

Abstract
Background
The epithelial cell‐derived danger signal mediators thymic stromal lymphopoietin (TSLP) and IL‐33 are consistently associated with adaptive Th2 immune responses in asthma. In addition, TSLP and IL‐33 synergistically promoted group 2 innate lymphoid cell (ILC2) activation to induce innate allergic inflammation. However, the mechanism of this synergistic ILC2 activation is unknown.
Methods
BALB/c WT and TSLP receptor‐deficient (TSLPR−/−) mice were challenged intranasally with Alternaria extract (Alt‐Ext) or PBS for 4 consecutive days to evaluate innate airway allergic inflammation. WT mice pre‐administered with rTSLP or vehicle, TSLPR−/− mice, and IL‐33 receptor‐deficient (ST2−/−) mice were challenged intranasally with Alt‐Ext or vehicle once or twice to evaluate IL‐33 release and TSLP expression in the lung. TSLPR and ST2 expression on lung ILC2 were measured by flow cytometry after treatment of rTSLP, rIL‐33, rTSLP + rIL‐33, or vehicle.
Results
Thymic stromal lymphopoietin receptor deficient mice had significantly decreased the number of lung ILC2 expressing IL‐5 and IL‐13 following Alt‐Ext‐challenge compared to WT mice. Further, eosinophilia, protein level of lung IL‐4, IL‐5, and IL‐13, and airway mucus score were also significantly decreased in TSLPR−/− mice compared to WT mice. Endogenous and exogenous TSLP increased Alt‐Ext‐induced IL‐33 release into BALF, and ST2 deficiency decreased Alt‐Ext‐induced TSLP expression in the lung. Further, rTSLP and rIL‐33 treatment reciprocally increased each other's receptor expression on lung ILC2 in vivo and in vitro.
Conclusion
Thymic stromal lymphopoietin and IL‐33 signaling reciprocally enhanced each other's protein release and expression in the lung following Alt‐Ext‐challenge and each other's receptor expression on lung ILC2 to enhance ILC2 activation.
Keywords: Alternaria‐extract (Alt‐Ext), Group 2 innate lymphoid cells (ILC2), IL‐33, TSLP
We found that TSLPR deficiency significantly decrease the number of lung ILC2, airway eosinophils, and mucus production in mouse challenged with Alternaria extract. TSLP‐TSLPR and IL‐33‐ST2 signaling augments each other's protein release and expression in the lung epithelial cells after aeroallergen challenge. TSLP and IL‐33 signaling increases each other's receptor expression on lung ILC2, and the IL‐33‐augmented TSLPR expression enhances pSTAT5 in the ILC2.
Abbreviations
- ab
antibody
- Alt‐Ext
Alternaria extract
- BALF
bronchoalveolar lavage fluid
- ECs
epithelial cells
- hBE33
human bronchial epithelial cells stably expressing IL‐33
- ILC2
group 2 innate lymphoid cells
- lin
lineage
- MFI
mean fluorescence intensity
- PAR‐2
protease‐activated receptor 2
- pSTAT5
phosphorylated signal transducer and activator of transcription 5
- rIL‐33
mouse recombinant IL‐33
- rTSLP
mouse recombinant TSLP
- ST2
IL‐33 receptor
- ST2−/−
ST2 deficient
- STAT5
signal transducer and activator of transcription 5
- TSLP
thymic stromal lymphopoietin
- TSLPR−/−
TSLP receptor deficient
- WT
wild type
1. INTRODUCTION
The epithelial cell‐derived danger signal mediators thymic stromal lymphopoietin (TSLP) and IL‐33 are consistently associated with adaptive Th2 immune responses in allergic diseases.1, 2
Genome‐wide association studies (GWAS) have shown the association of allergic diseases and genetic polymorphisms in the TSLP, IL33, and IL1RL1 (receptor for IL‐33).3
Thymic stromal lymphopoietin, an IL‐7‐like cytokine, is expressed mainly by epithelial cells (ECs) and epidermal keratinocytes (KCs). Previous animal studies reported that mice deficient in TSLPR (TSLPR−/−) had significantly decreased airway inflammation, serum IgE, and airway hyperresponsiveness (AHR) in chronic models of adaptive immunity‐mediated allergic airway inflammation with house dust mite antigen,4 OVA,5, 6 or a mixture of allergen extracts.2 In addition, subcutaneous TSLP or intranasal TSLP + OVA administration induced ear thickness or allergic airway inflammation with serum IgE induction.7, 8 IL‐33 normally resides in the nucleus of structural cells in the lung.9 Unlike the release of classic cytokines, IL‐33 is rapidly released from epithelial or endothelial cells upon cellular damage or stress.10 Previous studies reported that IL‐33 deficiency significantly decreased eosinophilia and AHR in the mouse model of OVA‐ or HDM‐induced adaptive allergic inflammation.11 However, the interaction of TSLP and IL‐33 in regulating the innate allergic responses is unknown.
Group 2 innate lymphoid cells (ILC2), as well as Th2 cells, are an important source of type‐2 cytokines to initiate and propagate allergic immune responses.12, 13 ILC2 are defined by the absence of lineage (lin) markers (for T cells, B cells, macrophages, dendritic cells, neutrophils, NK cells, or erythrocytes) and expression of CD45 (lymphocyte common antigen), ST2 (IL‐33 receptor), CD25 (IL‐2Rα), CD127 (IL‐7Rα), and ICOS.14, 15 ILC2 are resident in the lung, skin, gut, and the other organs16 and produce type‐2 cytokines in response to IL‐33, or a combination of IL‐33 and the other epithelium‐derived cytokines such as TSLP and IL‐25 in the peripheral tissues.14 These danger signals initiate ILC2 activation prior to adaptive immune responses.16, 17 While IL‐33 is an essential cytokine in the activation of ILC2,15 TSLP synergistically promoted the proliferation and type‐2 cytokine production by IL‐33‐stimulated ILC2 in vitro.14 Although TSLP itself did not induce IL‐5 and IL‐13 by ILC2, combined stimulation with TSLP and IL‐33 elicited an approximate 10‐fold increase in the cytokine production by ILC2 compared with stimulation by IL‐33 alone.14 However, the mechanisms of this synergistic ILC2 activation in the setting of the innate immune responses to an inhaled aeroallergen are not fully elucidated.
In this study, we hypothesized that TSLPR signaling increased T cell–independent innate allergic inflammation mediated by lung ILC2 activation. To test the hypothesis, we used a mouse model of Alternaria extract (Alt‐Ext)‐challenge for 4 consecutive days. Further, to determine the mechanisms of synergistic lung ILC2 activation by TSLP and IL‐33 during aeroallergen exposure, we evaluated the effects of TSLP and IL‐33 signaling on each other's protein release or expression in the lung after Alt‐Ext‐challenge. In addition, we evaluated the effects of TSLP and IL‐33 signaling to each other's receptor expression on lung ILC2.
