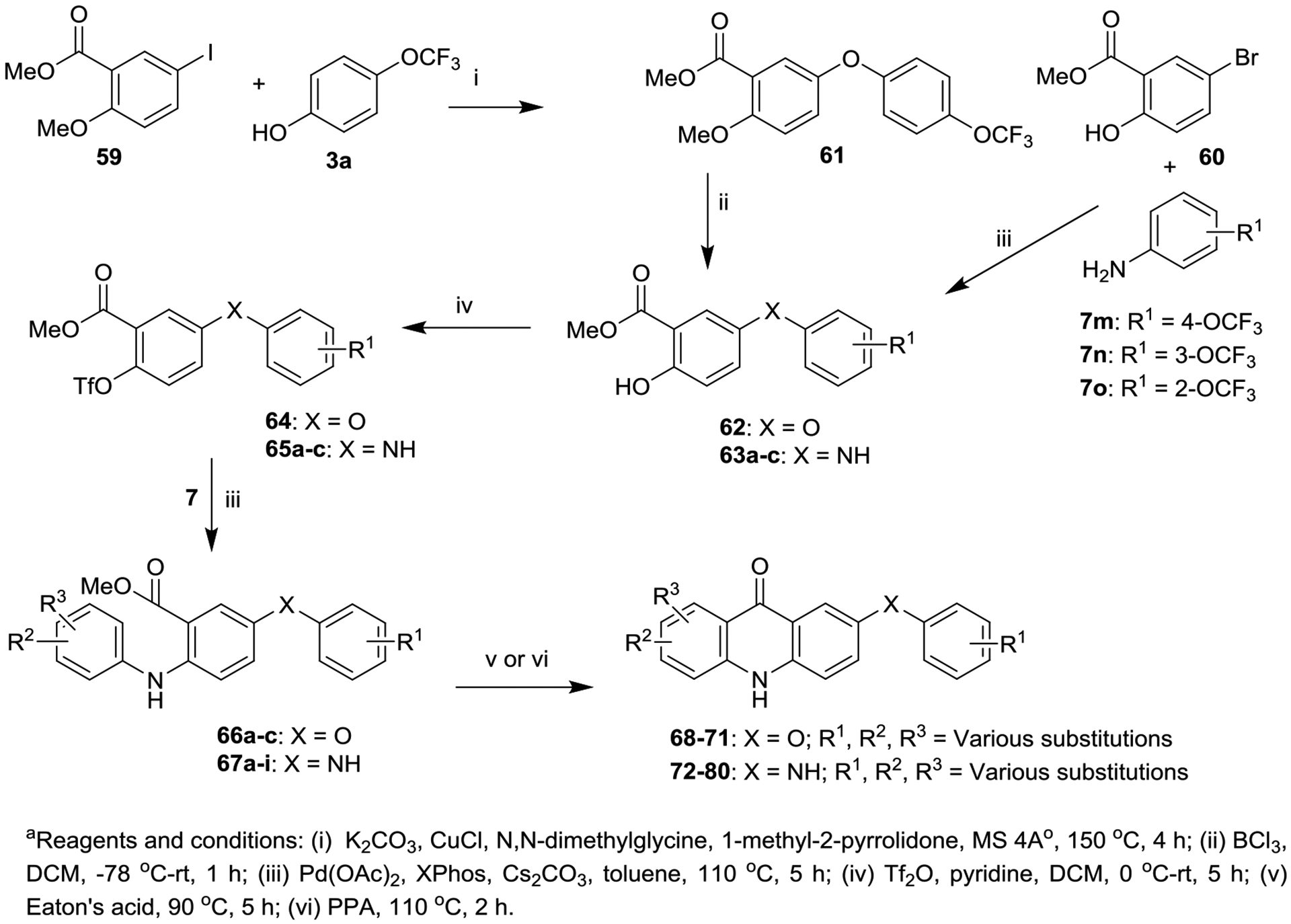
Synthesis of Acridones (68−80) with Aryloxy and Arylamino Groups at the 7 Position of Ring-Ba
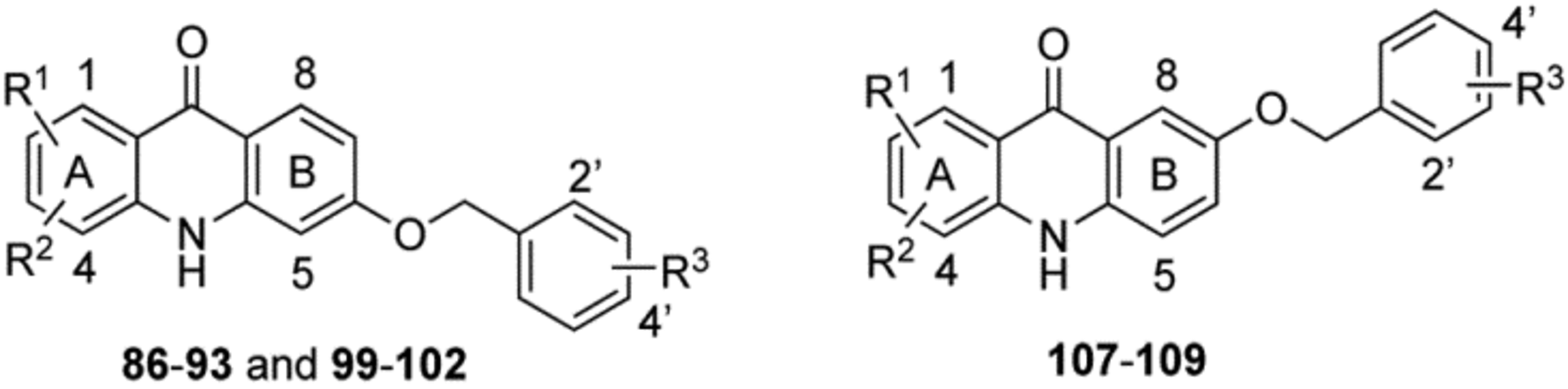
Synthesis of 6- and 7-benzyloxy-acridones (86–93, 99–102 and 107–109).
The third series of acridones 86−93, 99−102 and 107−109 with di-substitutions on ring-A and a variety of benzyloxy moieties at either the 6 or 7 position of ring-B were synthesized starting with commercially available methyl 2-hydroxy-4-methoxybenzoate (81), methyl 2,4-dihydroxybenzoate (94) and methyl 2-bromo-5-methoxybenzoate (103) (Scheme 3). The key hydroxy-acridone intermediates 85a−c and 106 were synthesized via the same synthetic methods that were employed in the previous Schemes 1 and 2. By treating with various benzyl bromides in the presence of anhydrous K2CO3, 85a−c and 106 provided the corresponding acridones 86−93 and 107−109 in good to excellent yields (top and bottom panels, Scheme 3). Conversely, a different strategy was employed for the synthesis of acridones 99−102 (Scheme 3, middle panel), in which the benzyloxy group was installed at the 6 position of ring-B before the Buchwald-Hartwig cross coupling reaction. Synthesis of 99−102 began with the O-benzylation of 94 in the presence of appropriate benzyl bromides and K2CO3 to provide the 4-benzyloxy intermediates 95a and 95b.19 Intermediates 95a and 95b were treated with Tf2O in the presence of pyridine to give the triflate intermediate 96a and 96b, which were further subjected to Buchwald-Hartwig cross-coupling with methoxy- anilines 7 to give the anthranilate intermediates 97a−d. BF3·Et2O-mediated cyclization of 97a−d under microwave irradiation conditions provided the desired acridones 99−102 as major products along with the 6-hydroxy-acridones 98a and 98b as minor products. These two products were separated by column chromatography using a mixture of ethyl acetate and MeOH as a mobile phase. Upon treatment with benzyl bromides in the presence of anhydrous K2CO3, 98a and 98b efficiently delivered the desired acridones 99−102 in excellent yields.19 It is noteworthy that this new synthetic strategy (Scheme 3, middle panel) efficiently generates acridones that contain a OMe group on ring-A and benzyloxy groups on ring-B.
Biological Activity.
The library of second-generation acridones with various mono- and disubstitutions on ring-A and aryloxy, arylamino, aryl or benzyloxy moieties at either the 6 or 7 position of ring-B were evaluated for both blood-stage and liver-stage antimalarial efficacy, safety, and metabolic/PK properties. The results are summarized and discussed in the following sections.
In Vitro Blood-Stage Activity and Structure–Activity Relationships (SAR) of 6-Aryloxy- Acridones (18–42).
In our previous work, a number of first-generation acridones with di-halogens on ring-A and (dialkylamino)alkoxy moieties at the 6 position on ring-B showed promising potency and efficacy against both blood-stage and liver-stage malarial infections.14 Considering that the alkyl moieties are probably easily metabolized in vivo, we hypothesized that the replacement of the (dialkylamino)alkoxy groups on ring-B with aryloxy groups might improve the metabolic stability and PK profiles. Therefore, we generated an initial series of acridones 18−42 bearing mono and di-substitutions on ring-A and aryloxy moieties at the 6 position of ring-B (Table 1). In vitro antimalarial blood-stage activity of acridone derivatives was tested against a panel of multidrug-resistant (MDR) P. falciparum (D6, Dd2, 7G8 and TM90-C2B) with different geographic and genetic backgrounds using a SYBR Green based assay20 and the results are listed in Table 1. Chloroquine (CQ) and atovaquone (ATV) were used as reference drugs in all experiments.
Table 1.
In Vitro Blood-Stage Antiplasmodial Activity and Selectivity of Acridones (18−42)
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | R1 | R2 | R3 | blood-stage IC50 (nM)a vs. P. falciparum |
cytotoxicity IC50 (nM)a vs. HepG2 Cells |
selectivity index vs. HepG2 IC50/D6 IC50 |
|||
| D6 | Dd2 | 7G8 | Tm90-C2B | ||||||
| T31 | - | - | - | 0.022 | 0.041 | 0.43 | 228 | >54510 | >2477727 |
| 18 | 1-Cl | 2-Cl | 4’-OCF3 | 3.2 | 6.8 | 14 | 1053 | 10992 | 3435 |
| 19 | 2-Cl | 3-Cl | 4’-OCF3 | 21 | 7.7 | 14 | 16 | >34418 | >1639 |
| 20 | 1-Cl | 3-Cl | 4’-OCF3 | 0.52 | 0.88 | 1.2 | 23 | >34418 | >66188 |
| 21 | H | 3-Cl | 4’-OCF3 | 2.6 | 2.4 | 14 | 142 | >37340 | >14361 |
| 22 | 1-F | 3-F | 4’-OCF3 | 0.0047 | 0.0030 | 0.0073 | 3.2 | >245700 | >52276595 |
| 23 | 1-F | 3-F | 4’-Cl | 0.010 | 0.28 | 0.68 | 73 | >27953 | >2795300 |
| 24 | 1-F | H | 4’-OCF3 | 0.24 | 0.06 | 0.43 | 12 | 196 | 816 |
| 25 | H | 3-F | 4’-OCF3 | 3.2 | 4.8 | 2.2 | 20 | 9432 | 2947 |
| 26 | 1-F | 3-Cl | 4’-OCF3 | 0.028 | 0.045 | 0.042 | 5.6 | >35754 | >1276928 |
| 27 | 1-Cl | 3-F | 4’-OCF3 | 0.050 | 0.18 | 0.28 | 31 | 798 | 15960 |
| 28 | 1-Cl | 3-OMe | 4’-OCF3 | 5.0 | 2.9 | 10 | 18 | >34763 | >6953 |
| 29 | 1-F | 3-OMe | 4’-OCF3 | 0.65 | 0.31 | 1.6 | 21 | 755 | 1161 |
| 30 | 1-F | 3-OMe | 4’-Cl | 0.51 | 0.39 | 2.8 | 47 | 548 | 1074 |
| 31 | 1-OMe | 3-Cl | 4’-OCF3 | 28 | 22 | 79 | 28 | >34768 | >1242 |
| 32 | 1-OMe | 3-F | 4’-OCF3 | 54 | 32 | 63 | 80 | 25903 | 480 |
| 33 | 1-OMe | 3-F | 4’-Cl | 18 | 15 | 82 | 150 | 22462 | 1248 |
| 34 | 1-Cl | 2-OMe | 4’-OCF3 | 17 | 24 | 51 | 151 | >34767 | >2045 |
| 35 | 2-OMe | 3-Cl | 4’-OCF3 | 157 | 221 | 410 | 137 | >34767 | >221 |
| 36 | 1-OMe | H | 4’-OCF3 | 40 | 57 | 72 | 52 | >100000 | >2500 |
| 37 | H | 3-OMe | 4’-OCF3 | 0.33 | 0.25 | 1.3 | 1.0 | >37751 | >114397 |
| 38 | 2-Cl | 3-OMe | 4’-OCF3 | 0.37 | 0.83 | 1.61 | 0.58 | >34767 | >93965 |
| 39 | 2-F | 3-OMe | 4’-OCF3 | 1.3 | 1.2 | 2.1 | 1.7 | 27908 | 21468 |
| 40 | 2-Cl | 3-OMe | 3’-OCF3 | 2.5 | 1.8 | 2.8 | 2.4 | >34767 | >13907 |
| 41 | 2-F | 3-OMe | 2’-OCF3 | 0.042 | 0.066 | 0.076 | 0.031 | 14838 | 353286 |
| 42 | 2-Cl | 3-OMe | 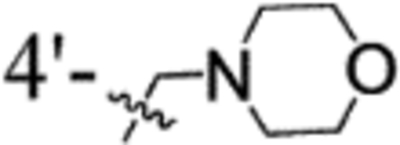 |
0.69 | 0.76 | 1.5 | 0.85 | >33600 | >48696 |
| CQ | - | - | - | 15 | 163 | 171 | 208 | 37577 | 2505 |
| ATV | - | - | - | 0.10 | 0.10 | 0.20 | 8256 | 23160 | 231600 |
IC50 values are the average of at least three determinations, each carried out in triplicate (±10%). CQ: chloroquine; ATV: atovaquone; D6: P. falciparum CQ sensitive strain; Dd2: MDR P. falciparum strain with Old World genetic background; 7G8: MDR P. falciparum strain with New World genetic background; Tm90-C2B: MDR P. falciparum clinical isolate, ATV resistant.
Starting from our first-generation lead acridone compound T31, the (dialkylamino)alkoxy group at the 6 position on ring-B of T31 was first replaced with 4-(trifluoromethoxy)phenyloxy moiety, leading to acridone 18. Although 18 exhibited respectable in vitro antiplasmodial potency with low nanomolar IC50 values against all tested P. falciparum strains (Table 1), it was less potent than T31 (18 IC50 = 3.2 nM vs. T31 IC50 = 0.022 nM against D6, Table 1). Notably, compound 18 showed reduced cross-resistance with ATV against Tm90-C2B (ATV resistant clinical isolate) compared to T31 and ATV (the ratio of Tm90-C2B IC50/D6 IC50 for 18 = 329 vs. T31 = 10363 and ATV = 82560). These findings demonstrated that the aryloxy moiety at the 6 position of ring-B is beneficial for overcoming ATV cross-resistance. Consequently, we retained the aryloxy moiety at the 6 position on ring-B in further modifications and introduced various mono- and di-substitutions at different positions on ring-A. Shifting the chlorine (Cl) of 18 from position 1 to the position 3 as in 19, led to 6-fold reduced antiplasmodial potency (19 IC50 = 21 nM vs. 18 IC50 = 3.2 nM against D6), whereas acridone 20, with Cl substitution of 18 shifting from position 2 to the position 3, showed 6-fold enhanced potency (20 IC50 = 0.52 nM vs. 18 IC50 = 3.2 nM against D6). Removal of Cl substitution at position 1 of 20, as in acridone compound 21, showed 80% decrease in antiplasmodial activity (21 IC50 = 2.6 nM vs. 20 IC50 = 0.52 nM against D6). Replacement of the dichloro substitutions at positions 1 and 3 of ring-A from 20 with difluoro substitutions, as in acridone compound 22, led to significant increase (>100-fold) in the antiplasmodial potency (22 IC50 = 0.0047 nM vs. 20 IC50 = 0.52 nM against D6) with excellent selectivity. Only a two-fold antiplasmodial potency loss was observed when the OCF3 group of 6-aryloxy moiety on ring-B of 22 was replaced with Cl as in 23 (23 IC50 = 0.010 nM vs. 22 IC50 = 0.0047 nM against D6). The greatest loss of antiplasmodial potency was observed with mono fluoro substituted acridones 24 and 25 (51-fold and 680-fold, respectively) when compared with the corresponding difluoro substituted acridones 22. Interestingly, the antiplasmodial potency was retained against all tested P. falciparum strains even after introducing a combination of F and Cl substitutions at both positions 1 and 3 on ring-A with acridones 26 (IC50 = 0.028 nM against D6) and 27 (IC50 = 0.050 nM against D6). These results demonstrated that the dihalogen substitutions on ring-A aryloxy moiety at the 6 position on ring-B are required for optimal potency. The cross-resistance against blood-stage parasites Tm90-C2B for mono and di-halogen substituted acridone compounds 18–27 was reduced compared to the first-generation acridones (Table 1).
To further diminish cross-resistance with ATV, we generated a series of acridones 28–42, with halogen and methoxy moieties on ring-A and aryloxy groups at the 6 position of ring-B, then tested for blood-stage antiplasmodial activity (Table 1). The acridones 28−30, which contain a halogen at position 1 and OMe group at position 3 on ring-A and an aryloxy moiety at position 6 on ring-B, retained the potency against all tested P. falciparum strains as compared to the corresponding di-halogenated acridones 20, 22, 23, 26 and 27. In contrast, the 6-aryloxy-acridones 31−33 containing a methoxy group at the 1 position and halogen at the 3 position on ring-A, showed decreased antiplasmodial activities compared to the corresponding positional isomers 28−30 (31 IC50 = 28 nM vs. 28 IC50 = 5.0 nM; 32 IC50 = 54 nM vs. 29 IC50 = 0.65 nM; 33 IC50 = 18 nM vs. 30 IC50 = 0.51 nM against D6). These observations demonstrated that the combination of halogen and OMe groups at position 1 and 3, respectively, are well tolerated on ring-A, regardless of the substitutions on aryloxy moiety at position 6 of ring-B. Shifting the OMe of 28 from position 3 to position 2 as in 34, led to a minimal reduction in antiplasmodial potency (34 IC50 = 17 nM vs. 28 IC50 = 5.0 nM against D6). A nearly 10-fold loss of activity occurred when the Cl of 34 shifted from position 1 to position 3 on ring-A as in acridone 35 (35 IC50 = 157 nM vs. 34 IC50 = 17 nM against D6). Complete removal of a halogen substitution at position 3 on ring-A of acridone 31 as with the analogue 36 showed a minimal effect on the antiplasmodial activity (36 IC50 = 40 nM vs. 31 IC50 = 28 nM against D6). Interestingly, acridone compound 37, in which the 3 position of ring-A is substituted with OMe group, exhibited significantly increased antiplasmodial potency compared to the corresponding positional isomer 36 (37 IC50 = 0.33 nM vs. 36 IC50 = 40 nM against D6). This work suggested that the OMe group at both positions 1 and 2 on ring-A had adverse effect on antiplasmodial activity, whereas the OMe group was well tolerated at the 3 position on ring-A. We next investigated the effect of halogen and OMe substitutions at positions 2 and 3 on ring-A. It is noteworthy that the 2,3-disubstituted-acridones 38−42 with different aryloxy moieties at the 6 position of ring-B, containing OMe group at position 3 on ring-A showed superior antiplasmodial potency with low nanomolar concentrations (IC50 < 2.8 nM across the entire test panel) and diminished cross-resistance with ATV against Tm90-C2B (ratio of Tm90-C2B IC50/D6 IC50 < 1.5). Most of the acridone compounds containing OMe group on ring-A (compounds 31, 32, and 35−42) also displayed diminished cross-resistance with ATV, compared to the first-generation lead candidate T31 and corresponding di-halogenated 6-aryloxy-acridones 18, 20, 22, 23, 26 and 27. Taken together, SAR analyses of acridone compounds 18−42 (Table 1) demonstrated that a halogen in position 1 or 2 and OMe group at position 3 of ring-A, along with an aryloxy moiety at the 6 position on ring-B are best tolerated.
In Vitro Blood-Stage Activity and SAR of 6-Arylamino-Acridones 43–52.