2. METHODS
2.1. Mice
Nine‐ to 12‐week old female wild‐type (WT) BALB/c mice were obtained from Charles River laboratories. TSLPR−/− mice and ST2‐deficient (ST2−/−) mice were generated on BALB/c background as previously described.5, 18 IL‐33Citrine/+ reporter mice were generated by crossbreeding WT BALB/c mice and IL‐33Citrine/Citrine mice which were the kind gift of Dr Andrew N. J. McKenzie (Cambridge, UK).19 Animal experiments were approved by the Institutional Animal Care and Use Committee at Vanderbilt University and were conducted according to the guidelines for the Care and Use of Laboratory Animals prepared by the Institute of Laboratory Animal Resources, National Research Council.
2.2. Alt‐Ext‐challenge in a mouse model of innate allergic airway inflammation
WT BALB/c and TSLPR−/− mice were challenged intranasally with 5 µg (protein amount) of Alt‐Ext (Stallergenes Greer) in 100 µL PBS or 100 µL PBS as the vehicle for 4 consecutive days. LPS contamination of the Alt‐Ext was measured by PierceTM Chromogenic Endotoxin Quant kit (Thermo fisher scientific). LPS was detected as 0.03 endotoxin unit (EU), approximately 3 pg in 5 µg (protein amount) of Alt‐Ext. The mice were killed, and bronchoalveolar lavage fluid (BALF) and whole lungs were harvested 24 hour after the last challenge of Alt‐Ext or PBS for cell differentials, cytokine measurement, and flow cytometry. In the other protocol, whole lungs were harvested 48 hour after the last Alt‐Ext‐challenge or PBS challenge to evaluate mucus production in histological section.
2.3. Endogenous TSLP neutralization
The 28F12 hybridoma was obtained from the Developmental Studies Hybridoma Bank (Iowa City, IA), and anti‐TSLP antibodies (ab) from the hybridoma were purified by the Vanderbilt Antibody and Protein Resource.20 WT BALB/c mice were injected intraperitoneally with the anti‐TSLP ab (200 µg) or isotype ab (200 µg) 1 hour prior to first Alt‐Ext‐challenge.
2.4. Statistical analysis
All data were analyzed with GraphPad Prism 8 (GraphPad Software). The P values were calculated by t test or one‐way analysis of variance (ANOVA) with Bonferroni‐multiple pair's comparisons test. Values of P < .05 were considered significant between two groups.
Further experimental details are provided in online Appendix S1. All antibodies used in this study are listed in Table S1.
3. RESULTS
3.1. TSLPR deficiency suppresses Alt‐Ext‐induced lung ILC2 activation and innate allergic airway inflammation
Previous studies reported that deficiency of TSLPR signaling decreased adaptive allergic immune responses,5, 7 but the effect of TSLPR deficiency on allergen‐induced T cell–independent innate immune responses has not been reported. To evaluate the effects of TSLPR signaling during aeroallergen‐induced innate type‐2 airway inflammation, we performed Alt‐Ext‐challenge for 4 consecutive days in WT and TSLPR−/− mice and killed them 24 or 48 hour after the last Alt‐Ext‐challenge (Figure 1A). Four consecutive days of Alt‐Ext‐challenge significantly increased the number of total BAL cells, macrophages, eosinophils, lymphocytes, and neutrophils compared with PBS‐challenged groups. TSLPR−/− mice challenged with Alt‐Ext had a significant decrease in the number of total BAL cells, eosinophils, and lymphocytes compared with WT mice (Figure 1B). In contrast, there was no change in the number of macrophages and neutrophils between WT and TSLPR−/− mice challenged with Alt‐Ext (Figure 1B). Next, we evaluated the lung ILC2 expressing IL‐5 and IL‐13. Figure S1 showed the gating strategy of identifying lung ILC2 after intracellular staining on flow cytometry. The lung ILC2 were identified as lin‐ CD3− CD4− CD45+ CD25+ ICOS+ cells to determine the IL‐5 and IL‐13 expression (Figure S1A,B). Alt‐Ext‐challenge significantly increased the number of lung ILC2 expressing IL‐5 and IL‐13 compared with PBS‐challenged groups (Figure 1C). There was a significant decrease in the number of lung ILC2 expressing IL‐5 and IL‐13 in TSLPR−/− mice compared with WT mice after the Alt‐Ext‐challenge (Figure 1C). Further, TSLPR−/− mice had a lower percentage of IL‐13+ cells in total ILC2 compared with WT mice after the Alt‐Ext‐challenge (Figure 1C). TSLPR−/− mice had a significant decrease in Alt‐Ext‐challenge‐induced protein expression of IL‐4, IL‐5, and IL‐13 in the lung homogenates to almost the same level as the PBS‐challenged mice (Figure 1D). TSLPR−/− mice had a significant decrease in the mucus score compared with WT mice after the Alt‐Ext‐challenge (Figure 1E). These findings revealed that TSLPR deficiency significantly decreased aeroallergen‐induced lung ILC2 activation and innate allergic airway inflammation.
Figure 1.
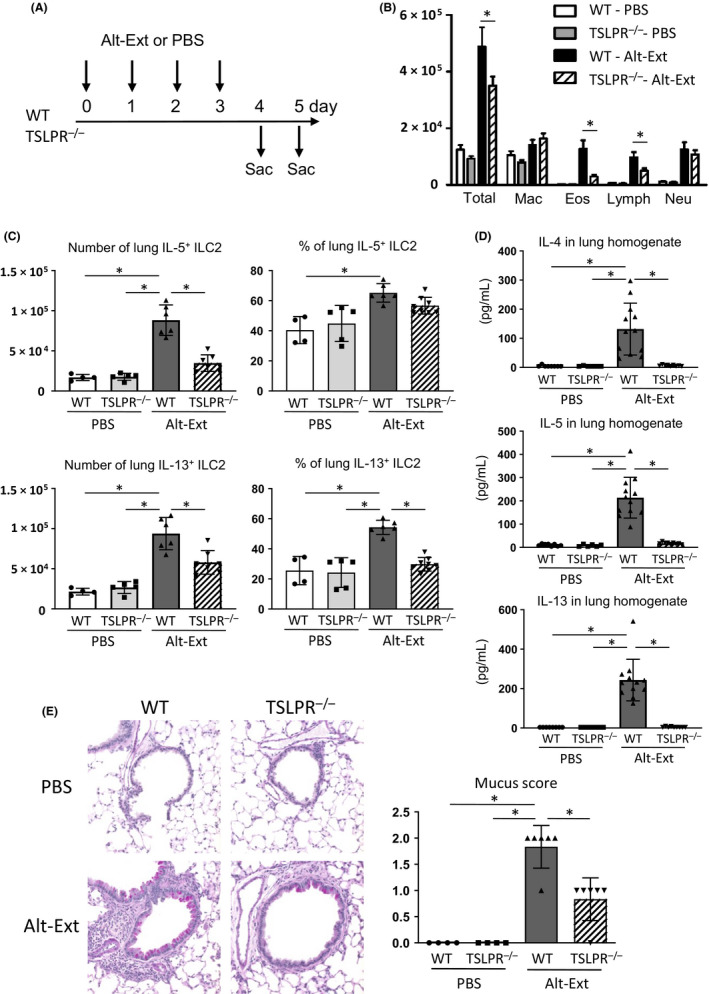
TSLPR deficiency decreased Alternaria extract (Alt‐Ext)‐induced type‐2 airway inflammation. A, WT and TSLPR−/− mice were challenged with Alt‐Ext intranasally for 4 consecutive days. Bronchoalveolar lavage fluid (BALF) and lung were harvested 24 h after the last Alt‐Ext‐challenge. Whole lungs for histological mucus detection were harvested 48 h after the last Alt‐Ext‐challenge. B, Cell differentials in BALF. C, The number and percentage of lung group 2 innate lymphoid cells (ILC2) expressing IL‐5 and IL‐13. D, The protein level of IL‐4, IL‐5, and IL‐13 in the lung homogenates. E, Representative sections and mucus score as determined by periodic acid‐Schiff (PAS) staining. The data are a combination of 2 independent experiments and shown as mean ± SD (n = 4‐9). *P < .05
3.2. Anti‐TSLP ab treatment suppresses Alt‐Ext‐induced innate allergic airway inflammation
To confirm the effect of TSLPR signaling for Alt‐Ext‐induced T cell–independent innate allergic inflammation, we performed anti‐TSLP ab treatment prior to Alt‐Ext‐challenge. WT mice were treated with anti‐TSLP ab or isotype ab 1 hour prior to the first Alt‐Ext‐challenge to neutralize the Alt‐Ext‐induced endogenous TSLP. Twenty‐four hours after the 4th daily Alt‐Ext‐challenge, BALF and lungs were harvested (Figure 2A) to evaluate innate allergic inflammation. Anti‐TSLP ab treatment significantly decreased the number of total BAL cells, macrophages, eosinophils, and lymphocytes, but not neutrophils compared with isotype ab treatment in Alt‐Ext‐challenged groups (Figure 2B). Further, anti‐TSLP ab treatment significantly decreased Alt‐Ext‐induced IL‐4, IL‐5, and IL‐13 in lung homogenates compared with isotype ab treatment (Figure 2C). While other studies have reported that TSLP antagonism reduced the adaptive allergic immune responses, to our knowledge, these results are the first to reveal that anti‐TSLP ab treatment suppressed Alt‐Ext‐induced innate allergic airway inflammation.