Having optimized the impact of various substituents on antimalarial activity of acridones that contain a combination of halogen and OMe substitutions on ring-A and an aryloxy moiety at the 6 position of ring-B, we next investigated the impact of the arylamino moiety in the place of aryloxy moiety at the 6 position of ring-B on in vitro blood-stage activity. 1,3-Dihalogenated acridones 43−45 with either 4-(trifluoromethoxy)phenylamino or 4-(chloro)phenylamino moiety at the 6 position of ring-B exhibited excellent potency with low IC50 values against D6 ranging from 0.0060 nM to 0.55 nM (Table 2). These results were comparable to the corresponding 6-aryloxy-acridones 20, 22 and 23 (43 IC50 = 0.55 nM vs. 20 IC50 = 0.52 nM; 44 IC50 = 0.20 nM vs. 22 IC50 = 0.0047 nM; 45 IC50 = 0.0060 nM vs. 23 IC50 = 0.010 nM against D6). Acridones 46−48 with a halogen at position 1 and OMe group at position 3 on ring-A and 4-(trifluoromethoxy)phenylamino and/or 4-(chloro)phenylamino moiety at position 6 of ring-B also retained the potency compared to the corresponding 6-aryloxy-acridones 28−30 (Tables 1 and 2). Significantly, 6-arylamino-acridones 49−52 containing a halogen at position 2 and OMe group at position 3 on ring-A exhibited optimal antiplasmodial activity at low nanomolar concentrations (IC50 = 0.81–5.7 nM) and most reduced cross-resistance with ATV against TM90-C2B. Antiplasmodial potency and cross-resistance pattern were similar to the corresponding 6-aryloxy-acridones 38−42 (Tables 1 and 2). Taken together, SAR of acridone compounds 43−52 demonstrated that a halogen substitution at either the 1 or 2 position and OMe group at the 3 position on ring-A and a substituted arylamino moiety at the 6 position on ring-B are also well tolerated.
Table 2.
In Vitro Blood-Stage Antiplasmodial Activity and Selectivity of Acridones (43−58)
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | R1 | R2 | R3 | blood-stage IC50 (nM)a vs. P. falciparum |
cytotoxicity IC50 (nM)a vs. HepG2 Cells |
selectivity index vs. HepG2 IC50/D6 IC50 |
|||
| D6 | Dd2 | 7G8 | Tm90-C2B | ||||||
| 43 | 1-Cl | 3-Cl | 4’-OCF3 | 0.55 | 1.2 | 4.6 | 71 | 10365 | 18845 |
| 44 | 1-F | 3-F | 4’-OCF3 | 0.20 | 0.12 | 0.73 | 35 | 5779 | 28895 |
| 45 | 1-F | 3-F | 4’-Cl | 0.0060 | 0.020 | 0.016 | 30 | 3036 | 506000 |
| 46 | 1-Cl | 3-OMe | 4’-OCF3 | 14 | 26 | 51 | 47 | >34846 | >2489 |
| 47 | 1-F | 3-OMe | 4’-OCF3 | 0.049 | 0.12 | 0.60 | 2.3 | >10000 | >204081 |
| 48 | 1-F | 3-OMe | 4’-Cl | 0.0070 | 0.0030 | 0.0055 | 2.9 | 3010 | 430000 |
| 49 | 2-F | 3-OMe | 4’-OCF3 | 1.8 | 2.0 | 4.0 | 3.3 | >36216 | >20120 |
| 50 | 2-F | 3-OMe | 3’-OCF3 | 0.81 | 1.5 | 2.2 | 5.4 | >36216 | >44711 |
| 51 | 2-F | 3-OMe | 2’-OCF3 | 1.2 | 2.4 | 5.4 | 2.8 | >36216 | >30180 |
| 52 | 2-Cl | 3-OMe | 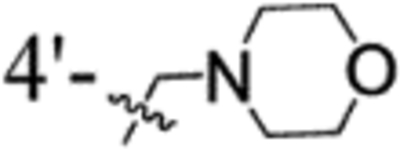 |
5.7 | 9.2 | 9.2 | 5.7 | 30722 | 5390 |
| 53 | 1-F | 3-F | Cl | 2.2 | 3.5 | 59 | 138 | 27266 | 12393 |
| 54 | 1-F | 3-F | OCF3 | 0.54 | 0.75 | 21 | 19 | >25556 | >47326 |
| 55 | 1-F | 3-F | H | 0.60 | 1.3 | 17 | 117 | 31972 | 53287 |
| 56 | 1-F | 3-OMe | Cl | 1.3 | 3.0 | 3.2 | 22 | >28266 | >21743 |
| 57 | 1-F | 3-OMe | OCF3 | 1.8 | 1.9 | 7.5 | 2.0 | 22223 | 12346 |
| 58 | 2-Cl | 3-OMe | 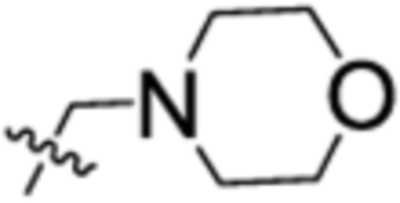 |
3.5 | 6.7 | 15 | 2.1 | 25614 | 7318 |
| CQ | - | - | - | 15 | 163 | 171 | 208 | 37577 | 2505 |
| ATV | - | - | - | 0.10 | 0.10 | 0.20 | 8256 | 23160 | 231600 |
IC50 values are the average of at least three determinations, each carried out in triplicate (±10%). CQ: chloroquine; ATV: atovaquone; D6: P. falciparum CQ sensitive strain; Dd2: MDR P. falciparum strain with Old World genetic background; 7G8: MDR P. falciparum strain with New World genetic background; Tm90-C2B: MDR P. falciparum clinical isolate, ATV resistant.
In Vitro Blood-Stage Activity and SAR of 6-Aryl-Acridones 53–58.
After establishing the halogen and OMe substitutions pattern at the 1, 3 and 2, 3 positions of ring-A, and aryloxy and arylamino groups at the 6 position of ring-B of acridones, we next investigated whether the aryl substitutions at the 6 position on ring-B are tolerated. We synthesized and screened a series of 6-aryl-acridones 53−58 (Table 2) with halogens at both positions 1 and 3, and a combination of halogen and OMe substitutions at either position 1 and 3 or positions 2 and 3 on ring-A. All the 6-aryl-acridones 53−58 showed great in vitro antiplasmodial potency with low nanomolar concentrations (IC50 = 0.54–3.5 nM against D6), similar to the SAR and cross-resistance trend observed with the corresponding 6-aryloxy-acridones and 6-arylamino-acridones. In general, SAR analyses of the in vitro blood-stage antiplasmodial activity with acridones 18−58 (Tables 1 and 2) unambiguously demonstrated that a halogen either at position 1 or 2 and OMe group at position 3 on ring-A and the aryloxy, arylamino and aryl moieties at position 6 on ring-B have an important role on potent antiplasmodial activity, as well as on mitigating cross-resistance with ATV.
In Vitro Blood-Stage Activity and SAR of 7-Aryloxy- and Arylamino-Acridones 68–80.
We next investigated whether the aryloxy and arylamino substitutions at position 7 on ring-B are tolerated for antiplasmodial activities. We synthesized a series of acridones 68−80, where ring-A is substituted with either two halogens or a combination of halogen and methoxy groups, and ring-B is substituted with various (trifluoromethoxy)phenyloxy and (trifluoromethoxy)phenylamino moieties and screened for their in vitro blood-stage antiplasmodial activity (Table 3). Of the 7-aryloxy-acridones 68−71, acridones 68 and 69 with di-fluoro substitutions at positions 1 and 3 and 1-fluoro and 3-methoxy substitutions on ring-A, respectively, exhibited the greatest antiplasmodial potency (IC50 < 0.69 nM against D6). However, the potency of compound 68 significantly declined compared to the corresponding 6-aryloxy-acridone 22 (68 IC50 = 0.12 nM vs. 22 IC50 = 0.0047 nM against D6). Interchange of fluorine and methoxy groups between positions 1 and 3 on ring-A as in 70 resulted in a 25-fold decrease in potency (70 IC50 = 17 nM vs. 69 IC50 = 0.69 nM against D6). Similarly, shifting of fluorine at position 1 of 69 to position 2, as in 71, the potency significantly declined (> 100-fold). It was observed that acridones 69−71 with a methoxy group on ring-A exhibited less cross-resistance with ATV than the di-fluoro substituted 7-aryloxy-acridone 68 (the ratio of Tm90-C2B IC50/D6 IC50 for 69−71 = 1.9–14 vs. 68 = 167), again suggesting the importance of the methoxy group on ring-A. Next, we prepared a series of acridones 72−80 with di-halogens at positions 1 and 3 and a combination of halogen and methoxy group at positions 2 and 3 on ring-A and various (trifluoromethoxy)phenylamino moieties at position 7 on ring-B. All of these 7-arylamino-acridones 72−80, with the exception of 74 (IC50 = 0.048 nM against D6), displayed moderate antiplasmodial activity (IC50 = 29–1474 nM against D6) than the corresponding 7-aryloxy-acridones and 6-arylamino-acridones. These findings indicate that the arylamino groups at position 7 of ring-B were detrimental to optimal antimalarial activities.
Table 3.
In Vitro Blood-Stage Antiplasmodial Activity and Selectivity of Acridones (68−80)
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | R1 | R2 | R3 | blood-stage IC50 (nM)a vs. P. falciparum |
cytotoxicity IC50 (nM)a vs. HepG2 Cells |
selectivity index vs. HepG2 IC50/D6 IC50 |
|||
| D6 | Dd2 | 7G8 | Tm90-C2B | ||||||
| 68 | 1-F | 3-F | 4’-OCF3 | 0.12 | 3.3 | 4.1 | 20 | >37199 | >309992 |
| 69 | 1-F | 3-OMe | 4’-OCF3 | 0.69 | 1.3 | 1.3 | 9.8 | >36131 | >52364 |
| 70 | 1-OMe | 3-F | 4’-OCF3 | 17 | 17 | 35 | 157 | >36131 | >2125 |
| 71 | 2-F | 3-OMe | 4’-OCF3 | 99 | 164 | 130 | 187 | >100000 | >1010 |
| 72 | 1-F | 3-F | 4’-OCF3 | 58 | 16 | 41 | 92 | >37289 | >643 |
| 73 | 1-F | 3-F | 3’-OCF3 | 29 | 17 | 32 | 41 | >37289 | >1286 |
| 74 | 1-F | 3-F | 2’-OCF3 | 0.048 | 0.047 | 0.73 | 23 | 18673 | 389021 |
| 75 | 1-Cl | 3-Cl | 4’-OCF3 | 385 | 1402 | 1733 | 748 | >34495 | >89 |
| 76 | 1-Cl | 3-Cl | 3’-OCF3 | 1474 | 1690 | 750 | 3017 | >34495 | >23 |
| 77 | 2-F | 3-OMe | 4’-OCF3 | 45 | 91 | 156 | 139 | >36216 | >805 |
| 78 | 2-F | 3-OMe | 3’-OCF3 | 100 | 145 | 286 | 130 | >36216 | >362 |
| 79 | 2-Cl | 3-OMe | 4’-OCF3 | 82 | 83 | 166 | 78 | >34846 | >425 |
| 80 | 2-Cl | 3-OMe | 3’-OCF3 | 78 | 56 | 141 | 55 | >34846 | >447 |
| CQ | - | - | - | 15 | 163 | 171 | 208 | 37577 | 2505 |
| ATV | - | - | - | 0.10 | 0.10 | 0.20 | 8256 | 23160 | 231600 |
IC50 values are the average of at least three determinations, each carried out in triplicate (±10%). CQ: chloroquine; ATV: atovaquone; D6: P. falciparum CQ sensitive strain; Dd2: MDR P. falciparum strain with Old World genetic background; 7G8: MDR P. falciparum strain with New World genetic background; Tm90-C2B: MDR P. falciparum clinical isolate, ATV resistant.
In Vitro Blood-Stage Activity and SAR of 6-and 7-Benzyloxy Acridones 86–93, 99–102 and 107–109.
Our detailed SAR explorations around ring-A and ring-B of acridones led to valuable insight of the structural features that are required for potent antimalarial activity. Last, we sought to explore whether the substitution of benzyloxy group in place of aryloxy group on ring-B is tolerated. A series of novel acridones 86−93, 99−102 and 107−109, in which ring-B is substituted with a variety of benzyloxy moieties at either position 6 or 7 and ring-A is substituted with either halogens or a combination of halogen and methoxy groups, were generated and tested for their in vitro blood-stage antimalarial activity (Table 4). Remarkably, all of these benzyloxy-acridones 86−93, 99−102 and 107−109 showed the highest antiplasmodial activities with many compounds exhibiting picomolar IC50 values against D6, regardless of the substitution pattern on the benzyloxy moieties of ring-B and halogen and methoxy substitutions on ring-A. These results unequivocally demonstrated that the benzyloxy moieties at positions 6 and 7 of ring-B play a vital role in enhancing antiplasmodial potency. Notably, the 6-benzyloxy-acridones 99−102 with OMe group on ring-A, exhibited equal or comparable potency across the entire P. falciparum test panel (Table 4) with diminished cross-resistance compared to the corresponding benzyloxy-acridones lacking the OMe group on ring-A. Overall, the detailed SAR of the second-generation acridones (Tables 1–4) suggested that aryl, aryloxy, arylamino and benzyloxy moieties at position 6 on ring-B, and halogens and OMe group at either the 1 and 3 or 2 and 3 positions on ring-A play key roles in enhancing in vitro blood-stage antimalarial potency with balanced cross-resistance pattern.
Table 4.
In Vitro Blood-Stage Antiplasmodial Activity and Selectivity of Acridones (86–93, 99–102 and 107–109)
 | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | R1 | R2 | R3 | blood-stage IC50 (nM)a vs. P. falciparum |
cytotoxicity IC50 (nM)a vs. HepG2 Cells |
selectivity index vs. HepG2 IC50/D6 IC50 |
|||
| D6 | Dd2 | 7G8 | Tm90-C2B | ||||||
| 86 | 1-F | 3-F | H | 0.031 | 0.046 | 0.54 | 45 | >29644 | >956258 |
| 87 | 1-F | 3-F | 4’-OCF3 | 0.0040 | 0.0065 | 0.020 | 7.2 | >23735 | >5933750 |
| 88 | 1-F | 3-F | 4’-SCF3 | 0.013 | 0.078 | 0.16 | 3.8 | 772 | 59384 |
| 89 | 1-F | 3-F | 4’-CF3 | 0.042 | 0.063 | 0.045 | 8.0 | 15617 | 371833 |
| 90 | 1-F | 3-F | 3’-OCF3 | 0.054 | 0.17 | 0.070 | 2.5 | >23735 | >439537 |
| 91 | 1-F | 3-F | 3’-CF3 | 0.084 | 0.21 | 0.13 | 15 | >24672 | >293714 |
| 92 | 1-F | 3-F | 2’-CF3 | 0.0030 | 0.0040 | 0.0063 | 4.3 | >24672 | >8224000 |
| 93 | 1-Cl | 2-Cl | 4’-Cl | 0.033 | 0.038 | 0.28 | 21 | >24711 | >748818 |
| 99 | 2-Cl | 3-OMe | 4’-OCF3 | 0.19 | 0.14 | 0.17 | 0.27 | >22232 | >117010 |
| 100 | 2-Cl | 3-OMe | 4’-CF3 | 0.08 | 0.14 | 0.45 | 0.43 | >23052 | >288150 |
| 101 | 2-F | 3-OMe | 4’-OCF3 | 1.5 | 1.9 | 2.9 | 2.8 | >100000 | >66667 |
| 102 | 2-F | 3-OMe | 4’-CF3 | 3.2 | 3.2 | 7.1 | 10.5 | >100000 | >31250 |
| 107 | 1-F | 3-F | 4’-OCF3 | 0.17 | 0.42 | 0.25 | 65 | >23735 | >139618 |
| 108 | 1-F | 3-F | 4’-CF3 | 0.41 | 0.60 | 0.48 | 189 | >24672 | >60175 |
| 109 | 1-F | 3-F | 4’-Cl | 0.069 | 0.19 | 2.4 | 235 | >26898 | >389826 |
| CQ | - | - | - | 15 | 163 | 171 | 208 | 37577 | 2505 |
| ATV | - | - | - | 0.10 | 0.10 | 0.20 | 8256 | 23160 | 231600 |
IC50 values are the average of at least three determinations, each carried out in triplicate (±10%). CQ: chloroquine; ATV: atovaquone; D6: P. falciparum CQ sensitive strain; Dd2: MDR P. falciparum strain with Old World genetic background; 7G8: MDR P. falciparum strain with New World genetic background; Tm90-C2B: MDR P. falciparum clinical isolate, ATV resistant.
In Vivo Blood-Stage Efficacy in a Rodent Malaria Model.
In vivo antimalarial blood-stage efficacy was determined using a well-established murine 4-day suppression model against P. yoelii.14, 21, 22 Sixty-two acridone compounds were tested in this in vivo model using CQ as a reference drug and the results are summarized in Table 5. The in vivo data are expressed as ED50 and ED90 values. The animals were considered cured if they survive 28 days after the infection without detectable bloodstream parasites. Most of the acridone derivatives exhibited excellent blood-stage oral efficacy (ED50 and ED90 values are shown Table 5). Most significantly, eighteen acridone compounds demonstrated superior blood-stage efficacy compared to first-generation lead compound T31, with ED50 values less than 1 mg/kg/d. Acridones 20, 22, 26, 27, 29 and 88 were curative out to day 28 in this model after treatment of either 10 mg/kg × 4 days or 30 mg/kg × 4 days, i.e., much lower dosages than required for T31 (80 mg/kg × 4 days) to achieve a cure. Notably, acridones 18 and 22 were curative out to day 28 in the same model at 5 and 3 mg/kg × 4 days, respectively. No overt clinical toxicity or behavior change was observed in mice treated with these acridones. In vitro blood-stage activity (Tables 1–4) mostly correlates well with in vivo blood-stage efficacy (Table 5), with the exceptions of compounds 23, 28, 41, 42, 47, 48, 54, 55, 56, 68, 74, 86, 87, 88, 92, 93, 99, and 109 where excellent in vitro blood-stage activity did not translate into superior in vivo blood-stage efficacy. Our down-selection process for lead candidates at this stage was primarily driven by superior in vivo efficacy.
Table 5.
In Vivo Blood-Stage Antimalarial Efficacy against P. yoelii in Mice
| compd | blood-stage efficacy vs. P. yoelii (4 days) | |
|---|---|---|
| ED50 (mg/kg/d) | ED90 (mg/kg/d) | |
| T31 | 1.0 | 1.6 |
| 18a | 0.52 | 0.82 |
| 19 | 1.8 | 1.9 |
| 21 | 4.5 | 6.8 |
| 20b | 1.8 | 3.6 |
| 22c,d | 0.18 | 0.32 |
| 23 | 5.0 | 9.8 |
| 24 | 0.55 | 0.86 |
| 25 | 3.7 | 6.3 |
| 26c | 0.045 | 0.098 |
| 27b | 2.1 | 7.2 |
| 28 | >30 | >30 |
| 29b,c | 0.28 | 0.32 |
| 30 | 3.6 | 12 |
| 31 | >30 | >30 |
| 32 | 23 | >30 |
| 33 | 5.4 | 8.9 |
| 34 | 2.8 | 7.5 |
| 35 | >30 | >30 |
| 36 | >30 | >30 |
| 37 | 1.8 | 14 |
| 38 | 0.75 | 3.9 |
| 39 | 1.3 | 7.3 |
| 40 | 0.16 | 3.6 |
| 41 | >30 | >30 |
| 42 | >30 | >30 |
| 43 | 0.23 | 9.8 |
| 44 | 0.23 | 0.79 |
| 45 | 0.96 | 2.2 |
| 46 | >30 | >30 |
| 47 | 1.1 | 2.3 |
| 48 | 4.1 | 7.4 |
| 49 | 3.7 | 6.7 |
| 50 | 6.8 | >30 |
| 51 | 6.1 | >30 |
| 52 | >30 | >30 |
| 53 | 10 | 19 |
| 54 | 27 | >30 |
| 55 | 15 | 28 |
| 56 | 17 | 32 |
| 57 | 9.2 | 14.7 |
| 58 | 8.9 | 16 |
| 68 | 1.6 | 1.8 |
| 69 | 0.30 | 1.0 |
| 70 | >30 | >30 |
| 73 | 4.6 | 6.6 |
| 74 | 3.9 | 5.8 |
| 77 | 5.4 | 6.9 |
| 86 | 7.2 | 18 |
| 87 | 0.34 | 0.42 |
| 88c | 0.36 | 0.45 |
| 89 | 0.28 | 7.7 |
| 90 | 0.17 | 0.63 |
| 91 | 0.15 | 0.56 |
| 92 | 0.38 | 0.47 |
| 93 | 8.0 | 15 |
| 99 | 6.1 | 12 |
| 100 | 2.8 | 13 |
| 101 | >10 | >10 |
| 102 | >10 | >10 |
| 107 | 0.15 | 1.5 |
| 108 | <5.0 | <5.0 |
| 109 | 9.5 | 16 |
| CQ | 1.5 | 3.3 |
curative at 5 mg/kg/d;
curative at 30 mg/kg/d;
curative at 10 mg/kg/d;
curative at 3.0 mg/kg/d.
In Vitro Liver-Stage Antimalarial Activity and SAR of Second-Generation Acridones.
In vitro liver-stage activity was assessed utilizing luciferase-expressing P. berghei sporozoite infected human hepatocyte HepG2 cells14, 23, 24 and the results are summarized in Table 6. A number of second-generation acridones exhibited potent activity against liver-stage parasites, without evident toxic effect on the host liver cells. Twenty-one compounds, including 6-aryloxy-acridones 18, 22, 24, 26, 29, 34, 38−41, 6-arylamino-acridones 44 and 48, 7-aryloxy-acridone 68, 7-arylamino-acridone 74, 6-benzyloxy-acridones 87, 88, 90−92 and 99, and 7-benzyloxy-acridone 107, showed the highest liver-stage antimalarial potency (IC50 = 0.12–5.9 nM) and were superior to the control drug ATV (IC50 = 6.5 nM). Surprisingly, the majority of the 6-aryl-acridones, and 6- and 7-arylamino-acridones exhibited inferior liver-stage activity, suggesting that aryl and arylamino substitutions on ring-B were detrimental to the liver-stage activity. It is noteworthy that all the 7-arylamino-acridones, with the exception of 74, exhibited inferior in vitro antiparasitic activities against both blood-stage and liver-stage parasites. It appears that aryloxy and benzyloxy moieties at either the 6 or 7 position on ring-B, and halogen and OMe groups at either the 1 and 3 or 2 and 3 positions on ring-A are optimal for liver-stage antimalarial activities.
Table 6.
In Vitro Liver-Stage Antimalarial Activity and In Vivo Liver-Stage Antimalarial Efficacy of Acridones.
| compd | in vitro vs. P. berghei IC50 (nM)a |
in vivo vs. P. berghei (IVIS) | ||
|---|---|---|---|---|
| dose (mg/kg/d)b | clearance rate at 48 hc | cured mice on day 31d | ||
| T31 | 0.50 | 1 | 76% | 0/5 |
| 4 | 99% | 5/5 | ||
| 10 | 100% | 5/5 | ||
| 40 | 100% | 5/5 | ||
| 18 | 5.9 | - | - | - |
| 19 | 66 | 40 | 55% | 0/5 |
| 20 | 37 | - | - | - |
| 21 | 66 | - | - | - |
| 22 | 1.6 | 1 | 88% | 0/5 |
| 4 | 96% | 0/5 | ||
| 10 | 99% | 1/5 | ||
| 40 | 100% | 3/5 | ||
| 23 | 7.4 | 40 | 60% | 0/5 |
| 24 | 0.46 | - | - | - |
| 25 | 36 | - | - | - |
| 26 | 4.5 | 1 | 97% | 1/5 |
| 4 | 100% | 5/5 | ||
| 10 | 100% | 5/5 | ||
| 40 | 100% | 5/5 | ||
| 27 | 32 | 40 | 64% | 0/5 |
| 28 | 41 | - | - | - |
| 29 | 0.45 | 10 | 15% | 0/5 |
| 40 | 99% | 0/5 | ||
| 30 | 14 | 40 | 63% | 0/5 |
| 31 | 73 | - | - | - |
| 32 | 59 | - | - | - |
| 33 | 824 | - | - | - |
| 34 | 5.6 | - | - | - |
| 35 | 126 | - | - | - |
| 37 | 190 | 40 | 71% | 0/5 |
| 38 | 2.2 | 10 | 40% | 0/5 |
| 40 | 29% | 0/5 | ||
| 39 | 4.4 | 10 | 45% | 0/5 |
| 40 | 86% | 0/5 | ||
| 40 | 0.41 | 40 | 98% | 0/5 |
| 41 | 0.60 | 40 | 0% | 0/5 |
| 42 | 7.4 | - | - | - |
| 43 | 6.8 | 1 | 0% | 0/5 |
| 4 | 12% | 0/5 | ||
| 10 | 0% | 0/5 | ||
| 44 | 2.4 | 1 | 0% | 0/5 |
| 4 | 95% | 0/5 | ||
| 10 | 71% | 0/5 | ||
| 45 | 86 | 40 | 23% | 0/5 |
| 46 | 66 | - | - | - |
| 48 | 4.7 | - | - | - |
| 49 | 86 | - | - | - |
| 50 | 109 | - | - | - |
| 51 | 69 | - | - | - |
| 52 | 48 | - | - | - |
| 53 | 816 | - | - | - |
| 54 | 84 | - | - | - |
| 55 | 1334 | - | - | - |
| 56 | 327 | - | - | - |
| 57 | 52 | - | - | - |
| 58 | 26 | - | - | - |
| 68 | 1.5 | 40 | 100% | 4/5 |
| 69 | 15 | 40 | 99% | 3/5 |
| 70 | 159 | 40 | 0% | 0/5 |
| 72 | 2461 | - | - | - |
| 73 | 1219 | - | - | - |
| 74 | 5.9 | - | - | - |
| 75 | 1429 | - | - | - |
| 76 | 2277 | - | - | - |
| 77 | 164 | - | - | - |
| 78 | 1411 | - | - | - |
| 79 | 98 | - | - | - |
| 80 | 418 | - | - | - |
| 86 | 8.0 | - | - | - |
| 87 | 0.64 | 10 | 95% | 0/5 |
| 40 | 100% | 3/5 | ||
| 88 | 1.7 | 40 | 81% | 0/5 |
| 89 | 7.5 | 40 | 67% | 0/5 |
| 90 | 0.51 | 4 | 99% | 4/5 |
| 10 | 100 | 5/5 | ||
| 40 | 100% | 5/5 | ||
| 91 | 0.12 | 10 | 100% | 1/5 |
| 40 | 100% | 5/5 | ||
| 92 | 0.20 | 40 | 56% | 0/5 |
| 93 | 17 | 40 | 6% | 0/5 |
| 100 | 113 | - | - | - |
| 107 | 0.70 | 10 | 100% | 1/5 |
| 40 | 100% | 5/5 | ||
| 108 | 8.2 | 40 | 10% | 0/5 |
| 109 | 29 | 40 | 50% | 0/5 |
| ATV | 6.5 | - | - | - |
| PQ | 13800 | 5 | 100% | 5/5 |
| CQ | > 31262 | - | - | - |
IC50 values are the average of at least three determinations, each carried out in triplicate (±10%).
oral doses of drugs were administered on day – 1, day 0, and day 1 with inoculation of sporozoites on day 0.
the clearance rate was calculated by the ratio of the mean luminescence signal intensity collected by IVIS in drug treated mice over vehicle-only control mice.
the blood parasitemia was measured by flow cytometry up to 31 days after inoculation and the cure was determined with negative parasitemia on day 31.
ATV: atovaquone; PQ: primaquine; CQ: chloroquine.
In Vivo Liver-Stage Efficacy in a Rodent Malaria Model.
In vivo antimalarial liver-stage efficacy was assessed with real-time in vivo imaging system (IVIS), using transgenic bioluminescent parasites in a rodent malaria model.14, 25–27 Luciferase-expressing P. berghei sporozoites were inoculated in mice on day 0, with oral doses of acridones administered on day −1, day 0, and day 1. After the inoculation, bioluminescent signals were measured at 24 h and 48 h for liver-stage development and at 72 h for blood-stage infection. As illustrated in Figure 2, strong bioluminescence signals were detected in untreated mice at both 24 h and 48 h in the liver region, followed by intense signals in the whole body at 72 h, resulting from infection in the peripheral blood circulation. Twenty-eight selected second-generation acridones were tested at various oral doses in this animal model and the results are summarized in Table 6. Acridones 26 (T111) and 90 (T122) provided full protection and cure at 10 mg/kg/d while acridones 91 (T124) and 107 (T129) provided full protection and cure at 40 mg/kg/d without any visible parasites in the liver at 24 h and 48 h (Figure 2), demonstration of true causal prophylactic efficacy. No blood-stage infections were developed at 72 h (Figure 2) and all treated mice were cured without blood-stage parasitemia up to day 31. Most significantly, acridone 26 (T111) provided full protection and sustained blood-stage cure even at 4 mg/kg/d (Figure 2). These results are comparable or superior to primaquine (full protection/cure at 25 mg/kg/d)28 and to the control drug 4-methyl-primaquine (full protection/cure at 5 mg/kg/d). Moreover, at 1 mg/kg/d acridone 26 (T111) offered partial parasite reduction with 97% suppression at 48 h and 1 out of 5 treated mice was cured out to day 31. In addition to these compounds, eight acridones (22, 29, 39, 40, 68, 69, 87, and 88) were able to provide substantial reduction of parasites (> 80% suppression) in the liver at 48 h at 40 mg/kg/d or less (Table 6).
Figure 2.
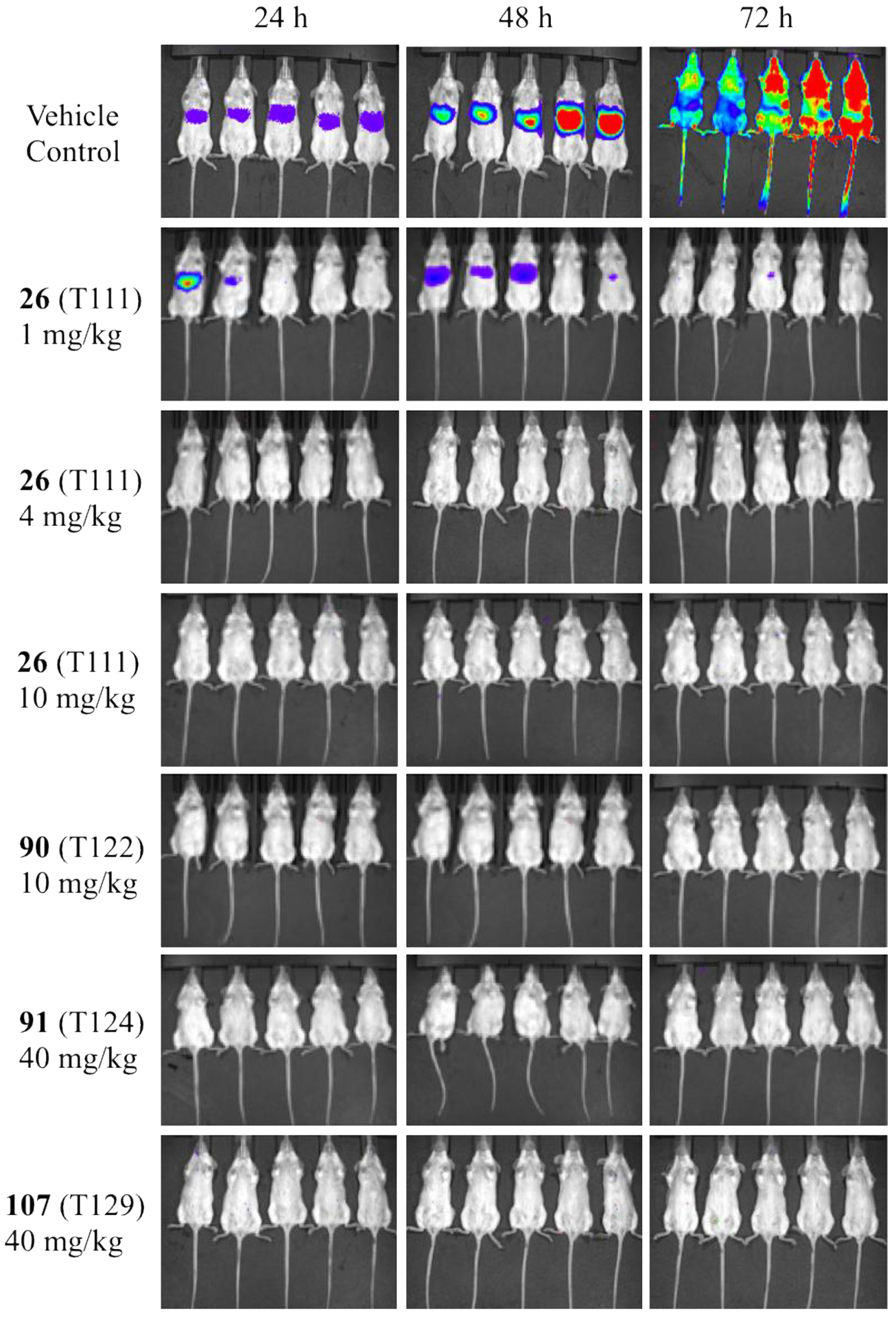
Bioluminescent and real-time in vivo imaging (IVIS) of parasite load in mice with and without acridone treatment. Mice were inoculated with 10000 luciferase-expressing P. berghei sporozoite intravenously (i.v.) on day 0 and oral doses of acridones were administered on day −1, day 0, and day 1. The bioluminescent level represents the parasite burden over the body surface area at 24, 48, and 72 h time points.
Ex Vivo Antimalarial Activity against Clinical Isolates.
Second-generation representative acridone compounds 38 and 39 were also evaluated with up to fifty P. falciparum field clinical isolates collected from malaria patients in Uganda, using an ex vivo IC50 assay.29 The IC50 values of 38 and 39 against these clinical isolates ranged from 1.1 to 8.6 nM (geometric mean = 3.9 nM; 95% CI, 3.5 – 4.5; N = 50) and 1.3 to 12 nM (geometric mean = 5.4 nM; 95% CI, 4.7 – 6.3; N = 49), respectively (Figure 3). The IC50 values for the control antimalarial drugs ranged from 4.1 to 727 nM (geometric mean = 34 nM; 95% CI, 22 – 52; N = 45) for chloroquine (CQ), reflecting the presence of CQ-resistant parasites circulating in eastern Uganda in 2016.29 For other standard antimalarials, IC50 values range from 1.0 to 27 nM (geometric mean = 5.9 nM; 95% CI, 4.8 – 7.3; N = 46) for piperaquine (PIP), 0.3 to 4.4 nM (geometric mean = 1.4 nM; 95% CI, 1.2 – 1.7; N = 41) for dihydroartemisinin (DHA), 0.1 to 0.6 nM (geometric mean = 0.27 nM; 95% CI, 0.20 – 0.35; N = 19) for atovaquone (ATV), and 0.6 to 15.0 nM (geometric mean = 4.2 nM; 95% CI, 3.4 – 5.2; N = 40) for lumefantrine (LUM). There was no statistical difference in median acridone IC50 values between isolates obtained from a clinical trial or from an outpatient center (data not shown). There was a strong positive correlation between responses to 38 and 39 (Spearman’s rank correlation coefficient = 0.8; p < 0.001.) and also between both compounds and ATV (correlation coefficient for 38 and ATV = 0.5, p < 0.05; between 39 and ATV = 0.68, p < 0.05). The latter result is likely due to a mutual target, as significant correlations between other antimalarials and the acridones was not observed.
Figure 3.
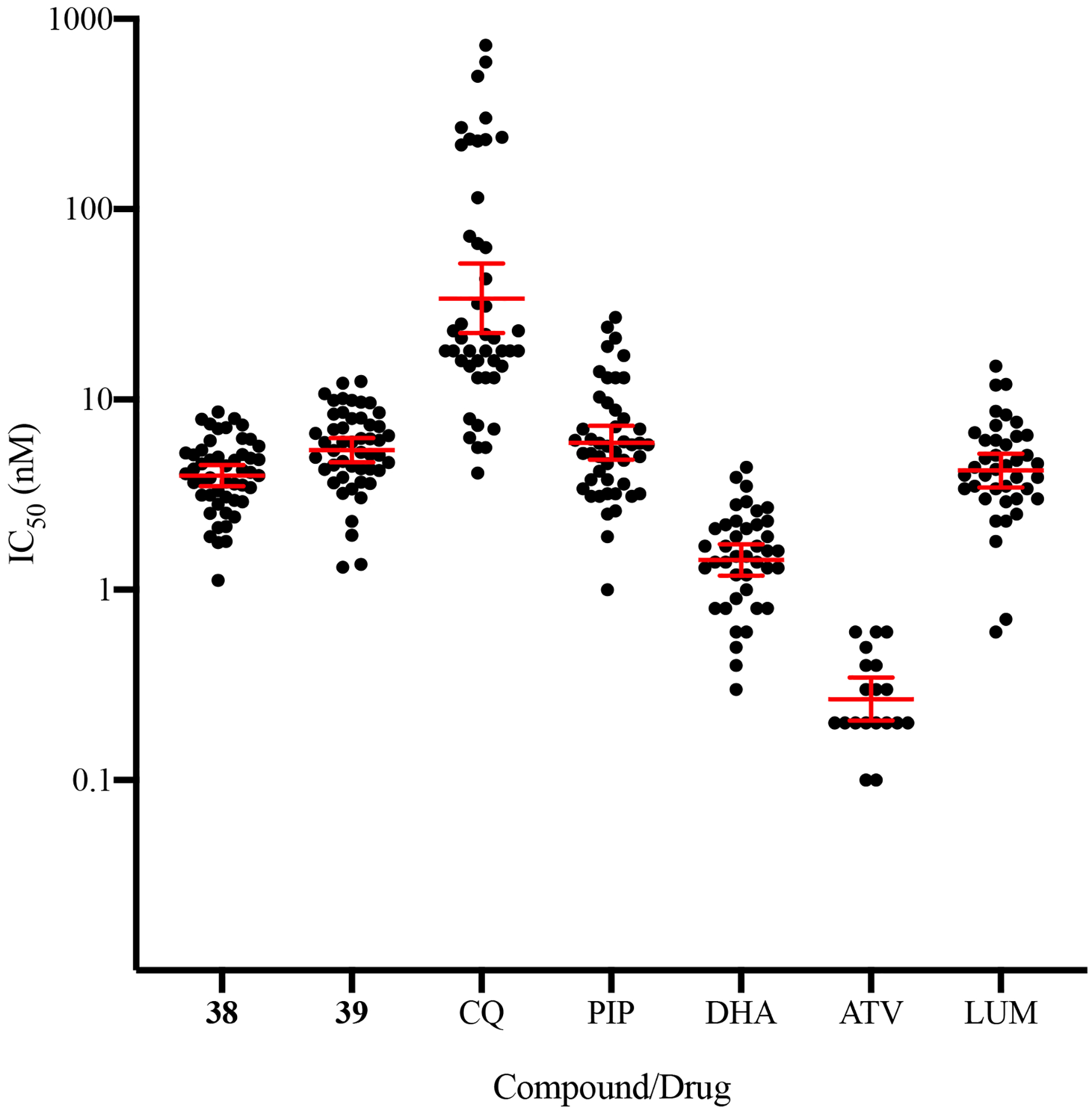
Ex vivo activity of acridones and control antimalarial drugs against P. falciparum clinical isolates in Uganda. Data are presented as geometric means ± 95% CI. CQ: chloroquine; PIP: piperaquine; DHA: dihydroartemisinin; ATV: atovaquone; LUM: lumefantrine.
In Vitro Cytotoxicity.
In vitro general cytotoxicity was tested using human hepatic HepG2 cells.30–32 There was no apparent in vitro mammalian cell cytotoxicity with IC50 values for most acridone compounds above 20 μM (Tables 1–4), indicating favorable therapeutic index (selectivity index >10000 nM).
In Vitro hERG Channel Inhibition.
The in vitro effect of second-generation acridones on the hERG (human-ether-a-go-go-related gene) potassium channel current expressed in mammalian cells was evaluated using an automatic parallel patch clamp system33 (Eurofins Inc.) and the results are listed in Table 7. All of the tested acridones demonstrated hERG inhibition level considerably lower than CQ with estimated IC50 values higher than 10 μM, which is a significant improvement compared to the first-generation acridones with similar or higher inhibition level as CQ.14
Table 7.
In Vitro Cardiotoxicity of Second-Generation Acridones
| compd | hERG (% inhibition at 10 μM) |
|---|---|
| T31 | 57 |
| 18 | 13 |
| 19 | 13 |
| 20 | 23 |
| 21 | 28 |
| 22 | 17 |
| 24 | 24 |
| 25 | 18 |
| 26 | 5.7 |
| 27 | 23 |
| 28 | 16 |
| 29 | 15 |
| 31 | 14 |
| 32 | 13 |
| 34 | 16 |
| 35 | 13 |
| 37 | 22 |
| 38 | 14 |
| 39 | 17 |
| 40 | 14 |
| 41 | 9.2 |
| 42 | 22 |
| 43 | 15 |
| 44 | 20 |
| 46 | 23 |
| 49 | 15 |
| 50 | 22 |
| 51 | 17 |
| 52 | 31 |
| 58 | 21 |
| 68 | 25 |
| 69 | 9.9 |
| 70 | 15 |
| 72 | 17 |
| 73 | 15 |
| 75 | 12 |
| 76 | 13 |
| 77 | 13 |
| 78 | 12 |
| 79 | 24 |
| 80 | 10 |
| CQ | 39 |
In Vitro Metabolic Stability.