Figure 2.
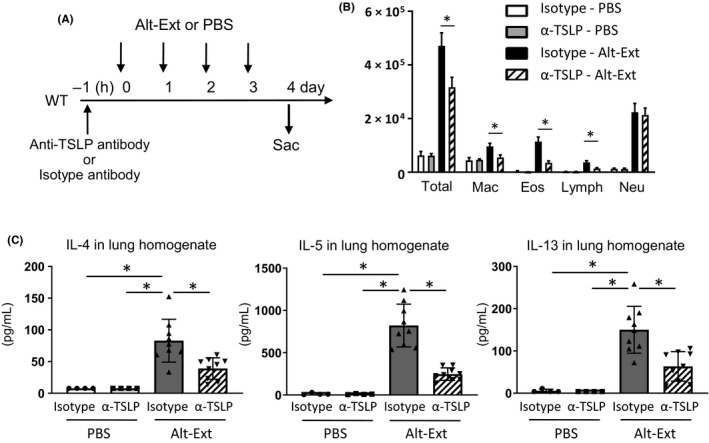
Endogenous TSLP neutralization decreased Alternaria extract (Alt‐Ext)‐induced type‐2 airway inflammation. A, Anti‐TSLP antibody (α‐TSLP) or the isotype antibody was administered subcutaneously 1 h prior to first Alt‐Ext‐challenge. Bronchoalveolar lavage fluid (BALF) and lung were harvested 24 h after the 4th Alt‐Ext‐challenge. B, Cell differentials in BALF. C, The protein level of IL‐4, IL‐5, and IL‐13 in the lung homogenates. The data are a combination of 2 independent experiments and shown as mean ± SD (n = 4‐9). *P < .05
3.3. TSLPR signaling increases Alt‐Ext‐induced IL‐33 release, and ST2 signaling increases Alt‐Ext‐induced TSLP expression in the lung
Previous studies reported that aeroallergen‐induced IL‐33 release from lung epithelial cell led to lung ILC2 activation.21, 22 We, therefore, investigated the effects of TSLPR signaling for Alt‐Ext‐induced IL‐33 release. First, we checked the time course of IL‐33 and TSLP in the BALF and lung homogenates after one Alt‐Ext‐challenge. IL‐33 protein in BALF peaked 1 hour after the Alt‐Ext‐challenge, but TSLP was not detected in the BALF (Figure 3A). Instead of BALF, TSLP protein peaked 6 hour after Alt‐Ext‐challenge in the lung homogenates (Figure 3A). These results are same as previous studies.21, 22 To determine the effect of exogenous TSLP on IL‐33 release into the BALF, mouse recombinant TSLP (rTSLP) was administered intranasally to WT mice 6 hour prior to Alt‐Ext‐challenge, and then, the BALF was harvested 1 hour after the Alt‐Ext‐challenge (Figure 3B). We found that rTSLP pretreatment enhanced the protein level of IL‐33 in the BALF 1 hour after the Alt‐Ext‐challenge compared with vehicle treatment (Figure 3C). The enhanced IL‐33 release by rTSLP pretreatment was abolished in TSLPR−/− mice (Figure S2).
Figure 3.