The assessments of in vitro metabolic stability of acridone derivatives using mouse and human liver microsomes using well-established methods.14, 32, 34, 35 Most of the second-generation acridones appear to be stable for up to 60 min or longer in both human and mouse microsomes (Table 8), strongly suggesting that the introduction of a variety of aryl moieties on ring-B of the acridone core significantly increased the metabolic stability.
Table 8.
In Vitro Metabolic Stability of Second-Generation Acridones
| compd | metabolic stability vs. microsomes (t1/2 in min) | |
|---|---|---|
| human | mouse | |
| T31 | 12 | 21 |
| 18 | >60 | >60 |
| 19 | >60 | >60 |
| 20 | 54 | >60 |
| 21 | 13 | 19 |
| 22 | >60 | >60 |
| 23 | >60 | >60 |
| 25 | >60 | 27 |
| 26 | >60 | 50 |
| 27 | 28 | 17 |
| 28 | >60 | >60 |
| 29 | >60 | 50 |
| 30 | >60 | >60 |
| 31 | >60 | >60 |
| 32 | 6.8 | 9.9 |
| 33 | >60 | >60 |
| 34 | >60 | >60 |
| 35 | >60 | >60 |
| 37 | 24 | 16 |
| 38 | >60 | >60 |
| 39 | >60 | >60 |
| 40 | >60 | >60 |
| 41 | 37 | 56 |
| 42 | 11 | 11 |
| 43 | >60 | >60 |
| 44 | >60 | >60 |
| 45 | >60 | >60 |
| 46 | >60 | >60 |
| 48 | 46 | 24 |
| 49 | >60 | >60 |
| 50 | >60 | >60 |
| 51 | >60 | >60 |
| 52 | 7.4 | 6.8 |
| 53 | >60 | 60 |
| 54 | >60 | 60 |
| 55 | >60 | 60 |
| 56 | >60 | >60 |
| 57 | >60 | 60 |
| 58 | 22 | 27 |
| 68 | >60 | >60 |
| 69 | >60 | >60 |
| 70 | 39 | >60 |
| 72 | >60 | 23 |
| 73 | >60 | >60 |
| 74 | >60 | >60 |
| 75 | >60 | >60 |
| 76 | >60 | >60 |
| 77 | >60 | >60 |
| 78 | >60 | >60 |
| 79 | >60 | >60 |
| 80 | >60 | >60 |
| 86 | >60 | >60 |
| 87 | >60 | >60 |
| 88 | 28 | >60 |
| 89 | >60 | >60 |
| 90 | >60 | >60 |
| 91 | >60 | >60 |
| 92 | >60 | >60 |
| 93 | >60 | >60 |
| 107 | >60 | >60 |
| 108 | >60 | >60 |
| 109 | >60 | >60 |
| CQ | >60 | >60 |
Pharmacokinetic (PK) Analysis.
In vivo pharmacokinetic (PK) studies of acridone lead candidates 26 (T111), 87 (T121), 90 (T122), 91 (T124) and 107 (T129) was conducted following a single intragastric (p.o.) administration in mice at 80 mg/kg, with blood and liver samples taken at the following time points: 0, 0.5, 1, 2, 4, 7, 24, 48 and 72 h.14, 28, 32 The key PK parameters of the acridone lead candidates in both plasma and liver are summarized in Table 9. Front-runner acridone compound 26 (T111) showed very rapid absorption and reached a plasma peak concentration (Tmax) at 0.5 h. The mean Cmax and AUCinf of 26 in plasma was 17.2 ng.h/mL and 508 ng.h/mL, respectively. The mean elimination half-life of 26 in plasma was 23.6 h and the total clearance rate was calculated to be 157510 mL/h/kg. Since liver-stage active drugs target hepatocytes, the PK profiles in the liver tissue provide important insight into the liver-stage efficacy and facilitate the lead optimization process. Tmax for compound 26 in liver was 7.0 h and the mean Cmax and AUCinf was 1732 ng/mL and 57122 ng.h/mL, respectively. The mean elimination half-life of 26 in liver was shown to be 14.0 h and the total clearance rate was calculated to be 1697 mL/h/kg. The ratio of the liver AUC to the plasma AUC in animals dosed with 26 was 112, whereas the liver/plasma ratio was only 21 for reference drug primaquine and 50 for the first-generation acridone lead candidate T3114. Acridone 26 has a half-life that is 4 times longer in plasma and 3 times longer in liver than that of T3114, further evidence for the improved metabolic stability of second-generation acridones. The PK data is supportive of the observed liver-stage efficacy, as it is evident that acridones demonstrating a strong prophylactic effect concentrate in the liver, with high liver/plasma ratio.
Table 9.
PK Parameters of Acridone Lead Candidates in Plasma and Liver Following Single Oral Dose of 80 mg/kg Administrations in Mice
| compd | matrix |
Cmax (ng/mL) |
Tmax (h) | AUClast (ng.h/mL) |
AUCinf (ng.h/mL) |
t1/2 (h) |
CL/F (mL/h/kg) |
Vz/F (mL/kg) |
MRT b (h) |
|---|---|---|---|---|---|---|---|---|---|
| 26 (T111) | plasma | 17.2 | 0.50 | 392 | 508 | 23.6 | 157510 | 5370641 | 17.6 |
| liver | 1732 | 7.0 | 46232 | 57122 | 14.0 | 1697 | 34167 | 19.9 | |
| 87 (T121) | plasma | 42.0 | 2.0 | 105 | 518 | 8.70 | 154543 | 1940321 | 7.20 |
| liver | 1193 | 4.0 | 20416 | 20494 | 6.00 | 3903 | 33802 | 10.0 | |
| 90 (T122) | plasma | 21.5 | 1.0 | 225 | 236 | 4.90 | 339065 | 2402651 | 10.18 |
| liver | 1967 | 0.50 | 19188 | 19271 | 6.10 | 4151 | 36779 | 7.60 | |
| 91 (T124) | plasma | 19.5 | 1.0 | 175 | 187 | 5.90 | 428286 | 3648089 | 6.20 |
| liver | 925 | 1.0 | 11485 | 11570 | 6.80 | 6914 | 67596 | 8.30 | |
| 107 (T129) | plasma | 170 | 0.5 | 1680 | 1705 | 4.30 | 46906 | 290091 | 5.70 |
| liver | 684 | 2.0 | 8547 | 8674 | 4.00 | 9223 | 53720 | 5.80 | |
| Primaquine | plasma | 531 | 0.50 | 1256 | 1314 | 1.84 | 16415 | 43801 | 2.30 |
| liver | 8148 | 0.50 | 27450 | 27665 | 4.27 | 724 | 4418 | 4.50 |
Cmax: maximum plasma or hepatic concentration; Tmax: time to Cmax; AUXlast: area under the concentration-time curve from 0 up to the last sampling time at which a quantifiable concentration is found; AUCinf: area under the concentration-time curve from 0 up to infinity; t1/2: apparent elimination half-life; CL/F: apparent oral clearance; Vz/F: apparent volume of distribution after oral dose; MRT b: mean residence time, body.
CONCLUSIONS
Building from our first-generation acridone structural optimization, a large library of second-generation acridones was designed and synthesized and extensive SAR profiling demonstrated that the replacement of (dialkylamino)alkoxy pendants at both the 6 and 7 positions on ring-B of first-generation acridones with a variety of aryl, aryloxy, arylamino and benzyloxy moieties led to enhanced antimalarial activity, improved metabolic stability, PK and safety profiles, as well as diminished cross-resistance with ATV. The lead optimization strategy of varying the substitutions on ring-A and ring-B produced a new lead acridone candidate 26 (T111) with the following attributes: 1) inhibition of in vitro P. falciparum blood-stage growth at low concentration against MDR (including ATV-resistant) parasites with a highly favorable therapeutic index; 2) in vivo curative efficacy via oral administration in erythrocytic P. yoelii murine model; 3) prevention of in vitro P. berghei sporozoite-induced development in human hepatocytes at low nanomolar concentration; 4) full in vivo protection and cure via oral administration against liver-stage P. berghei sporozoite-induced infection in mice; 5) mitigated cardiotoxicity with in vitro hERG inhibition level considerably lower than CQ and the first-generation acridones; 6) increased in vitro metabolic stability in both mouse and human liver microsomes; and 7) improved in vivo PK profiles with rapid absorption, high level drug concentration in the liver, and longer half-time than the first-generation acridones. Having achieved these promising target product profiles from the first acridone analogue with hetero-halogen substituents on ring-A (compound 26), we have launched further structural modifications focusing on hetero-halogen containing compounds for the next generation optimization studies and we will ultimately select a late lead candidate for extensive preclinical development.
True causal prophylactic drugs targeting liver-stage malaria offer many advantages,36–39 such as preventing the onset of symptoms as malaria associated pathology only occurs during the blood-stage infection, reducing the likelihood of emergence of drug-resistant parasites given the low number of hepatic forms, interrupting transmission as it depends on maturation of gametocytes in the blood-stage, which is after the liver-stage. Most importantly, only liver-stage active drugs could provide the ability for radical cure to prevent P. vivax and P. ovale relapses, caused by dormant hypnozoites hibernating in host liver cells for long periods of time.13, 40, 41 If a global effort to eradicate malaria is to be successful and sustainable, then it must address the gaps and weaknesses in the armamentarium of currently available prevention and treatment therapies. Ongoing needs include affordability, safety in the most vulnerable patients, low susceptibility to drug resistance adaptations, single-dose treatment, aptitude to kill liver-stage parasites with relapses prevention, and ability to block transmission. The novel chemotype described herein represents a broad-spectrum approach with the potential to overcome these challenges.
EXPERIMENTAL SECTION
General.
NMR spectra were recorded on the Bruker AMX-400 spectrometer at 400 MHz. NMR experiments were recorded in CDCl3 and DMSO-d6 at 25 °C. Chemical shifts are given in parts per million (ppm) downfield from internal standard Me4Si (TMS). High-resolution mass spectrometry (HRMS) (electrospray ionization (ESI)) were recorded on a vanquish UHPLC/HPLC system coupled with high-resolution (35000) Q Exactive Orbitrap mass spectrometer. The microwave reactions were conducted using Biotage® Initiator+ microwave synthesizer. Unless otherwise stated, all reagents and solvents were purchased from commercial suppliers and used without further purification. Reactions which required in anhydrous conditions were carried out under an atmosphere of argon/nitrogen. Chromatography was executed on Biotag-Isolera and/or CombiFlash instruments, using silica gel (230–400 mesh) and/or neutral alumina as the stationary phase and mixtures of ethyl acetate (EtOAc) and hexanes or dichloromethane (DCM) and methanol as eluents. Analytical HPLC analysis was performed on a Supelco Discovery HS C18 column (4.6 mm × 250 mm) with a linear elution gradient of water/methanol (containing 10 mM ammonium acetate) ranging from 80:20 to 0:100 for 50 min at flow rate of 0.9 mL/min, at 254 nm. A purity of > 95% has been established for all tested compounds.
Representative procedure for the synthesis of methyl 2-methoxy-4-(4-(trifluoromethoxy)phenoxy)benzoate (4a).
Methyl 4-iodo-2-methoxybenzoate (1) (10.0 g, 34.2 mmol), 4-(trifluoromethoxy)phenol (3a) (6.1g, 34.2 mmol), copper(I) chloride (CuCl) (678 mg, 6.8 mmol), N,N-dimethylglycine (706 mg, 6.8 mmol), K2CO3 (9.46 g, 68.5 mmol) and 1-methyl-2-pyrrolidone (50 mL) were placed in a sealed tube with a teflon-lined cap under an argon atmosphere at room temperature. The reaction mixture was stirred and heated for 4 h at 150 °C. After cooling to room temperature, the reaction mixture was added to water (300 mL) and extracted with ethyl acetate (3 × 100 mL). The combined organic layers were dried over anhydrous Na2SO4, and the solvent was evaporated under reduced pressure. The crude product was chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afford the pure diaryl ether intermediate 4a (10.7 g, 91%). 1H NMR (CDCl3, 400 MHz) δ 7.83 (d, J = 8.6 Hz, 1H), 7.23 (d, J = 8.7 Hz, 2H), 7.07 (d, J = 8.7 Hz, 2H), 6.63 (d, J = 1.7 Hz, 1H), 6.50 (dd, J = 8.6, 1.7 Hz, 1H), 3.88 (s, 3H), 3.86 (s, 3H). Diaryl ether intermediates 4b–e were synthesized and purified in good yields (56–94%) by the same procedure as described for 4a from 1 and appropriate phenols 3 and their identity was confirmed by GC-MS analysis and carried forward into the next reaction.
Synthesis of methyl 2-hydroxy-4-(4-(trifluoromethoxy)phenoxy)benzoate (5a).
To a stirred solution of 4a (2.0 g, 5.84 mmol) in anhydrous dichloromethane (DCM) (50 mL) maintained under an argon atmosphere was added dropwise boron trichloride solution (BCl3) (7.0 mL, 7.01 mmol, 1.0 M in DCM) at −78 °C. Then the reaction mixture was stirred for 1 h while it was allowed to warm to room temperature. The reaction was quenched with water (100 mL) and extracted with DCM (2 × 50 mL). The combined organic layers were washed with brine and dried over anhydrous Na2SO4. The organic solvent was evaporated under reduced pressure and the product was chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afforded the title compound 5a (1.74 g, 91%). 1H NMR (CDCl3, 400 MHz) δ 10.94 (s, 1H), 7.80 (d, J = 8.8 Hz, 1H), 7.24 (d, J = 8.7 Hz, 2H), 7.09 (d, J = 8.7 Hz, 2H), 6.51 (dd, J = 8.8, 2.1 Hz, 1H), 6.46 (d, J = 2.1 Hz, 1H), 3.93 (s, 3H). Compounds 5b–e were synthesized and purified in excellent yields (90–95%) by the same procedure as described for 5a from 4b–e and their identity was confirmed by GC-MS analysis and carried forward into the next reaction.
Representative procedure for the synthesis of methyl 4-(4-(trifluoromethoxy)phenoxy)-2-(((trifluoromethyl)sulfonyl)oxy)benzoate (6a).
To a stirred solution of 5a (1.50 g, 4.57 mmol) and pyridine (5.0 mL) in anhydrous DCM (25 mL) at 0 °C was added dropwise triflic anhydride (Tf2O) (1.93 g, 6.85 mmol). Then the reaction mixture was stirred for 5 h while it was allowed to warm to room temperature. The mixture was poured into ice-cold water (50 mL) and extracted with diethyl ether (2 × 25 mL). The combined organic layers were washed with 2N HCl (2 × 25 mL), brine and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure to afford the desired product 6a (2.0 g, 95%). The product 6a was carried forward into the next reaction without further purification. 1H NMR (CDCl3, 400 MHz) δ 8.08 (d, J = 8.7 Hz, 1H), 7.29 (d, J = 8.8 Hz, 2H), 7.12 (d, J = 8.8 Hz, 2H), 6.99 (dd, J = 8.7, 2.1 Hz, 1H), 6.86 (d, J = 2.1 Hz, 1H), 3.95 (s, 3H). Compounds 6b–e were synthesized in excellent yields (93–96%) by the same procedure as described for 6a from 5b–e and their identity was confirmed by GC-MS analysis and carried forward into the next reaction.
Representative procedure for the synthesis of methyl 2-((3,4-dichlorophenyl)amino)-4-(4-(trifluoromethoxy)phenoxy)benzoate (8a).
To a degassed solution of 6a (1.8 g, 3.91 mmol) and 3,4-dichloroaniline (7a) (760 mg, 4.69 mmol) in toluene (50 mL) at room temperature were added palladium(II) acetate (Pd(OAc)2) (53 mg, 0.23 mmol), XPhos (223 mg, 0.47 mmol) and Cs2CO3 (2.54 g, 7.82 mmol). The reaction mixture was again degassed and heated at 110 °C for 5 h. After cooling to room temperature, the mixture was filtered through a pad of Celite and washed with DCM (100 mL). The filtrate was concentrated under reduced pressure and the crude product was chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afford the desired product 8a (1.03 g, 56%). 1H NMR (CDCl3, 400 MHz) δ 9.66 (s, 1H), 7.95 (d, J = 8.9 Hz, 1H), 7.33 (d, J = 8.6 Hz, 1H), 7.29 (d, J = 2.5 Hz, 1H), 7.23 (d, J = 8.9 Hz, 2H), 7.06 (d, J = 8.9 Hz, 2H), 7.01 (dd, J = 8.6, 2.5 Hz, 1H), 6.75 (d, J = 2.3 Hz, 1H), 6.37 (dd, J = 8.9, 2.3 Hz, 1H), 3.89 (s, 3H). Compounds 8b–q were synthesized and purified in good yields (52%–65%) by the same procedure as described for 8a from 6a–e and appropriate anilines 7 and their identity was confirmed by GC-MS analysis and carried forward into the next reaction.
Synthesis of intermediates 9a–e.
Compounds 9a–e were synthesized and purified in good yields (67–79%) by the same procedure as described for 8a from methyl 4-bromo-2-methoxybenzoate (2) and appropriate anilines 7 and their identity was confirmed by GC-MS analysis and carried forward into the next reaction.
Synthesis of intermediates 10a–e.
Compounds 10a–e were synthesized and purified in excellent yields (90–93%) by the same procedure as described for 5a from 9a–e and their identity was confirmed by GC-MS analysis and carried forward into the next reaction.
Synthesis of intermediates 11a–e.
Compounds 11a–e were synthesized in excellent yields (92–95%) by the same procedure as described for 6a from 10a–e and their identity was confirmed by GC-MS analysis and carried forward into the next reaction without further purification.
Synthesis of intermediates 12a–j.
Compounds 12a–j were synthesized and purified in good yields (50–65%) by the same procedure as described for 8a from 11a–e and appropriate anilines 7 and their identity was confirmed by GC-MS analysis and carried forward into the next reaction.
Representative procedure for the synthesis of methyl 3-methoxy-4’-(trifluoromethoxy)-[1,1’-biphenyl]-4-carboxylate (14a).
To a degassed stirred solution of 2 (3.0 g, 12.24 mmol) and 4-chlorophenylboronic acid (13a) (2.86 g, 18.36 mmol) in 10% water/1,4-dioxane (100 mL) were added Pd(PPh3)4 (707 mg, 0.61 mmol) and Na2CO3 (3.89 g, 36.73 mmol). The reaction mixture was stirred for 5 h at 100 °C and poured onto water (100 mL). The pH of the solution was lowered to pH 7 with 2 N HCl and extracted with ethyl acetate (3 × 75 mL). The combined organic layers were washed with water and brine and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the product was chromatographed on silica gel, with ethyl acetate/hexanes as eluent, to afford the pure product 14a (2.53 g, 75%). 1H NMR (CDCl3, 400 MHz) δ 7.88 (d, J = 8.2 Hz, 1H), 7.13 (d, J = 8.5 Hz, 2H), 7.43 (d, J = 8.5 Hz, 2H), 7.11 (dd, J = 8.2, 1.4 Hz, 1H), 7.11 (d, J = 1.4 Hz, 1H), 3.97 (s, 3H), 3.91 (s, 3H). Compounds 14b–d were synthesized and purified in good yields (72–80%) by the same procedure as described for 14a from 2 and 13b–d and their identity was confirmed by GC-MS analysis and carried forward into the next reaction.
Synthesis of intermediates 15a–d.
Compounds 15a–d were synthesized and purified in excellent yields (90–94%) by the same procedure as described for 5a from 14a–d and their identity was confirmed by GC-MS analysis and carried forward into the next reaction.
Synthesis of intermediates 16a–d.
Compounds 16a–d were synthesized in excellent yields (92–95%) by the same procedure as described for 6a from 15a–d and their identity was confirmed by GC-MS analysis and carried forward into the next reaction without further purification.
Synthesis of intermediates 17a–f.
Compounds 17a–f were synthesized and purified in good yields (65–72%) by the same procedure as described for 8a from 16a–d and appropriate anilines 7 and their identity was confirmed by GC-MS analysis and carried forward into the next reaction.
Representative procedure for the synthesis of 1,2-dichloro-6-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (18).
BF3.Et2O (603 μL, 4.24 mmol) was added to a microwave reaction vial containing 8a (1.0 g, 2.12 mmol). Then the reaction mixture was exposed to microwave irradiation for 1.0 min at 150 °C. After cooling to room temperature, the reaction mixture was poured into water (25 mL) and allowed to stir for 5.0 min. The solid material was filtered by a sintered funnel and washed with water (50 mL) to afford the mixture of the acridones 18 and 19. The pure acridones 18 (451 mg, 48%) and 19 (111 mg, 12%) were obtained by recrystallization from a mixture of ethyl acetate and MeOH and DCM and acetonitrile, respectively.
1,2-Dichloro-6-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (18).
1H NMR (DMSO-d6, 400 MHz) δ 11.77 (s, 1H), 8.19 (d, J = 8.9 Hz, 1H), 7.82 (d, J = 9.0 Hz, 1H), 7.52 (d, J = 8.5 Hz, 2H), 7.39 (m, 3H), 7.01 (dd, J = 9.0, 2.3 Hz, 1H), 6.76 (d, J = 2.2 Hz, 1H); HRMS (ESI) calcd for C20H11Cl2F3NO3 (M + H)+ 440.0063, found 440.0059.
2,3-Dichloro-6-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (19).
1H NMR (DMSO-d6, 400 MHz) δ 11.80 (d, J = 0.4 Hz, 1H), 8.26 (s, 1H), 8.22 (d, J = 9.0 Hz, 1H), 7.63 (s, 1H), 7.53 (d, J = 8.4 Hz, 2H), 7.39 (d, J = 8.9 Hz, 2H), 7.03 (dd, J = 8.8, 2.2 Hz, 1H), 6.81 (d, J = 2.1 Hz, 1H); HRMS (ESI) calcd for C20H11Cl2F3NO3 (M + H)+ 440.0063, found 440.0062.
1,3-Dichloro-6-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (20).
1H NMR (DMSO-d6, 400 MHz) δ 11.70 (s, 1H), 8.16 (d, J = 9.0 Hz, 1H), 7.53 (d, J = 8.6 Hz, 2H), 7.39–7.35 (m, 3H), 7.28 (s, 1H), 7.00 (dd, J = 8.9, 2.0 Hz, 1H), 6.73 (d, J = 1.9 Hz, 1H); HRMS (ESI) calcd for C20H11Cl2F3NO3 (M + H)+ 440.0063, found 440.0060.
3-Chloro-6-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (21).
1H NMR (DMSO-d6, 400 MHz) δ 11.70 (s, 1H), 8.24 (d, J = 8.9 Hz, 1H), 8.19 (d, J = 8.7 Hz, 1H), 7.53 (d, J = 8.6 Hz, 2H), 7.44 (d, J = 1.9 Hz, 1H), 7.38 (d, J = 9.0 Hz, 2H), 7.26 (dd, J = 8.6, 2.0 Hz, 1H), 7.02 (dd, J = 8.9, 2.4 Hz, 1H), 6.82 (d, J = 2.3 Hz, 1H); HRMS (ESI) calcd for C20H12ClF3NO3 (M + H)+ 406.0452, found 406.0450.
1,3-Difluoro-6-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (22).
1H NMR (DMSO-d6, 400 MHz) δ 11.77 (s, 1H), 8.19–8.17 (m, 1H), 7.52 (d, J = 8.6 Hz, 2H), 7.37 (d, J = 8.9 Hz, 2H), 6.98 (m, 3H), 6.77 (d, J = 1.6 Hz, 1H); HRMS (ESI) calcd for C20H11F5NO3 (M + H)+ 408.0654, found 408.0649.
6-(4-Chlorophenoxy)-1,3-difluoroacridin-9(10H)-one (23).
1H NMR (DMSO-d6, 400 MHz) δ 11.74 (d, J = 0.5 Hz, 1H), 8.16 (d, J = 8.9 Hz, 1H), 7.57 (d, J = 7.8 Hz, 2H), 7.29 (d, J = 7.7 Hz, 2H), 6.96 (m, 3H), 6.73 (s, 1H); HRMS (ESI) calcd for C20H11ClF2NO2 (M + H)+ 358.0441, found 358.0435.
1-Fluoro-6-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (24).
1H NMR (DMSO-d6, 400 MHz) δ 11.67 (d, J = 0.3 Hz, 1H), 8.19 (d, J = 8.9 Hz, 1H), 7.63 (m, 1H), 7.52 (d, J = 8.6 Hz, 2H), 7.38 (d, J = 9.0 Hz, 2H), 7.21 (dd, J = 8.5, 0.2 Hz, 1H), 6.98 (dd, J = 8.9, 2.2 Hz, 1H), 6.92 (dd, J = 11.8, 8.0 Hz, 1H), 6.80 (d, J = 2.3 Hz, 1H); HRMS (ESI) calcd for C20H12F4NO3 (M + H)+ 390.0748, found 390.0740.
3-Fluoro-6-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (25).
1H NMR (DMSO-d6, 400 MHz) δ 11.69 (s, 1H), 8.27–8.22 (m, 2H), 7.52 (d, J = 8.3 Hz, 2H), 7.38 (d, J = 9.1 Hz, 2H), 7.12 (m, 2H), 7.00 (dd, J = 8.9, 2.4 Hz, 1H), 6.82 (d, J = 2.3 Hz, 1H); HRMS (ESI) calcd for C20H12F4NO3 (M + H)+ 390.0748, found 390.0743.
3-Chloro-1-fluoro-6-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (26).
1H NMR (DMSO-d6, 400 MHz) δ 11.75 (s, 1H), 8.17 (d, J = 8.9 Hz, 1H), 7.52 (d, J = 8.2 Hz, 2H), 7.38 (d, J = 8.7 Hz, 2H), 7.22 (s, 1H), 7.11 (d, J = 11.2 Hz, 1H), 7.01 (dd, J = 9.1, 1.4 Hz, 1H), 6.77 (d, J = 1.6 Hz, 1H); HRMS (ESI) calcd for C20H11ClF4NO3 (M + H)+ 424.0358, found 424.0353.
1-Chloro-3-fluoro-6-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (27).
1H NMR (DMSO-d6, 400 MHz) δ 11.72 (s, 1H), 8.17 (d, J = 8.9 Hz, 1H), 7.52 (d, J = 8.5 Hz, 2H), 7.38 (d, J = 8.9 Hz, 2H), 7.18 (dd, J = 8.9, 2.3 Hz, 1H), 7.09 (dd, J = 9.7, 2.1 Hz, 1H), 6.99 (dd, J = 8.9, 2.1 Hz, 1H), 6.74 (d, J = 2.0 Hz, 1H); HRMS (ESI) calcd for C20H11ClF4NO3 (M + H)+ 424.0358, found 424.0352.
1-Chloro-3-methoxy-6-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (28).
1H NMR (DMSO-d6, 400 MHz) δ 11.52 (s, 1H), 8.15 (d, J = 8.9 Hz, 1H), 7.51 (d, J = 8.3 Hz, 2H), 7.36 (d, J = 9.0 Hz, 2H), 6.94 (dd, J = 8.9, 2.4 Hz, 1H), 6.84 (d, J = 2.4 Hz, 1H), 6.74 (m, 2H), 3.87 (s, 3H); HRMS (ESI) calcd for C21H14ClF3NO4 (M + H)+ 436.0558, found 436.0555.
1-Fluoro-3-methoxy-6-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (29).
1H NMR (DMSO-d6, 400 MHz) δ 11.54 (s, 1H), 8.14 (d, J = 8.9 Hz, 1H), 7.51 (d, J = 8.5 Hz, 2H), 7.36 (d, J = 9.0 Hz, 2H), 6.94 (dd, J = 8.9, 2.3 Hz, 1H), 6.76 (d, J = 2.3 Hz, 1H), 6.63–6.59 (m, 2H), 3.86 (s, 3H); HRMS (ESI) calcd for C21H14F4NO4 (M + H)+ 420.0853, found 420.0847.
6-(4-Chlorophenoxy)-1-fluoro-3-methoxyacridin-9(10H)-one (30).
1H NMR (DMSO-d6, 400 MHz) δ 11.52 (s, 1H), 8.13 (d, J = 8.9 Hz, 1H), 7.56 (d, J = 8.9 Hz, 2H), 7.27 (d, J = 8.9 Hz, 2H), 6.92 (dd, J = 8.9, 2.4 Hz, 1H), 6.73 (d, J = 2.3 Hz, 1H), 6.63–6.58 (m, 2H), 3.86 (s, 3H); HRMS (ESI) calcd for C20H14ClFNO3 (M + H)+ 370.0641, found 370.0635.
3-Chloro-1-methoxy-6-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (31).
1H NMR (DMSO-d6, 400 MHz) δ 11.41 (s, 1H), 8.13 (d, J = 8.9 Hz, 1H), 7.51 (d, J = 8.6 Hz, 2H), 7.36 (d, J = 9.0 Hz, 2H), 6.93 (m, 2H), 6.72 (m, 2H), 3.87 (s, 3H); HRMS (ESI) calcd for C21H14ClF3NO4 (M + H)+ 436.0558, found 436.0553.
3-Fluoro-1-methoxy-6-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (32).
1H NMR (DMSO-d6, 400 MHz) δ 11.45 (s, 1H), 8.13 (d, J = 8.9 Hz, 1H), 7.50 (d, J = 8.3 Hz, 2H), 7.35 (d, J = 9.1 Hz, 2H), 6.93 (dd, J = 8.9, 2.4 Hz, 1H), 6.74 (d, J = 2.3 Hz, 2H), 6.62 (m, 2H), 3.86 (s, 3H); HRMS (ESI) calcd for C21H14F4NO4 (M + H)+ 420.0853, found 420.0847.
6-(4-Chlorophenoxy)-3-fluoro-1-methoxyacridin-9(10H)-one (33).
1H NMR (DMSO-d6, 400 MHz) δ 11.52 (s, 1H), 8.13 (d, J = 8.9 Hz, 1H), 7.56 (d, J = 8.8 Hz, 2H), 7.27 (d, J = 8.8 Hz, 2H), 6.92 (dd, J = 8.9, 2.1 Hz, 1H), 6.73 (d, J = 2.0 Hz, 1H), 6.63–6.59 (m, 2H), 3.86 (s, 3H); HRMS (ESI) calcd for C20H14ClFNO3 (M + H)+ 370.0641, found 370.0636.
1-Chloro-2-methoxy-6-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (34).
1H NMR (DMSO-d6, 400 MHz) δ 11.51 (s, 1H), 8.18 (d, J = 8.9 Hz, 1H), 7.62 (d, J = 9.2 Hz, 1H), 7.51 (d, J = 8.5 Hz, 2H), 7.38 (m, 3H), 6.94 (dd, J = 8.8, 1.7 Hz, 1H), 6.74 (d, J = 1.9 Hz, 1H), 3.89 (s, 3H); HRMS (ESI) calcd for C21H14ClF3NO4 (M + H)+ 436.0558, found 436.0553.
3-Chloro-2-methoxy-6-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (35).
1H NMR (DMSO-d6, 400 MHz) δ 11.62 (s, 1H), 8.25 (d, J = 8.9 Hz, 1H), 7.73 (s, 1H), 7.52 (m, 3H), 7.38 (d, J = 9.0 Hz, 2H), 6.99 (dd, J = 8.9, 2.1 Hz, 1H), 6.80 (d, J = 2.0 Hz, 1H), 3.95 (s, 3H); HRMS (ESI) calcd for C21H14ClF3NO4 (M + H)+ 436.0558, found 436.0554.
1-Methoxy-6-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (36).
1H NMR (DMSO-d6, 400 MHz) δ 11.32 (s, 1H), 8.13 (d, J = 8.9 Hz, 1H), 7.54 (d, J = 8.2 Hz, 1H), 7.50 (d, J = 8.4 Hz, 2H), 7.35 (d, J = 9.1 Hz, 2H), 6.92–6.90 (m, 2H), 6.76 (d, J = 2.3 Hz, 1H), 6.68 (dd, J = 8.3, 0.7 Hz, 1H), 3.84 (s, 3H); HRMS (ESI) calcd for C21H15F3NO4 (M + H)+ 402.0948, found 402.0942.
3-Methoxy-6-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (37).
1H NMR (DMSO-d6, 400 MHz) δ 11.51 (s, 1H), 8.21 (d, J = 8.8 Hz, 1H), 8.11 (d, J = 8.9 Hz, 1H), 7.51 (d, J = 8.4 Hz, 2H), 7.36 (d, J = 8.8 Hz, 2H), 6.94 (dd, J = 8.8, 1.4 Hz, 1H), 6.86–6.79 (m, 3H), 3.87 (s, 3H); HRMS (ESI) calcd for C21H15F3NO4 (M + H)+ 402.0948, found 402.0942.
2-Chloro-3-methoxy-6-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (38).
1H NMR (DMSO-d6, 400 MHz) δ 11.68 (s, 1H), 8.20 (d, J = 8.9 Hz, 1H), 8.11 (s, 1H), 7.52 (d, J = 8.5 Hz, 2H), 7.37–7.35 (m, 2H), 6.97 (dd, J = 8.9, 2.3 Hz, 1H), 6.92 (s, 1H), 6.83 (d, J = 2.3 Hz, 1H), 3.97 (s, 3H); HRMS (ESI) calcd for C21H14ClF3NO4 (M + H)+ 436.0558, found 436.0554.
2-Fluoro-3-methoxy-6-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (39).
1H NMR (DMSO-d6, 400 MHz) δ 11.67 (s, 1H), 8.20 (d, J = 8.8 Hz, 1H), 7.83 (d, J = 11.7 Hz, 1H), 7.51 (d, J = 8.6 Hz, 2H), 7.36 (d, J = 9.0 Hz, 2H), 6.97 (m, 2H), 6.84 (d, J = 2.2 Hz, 1H), 3.96 (s, 3H); HRMS (ESI) calcd for C21H14F4NO4 (M + H)+ 421.0853, found 421.0847.
2-Chloro-3-methoxy-6-(3-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (40).
1H NMR (DMSO-d6, 400 MHz) δ 11.73 (s, 1H), 8.21 (d, J = 8.9 Hz, 1H), 8.12 (s, 1H), 7.63 (t, J = 8.2 Hz, 1H), 7.29 (m, 3H), 6.98 (dd, J = 8.9, 2.2 Hz, 1H), 6.94 (s, 1H), 6.89 (d, J = 2.1 Hz, 1H), 3.98 (s, 3H); HRMS (ESI) calcd for C21H14ClF3NO4 (M + H)+ 436.0558, found 436.0552.
2-Chloro-3-methoxy-6-(2-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (41).
1H NMR (DMSO-d6, 400 MHz) δ 11.68 (s, 1H), 8.21 (d, J = 8.9 Hz, 1H), 8.11 (s, 1H), 7.65–7.62 (m, 1H), 7.57–7.53 (m, 1H), 7.45 (m, 2H), 6.95 (dd, J = 8.9, 2.4 Hz, 1H), 6.91 (s, 1H), 6.81 (d, J = 2.4 Hz, 1H), 3.97 (s, 3H); HRMS (ESI) calcd for C21H14ClF3NO4 (M + H)+ 436.0558, found 436.0553.
2-Chloro-3-methoxy-6-(4-(morpholinomethyl)phenoxy)acridin-9(10H)-one (42).
1H NMR (DMSO-d6, 400 MHz) δ 11.66 (s, 1H), 8.18 (d, J = 8.9 Hz, 1H), 8.11 (s, 1H), 7.43 (d, J = 8.4 Hz, 2H), 7.18 (d, J = 8.4 Hz 2H), 6.95–6.92 (m, 2H), 6.79 (d, J = 2.1 Hz, 1H), 3.96 (s, 3H), 3.60 (t, J = 4.3 Hz, 4H), 3.50 (s, 2H), 2.39 (m, 4H); HRMS (ESI) calcd for C25H24ClN2O4 (M + H)+ 451.1419, found 451.1413.
Representative procedure for the synthesis of 1,3-dichloro-6-((4-(trifluoromethoxy)phenyl)amino)acridin-9(10H)-one (43).
Eaton’s acid (25 mL) was added to a flask containing 12a (500 mg, 1.06 mmol) and the reaction mixture was then stirred at 90 °C for 5 h. After cooling to room temperature, the reaction mixture was added slowly to water, allowed to stir for 15 min, then neutralized with 2 N NaOH. The resulting solid material was filtered by a sintered funnel and purified by column chromatography using ethyl acetate with 10% methanol as a mobile phase to obtain the pure compound 43 (266 mg, 57%). The acridones 44–52 were synthesized and purified in good yields (55–62%) by the same procedure as described for 43 from 12b–j.
1,3-Dichloro-6-((4-(trifluoromethoxy)phenyl)amino)acridin-9(10H)-one (43).
1H NMR (DMSO-d6, 400 MHz) δ 11.50 (s, 1H), 9.08 (s, 1H), 8.00 (d, J = 8.7 Hz, 1H), 7.39–7.35 (m, 4H), 7.22 (d, J = 1.8 Hz, 1H), 6.93–6.89 (m, 3H); HRMS (ESI) calcd for C20H12Cl2F3N2O4 (M + H)+ 439.0222, found 439.0219.
1,3-Difluoro-6-((4-(trifluoromethoxy)phenyl)amino)acridin-9(10H)-one (44).
1H NMR (DMSO-d6, 400 MHz) δ 11.54 (s, 1H), 9.06 (s, 1H), 8.00 (d, J = 8.8 Hz, 1H), 7.39–7.34 (m, 4H), 6.96–6.88 (m, 4H); HRMS (ESI) calcd for C20H12F5N2O2 (M + H)+ 407.0813, found 407.0807.
6-((4-Chlorophenyl)amino)-1,3-difluoroacridin-9(10H)-one (45).
1H NMR (DMSO-d6, 400 MHz) δ 11.53 (s, 1H), 9.01 (s, 1H), 7.99 (d, J = 8.8 Hz, 1H), 7.41 (d, J = 8.5 Hz, 2H), 7.28 (d, J = 8.5 Hz, 2H), 6.91 (m, 4H); HRMS (ESI) calcd for C19H12ClF2N2O (M + H)+ 357.0601, found 357.0596.
1-Chloro-3-methoxy-6-((4-(trifluoromethoxy)phenyl)amino)acridin-9(10H)-one (46).
1H NMR (DMSO-d6, 400 MHz) δ 8.94 (s, 1H), 7.97 (d, J = 8.8 Hz, 1H), 7.37–7.32 (m, 4H), 6.94 (d, J = 2.0 Hz, 1H), 6.84 (dd, J = 8.8, 2.0 Hz, 1H), 6.75–6.74 (m, 2H), 3.85 (s, 3H); HRMS (ESI) calcd for C21H15ClF3N2O3 (M + H)+ 435.0718, found 435.0714.
1-Fluoro-3-methoxy-6-((4-(trifluoromethoxy)phenyl)amino)acridin-9(10H)-one (47).
1H NMR (DMSO-d6, 400 MHz) δ 11.31 (s, 1H), 8.98 (s, 1H), 7.97 (d, J = 8.8 Hz, 1H), 7.37–7.32 (m, 4H), 6.94 (d, J = 1.2 Hz, 1H), 6.86 (dd, J = 8.7, 1.3 Hz, 1H), 6.60 (d, J = 1.1 Hz, 1H), 6.53 (dd, J = 13.6, 1.9 Hz, 1H), 3.86 (s, 3H); HRMS (ESI) calcd for C21H15F4N2O3 (M + H)+ 419.1013, found 419.1010.
6-((4-Chlorophenyl)amino)-1-fluoro-3-methoxyacridin-9(10H)-one (48).
1H NMR (DMSO-d6, 400 MHz) δ 11.31 (s, 1H), 8.94 (s, 1H), 7.96 (d, J = 8.8 Hz, 1H), 7.40 (d, J = 8.8 Hz, 2H), 7.26 (d, J = 8.8 Hz, 2H), 6.92 (d, J = 2.0 Hz, 1H), 6.85 (dd, J = 8.8, 2.1 Hz, 1H), 6.60 (d, J = 2.0 Hz, 1H), 6.53 (dd, J = 13.5, 2.3 Hz, 1H), 3.86 (s, 3H); HRMS (ESI) calcd for C20H15ClFN2O2 (M + H)+ 369.0801, found 369.0791.
2-Fluoro-3-methoxy-6-((4-(trifluoromethoxy)phenyl)amino)acridin-9(10H)-one (49).