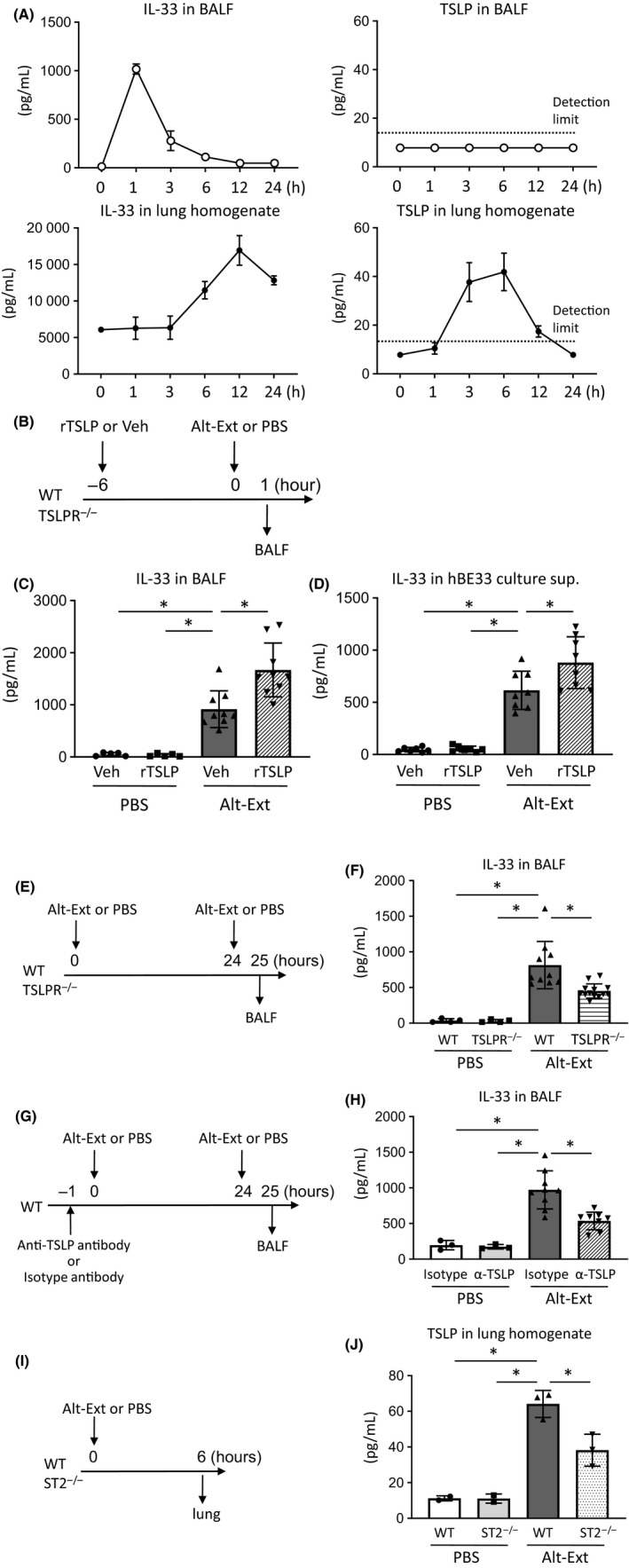
TSLPR signaling increased IL‐33 release and IL‐33 receptor (ST2) signaling increased TSLP expression in the lung. A, The time course of TSLP and IL‐33 in bronchoalveolar lavage fluid (BALF) and lung homogenates after one Alternaria extract (Alt‐Ext)‐challenge. B, E, G, I, Diagrams of experimental design using mouse model to detect IL‐33 in BALF and TSLP in the lung. C, IL‐33 protein level in the BALF from wild‐type (WT) mice challenged with Alt‐Ext or PBS in the presence or absence of recombinant TSLP (rTSLP) treatment. D, IL‐33 protein level in cell‐free culture supernatant (sup) from human bronchial epithelial cells stably expressing IL‐33 (hBE33) challenged with Alt‐Ext or PBS in the presence or absence of human recombinant TSLP treatment. F, IL‐33 protein level in the BALF from WT and TSLPR−/− mice challenged with Alt‐Ext or PBS. H, IL‐33 protein level in the BALF from WT mice challenged with Alt‐Ext or PBS in the presence or absence of anti‐TSLP antibody (α‐TSLP) treatment. J, TSLP protein level in the lung homogenates from WT and ST2−/− mice challenged with Alt‐Ext or PBS. The data are a combination of 2 independent experiments and shown as mean ± SD (n = 3‐9). *P < .05
To determine whether rTSLP treatment increases IL‐33 expression to enhance Alt‐Ext‐induced IL‐33 release, we measured IL‐33/citrine expression in the lung cells from IL‐33Citrine/+ reporter mice treated with rTSLP or vehicle for 6 hour. Although a portion of lung ECs (CD45− EpCAM+) expresses IL‐33/citrine (Figure S3A), there was no difference in the mean fluorescence intensity (MFI) of IL‐33/citrine between rTSLP treatment and vehicle treatment (Figure S3B,C). In contrast, IL‐33/citrine was not detected on lung T cells, B cells, neutrophils, eosinophils, CD11c+ macrophages, and dendritic cells from IL‐33Citrine/+ reporter mice treated with rTSLP or vehicle (Figure S3D‐G).
Functional IL‐33 is released from damaged or necrotic epithelial cells.10 Therefore, we measured IL‐33 release in human bronchial epithelial cells stably expressing IL‐33 (hBE33) culture supernatant to confirm whether human recombinant TSLP treatment increases IL‐33 release after Alt‐Ext‐challenge in an in vitro model. Alt‐Ext stimulation induced IL‐33 release from hBE33 into the culture supernatant (Figure 3D). Human recombinant TSLP treatment significantly increased Alt‐Ext‐induced IL‐33 release compared with vehicle treatment (Figure 3D). To determine whether rTSLP treatment increases IL‐33 expression in hBE33 to enhance IL‐33 release, we measured GFP co‐expressed with IL‐33 in hBE3323 after rTSLP or vehicle treatment. There was no difference in the MFI of IL‐33/GFP between rTSLP‐treated cells and vehicle‐treated cells (Figure S4A,B). Further, we tested treatment with the signal transducer and activator of transcription 5 (STAT5) inhibitor pimozide with rTSLP to investigate the effect of TSLPR‐STAT5 cascade for IL‐33 release from hBE33. Pimozide + rTSLP treatment significantly decreased the Alt‐Ext‐induced IL‐33 release compared with rTSLP treatment alone (Figure S4C). In addition, we found that rTSLP treatment increased lactate dehydrogenase (LDH) in the culture supernatant compared with vehicle treatment (Figure S4D). Pimozide + rTSLP treatment significantly decreased LDH compared with rTSLP treatment alone (Figure S4D). These in vivo and in vitro results suggested that exogenous TSLP enhanced Alt‐Ext‐induced IL‐33 release from lung epithelial cells by promoting the process of necrosis.
Next, we tested the effect of endogenous TSLP on IL‐33 release. Since there is no endogenous TSLP in the lung 1 hour after the first Alt‐Ext‐challenge (Figure 3A), a second Alt‐Ext‐challenge is necessary to induce TSLP expression in order to determine the effect of endogenous TSLP on IL‐33 release. Therefore, we performed the second Alt‐Ext‐challenge 24 hour after the first challenge to allow the TSLP expression after the first challenge to exert its biological effect, and then, we harvested the BALF from WT and TSLPR−/− mice 1 hour after second Alt‐Ext‐challenge (Figure 3E). The second Alt‐Ext‐challenge significantly induced IL‐33 protein in BALF compared with PBS challenge, and the Alt‐Ext‐induced IL‐33 was decreased in TSLPR−/− mice compared with WT mice (Figure 3F). To confirm the effect of endogenous TSLP, WT mice were treated with anti‐TSLP ab or isotype ab 1 hour prior to first Alt‐Ext‐challenge to neutralize the Alt‐Ext‐induced endogenous TSLP. We then harvested BALF 1 hour after a second Alt‐Ext‐challenge (Figure 3G) to evaluate whether endogenous TSLP neutralization suppresses the Alt‐Ext‐induced IL‐33 release. Anti‐TSLP ab treatment significantly decreased the second Alt‐Ext‐challenge‐induced IL‐33 release in the BALF compared with isotype ab treatment (Figure 3H).
In addition, to evaluate an effect of endogenous ST2 signaling for Alt‐Ext‐induced TSLP expression in the lung, WT and ST2−/− mice were challenged with Alt‐Ext once, and 6 hour later, the lungs were harvested to detect TSLP protein (Figure 3I). Alt‐Ext‐challenge significantly induced TSLP protein in the lung homogenates compared with PBS challenge. The Alt‐Ext‐induced TSLP was significantly decreased in ST2−/− mice compared with WT mice (Figure 3J). To confirm the TSLP expression on lung ECs, we isolated CD45‐ EpCAM+ cells from naïve WT mice to detect mRNA expression of TSLP. Flow cytometry analysis indicated that CD45‐ EpCAM+ cell fraction by magnetic isolation had 85.1% purity (Figure S5A). TSLP mRNA expression was detected in the isolated CD45‐ EpCAM+ cells (Figure S5B). These results revealed that TSLPR signaling and ST2 signaling reciprocally induced the expression of each other's receptor cytokine after Alt‐Ext challenge in the lung.