1H NMR (DMSO-d6, 400 MHz) δ 11.41 (s, 1H), 9.00 (s, 1H), 8.03 (d, J = 8.7 Hz, 1H), 7.78 (d, J = 11.7 Hz, 1H), 7.36 (m, 4H), 7.01–6.89 (m, 3H), 3.96 (s, 3H); HRMS (ESI) calcd for C21H15F4N2O3 (M + H)+ 419.1013, found 419.1007.
2-Fluoro-3-methoxy-6-((3-(trifluoromethoxy)phenyl)amino)acridin-9(10H)-one (50).
1H NMR (DMSO-d6, 400 MHz) δ 11.44 (s, 1H), 9.13 (s, 1H), 8.05 (d, J = 8.8 Hz, 1H), 7.79 (d, J = 11.8 Hz, 1H), 7.47 (d, J = 8.2 Hz, 1H), 7.29 (dd, J = 8.2, 1.3 Hz, 1H), 7.16 (m, 1H), 7.08 (d, J = 2.1 Hz, 1H), 6.99–6.92 (m, 3H), 3.97 (s, 3H); HRMS (ESI) calcd for C21H15F4N2O3 (M + H)+ 419.1013, found 419.1010.
2-Fluoro-3-methoxy-6-((2-(trifluoromethoxy)phenyl)amino)acridin-9(10H)-one (51).
1H NMR (DMSO-d6, 400 MHz) δ 11.36 (s, 1H), 8.66 (s, 1H), 8.01 (d, J = 8.7 Hz, 1H), 7.78 (d, J = 11.5 Hz, 1H), 7.57 (dd, J = 8.0, 1.5 Hz, 1H), 7.48–7.40 (m, 2H), 7.24 (t, J = 7.4 Hz, 1H), 6.95 (d, J = 7.2 Hz, 1H), 6.90 (dd, J = 8.9, 1.6 Hz, 1H), 6.79 (d, J = 1.8 Hz, 1H), 3.95 (s, 3H); HRMS (ESI) calcd for C21H15F4N2O3 (M + H)+ 419.1013, found 419.1005.
2-Chloro-3-methoxy-6-((4-(morpholinomethyl)phenyl)amino)acridin-9(10H)-one (52).
1H NMR (DMSO-d6, 400 MHz) δ 11.03 (br s, 1H), 8.43 (br s, 1H), 8.10 (d, J = 2.0 Hz, 1H), 8.01 (dd, J = 8.7, 1.7 Hz, 1H), 7.84–7.82 (m, 1H), 7.24–7.16 (m, 3H), 6.91 (s, 1H), 6.82–6.80 (m, 2H), 3.91 (s, 3H), 3.60 (m, 4H), 3.41 (m, 2H), 2.38 (m, 4H); HRMS (ESI) calcd for C25H25ClN3O3 (M + H)+ 450.1579, found 450.1572.
Synthesis of acridones 53–58.
Acridones 53–58 were synthesized and purified in good yields (65–74%) by the same procedure as described for 18 from 17a–f.
6-(4-Chlorophenyl)-1,3-difluoroacridin-9(10H)-one (53).
1H NMR (DMSO-d6, 400 MHz) δ 11.98 (s, 1H), 8.23 (d, J = 8.4 Hz, 1H), 7.80 (dd, J = 6.6, 1.9 Hz, 2H), 7.66 (d, J = 1.6 Hz, 1H), 7.63–7.57 (m, 3H), 7.09 (d, J = 10.0 Hz, 1H), 7.05–6.99 (m, 1H); HRMS (ESI) calcd for C19H11ClF2NO (M + H)+ 342.0492, found 342.0501.
1,3-Difluoro-6-(4-(trifluoromethoxy)phenyl)acridin-9(10H)-one (54).
1H NMR (DMSO-d6, 400 MHz) δ 11.99 (s, 1H), 8.25 (d, J = 8.4 Hz, 1H), 7.90 (d, J = 8.6 Hz, 2H), 7.68 (s, 1H),7.60 (d, J = 8.4 Hz, 1H), 7.55 (d, J = 8.4 Hz, 2H), 7.09 (dd, J = 9.7 Hz, 1H), 7.05–7.00 (m, 1H); HRMS (ESI) calcd for C20H11F5NO2 (M + H)+ 392.0704, found 392.0713.
1,3-Difluoro-6-phenylacridin-9(10H)-one (55).
1H NMR (DMSO-d6, 400 MHz) δ 11.97 (s, 1H), 8.23 (d, J = 8.4 Hz, 1H), 7.78 (d, J = 8.5 Hz, 2H), 7.67 (d, J = 1.5 Hz, 1H), 7.61–7.54 (m, 3H), 7.49 (d, J = 7.3 Hz, 1H), 7.09 (d, J = 10.0 Hz, 1H), 7.04–6.98 (m, 1H); HRMS (ESI) calcd for C19H12F2NO (M + H)+ 308.0881, found 308.0893.
6-(4-Chlorophenyl)-1-fluoro-3-methoxyacridin-9(10H)-one (56).
1H NMR (DMSO-d6, 400 MHz) δ 11.83 (s, 1H), 8.20 (d, J = 8.4 Hz, 1H), 7.78 (d, J = 7.9 Hz, 2H), 7.64 (s, 1H), 7.61 (d, J = 7.9 Hz, 2H), 7.52 (d, J = 8.4 Hz, 1H), 6.75 (s, 1H), 6.63 (d, J = 13.4 Hz, 1H), 3.90 (s, 3H); HRMS (ESI) calcd for C20H14ClFNO2 (M + H)+ 354.0692, found 354.0685.
1-Fluoro-3-methoxy-6-(4-(trifluoromethoxy)phenyl)acridin-9(10H)-one (57).
1H NMR (DMSO-d6, 400 MHz) δ 11.73 (s, 1H), 8.21 (d, J = 8.4 Hz, 1H), 7.87 (d, J = 8.6 Hz, 2H), 7.62 (s, 1H), 7.54 (m, 3H), 6.72 (s, 1H), 6.63 (dd, J = 13.4, 1.8 Hz, 1H), 3.90 (s, 3H); HRMS (ESI) calcd for C21H14F4NO3 (M + H)+ 404.0904, found 404.0914.
2-Chloro-3-methoxy-6-(4-(morpholinomethyl)phenyl)acridin-9(10H)-one (58).
1H NMR (DMSO-d6, 400 MHz) δ 11.85 (s, 1H), 8.25 (d, J = 8.4 Hz, 1H), 8.15 (s, 1H), 7.74–7.69 (m, 3H), 7.56 (dd, J = 8.4, 1.2 Hz, 1H), 7.48 (d, J = 8.0 Hz, 2H), 7.06 (s, 1H), 4.02 (s, 3H), 3.60 (t, J = 4.6 Hz, 4H), 3.54 (s, 2H), 2.40 (t, J = 4.0 Hz, 4H); HRMS (ESI) calcd for C25H24ClN2O3 (M + H)+ 435.1470, found 435.1466.
Synthesis of intermediate 61.
Compound 61 was synthesized and purified in good yield (3.35 g, 57%) by the same procedure as described for 4a from methyl 5-iodo-2-methoxybenzoate (59) and 3a and its identity was confirmed by GC-MS analysis and carried forward into the next reaction.
Synthesis of intermediate 62.
Compound 62 was synthesized and purified in good yield (2.69 g, 91%) by the same procedure as described for 5a from 61 and its identity was confirmed by GC-MS analysis and carried forward into the next reaction.
Synthesis of intermediates 63a–c.
Compounds 63a–c were synthesized and purified in good yields (52–63%) by the same procedure as described for 8a from methyl 5-bromo-2-hydroxybenzoate (60) and appropriate anilines 7 and their identity was confirmed by GC-MS analysis and carried forward into the next reaction.
Synthesis of intermediates 64 and 65a–c.
Compounds 64 and 65a–c were synthesized in excellent yields (93–96%) by the same procedure as described for 6a from 62 and 63a–c, respectively, and their identity was confirmed by GC-MS analysis and carried forward into the next reaction without further purification.
Synthesis of intermediates 66a–c and 67a–i.
Compounds 66a–c and 67a–i were synthesized and purified in good yields (64–71%) by the same procedure as described for 8a from 64 and 65a–c and their identity was confirmed by GC-MS analysis and carried forward into the next reaction.
Synthesis of acridones 68–71.
The acridones 68–71 were synthesized and purified in good yields (57–65%) by the same procedure as described for 43 from 66a–c.
1,3-Difluoro-7-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (68).
1H NMR (DMSO-d6, 400 MHz) δ 12.03 (br s, 1H), 7.64 (d, J = 1.2 Hz, 1H), 7.63 (d, J = 1.2 Hz, 1H), 7.58 (d, J = 2.2 Hz, 1H), 7.42 (d, J = 8.4 Hz, 2H), 7.17 (m, 2H), 7.07–6.97 (m, 2H); HRMS (ESI) calcd for C20H11F5NO3 (M + H)+ 408.0654, found 408.0665.
1-Fluoro-3-methoxy-7-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (69).
1H NMR (DMSO-d6, 400 MHz) δ 11.80 (s, 1H), 7.63 (d, J = 2.0 Hz, 1H), 7.54 (m, 2H), 7.42 (d, J = 8.8 Hz, 2H), 7.16 (d, J = 9.0 Hz, 2H), 6.71 (d, J = 1.9 Hz, 1H), 6.62 (dd, J = 13.4, 2.3 Hz, 1H), 3.89 (s, 3H); HRMS (ESI) calcd for C21H14F4NO4 (M + H)+ 420.0853, found 420.0847.
3-Fluoro-1-methoxy-7-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (70).
1H NMR (DMSO-d6, 400 MHz) δ 11.67 (s, 1H), 7.62 (m, 1H), 7.50 (m, 2H), 7.41 (d, J = 8.4 Hz, 2H), 7.15 (d, J = 9.1 Hz, 2H), 6.74 (dd, J = 10.2, 2.3 Hz, 1H), 6.61 (dd, J = 11.9, 2.3 Hz, 1H), 3.86 (s, 3H); HRMS (ESI) calcd for C21H14F4NO4 (M + H)+ 420.0853, found 420.0847.
2-Fluoro-3-methoxy-7-(4-(trifluoromethoxy)phenoxy)acridin-9(10H)-one (71).
1H NMR (DMSO-d6, 400 MHz) δ 11.90 (s, 1H), 7.82 (d, J = 11.7 Hz, 1H), 7.66 (d, J = 2.8 Hz, 1H), 7.60 (m, 1H), 7.56 (m, 1H), 7.41 (d, J = 8.6 Hz, 2H), 7.16 (d, J = 8.6 Hz, 2H), 7.07 (d, J = 7.3 Hz, 1H), 3.99 (s, 3H); HRMS (ESI) calcd for C21H14F4NO4 (M + H)+ 420.0853, found 420.0864.
Representative procedure for the synthesis of 1,3-difluoro-7-((4-(trifluoromethoxy)-phenyl)amino)acridin-9(10H)-one (72).
Polyphosphoric acid (PPA) (5.0 g) was added to a flask containing 67a (500 mg, 1.14 mmol) and the reaction mixture was then stirred at 110 °C for 2 h. After cooling to room temperature, water was added slowly to the flask and allowed to stir for 15 min. Then the reaction mixture was neutralized with 2 N NaOH and the solid material was filtered through a sintered funnel and washed with water (100 mL) and methanol (10 mL), to obtain the pure compound 72 (287 mg, 62%). The acridones 73–80 were synthesized and purified in good yields (60–65%) by the same procedure as described for 72 from 67b–i.
1,3-Difluoro-7-((4-(trifluoromethoxy)phenyl)amino)acridin-9(10H)-one (72).
1H NMR (DMSO-d6, 400 MHz) δ 11.91 (br s, 1H), 8.55 (s, 1H), 7.84 (d, J = 2.3 Hz, 1H), 7.55 (dd, J = 8.8, 2.3 Hz, 1H), 7.47 (d, J = 8.8 Hz, 1H), 7.24 (d, J = 8.6 Hz, 2H), 7.13 (d, J = 8.6 Hz, 2H), 7.02 (d, J = 10.0 Hz, 1H), 6.93 (m, 1H); HRMS (ESI) calcd for C20H12F5N2O2 (M + H)+ 407.0813, found 407.0814.
1,3-Difluoro-7-((3-(trifluoromethoxy)phenyl)amino)acridin-9(10H)-one (73).
1H NMR (DMSO-d6, 400 MHz) δ 11.89 (s, 1H), 8.69 (s, 1H), 7.88 (s, 1H), 7.56 (d, J = 7.3 Hz, 1H), 7.49 (d, J = 8.8 Hz, 1H), 7.34 (d, J = 8.1 Hz, 1H), 7.07–6.94 (m, 4H), 6.76 (d, J = 7.6 Hz, 1H); HRMS (ESI) calcd for C20H12F5N2O2 (M + H)+ 407.0813, found 407.0816.
1,3-Difluoro-7-((2-(trifluoromethoxy)phenyl)amino)acridin-9(10H)-one (74).
1H NMR (DMSO-d6, 400 MHz) δ 11.88 (s, 1H), 8.18 (s, 1H), 7.81 (d, J = 2.2 Hz, 1H), 7.57 (m, 1H), 7.46 (d, J = 8.8 Hz, 1H), 7.35 (d, J = 8.2 Hz, 1H), 7.30 (m, 2H), 7.03 (d, J = 10.2 Hz, 1H), 6.97 (m, 2H); HRMS (ESI) calcd for C20H12F5N2O2 (M + H)+ 407.0813, found 407.0802.
1,3-Dichloro-7-((4-(trifluoromethoxy)phenyl)amino)acridin-9(10H)-one (75).
1H NMR (DMSO-d6, 400 MHz) δ 11.96 (s, 1H), 8.57 (s, 1H), 7.85 (d, J = 1.7 Hz, 1H), 7.53 (d, J = 1.9 Hz, 1H), 7.48 (d, J = 8.8 Hz, 2H), 7.25 (m, 3H), 7.14 (d, J = 8.8 Hz, 2H); HRMS (ESI) calcd for C20H12Cl2F3N2O2 (M + H)+ 439.0222, found 439.0225.
1,3-Dichloro-7-((3-(trifluoromethoxy)phenyl)amino)acridin-9(10H)-one (76).
1H NMR (DMSO-d6, 400 MHz) δ 11.86 (s, 1H), 8.70 (s, 1H), 7.88 (s, 1H), 7.56 (d, J = 7.2 Hz, 1H), 7.48 (d, J = 7.4 Hz, 2H), 7.34 (d, J = 7.8 Hz, 1H), 7.25 (s, 1H), 7.08 (d, J = 7.2 Hz, 1H), 6.94 (s, 1H), 6.76 (d, J = 6.4 Hz, 1H); HRMS (ESI) calcd for C20H12Cl2F3N2O2 (M + H)+ 439.0222, found 439.0226.
2-Fluoro-3-methoxy-7-((4-(trifluoromethoxy)phenyl)amino)acridin-9(10H)-one (77).
1H NMR (DMSO-d6, 400 MHz) δ 11.81 (s, 1H), 8.49 (br s, 1H), 7.87 (d, J = 1.8 Hz, 1H), 7.82 (d, J = 11.8 Hz, 1H), 7.52 (m, 2H), 7.23 (d, J = 8.6 Hz, 2H), 7.12 (m, 2H), 7.07 (m, 1H), 3.98 (s, 3H); HRMS (ESI) calcd for C21H15F4N2O3 (M + H)+ 419.1013, found 419.1015.
2-Fluoro-3-methoxy-7-((3-(trifluoromethoxy)phenyl)amino)acridin-9(10H)-one (78).
1H NMR (DMSO-d6, 400 MHz) δ 11.78 (s, 1H), 8.64 (s, 1H), 7.91 (s, 1H), 7.82 (d, J = 11.8 Hz, 1H), 7.53 (m, 2H), 7.34 (d, J = 8.1 Hz, 1H), 7.05 (d, J = 7.4 Hz, 2H), 6.92 (s, 1H), 6.73 (d, J = 7.8 Hz, 1H), 3.99 (s, 3H); HRMS (ESI) calcd for C21H15F4N2O3 (M + H)+ 419.1013, found 419.1016.
2-Chloro-3-methoxy-7-((4-(trifluoromethoxy)phenyl)amino)acridin-9(10H)-one (79).
1H NMR (DMSO-d6, 400 MHz) δ 8.49 (s, 1H), 8.11 (s, 1H), 7.88 (s, 1H), 7.51 (s, 2H), 7.23 (d, J = 8.6 Hz, 2H), 7.11 (d, J = 8.6 Hz, 2H), 7.04 (s, 1H), 3.98 (s, 3H); HRMS (ESI) calcd for C21H15ClF3N2O3 (M + H)+ 435.0718, found 435.0720.
2-Chloro-3-methoxy-7-((3-(trifluoromethoxy)phenyl)amino)acridin-9(10H)-one (80).
1H NMR (DMSO-d6, 400 MHz) δ 11.80 (s, 1H), 8.65 (s, 1H), 8.12 (s, 1H), 7.91 (d, J = 1.6 Hz, 1H), 7.54 (m, 2H), 7.34 (d, J = 8.2 Hz, 1H), 7.05 (d, J = 8.2 Hz, 2H), 7.02 (s, 1H), 6.92 (s, 1H), 6.74 (d, J = 7.8 Hz, 1H), 3.99 (s, 3H); HRMS (ESI) calcd for C21H15ClF3N2O3 (M + H)+ 435.0718, found 435.0720.
Synthesis of methyl 4-methoxy-2-(((trifluoromethyl)sulfonyl)oxy)benzoate (82).
Compound 82 was synthesized in excellent yield (16.56 g, 96%) by the same procedure as described for 6a from methyl 2-hydroxy-4-methoxybenzoate (81) and its identity was confirmed by GC-MS analysis and carried forward into the next reaction without further purification.
Synthesis of compounds 83a–c.
Compounds 83a–c were synthesized and purified in good yields (65–75%) by the same procedure as described for 8a from 82 and appropriate anilines 7 and their identity was confirmed by GC-MS analysis and carried forward into the next reaction.
Synthesis of acridone intermediates (84a–c).
Compounds 84a–c were synthesized and purified in good yields (93–95%) by the same procedure as described for 18 from 83a–c and carried forward into the next reaction.
Representative procedure for the synthesis of 1,3-difluoro-6-hydroxyacridin-9(10H)-one (85a).
To a stirred solution of 84a (3.0 g, 11.49 mmol) in 75 mL of 57% hydriodic acid (HI) at room temperature was added phenol (17.3 g, 183.9 mmol), and the reaction mixture was stirred at reflux for 3 h. After cooling to room temperature, water was added slowly to the flask and the resulting solid was filtered. Trituration with acetone and water yielded the title compound 85a (2.58 g, 91%). Compounds 85b and 85c were synthesized in excellent yields 87% and 90%, respectively, by the same procedure as described for 85a, and carried forward into the next reaction.
Representative procedure for the synthesis of 1,3-difluoro-6-((4-(trifluoromethoxy)benzyl)oxy)acridin-9(10H)-one (86).
To a stirred solution of 85a (200 mg, 0.80 mmol) in anhydrous acetone (100 mL) at room temperature were added K2CO3 (1.67 g, 12.14 mmol) and benzyl bromide (152 mg, 0.89 mmol), and the reaction mixture was stirred at reflux for 3 h. The hot reaction mixture was then filtered and the solvent was removed by rotary evaporation. The obtained product was washed with methanol (25 mL), to afford the title compound 86 (202 mg, 74%). Acridones 87–93 were synthesized and purified in excellent yields (80–86%) by the same procedure as described for 86.
6-(Benzyloxy)-1,3-difluoroacridin-9(10H)-one (86).
1H NMR (DMSO-d6, 400 MHz) δ 11.77 (br s, 1H), 8.07 (d, J = 8.9 Hz, 1H), 7.50 (d, J = 7.2 Hz, 2H), 7.43 (d, J = 7.2 Hz, 2H), 7.37 (d, J = 7.0 Hz, 1H), 7.10 (d, J = 10.9 Hz, 1H), 6.96 (d, J = 6.8 Hz, 2H), 6.89 (s, 1H), 5.25 (s, 2H); HRMS (ESI) calcd for C20H14F2NO2 (M + H)+ 338.0987, found 338.0999.
1,3-Difluoro-6-((4-(trifluoromethoxy)benzyl)oxy)acridin-9(10H)-one (87).
1H NMR (DMSO-d6, 400 MHz) δ 11.77 (br s, 1H), 8.08 (d, J = 8.9 Hz, 1H), 7.64 (d, J = 8.4 Hz, 2H), 7.43 (d, J = 8.4 Hz, 2H), 7.02–6.93 (m, 3H), 6.88 (d, J = 2.0 Hz, 1H), 5.29 (s, 2H); HRMS (ESI) calcd for C21H13F5NO3 (M + H)+ 422.0810, found 422.0815.