3.4. rTSLP treatment does not induce PAR‐2 expression on IL‐33+ lung ECs
A previous study reported that the Alt‐Ext‐induced IL‐33 release from lung ECs was mediated by protease‐activated receptor 2 (PAR‐2) signaling.22 Therefore, to determine whether TSLPR signaling increases PAR‐2 expression, we measured PAR‐2 expression on lung ECs expressing IL‐33 using IL‐33Citrine/+ mice following intranasal rTSLP treatment for 6 hour. The ECs expressing IL‐33 were identified as CD45‐ EpCAM+ citrine+ cells (Figure S6A). TSLPR and PAR‐2 were expressed on the lung ECs expressing IL‐33 (Figure S6B); however, rTSLP treatment did not change the MFI of PAR‐2 (Figure S6C).
3.5. Alt‐Ext‐induced IL‐33 does not increase TSLPR expression on lung ECs
To determine whether Alt‐Ext‐induced IL‐33 enhances TSLPR expression on lung ECs, we performed flow cytometry using WT and ST2−/− mice challenged with Alt‐Ext or PBS for 24 hour. The gating strategy of lung ECs is shown in Figure S7A. TSLPR was expressed on lung ECs from both WT and ST2−/− mice; however, there was no difference in the MFI of TSLPR between WT and ST2−/− mice challenged with PBS (Figure S7B,C). Alt‐Ext‐challenge did not alter the MFI of TSLPR on the lung ECs from both WT and ST2−/− mice (Figure S7B,C).
3.6. rTSLP increases ST2, and rIL‐33 increases TSLPR on lung ILC2 in vivo and in vitro
Thymic stromal lymphopoietin synergistically activates ILC2 with IL‐33‐ST2 signaling.14 However, the mechanism of the synergistic effect of TSLP and IL‐33 is still unknown. To determine the effect of TSLPR and ST2 signaling on the expression of each other's receptor on ILC2, we first measured ST2 and TSLPR expression on lung ILC2 in vivo. WT mice were treated intranasally with rTSLP, mouse recombinant IL‐33 (rIL‐33), or vehicle, and then 18 hour later, ST2 and TSLPR expression on lung ILC2 was measured by flow cytometry. Lung ILC2 were identified as lineage‐ CD45+ CD3− CD25+ CD127+ (Figure S8). We found that rTSLP treatment increased the MFI of ST2 on lung ILC2 compared with vehicle treatment (Figure 4A,4). However, rTSLP treatment decreased the MFI of TSLPR compared with vehicle treatment (Figure 4C,4). Meanwhile, rIL‐33 treatment significantly increased the MFI of both TSLPR and ST2 on lung ILC2 compared with vehicle treatment (Figure 4A‐D). To confirm the results in this in vivo model, we performed ILC2 treatment in an in vitro model. Isolated lung ILC2 were treated with rTSLP, rIL‐33, rTSLP + rIL‐33, or vehicle for 40 hour, and then, ST2 and TSLPR expression on the treated ILC2 was measured by flow cytometry. We found that rTSLP treatment increased the MFI of ST2, but decreased the MFI of TSLPR on the isolated lung ILC2 compared with vehicle treatment (Figure 4E‐H). rIL‐33 treatment significantly increased the MFI of TSLPR and ST2 on the isolated lung ILC2 compared with vehicle treatment (Figure 4E‐H). rTSLP + rIL‐33 treatment further enhanced the MFI of ST2 compared with rTSLP or rIL‐33 treatment alone (Figure 4E,4).
Figure 4.
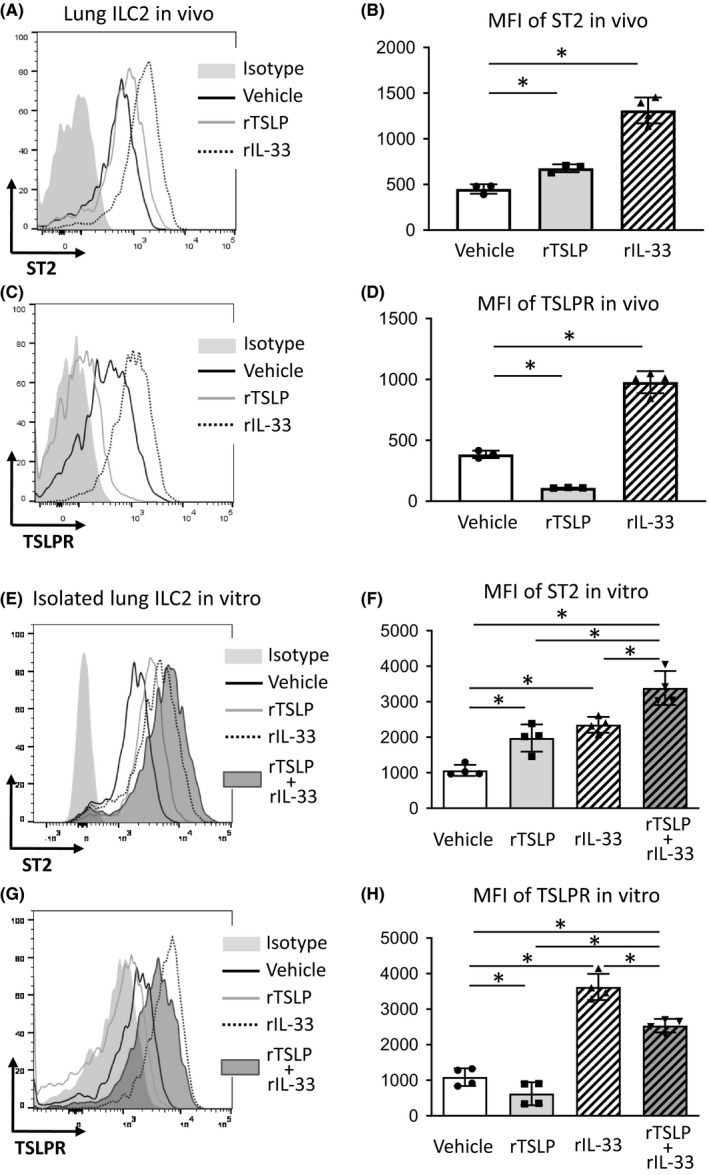
Recombinant TSLP (rTSLP) and recombinant IL‐33 (rIL‐33) treatment reciprocally increased IL‐33 receptor (ST2) and TSLPR on lung group 2 innate lymphoid cells (ILC2). A, C, Representative histograms of ST2 and TSLPR expression on lung ILC2 from wild‐type (WT) mice treated with rTSLP, rIL‐33 or the vehicle in an in vivo model. B, D, Mean fluorescence intensity (MFI) of ST2 and TSLPR on lung ILC2 in vivo. E, G, Representative histograms of ST2 and TSLPR expression on isolated lung ILC2 treated with rTSLP, rIL‐33, rTSLP + rIL‐33, or the vehicle in an in vitro model. F, H, MFI of ST2 and TSLPR on isolated lung ILC2 in vitro. The data are a combination of 2 independent experiments and shown as mean ± SD. (n = 3‐4). *P < .05
Based on our results that TSLP augmented ST2 expression on ILC2 and IL‐33 increased TSLPR expression on ILC2, we measured the protein levels of IL‐5 and IL‐13 in the isolated ILC2 culture supernatant to confirm the synergistic effect of rTSLP and rIL‐33. rTSLP treatment did not statistically induce IL‐5 and IL‐13 compared with vehicle treatment (Figure S5). rIL‐33 treatment significantly induced IL‐5 and IL‐13 compared with vehicle treatment. Further, rTSLP + rIL‐33 treatment synergistically increased IL‐5 and IL‐13 at, respectively, 10‐fold and 8‐fold greater than the treatment of rIL‐33 alone (Figure S9).