1,3-Difluoro-6-((4-((trifluoromethyl)thio)benzyl)oxy)acridin-9(10H)-one (88).
1H NMR (DMSO-d6, 400 MHz) δ 11.80 (br s, 1H), 8.09 (d, J = 8.9 Hz, 1H), 7.79 (d, J = 8.2 Hz, 2H), 7.66 (d, J = 8.2 Hz, 2H), 7.07–6.96 (m, 3H), 6.89 (d, J = 2.3 Hz, 1H), 5.36 (s, 2H); HRMS (ESI) calcd for C21H13F5NO2S (M + H)+ 438.0582, found 438.0596.
1,3-Difluoro-6-((4-(trifluoromethyl)benzyl)oxy)acridin-9(10H)-one (89).
1H NMR (DMSO-d6, 400 MHz) δ 11.78 (br s, 1H), 8.09 (d, J = 8.9 Hz, 1H), 7.81 (d, J = 8.2 Hz, 2H), 7.72 (d, J = 8.2 Hz, 2H), 7.02–6.95 (m, 3H), 6.88 (d, J = 2.0 Hz, 1H), 5.39 (s, 2H); HRMS (ESI) calcd for C21H13F5NO2 (M + H)+ 406.0861, found 406.0870.
1,3-Difluoro-6-((3-(trifluoromethoxy)benzyl)oxy)acridin-9(10H)-one (90).
1H NMR (DMSO-d6, 400 MHz) δ 11.78 (br s, 1H), 8.08 (d, J = 8.9 Hz, 1H), 7.60–7.53 (m, 2H), 7.50 (s, 1H), 7.37 (d, J = 7.5 Hz, 1H), 7.02–6.93 (m, 3H), 6.88 (d, J = 2.0 Hz, 1H), 5.32 (s, 2H); HRMS (ESI) calcd for C21H13F5NO3 (M + H)+ 422.0810, found 422.0818.
1,3-Difluoro-6-((3-(trifluoromethyl)benzyl)oxy)acridin-9(10H)-one (91).
1H NMR (DMSO-d6, 400 MHz) δ 11.79 (br s, 1H), 8.09 (d, J = 8.9 Hz, 1H), 7.87 (s, 1H), 7.82 (d, J = 7.6 Hz, 1H), 7.75 (d, J = 7.7 Hz, 1H), 7.69 (d, J = 7.6 Hz, 1H), 7.02–6.93 (m, 3H), 6.90 (d, J = 2.0 Hz, 1H), 5.36 (s, 2H); HRMS (ESI) calcd for C21H13F5NO2 (M + H)+ 406.0861, found 406.0874.
1,3-Difluoro-6-((2-(trifluoromethyl)benzyl)oxy)acridin-9(10H)-one (92).
1H NMR (DMSO-d6, 400 MHz) δ 11.80 (br s, 1H), 8.10 (d, J = 8.9 Hz, 1H), 7.84 (d, J = 8.3 Hz, 1H), 8.81 (s, 1H), 7.76 (d, J = 7.4 Hz, 1H), 7.62 (d, J = 7.6 Hz, 1H), 7.02–6.93 (m, 3H), 6.89 (d, J = 2.3 Hz, 1H), 5.39 (s, 2H); HRMS (ESI) calcd for C21H13F5NO2 (M + H)+ 406.0861, found 406.0873.
1,2-Dichloro-6-((4-chlorobenzyl)oxy)acridin-9(10H)-one (93).
1H NMR (DMSO-d6, 400 MHz) δ 11.80 (s, 1H), 8.11 (d, J = 9.0 Hz, 1H), 7.83 (d, J = 9.0 Hz, 1H), 7.50 (m, 5H), 6.97 (dd, J = 9.0, 1.3 Hz, 1H), 6.89 (s, 1H), 5.27 (s, 2H); HRMS (ESI) calcd for C20H13Cl3NO2 (M + H)+ 404.0006, found 404.0010.
Synthesis of intermediates 95a and 95b.
Compounds 95a and 95b were synthesized and purified in good yields (6.82 g, 67% and 6.88 g, 71%, respectively), by the same procedure as described for 86 from methyl 2,4-dihydroxybenzoate (94) and appropriate benzyl bromides, and their identity was confirmed by GC-MS analysis and carried forward into the next reaction.
Synthesis of intermediates 96a and 96b.
Compounds 96a and 96b were synthesized and purified in excellent yields (6.37 g, 92% and 6.32 g, 90%) by the same procedure as described for 6a from 95a and 95b, respectively, and their identity was confirmed by GC-MS analysis and carried forward into the next reaction.
Synthesis of intermediates 97a–d.
Compounds 97a–d were synthesized and purified in good yields (59–67%) by the same procedure as described for 8a from 96a and 96b, and appropriate anilines 7 and their identity was confirmed by GC-MS analysis and carried forward into the next reaction.
Synthesis of acridones 99–102.
The acridones 99−102 were synthesized (9−50%) by the same procedure as described for 18 from 97a–d. The 6-hydroxy acridones 98a and 98b were also isolated as minor products (23–48%). These two products were separated by column chromatography using a mixture of DCM and MeOH as a mobile phase. Upon treatment with appropriate benzyl bromides and K2CO3, 6-hydroxy acridones 98a and 98b furnished the acridones 99−102 in excellent yields (76–81%).
2-Chloro-3-methoxy-6-((4-(trifluoromethoxy)benzyl)oxy)acridin-9(10H)-one (99).
1H NMR (DMSO-d6, 400 MHz) δ 11.67 (s, 1H), 8.13–8.10 (m, 2H), 7.65 (d, J = 8.7 Hz, 2H), 7.44–7.42 (m, 2H), 6.99 (s, 1H), 6.95 (dd, J = 8.9, 2.3 Hz, 1H), 6.92 (d, J = 2.3 Hz, 1H), 5.30 (s, 2H), 3.99 (s, 3H); HRMS (ESI) calcd for C22H16ClF3NO4 (M + H)+ 450.0714, found 450.0715.
2-Chloro-3-methoxy-6-((4-(trifluoromethyl)benzyl)oxy)acridin-9(10H)-one (100).
1H NMR (DMSO-d6, 400 MHz) δ 11.66 (s, 1H), 8.13–8.10 (m, 2H), 7.80 (d, J = 7.9 Hz, 2H), 7.73 (d, J = 7.7 Hz, 2H), 6.99–6.91 (m, 3H), 5.39 (s, 2H), 3.98 (s, 3H); HRMS (ESI) calcd for C22H16ClF3NO3 (M + H)+ 434.0766, found 434.0769.
2-Fluoro-3-methoxy-6-((4-(trifluoromethoxy)benzyl)oxy)acridin-9(10H)-one (101).
1H NMR (DMSO-d6, 400 MHz) δ 11.63 (s, 1H), 8.11 (d, J = 8.8 Hz, 1H), 7.81 (d, J = 11.8 Hz, 1H), 7.65 (d, J = 8.6 Hz, 2H), 7.43 (d, J = 8.2 Hz, 2H), 7.03 (d, J = 7.2 Hz, 1H), 6.96–6.92 (m, 2H), 5.30 (s, 2H), 3.98 (s, 3H); HRMS (ESI) calcd for C22H16F4NO4 (M + H)+ 434.1010, found 434.1011.
2-Fluoro-3-methoxy-6-((4-(trifluoromethyl)benzyl)oxy)acridin-9(10H)-one (102).
1H NMR (DMSO-d6, 400 MHz) δ 11.63 (s, 1H), 8.12 (d, J = 8.8 Hz, 1H), 7.83–7.80 (m, 3H), 7.73 (d, J = 8.0 Hz, 2H), 7.03 (d, J = 7.1 Hz, 1H), 6.97 (d, J = 9.1 Hz, 1H), 6.91 (s, 1H), 5.39 (s, 2H), 3.98 (s, 3H); HRMS (ESI) calcd for C22H16F4NO3 (M + H)+ 418.1061, found 418.1064.
Synthesis of methyl 2-((3,5-difluorophenyl)amino)-5-methoxybenzoate (104).
Compound 104 was synthesized and purified in good yield (2.46 g, 68%) by the same procedure as described for 8a from methyl 2-bromo-5-methoxybenzoate (103), and 3,5-difluoro aniline (7d) and its identity was confirmed by GC-MS analysis and carried forward into the next reaction.
Synthesis of 1,3-difluoro-7-methoxyacridin-9(10H)-one (105).
Compound 105 was synthesized and purified in good yield (1.25 g, 70%) by the same procedure as described for 43 from 104 and carried forward into the next reaction.
Synthesis of 1,3-difluoro-7-hydroxyacridin-9(10H)-one (106).
Compound 106 was synthesized and purified in excellent yield (987 mg, 87%) by the same procedure as described for 85a from 105 and carried forward into the next reaction.
Synthesis of acridones 107–109.
The acridones 107−109 were synthesized in excellent yield (81–84%) by the same procedure as described for 86 from 106 and appropriate benzyl bromides.
1,3-Difluoro-7-((4-(trifluoromethoxy)benzyl)oxy)acridin-9(10H)-one (107).
1H NMR (DMSO-d6, 400 MHz) δ 11.91 (br s, 1H), 7.65–7.63 (m, 3H), 7.49–7.42 (m, 4H), 7.04–6.96 (m, 2H), 5.24 (s, 2H); HRMS (ESI) calcd for C21H13F5NO3 (M + H)+ 422.0810, found 422.0822.
1,3-Difluoro-7-((4-(trifluoromethyl)benzyl)oxy)acridin-9(10H)-one (108).
1H NMR (DMSO-d6, 400 MHz) δ 11.89 (br s, 1H), 7.79 (d, J = 8.3 Hz, 2H), 7.12 (d, J = 8.3 Hz, 2H), 7.66 (d, J = 1.8 Hz, 1H), 7.50 (m, 2H), 7.03 (d, J = 10.3 Hz, 1H), 6.99–6.93 (m, 1H), 5.33 (s, 2H); HRMS (ESI) calcd for C21H13F5NO2 (M + H)+ 406.0861, found 406.0873.
7-((4-Chlorobenzyl)oxy)-1,3-difluoroacridin-9(10H)-one (109).
1H NMR (DMSO-d6, 400 MHz) δ 11.90 (s, 1H), 7.65 (s, 1H), 7.54–7.48 (m, 6H), 7.04 (d, J = 10.1 Hz, 1H), 6.96 (t, J = 10.6 Hz, 1H), 5.21 (s, 2H); HRMS (ESI) calcd for C20H13ClF2NO2 (M + H)+ 372.0597, found 372.0595.
Biological Experiments.
Culture Conditions.
P. falciparum strains D6, Dd2, 7G8, and TM90-C2B were cultured in human erythrocytes at 2% hematocrit in RPMI 1640 containing 0.5% Albumax, 45 μg/L hypoxanthine, and 50 μg/L gentamicin, as previously described.20, 42
In Vitro Blood-Stage Antimalarial Activity.
In vitro antimalarial activity was determined by the Malaria SYBR Green I-based Fluorescence (MSF) assay described previously with minor modifications.20 Acridones were evaluated for antiplasmodial activity with CQ and ATV as controls. Experiments were set up in triplicate in 96-well plates with a total 100 μL total volume per well with 2% hematocrit, 0.2% parasitemia, and drug dilutions in complete culture medium described above. Initial drug dilutions were set between 2.5 and 2500 nM and subsequently adjusted to either a lower or higher range in order to achieve the most accurate IC50 values. The plates were incubated at 37 °C in low-oxygen conditions described previously for 72 h, at which point 100 μL of fluorescent dye-detergent mixture (0.2 μL SYBR Green I: 1 mL lysis buffer) was added and the plates incubated in the dark at room temperature for one hour. After incubation with the fluorescent dye, a 96-well plate reader with excitation wavelength set at 497 nm, and emission at 520 nm was used to measure fluorescence of each well. Fluorescence readings were plotted as a function of drug concentration, and the IC50 values (50% inhibitory concentrations) for each compound were calculated by nonlinear regression analysis (GraphPad Prism software) using calculated drug concentration that gave 50% reduction in fluorescence compared to drug-free controls.
In Vitro Liver-Stage Antimalarial Activity.
In vitro liver-stage activity of each new acridone was assessed utilizing luciferase-expressing P. berghei sporozoite infected HepG2 hepatocyte cells14, 23, 24 (a hepatoma cell line that allows higher rates of sporozoites invasion and full development of P. berghei). Briefly, luciferase-expressing P. berghei (ANKA-676m1cl1) sporozoites were extracted from salivary glands of infected Anopheles stephensi mosquitoes. HepG2-A16 cells were cultivated and seeded in 96-well plates and grown for 24 h then 10000 sporozoites were incubated with the HepG2 cells for 3 h; after which, the wells were washed with media to remove non-invaded sporozoites. Drug solutions then were added and plates were placed a 5% CO2 incubator at 37 °C and for 45 h. Luciferin was added and the plates were incubated for 30 min; after which, luciferase activity was determined in a luminometer. The IC50 values (50% inhibitory concentrations) for each compound were calculated by nonlinear regression analysis (GraphPad Prism software).
In Vivo Blood-Stage Efficacy in a Rodent Malaria Model.
In vivo antimalarial blood-stage efficacy was determined using a well-established modified Thompson 4-day suppression rodent model against P. yoelii.14, 21, 22 A 4- to 5-week-old female CF1 mice (Charles River Laboratories) were infected intravenously with 2.5 × 105 P. yoelii (Kenya strain, MR4 MRA-428) parasitized erythrocytes from a donor animal. Drug administration commenced the day after the animals were inoculated (day 1). The test compounds were dissolved in PEG-400 and administered by oral gavage once daily for four successive days; CQ phosphate was used as a positive control. Blood for blood film analysis and body weights were obtained on the day following the last dose and then at weekly intervals through day 28. Blood films were Giemsa stained and examined microscopically to determine the levels of parasitemia. These blood samples were collected from the tail vein with the aid of a syringe-needle. All mice were observed daily to assess their clinical signs, which were recorded. Animals with observable parasitemia following the experiment were euthanized; animals cleared of parasites from their bloodstream were observed daily with assessment of parasitemia performed weekly until day 28 at which point we score the animal(s) are scored as cured of infection and then euthanized. All treated mice with a negative smear on day 28 were considered cured (100% protection). ED50 and ED90 values (mg/kg/day) were derived graphically from the dose required to reduce parasite burden by 50% and 90%, respectively, relative to drug-free controls.
Animals and Ethic Statement at Portland VA Medical Center (PVAMC).
In vivo blood-stage testing was carried out at PVAMC, and the procedures were conducted under protocols approved by PVAMC Institutional Animal Care and Use Committee (Protocol number 3147). Research was conducted in an AAALAC accredited facility in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adheres to principles stated in the Guide for the Care and Use of Laboratory Animals, NRC Publication, 2011 edition.
In Vivo Liver-Stage Efficacy in a Rodent Malaria Model.
Real-time in vivo imaging system (IVIS) was used to determine the in vivo liver-stage antimalarial efficacy of selected acridone candidates.14, 25–27 Luciferase-expressing P. berghei sporozoites were extracted from salivary glands of A. stephensi mosquitoes. C57BL/6 male albino mice were infected i.v. in the tail vein with 10000 sporozoites on day 0. Drugs were administered orally on days −1, 0, +1. All drug solutions were prepared fresh before drug administration by dissolving the needed quantity of drug in cold (4 °C) 0.5% (w/v) hydroxyethyl cellulose and 0.2% (v/v) Tween-80 (0.5% HECT) or PEG400. If needed, drugs were ground using a ProScientific 300D homogenizer and the particle size was measured using a Horiba LA-950V2 particle size analyzer. Formulations of acridones were prepared in PEG 400 and administered orally (PO) at 1.0 or 4.0 or 10 or 40 mg/kg on days −1, 0, and 1. A vehicle control (PEG 400) group was included as a negative control in all studies. Positive control consisted of three doses of 4-methyl-primaquine which was dissolved in 0.5% HECT and was given at 5 mg/kg on days −1, 0, and 1 post-infections. All dosing solutions were given based on mice body weight measured on the day before infection took place. P. berghei parasite load in the liver and blood (approximately at 24 and 48 and 72 h post-animal infections) was assessed using an IVIS Spectrum instrument. Mice were injected IP with 150 μL D-luciferin solution (Gold Biotechnology, St. Louis, MO) at a concentration of 200 mg/kg body weight. Three min after luciferin injection, the mice were placed in the induction chamber and anesthetized. As soon as the mice were asleep, they were transferred in the IVIS chamber and positioned ventral side down on the heated platform with their noses positioned inside the nose cones. The IVIS camera exposure times were 1 and 5 min for the 24, 48, and 72 h time points with a large binning setting and a f-stop = 1. The IVIS Living Image software (version 4.0) was used to quantitate the bioluminescence in photons per second observed from the liver region or whole animal surface area respectively, for the liver-stage and blood-stage efficacy studies. Starting at 6 days after infection, all mice were assessed for blood-stage parasitemia which was quantitated using flow cytometry conducted using a FC500 MPL flow cytometer (Beckman Coulter, Fullerton, CA). The green photomultiplier tube and filter setting were used in these studies (520–555 nm filter settings for the green PMT and greater than 580 nm settings for the red PMT). Infected erythrocytes, uninfected erythrocytes, and leukocytes were gated on logarithmic forward/side dot plots. The method of FCM sample preparation has been previously described previously.43 In brief, a small 3 μL sample of blood was obtained from the tails of all mice. The blood was transferred into 0.3 mL of 1% heparinized isotonic buffer (PBS saline). 1 mM of 0.04% of glutaraldehyde was added to fix each sample, and samples were incubated for 60 min at 4 °C followed by centrifugation at 450 g for 5 min. The supernatant was removed by aspiration, and the cells were re-suspended in 0.5 mL PBS buffer supplemented with 0.25% (v/v) Triton X-100 for 10-min incubation at room temperature. After centrifugation, the permeabilized cells were re-suspended in 0.5 mL of RNAase at 1 mg/mL concentrations and incubated for at least 2 h at 37 °C to ensure complete digestion of reticulocytes which are at high concentrations due to anemia associated with P. berghei infection. 20 μL of YOYO-1 dye at a concentration of 2500 ng/mL was added to the 0.5 mL sample volume to create a final dye concentration of 100 ng/mL of YOYO-1. Parasitemia was monitored for up to 31 days post-infections. Blood samples for parasitemia determination were taken every week or prior to euthanizing the animals sick with malaria that were being removed from the study. Mice that tested negative for parasitemia on day 31 post-infection were considered cured. The minimum curative dose (ED100) in 100% of animals dosed was defined as the lowest dose which cured all animals for the entire 31-day follow-up period.
Animals and Ethic Statement at Walter Reed Army Institute of Research (WRAIR).
Male C57BL/6 Albino mice aged 6 weeks old (Charles River) were used in this study. The mice were left to acclimatize for 7 days prior to the beginning of research studies. All mice were assigned a study number with an individual ear tag. Cards attached to each mouse cage were also used to identify the study groups. All animals were quarantined for stabilization for 7 days prior to infection. Mice were housed in a designated room with food and water supplied ad libitum and a 12:12 light:dark cycle.
The animal protocol for this study was approved by the Walter Reed Army Institute of Research, Institutional Animal Care and Use Committee (Protocol number 18-ET-12) in accordance with national and Department of Defense guidelines. Research was conducted in an AAALAC accredited facility in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adheres to principles stated in the Guide for the Care and Use of Laboratory Animals, NRC Publication, 2011 edition.
Ex Vivo Antimalarial Activity against Clinical Isolates in Uganda.
The activity of 38, 39 and the standard antimalarials CQ, PIP, DHA, ATV and LUM (standard antimalarials from Medicines for Malaria Venture, Geneva, Switzerland) were tested, as previously described, against P. falciparum isolates using a 72 h growth inhibition assay with parasite DNA readout by SYBR Green detection.20, 29 These isolates were collected in May to July 2016, from two sources in the Tororo District of Uganda. First, children and adults presenting at the Tororo District Hospital outpatient clinic with malaria (temperature of ≥ 37.5°C axillary or history of fever in the previous 24 h and a positive Giemsa-stained blood smear for P. falciparum) and without signs of severe disease were enrolled after informed consent. Second, children enrolled in a clinical trial (registered at ClinicalTrials.gov [NCT02163447]) comparing monthly versus every 3-month intermittent therapy with DHA-PIP to prevent malaria provided samples if they presented to the study clinic with uncomplicated malaria, defined as above.29 The relevant clinical trials and analyses of cultured parasites were approved by the Uganda National Council of Science and Technology, the Makerere University Research and Ethics Committee, and the University of California, San Francisco Committee on Human Research. Comparison of median acridone IC50 values obtained from patients between sites was performed using a Mann-Whitney U test. Correlations between the acridones and with other standard antimalarials were assessed with a Spearman’s rank correlation coefficient. All tests were two-tailed and p-values <0.05 considered statistically significant.