To confirm the synergistic effect of TSLPR and ST2 signaling for ILC2 activation, we isolated lung ILC2 from WT, TSLPR−/−, and ST2−/− mice. The cells were treated with rTSLP, rIL‐33, rTSLP + rIL‐33, or vehicle for 40 hour. rIL‐33 treatment increased TSLPR expression on WT lung ILC2, and the rIL‐33‐enhanced TSLPR expression was abolished on ST2−/− lung ILC2 (Figure S10A). rTSLP treatment decreased TSLPR expression on ST2−/− lung ILC2 as well as WT lung ILC2 (Figure S10A). rTSLP + rIL‐33 treatment synergistically increased ST2 expression on WT lung ILC2, and the synergistic enhanced‐ST2 expression was abolished on TSLPR−/− lung ILC2 (Figure S10B). Further, the synergistic increase of IL‐5 and IL‐13 in WT lung ILC2 by rTSLP + rIL‐33 treatment was abolished in TSLPR−/− lung ILC2 (Figure S10C). Meanwhile, there was no cytokine detection in all of ST2−/− lung ILC2 (Figure S10C). These findings suggest that TSLP and IL‐33 reciprocally enhance the expression of receptors, and this may be one of the mechanisms by which these cytokines induce synergistic ILC2 activation.
3.7. IL‐33 treatment enhanced TSLP‐induced pSTAT5
To confirm the effect of enhanced TSLPR expression on lung ILC2 by IL‐33 treatment, we measured phosphorylated STAT5 (pSTAT5) which is down stream of TSLPR signaling.24 Isolated lung ILC2 were treated with IL‐33 or vehicle for 24 hour, and then, the cells were treated with rTSLP or vehicle for 1 hour to detect pSTAT5 protein by Western blotting. There was no pSTAT5 in the ILC2 treated with vehicle or IL‐33. In contrast, pSTAT5 was detected in the ILC2 treated with rTSLP. rIL‐33 pretreatment increased rTSLP‐induced pSTAT5 (Figure 5A,5). To confirm the synergistic effect of ILC2 activation mediated by STAT5 cascade, the isolated lung ILC2 was treated with rIL‐33, rTSLP + rIL‐33, or vehicle in the presence or absence of 5 μmol/L pimozide for 40 hour. Pimozide significantly decreased IL‐5 and IL‐13 from ILC2 treated with rTSLP + rIL‐33 compared with vehicle treatment (Figure 5C). In contrast, pimozide slightly enhanced rIL‐33 treatment‐induced IL‐5 and IL‐13 from ILC2 (Figure 5C). These results revealed that IL‐33 treatment augmented TSLP‐induced phosphorylation of STAT5 in lung ILC2, and the STAT5 cascade elicited a synergistic effect in ILC2 activation.
Figure 5.
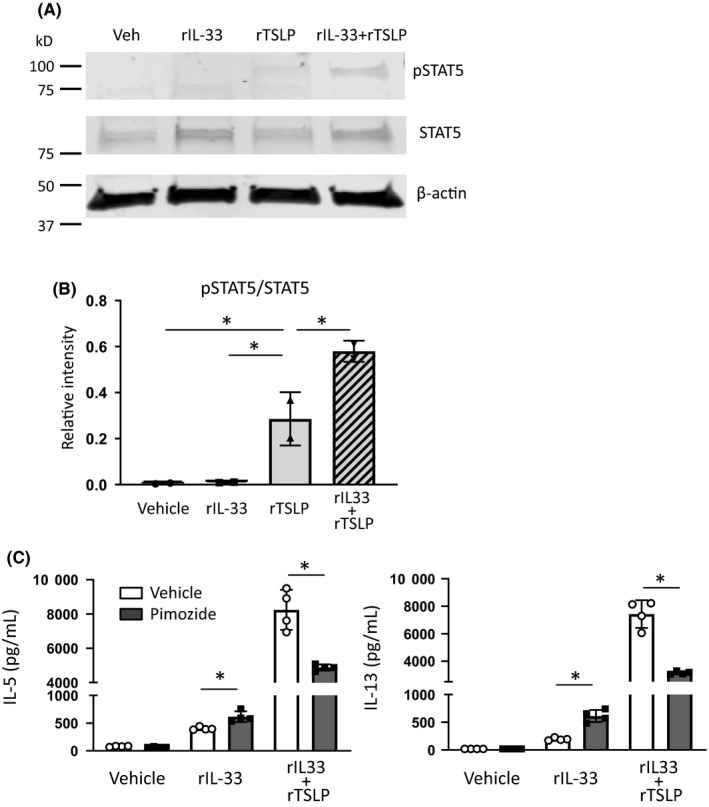
Recombinant IL‐33 (rIL‐33) treatment enhanced TSLP‐induced signal transducer and activator of transcription 5 (STAT5) signaling. A, Representative image of Western blotting. Isolated lung group 2 innate lymphoid cells (ILC2) were stimulated for 24 h with rIL‐33 (10 ng/mL) or vehicle; then, the cells were stimulated for 1 h with recombinant TSLP (rTSLP) (10 ng/mL) or vehicle. Cell lysates were separated by SDS‐PAGE and analyzed by Western blotting with antibodies to phosphorylated STAT5 (pSTAT5), STAT5, and β‐actin. B, Relative intensity of pSTAT5/STAT5 was analyzed by densitometry. C, Isolated lung ILC2 were stimulated with rIL‐33, rTSLP + rIL‐33, or vehicle in presence or absence of pimozide (5 μmol/L) for 40 h. IL‐5 and IL‐13 protein in the ILC2 culture supernatant were measured by ELISA. The data are a combination of 2 independent experiments. *P < .05
4. DISCUSSION
Previous studies reported that TSLPR and ST2 signaling promotes adaptive immunity‐driven allergic inflammation, and as a result, TSLP and IL‐33 have been identified as therapeutic targets for allergic disorders.3, 25, 26 This is the first report of the contribution of TSLPR signaling to the T cell–independent innate immunity‐driven allergic inflammatory response. We found that TSLPR deficiency significantly decreased the number of lung ILC2 expressing IL‐5 and IL‐13, airway eosinophils and lymphocytes, and mucus production compared with WT mice in an innate allergic inflammation model by which the mice were challenged with Alt‐Ext for 4 consecutive days. Neutralization of endogenous TSLP also decreased the Alt‐Ext induced eosinophilia, IL‐5 and IL‐13 expression in the lung. Regarding the mechanisms of lung ILC2 activation during Alt‐Ext‐challenge, we found that TSLPR and ST2 signaling reciprocally enhanced Alt‐Ext‐induced IL‐33 release and TSLP expression in the lung. Further, rTSLP and rIL‐33 stimulation reciprocally induced each other's receptor expression on lung ILC2 both in vivo and in vitro.