In Vitro Cytotoxicity Assessment.
To identify toxicity to mammalian cells, an MTT assay using hepatic HepG2 cells was employed.30–32 Human hepatocarcinoma cells (HepG2) were cultured in complete Minimal Essential Medium (Gibco-Invitrogen, No.11090–099) prepared by supplementing MEM with 0.19% sodium bicarbonate (Gibco-BRL No. 25080–094), 10% heat inactivated FBS (Gibco-Invitrogen No. 16000–036), 2 mM L-glutamine (Gibco-Invitrogen No. 25030–081), 0.1 mM MEM nonessential amino acids (Gibco- Invitrogen No. 11140–050), 0.009 mg/mL insulin (Sigma No. I1882), 1.76 mg/mL bovine serum albumin (Sigma No. A1470). Cells viability were assessed using trypan blue. Ninety-six-well plates were seeded with 2.5 × 104 cells in 170 μL of culture medium per well and incubate at 37 °C overnight in a humidified 5% CO2 atmosphere. Drug plates were prepared with a Biomek 4000 automated laboratory station and 96 well plates containing 11 duplicate 1.6-fold serial dilutions of each test compound suspended in DMSO. A 30 μL sample of diluted test compound was then added to 170 μL of media per well. Cells were exposed to drug concentrations ranging from 10 ug/mL to 0.15 ug/mL for 48 h. After incubation, 30 μL of a 1.5 mg/mL solution of MTT diluted in complete MEM media was added to each well, all plates were subsequently incubated in the dark for 1 h at room temperature. Next, the media and drugs in each well were removed and plates were then left to dry in a hood for 15 min. 60 μL of acidified isopropyl alcohol was added to dissolve the formazan dye crystals created by the reduction of MTT. Plates were placed on a 3-D rotator for 15–30 min. Absorbance was determined with the Perkin Elmer EnSight plate reader. All experiments were run in duplicates and the fifty-percent inhibitory concentrations (IC50’s) were determined using GraphPad Prism software using the nonlinear regression equation (sigmoidal dose-response, variable slope).
In Vitro hERG Channel Inhibition.
The measurement of hERG channel inhibitory activity of the test compounds was performed by Eurofins Inc. Basic principles and operation of the IonWorks platform have been described by Schroeder et al.33
In Vitro Metabolic Profile.
The half-life of lead acridones in mouse and human liver microsomes were determined as described previously.14, 32, 34, 35 All samples were tested in human and mouse liver microsomal preparations. Sample stocks at 10 or 20 μM (depending on solubility) in DMSO were diluted to a final concentration of 1 μM with a mixture containing 0.5 mg/mL of prewarmed pooled human or mouse liver microsomes (BD Gentest), 1.3 μM NADP (Sigma), 3.3 μM MgCl2 (Sigma), and 0.1 M pH 7.4 PBS using a TECAN Genesis robotic liquid handler. The reaction was started with the addition of 1U/mL glucose-6-phosphate dehydrogenase (G6PD). The mixture was incubated on a shaking platform at 37 °C, and aliquots were taken and quenched with the addition of an equal volume of cold acetonitrile at 0, 10, 20, 30, and 60 min. Samples were centrifuged at 3700 rpm for 10 min at 20 °C to remove debris. Sample quantification was carried out by LC/MS, and metabolic half-life was calculated by log plots of the total ion chromatograph area remaining.
In Vivo Pharmacokinetic (PK) Studies.
PK studies were conducted using the previously established methods.14, 28, 32 Male 7-week-old ICR mice weighing from 23 to 28 g (Charles River Laboratories. Inc. Raleigh, NC) were used for the PK evaluations. On arrival, the animals were acclimated for 7 days in quarantine. The animals were housed in a cage maintained in a room with a temperature range of 64–79 °F, 34–68% relative humidity and a 12-h light/dark cycle. Food and water were provided ad libitum during quarantine and throughout the study. The animals were fed a standard rodent maintenance diet. All animal studies were performed under IACUC-approved protocols. These protocols detail the experimental procedures and designs as well as the number of animals that were used. All animal use, care, and handling were performed in accordance with the current “Guide for the Care and Use of Laboratory Animals” (8th Edition, 2011). PK studies were performed using intragastric (IG) administration. For each time point to be acquired, three male ICR mice per time-point were dosed at single p.o. dosing of 80 mg/kg. The drug vehicle was DD water, administered at 100 μL/20 g. At each time point, blood and plasma samples were collected. Whole blood was collected by cardiac puncture. Blood samples were collected in lithium heparin tubes. Following the separation of appropriate aliquots, plasma was obtained from the whole blood via centrifugation. Liver tissues were isolated. All liquid and tissue samples were immediately preserved on dry ice and later stored at −80 °C until analytical work was performed. Drug concentrations were generated for each sample taken from animals dosed with test compounds. A measured plasma or liver drug concentration vs. time curve was produced, in graphic and tabular form, for each subject on both linear/linear and log/linear scales, for the parent compound. Mean plasma drug concentration vs. time curves were also prepared separately. Maximum plasma concentration (Cmax), and time to maximum concentration (tmax) were obtained from the plasma drug concentration–time curves. The elimination half-life (t1/2) was calculated from (ln 2)/kel, which is the elimination rate constant calculated from the log concentration–time plot. The area under the curve (AUC) was determined by the linear trapezoidal rule with extrapolation to infinity based on the concentration of the last time point divided by the terminal rate constant. Mean residence time (MRT) was determined by dividing the area under the first moment curve (AUMC) by AUC. The volume of the central compartment (Vz) and volume of the tissue compartment (Vz/F) were calculated as the product of CL and MRT.
Supplementary Material
ACKNOWLEDGMENT
This project was supported by NIH/NIAID (award 1R01AI093784) and DOD/PRMRP (award PR160693/W81XWH-17-2-0041). Material has been reviewed by the Walter Reed Army Institute of Research (WRAIR). There is no objection to its presentation and/or publication. The opinions or assertions contained herein are the private views of the author, and are not to be construed as official, or as reflecting true views of the Department of the Army or the Department of Defense. Research was conducted in AAALAC accredited facilities in compliance with the Animal Welfare Act and other federal statutes and regulations relating to animals and experiments involving animals and adheres to principles stated in the Guide for the Care and Use of Laboratory Animals, NRC Publication.
ABBREVIATIONS
- CQ
chloroquine
- ATV
atovaquone
- PIP
piperaquine
- DHA
dihydroartemisinin
- LUM
lumefantrine
- SAR
structure–activity relationship
- IC50
half maximal inhibitory concentration
- nM
nanomolar
- MDR
multidrug-resistant
- ED50
50% effective dose
- ED90
90% effective dose
- PK
pharmacokinetic
- IVIS
in vivo imaging system
- i.v
intravenously
- p.o.
oral route
- hERG
human-ether-a-go-go-related gene
Footnotes
The authors declare no competing financial interest.
SUPPORTING INFORMATION
The Supporting Information is available free of charge on the ACS Publications website.
1H NMR spectra of all final compounds and 19F NMR of 26 and 27 (PDF); HPLC chromatograms of lead candidates (PDF).
Molecular formula strings and in vitro data (CSV).
REFERENCES
- (1).WHO. World Malaria Report; WHO, 2019. [Google Scholar]
- (2).Phillips MA; Burrows JN; Manyando C; van Huijsduijnen RH; Van Voorhis WC; Wells TNC Malaria. Nat. Rev. Dis. Primers 2017, 3, 17050. [DOI] [PubMed] [Google Scholar]
- (3).Ashley EA; Phyo AP Drugs in development for malaria. Drugs 2018, 78, 861–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Hooft van Huijsduijnen R; Wells TN The antimalarial pipeline. Curr. Opin. Pharmacol 2018, 42, 1–6. [DOI] [PubMed] [Google Scholar]
- (5).Okombo J; Chibale K Recent updates in the discovery and development of novel antimalarial drug candidates. Medchemcomm. 2018, 9, 437–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Ashley EA; Dhorda M; Fairhurst RM; Amaratunga C; Lim P; Suon S; Sreng S; Anderson JM; Mao S; Sam B; Sopha C; Chuor CM; Nguon C; Sovannaroth S; Pukrittayakamee S; Jittamala P; Chotivanich K; Chutasmit K; Suchatsoonthorn C; Runcharoen R; Hien TT; Thuy-Nhien NT; Thanh NV; Phu NH; Htut Y; Han KT; Aye KH; Mokuolu OA; Olaosebikan RR; Folaranmi OO; Mayxay M; Khanthavong M; Hongvanthong B; Newton PN; Onyamboko MA; Fanello CI; Tshefu AK; Mishra N; Valecha N; Phyo AP; Nosten F; Yi P; Tripura R; Borrmann S; Bashraheil M; Peshu J; Faiz MA; Ghose A; Hossain MA; Samad R; Rahman MR; Hasan MM; Islam A; Miotto O; Amato R; MacInnis B; Stalker J; Kwiatkowski DP; Bozdech Z; Jeeyapant A; Cheah PY; Sakulthaew T; Chalk J; Intharabut B; Silamut K; Lee SJ; Vihokhern B; Kunasol C; Imwong M; Tarning J; Taylor WJ; Yeung S; Woodrow CJ; Flegg JA; Das D; Smith J; Venkatesan M; Plowe CV; Stepniewska K; Guerin PJ; Dondorp AM; Day NP; White NJ Spread of artemisinin resistance in Plasmodium falciparum malaria. N. Engl. J. Med 2014, 371, 411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Duru V; Witkowski B; Menard D Plasmodium falciparum resistance to artemisinin derivatives and piperaquine: a major challenge for malaria elimination in Cambodia. Am. J. Trop. Med. Hyg 2016, 95, 1228–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Tilley L; Straimer J; Gnadig NF; Ralph SA; Fidock DA Artemisinin action and resistance in Plasmodium falciparum. Trends Parasitol. 2016, 32, 682–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Thomas D; Tazerouni H; Sundararaj KG; Cooper JC Therapeutic failure of primaquine and need for new medicines in radical cure of Plasmodium vivax. Acta. Trop 2016, 160, 35–38. [DOI] [PubMed] [Google Scholar]
- (10).Val F; Costa FT; King L; Brito-Sousa JD; Bassat Q; Monteiro WM; Siqueira AM; Luzzatto L; Lacerda MV Tafenoquine for the prophylaxis, treatment and elimination of malaria: eagerness must meet prudence. Future Microbiol. 2019, 14, 1261–1279. [DOI] [PubMed] [Google Scholar]
- (11).Ashton TD; Devine SM; Mohrle JJ; Laleu B; Burrows JN; Charman SA; Creek DJ; Sleebs BE The development process for discovery and clinical advancement of modern antimalarials. J. Med. Chem 2019, 62, 10526–10562. [DOI] [PubMed] [Google Scholar]
- (12).Burrows JN; Duparc S; Gutteridge WE Hooft van Huijsduijnen R; Kaszubska W; Macintyre F; Mazzuri S; Mohrle JJ; Wells TNC New developments in anti-malarial target candidate and product profiles. Malar. J 2017, 16, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Wells TN; Burrows JN; Baird JK Targeting the hypnozoite reservoir of Plasmodium vivax: the hidden obstacle to malaria elimination. Trends Parasitol. 2010, 26, 145–151. [DOI] [PubMed] [Google Scholar]
- (14).Dodean RA; Kancharla P; Li Y; Melendez V; Read L; Bane CE; Vesely B; Kreishman-Deitrick M; Black C; Li Q; Sciotti RJ; Olmeda R; Luong TL; Gaona H; Potter B; Sousa J; Marcsisin S; Caridha D; Xie L; Vuong C; Zeng Q; Zhang J; Zhang P; Lin H; Butler K; Roncal N; Gaynor-Ohnstad L; Leed SE; Nolan C; Huezo SJ; Rasmussen SA; Stephens MT; Tan JC; Cooper RA; Smilkstein MJ; Pou S; Winter RW; Riscoe MK; Kelly JX Discovery and structural optimization of acridones as broad-spectrum antimalarials. J. Med. Chem 2019, 62, 3475–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Ma D; Cai QN,N-dimethyl glycine-promoted Ullmann coupling reaction of phenols and aryl halides. Org. Lett 2003, 5, 3799–3802. [DOI] [PubMed] [Google Scholar]
- (16).Kaisalo L; Latvala A; Hase T Selective demethylations in 2, 3, 4-trimethoxyaryl carbonyl compounds. Synth. Commun 1986, 16, 645–648. [Google Scholar]
- (17).Schuster C; Borger C; Julich-Gruner KK; Hesse R; Jager A; Kaufmann G; Schmidt AW; Knolker H Synthesis of 2‐hydroxy‐7‐methylcarbazole, glycozolicine, mukoline, mukolidine, sansoakamine, clausine‐H, and clausine‐K and structural revision of clausine‐TY. Eur. J. Org. Chem 2014, 4741–4752. [Google Scholar]
- (18).Kancharla P; Dodean RA; Kelly JX Boron trifluoride etherate promoted microwave-assisted synthesis of antimalarial acridones. RSC Adv. 2019, 42284–42293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Kelly JX; Smilkstein MJ; Brun R; Wittlin S; Cooper RA; Lane KD; Janowsky A; Johnson RA; Dodean RA; Winter R; Hinrichs DJ; Riscoe MK Discovery of dual function acridones as a new antimalarial chemotype. Nature 2009, 459, 270–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Smilkstein M; Sriwilaijaroen N; Kelly JX; Wilairat P; Riscoe M Simple and inexpensive fluorescence-based technique for high-throughput antimalarial drug screening. Antimicrob. Agents Chemother 2004, 48, 1803–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Nilsen A; LaCrue AN; White KL; Forquer IP; Cross RM; Marfurt J; Mather MW; Delves MJ; Shackleford DM; Saenz FE; Morrisey JM; Steuten J; Mutka T; Li Y; Wirjanata G; Ryan E; Duffy S; Kelly JX; Sebayang BF; Zeeman AM; Noviyanti R; Sinden RE; Kocken CH; Price RN; Avery VM; Angulo-Barturen I; Jimenez-Diaz MB; Ferrer S; Herreros E; Sanz LM; Gamo FJ; Bathurst I; Burrows JN; Siegl P; Guy RK; Winter RW; Vaidya AB; Charman SA; Kyle DE; Manetsch R; Riscoe MK Quinolone-3-diarylethers: a new class of antimalarial drug. Sci. Transl. Med 2013, 5, 177ra37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Thompson PE; Bayles A; Olszewski B PAM 1392 (2,4-diamino-6-(3,4-dichlorobenzylamino) quinazoline) as a chemotherapeutic agent: Plasmodium berghei, P. cynomolgi, P. knowlesi, and Trypanosoma cruzi. Exp. Parasitol 1969, 25, 32–49. [DOI] [PubMed] [Google Scholar]
- (23).Janse CJ; Franke-Fayard B; Mair GR; Ramesar J; Thiel C; Engelmann S; Matuschewski K; van Gemert GJ; Sauerwein RW; Waters AP High efficiency transfection of Plasmodium berghei facilitates novel selection procedures. Mol. Biochem. Parasitol 2006, 145, 60–70. [DOI] [PubMed] [Google Scholar]
- (24).Wang H; Li Q; Reyes S; Zhang J; Zeng Q; Zhang P; Xie L; Lee PJ; Roncal N; Melendez V; Hickman M; Kozar MP Nanoparticle formulations of decoquinate increase antimalarial efficacy against liver stage Plasmodium infections in mice. Nanomedicine 2014, 10, 57–65. [DOI] [PubMed] [Google Scholar]
- (25).Li Q; Xie L; Caridha D; Roncal N; Zeng Q; Zhang J; Zhang P; Hickman M; Read L Comparative susceptibility of different mouse strains to liver-stage infection with Plasmodium berghei sporozoites assessed using in vivo imaging. Mil. Med 2017, 182, 360–368. [DOI] [PubMed] [Google Scholar]
- (26).Ploemen IH; Prudencio M; Douradinha BG; Ramesar J; Fonager J; van Gemert GJ; Luty AJ; Hermsen CC; Sauerwein RW; Baptista FG; Mota MM; Waters AP; Que I; Lowik CW; Khan SM; Janse CJ; Franke-Fayard BM Visualisation and quantitative analysis of the rodent malaria liver stage by real time imaging. PLoS One 2009, 4, e7881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Thiberge S; Blazquez S; Baldacci P; Renaud O; Shorte S; Menard R; Amino R In vivo imaging of malaria parasites in the murine liver. Nat. Protoc 2007, 2, 1811–1818. [DOI] [PubMed] [Google Scholar]
- (28).Li Q; O’Neil M; Xie L; Caridha D; Zeng Q; Zhang J; Pybus B; Hickman M; Melendez V Assessment of the prophylactic activity and pharmacokinetic profile of oral tafenoquine compared to primaquine for inhibition of liver stage malaria infections. Malar. J 2014, 13, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Rasmussen SA; Ceja FG; Conrad MD; Tumwebaze PK; Byaruhanga O; Katairo T; Nsobya SL; Rosenthal PJ; Cooper RA Changing antimalarial drug sensitivities in Uganda. Antimicrob. Agents Chemother 2017, 61, e01516–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Caridha D; Yourick D; Cabezas M; Wolf L; Hudson TH; Dow GS Mefloquine-induced disruption of calcium homeostasis in mammalian cells is similar to that induced by ionomycin. Antimicrob. Agents Chemother 2008, 52, 684–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ferrari M; Fornasiero MC; Isetta AM MTT colorimetric assay for testing macrophage cytotoxic activity in vitro. J. Immunol. Methods 1990, 131, 165–172. [DOI] [PubMed] [Google Scholar]
- (32).Korotchenko V; Sathunuru R; Gerena L; Caridha D; Li Q; Kreishman-Deitrick M; Smith PL; Lin AJ Antimalarial activity of 4-amidinoquinoline and 10-amidinobenzonaphthyridine derivatives. J. Med. Chem 2015, 58, 3411–3431. [DOI] [PubMed] [Google Scholar]
- (33).Schroeder K; Neagle B; Trezise DJ; Worley J Ionworks HT: a new high-throughput electrophysiology measurement platform. J. Biomol. Screen 2003, 8, 50–64. [DOI] [PubMed] [Google Scholar]
- (34).Dow GS; Chen Y; Andrews KT; Caridha D; Gerena L; Gettayacamin M; Johnson J; Li Q; Melendez V; Obaldia N 3rd; Tran TN; Kozikowski AP Antimalarial activity of phenylthiazolyl-bearing hydroxamate-based histone deacetylase inhibitors. Antimicrob. Agents Chemother 2008, 52, 3467–3477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Zhang L; Sathunuru R; Luong T; Melendez V; Kozar MP; Lin AJ New imidazolidinedione derivatives as antimalarial agents. Bioorg. Med. Chem 2011, 19, 1541–1549. [DOI] [PubMed] [Google Scholar]
- (36).Lindner SE; Miller JL; Kappe SH Malaria parasite pre-erythrocytic infection: preparation meets opportunity. Cell Microbiol. 2012, 14, 316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Mazier D; Renia L; Snounou G A pre-emptive strike against malaria’s stealthy hepatic forms. Nat. Rev. Drug. Discov 2009, 8, 854–864. [DOI] [PubMed] [Google Scholar]
- (38).Rodrigues T; Prudencio M; Moreira R; Mota MM; Lopes F Targeting the liver stage of malaria parasites: a yet unmet goal. J. Med. Chem 2012, 55, 995–1012. [DOI] [PubMed] [Google Scholar]
- (39).Vaughan AM; Kappe SHI Malaria parasite liver infection and exoerythrocytic biology. Cold Spring Harb. Perspect Med 2017, 7, a025486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Adams JH; Mueller I The biology of Plasmodium vivax. Cold Spring Harb Perspect Med. 2017, 7, a025585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Campo B; Vandal O; Wesche DL; Burrows JN Killing the hypnozoite-drug discovery approaches to prevent relapse in Plasmodium vivax. Pathog. Glob. Health 2015, 109, 107–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Schuster FL Cultivation of Plasmodium spp. Clin. Microbiol. Rev 2002, 15, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Xie L; Li Q; Johnson J; Zhang J; Milhous W; Kyle D Development and validation of flow cytometric measurement for parasitaemia using autofluorescence and YOYO-1 in rodent malaria. Parasitology 2007, 134, 1151–1162. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.