In this mouse model, 4 consecutive days of Alt‐Ext‐challenge induced allergic inflammation without CD4 T‐cell development, total IgE, and the Alternaria antigen‐specific IgG1 induction.27 However, the number of lung ILC2 expressing IL‐5 and IL‐13 was significantly greater than lung CD4 T cells expressing IL‐5 and IL‐13.27, 28 Based on these reports, ILC2 are a primary driver of the innate type‐2 inflammation induced by Alt‐Ext‐challenge. We detected LPS contamination as 0.03 EU, approximately 3 pg in 5 µg protein amount of Alt‐Ext. Alternaria alternata is a genus of ascomycete fungi, and the Alt‐Ext includes large amount of fungus components, such as lipoprotein, β‐glucan, chitin, and nucleotides to stimulate TLR2/TLR6, TLR3, TLR7, and TLR9 for inflammatory responses.29, 30 Therefore, we did not distinguish between effects of contaminated LPS and effects of fungal pathogens.
We found that TSLPR and PAR‐2 were expressed on lung ECs expressing IL‐33. While rTSLP treatment increased Alt‐Ext‐induced IL‐33 release, PAR‐2 expression was not changed by rTSLP treatment on lung ECs. Previous studies reported that Alternaria‐induced rapid IL‐33 release from normal human bronchial epithelial cells (NHBE) was mediated by ATP autocrine and consequent Ca2+ influx through P2Y2 or P2X7 receptor,21 and the consequent NADPH oxidase dual oxidase 1 (DUOX1) activation through the Ca2+ influx.31 Further, Wilson et al reported that TSLP treatment of dorsal root ganglia neurons resulted in Ca2+ influx to induce itch.32 These previous studies may suggest the possibility that TSLPR signaling has an influence on internal signaling, but not PAR‐2 expression on lung epithelial cells. While intracellular caspase during apoptosis cleaved IL‐33 to inhibit biological activity,33 full length of IL‐33 molecule is released from damaged or necrotic epithelial cells.10 Although we could not find an increase of IL‐33 expression in lung ECs by rTSLP treatment in both in vivo and in vitro, the rTSLP treatment enhanced Alt‐Ext‐induced IL‐33 and LDH release from hBE33. This suggests that TSLPR‐STAT5 cascade may participate in the cell damage that leads to IL‐33 release.
The mechanisms of the synergistic activation of ILC2 by IL‐33 and TSLP are not fully understood. Previous studies reported that ST2 expression level was significantly increased in the bone marrow ILC2 by IL‐33 treatment,34, 35 and it suggests a positive feedback loop amplifying ILC2 activation by IL‐33. In this study, we newly found that TSLP and IL‐33 treatment reciprocally enhanced the expression of each other's receptor on lung ILC2. Further, IL‐33 treatment enhanced phosphorylation of STAT5 following TSLPR signaling. Although pimozide, STAT5 inhibitor treatment significantly decreased the rTSLP + rIL‐33 enhanced IL‐5 and IL‐13, but slightly increased rIL‐33‐induced IL‐5 and IL‐13. These findings indicate one of mechanisms of the synergistic ILC2 activation by which IL‐33‐ST2 signaling increased TSLPR expression and augmented consequent STAT5 phosphorylation and the signal transduction.
Development of human monoclonal antibodies is progressing as promising biological therapeutic agents for allergic diseases.36 Tezepelumab (AMG 157/MEDI9929), an anti‐human TSLP antibody, attenuated allergen‐induced bronchoconstriction in both early and late asthmatic responses and decreased in levels of blood and sputum eosinophils.37 Further, Tezepelumab reduced the rate of asthma exacerbations in adult patients whose asthma remained uncontrolled despite treatment with medium‐to‐high doses of inhaled glucocorticoids.38 In addition, anti‐IL‐33 monoclonal antibodies (ANB020, AMG282, and REGN3500) and an anti‐ST2 monoclonal antibody (GSK3772847) are currently in ongoing clinical trials.39 Our findings highlight a synergistic suppressive effect by TSLP deficiency for allergic inflammation mediated by down‐regulation of IL‐33 release and consequent ILC2 activation. Therefore, this study suggests that therapeutic treatments that inhibit TSLP signaling may have the added bonus of inhibiting IL‐33‐mediated inflammation, while IL‐33 antagonism may also reduce TSLP‐mediated allergic responses. Thus, targeting the antagonism of one of these cytokines may be an effective and efficient method to decrease allergic inflammation mediated by both cytokines in allergic diseases such as asthma.
CONFLICT OF INTEREST
The authors that they have no conflicts of interest.
AUTHOR CONTRIBUTIONS
ST designed and performed experiments, analyzed data, and wrote the manuscript. KG and JZ contributed to the acquisition of data. WZ and DCN advised and contributed to the overall development of this study. BZ contributed to TSLPR−/− mice study. HK contributed to hBE33 in vitro study. KLB contributed to the analysis interpretation of histopathology data. RSP designed and supervised the research and edited the manuscript.
Supporting information
ACKNOWLEDGMENTS
The authors are grateful to Dr Andrew N. J. McKenzie, MRC Laboratory of Molecular Biology, Cambridge University, Cambridge, UK, for providing IL‐33Citrine/Citrine mice. This work was supported by NIH R01AI145265, R01AI124456, R01AI111820, R21AI145397, U19AI095227 (to RSP), and US Department of Veterans Affairs I01BX004299 (to RSP).
Toki S, Goleniewska K, Zhang J, et al. TSLP and IL‐33 reciprocally promote each other's lung protein expression and ILC2 receptor expression to enhance innate type‐2 airway inflammation. Allergy. 2020;75:1606–1617. 10.1111/all.14196
REFERENCES
- 1. Saenz SA, Taylor BC, Artis D. Welcome to the neighborhood: epithelial cell‐derived cytokines license innate and adaptive immune responses at mucosal sites. Immunol Rev. 2008;226:172‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Iijima K, Kobayashi T, Hara K, et al. IL‐33 and thymic stromal lymphopoietin mediate immune pathology in response to chronic airborne allergen exposure. J Immunol. 2014;193:1549‐1559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ober C, Yao TC. The genetics of asthma and allergic disease: a 21st century perspective. Immunol Rev. 2011;242:10‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Togbe D, Fauconnier L, Madouri F, et al. Thymic stromal lymphopoietin enhances Th2/Th22 and reduces IL‐17A in protease‐allergen‐induced airways inflammation. ISRN Allergy. 2013;2013:1‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Zhou B, Comeau MR, De Smedt T, et al. Thymic stromal lymphopoietin as a key initiator of allergic airway inflammation in mice. Nat Immunol. 2005;6:1047‐1053. [DOI] [PubMed] [Google Scholar]
- 6. Al‐Shami A, Spolski R, Kelly J, Keane‐Myers A, Leonard WJ. A role for TSLP in the development of inflammation in an asthma model. J Exp Med. 2005;202:829‐839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Seshasayee D, Lee WP, Zhou M, et al. In vivo blockade of OX40 ligand inhibits thymic stromal lymphopoietin driven atopic inflammation. J Clin Invest. 2007;117:3868‐3878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Headley MB, Zhou B, Shih WX, Aye T, Comeau MR, Ziegler SF. TSLP conditions the lung immune environment for the generation of pathogenic innate and antigen‐specific adaptive immune responses. J Immunol. 2009;182:1641‐1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Smith DE. IL‐33 meets allergens at the gate. Nature Immunol. 2018;19:318‐320. [DOI] [PubMed] [Google Scholar]
- 10. Cayrol C, Girard JP. IL‐33: an alarmin cytokine with crucial roles in innate immunity, inflammation and allergy. Curr Opin Immunol. 2014;31:31‐37. [DOI] [PubMed] [Google Scholar]
- 11. Oboki K, Ohno T, Kajiwara N, et al. IL‐33 is a crucial amplifier of innate rather than acquired immunity. Proc Natl Acad Sci USA. 2011;107:18581‐18586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Neill DR, Wong SH, Bellosi A, et al. Nuocytes represent a new innate effector leukocyte that mediates type‐2 immunity. Nature 2010;464:1367‐1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Moro K, Yamada T, Tanabe M, et al. Innate production of TH2 cytokines by adipose tissue‐associated c‐Kit+Sca‐1+ lymphoid cells. Nature 2010;463:540‐544. [DOI] [PubMed] [Google Scholar]
- 14. Halim TY, Krauss RH, Sun AC, Takei F. Lung natural helper cells are a critical source of Th2 cell‐type cytokines in protease allergen‐induced airway inflammation. Immunity 2012;36:451‐463. [DOI] [PubMed] [Google Scholar]
- 15. Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL‐33‐responsive lineage‐CD25+CD44hi lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol. 2012;188:1503‐1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Nussbaum JC, Van Dyken SJ, von Moltke J, et al. Type 2 innate lymphoid cells control eosinophil homeostasis. Nature 2013;502:245‐248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim HY, Chang YJ, Subramanian S, et al. Innate lymphoid cells responding to IL‐33 mediate airway hyperreactivity independently of adaptive immunity. J Allergy Clin Immunol. 2012;129:216‐227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Townsend MJ, Fallon PG, Matthews DJ, Jolin HE, McKenzie AN. T1/ST2‐deficient mice demonstrate the importance of T1/ST2 in developing primary T helper cell type 2 responses. J Exp Med. 2000;191:1069‐1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Hardman CS, Panova V, McKenzie AN. IL‐33 citrine reporter mice reveal the temporal and spatial expression of IL‐33 during allergic lung inflammation. Eur J Immunol. 2013;43:488‐498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stier MT, Bloodworth MH, Toki S, et al. Respiratory syncytial virus infection activates IL‐13‐producing group 2 innate lymphoid cells through thymic stromal lymphopoietin. J Allergy Clin Immunol. 2016;138:814‐824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kouzaki H, Iijima K, Kobayashi T, O'Grady SM, Kita H. The danger signal, extracellular ATP, is a sensor for an airborne allergen and triggers IL‐33 release and innate Th2‐type responses. J Immunol. 2011;186:4375‐4387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Snelgrove RJ, Gregory LG, Peiró T, et al. Alternaria‐derived serine protease activity drives IL‐33‐mediated asthma exacerbations. J Allergy Clin Immunol. 2014;134:583‐592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Uchida M, Anderson EL, Squillace DL, et al. Oxidative stress serves as a key checkpoint for IL‐33 release by airway epithelium. Allergy 2017;72:1521‐1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Isaksen DE, Baumann H, Zhou B, et al. Uncoupling of proliferation and Stat5 activation in thymic stromal lymphopoietin‐mediated signal transduction. J Immunol. 2002;168:3288‐3294. [DOI] [PubMed] [Google Scholar]
- 25. Gao PS, Rafaels NM, Mu D, et al. Genetic variants in thymic stromal lymphopoietin are associated with atopic dermatitis and eczema herpeticum. J Allergy Clin Immunol. 2010;125:1403‐1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Smith D, Helgason H, Sulem P, et al. A rare IL33 loss‐of‐function mutation reduces blood eosinophil counts and protects from asthma. PLoS Genet. 2017;13:e1006659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhou W, Toki S, Zhang J, et al. Prostaglandin I2 signaling and inhibition of group 2 innate lymphoid cell responses. Am J Respir Crit Care Med. 2016;193:31‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Toki S, Goleniewska K, Reiss S, et al. The histone deacetylase inhibitor trichostatin A suppresses murine innate allergic inflammation by blocking group 2 innate lymphoid cell (ILC2) activation. Thorax 2016;71:633‐645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bourgeois C, Kuchler K. Fungal pathogens‐a sweet and sour treat for toll‐like receptors. Front Cell Infect Microbiol. 2012;2:142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Patin EC, Thompson A, Orr SJ. Pattern recognition receptors in fungal immunity. Semin Cell Dev Biol. 2019;89:24‐33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hristova M, Habibovic A, Veith C, et al. Airway epithelial dual oxidase 1 mediates allergen‐induced IL‐33 secretion and activation of type 2 immune responses. J Allergy Clin Immunol. 2016;137:1545‐1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Wilson SR, Thé L, Batia LM, et al. The epithelial cell‐derived atopic dermatitis cytokine TSLP activates neurons to induce itch. Cell 2013;155:285‐295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lüthi AU, Cullen SP, McNeela EA, et al. Suppression of interleukin‐33 bioactivity through proteolysis by apoptotic caspases. Immunity 2009;31:84‐98. [DOI] [PubMed] [Google Scholar]
- 34. Brickshawana A, Shapiro VS, Kita H, Pease LR. Lineage‐Sca1+c‐Kit‐CD25+ cells are IL‐33‐responsive type 2 innate cells in the mouse bone marrow. J Immunol. 2011;187:5795‐5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Spooner CJ, Lesch J, Yan D, et al. Specification of type 2 innate lymphocytes by the transcriptional determinant Gfi1. Nat Immunol. 2013;14:1229‐1236. [DOI] [PubMed] [Google Scholar]
- 36. Mitchell PD, El‐Gammal AI, O'Byrne PM. Emerging monoclonal antibodies as targeted innovative therapeutic approaches to asthma. Clin Pharmacol Ther. 2016;99:38‐48. [DOI] [PubMed] [Google Scholar]
- 37. Gauvreau GM, O'Byrne PM, Boulet LP, et al. Effects of an anti‐TSLP antibody on allergen‐induced asthmatic responses. N Engl J Med. 2014;370:2102‐2110. [DOI] [PubMed] [Google Scholar]
- 38. Corren J, Parnes JR, Wang L, et al. Tezepelumab in adults with uncontrolled asthma. N Engl J Med. 2017;377:936‐946. [DOI] [PubMed] [Google Scholar]
- 39. Takatori H, Makita S, Ito T, Matsuki A, Nakajima H. Regulatory mechanisms of IL‐33‐ST2‐mediated allergic inflammation. Front Immunol. 2018;9:2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials