

Abstract
Background
Nanosized drug delivery systems (NDDSs) have shown excellent prospects in tumor therapy. However, insufficient penetration of NDDSs has significantly impeded their development due to physiological instability and low passive penetration efficiency.
Methods
Herein, we prepared a core cross-linked pullulan-modified nanosized system, fabricated by visible-light-induced diselenide bond cross-linked method for transporting β-Lapachone and doxorubicin prodrug (boronate-DOX, BDOX), to improve the physiological stability of the NDDSs for efficient passive accumulation in tumor blood vessels (β-Lapachone/BDOX-CCS). Additionally, ultrasound (US) was utilized to transfer β-Lapachone/BDOX-CCS around the tumor vessel in a relay style to penetrate the tumor interstitium. Subsequently, β-Lapachone enhanced ROS levels by overexpressing NQO1, resulting in the transformation of BDOX into DOX. DOX, together with abundant levels of ROS, achieved synergistic tumor therapy.
Results
In vivo experiments demonstrated that ultrasound (US) + cross-linked nanosized drug delivery systems (β-Lapachone/BDOX-CCS) group showed ten times higher DOX accumulation in the tumor interstitium than the non-cross-linked (β-Lapachone/BDOX-NCS) group.
Conclusion
Thus, this strategy could be a promising method to achieve deep penetration of NDDSs into the tumor.
Keywords: nanosized drug delivery system, core cross-linked, physiological stability, ultrasound, deep tumor penetration
Introduction
Recent advances in the field of nanosized drug delivery systems (NDDSs) have resulted in the development of effective therapies for the treatment of malignant tumors.1–4 Several NDDSs, such as Abraxane®, Vyxeos, Doxil™, Onivyde, Genexol-PM, and Lonquex, exhibit minimal side effects and have been approved for clinical use. Only Vyxeos, a newly approved drug for hematological malignancy, has shown a 3.7-month increase in overall survival (OS); others have shown no significant benefits over the use of conventional parent drugs in terms of the OS of the patients.5–7 One of the important reasons was the limited tumor penetration capability of NDDSs.8–13
Low tumor penetration capability of NDDSs is mainly related to their physiological stability and low efficiency of passive penetration.14–16 Insufficient stability of NDDSs leads to agglomeration or dispersion in vivo.14–18 Due to the particle size of NDDSs, which is either too large (greater than 200 nm) to be removed quickly by the reticuloendothelial system (RES) after agglomeration or is too small (less than 10 nm) to be removed from the tissue by extravasation after dispersion, an inadequate enhanced permeability retention (EPR) effect is observed.8,19,20 Several strategies have been employed to enhance the stability of the NDDSs, such as cross-linking techniques,21–27 including the addition of reagents28,29 (homobifunctional reagents, DTSSP (3, 3´-dithiobis (sulfosuccinimidyl propionate), poly thiol), polymerization reactions,30 and photo-cross-linking methods31 (UV light).32 However, biomedical safety concerns, such as unreacted reagents, side reactions, phototoxicity,33 and payload drug damage, may impact the use of these techniques. The use of visible light for mild cross-linking could avoid biosafety risk and maximize the efficiency by inducing the recombination and rupture of diselenide bonds (Se-Se).34 Therefore, visible-light-induced core cross-linking of diselenide bonds is expected to provide a new direction for preparing physiologically stable cross-linked NDDSs and achieve enhanced EPR effect in tumors.
The core cross-linked NDDSs have the potential to passively accumulate in tumor blood vessels due to the EPR effect owning to their excellent stability.35,36 However, previous studies have reported that poor diffusion in the dense interstitial collagen environment and vascular permeability resulted in restrictive accumulation of NDDSs around the tumor vasculature and minimal deep tumor parenchymal penetration.37,38 The shock waves, acoustic radiation force, and ultrasound-induced micro streaming could reversibly open the vascular endothelial tight junctions and drive the NDDSs into the tumor tissues simultaneously,31,39-41 enhancing the ability of NDDSs to extravasate from the vasculature to penetrate the interstitium of the tumor. Therefore, ultrasound is expected to effectively improve NDDSs to break for the inefficient penetration.
Here, a core cross-linked pullulan-modified nanosized system was developed to load both β-Lapachone and ROS (reactive oxygen species)-responsive BDOX. Next, the use of visible-light-induced dynamic diselenide bond exchange within the hydrophobic core facilitated the formation of core cross-linked nanosized drug delivery system formation, β-Lapachone/BDOX-CCS (Figure 1). When β-Lapachone/BDOX-CCS was intravenously administered into the blood system, it maintained circulation stability and improved its passive accumulation in tumor blood vessels via the EPR effect. Ultrasound enhanced the capability of β-Lapachone/BDOX-CCS to extravasate from blood vessels to penetrate the interstitial environment of the tumor. Subsequently, β-Lapachone/BDOX-CCS was effectively internalized by the tumor cells, and β-Lapachone and BDOX were released in the intracellular environment, exhibiting excellent biosafety and synergistically inducing apoptosis of the tumor cells. The combined US + β-Lapachone/BDOX-CCS treatment showed no side effects and resulted in reduced tumor growth and enhanced survival. Therefore, this technique could be adopted to achieve deep tumor penetration of β-Lapachone/BDOX-CCS for tumor therapy.
Figure 1.

Schematic illustrations of visible-light-induced core cross-linked nanosized drug delivery system combined with ultrasound for effective tumor therapy. (A) a core cross-linked nanosized system was fabricated to encapsulate β-Lapachone and BDOX. Then, visible-light was used to promote the formation of core cross-linked nanosized drug delivery system via diselenide bond exchange (β-Lapachone/BDOX-CCS), endowing excellent physiological stability. (B) β-Lapachone/BDOX-CCS efficiently penetrated the tumor interstitium after ultrasound irradiation. (C) In tumor cells, β-Lapachone was first released from the β-Lapachone/BDOX-CCS and promoted reactive oxygen species (ROS) generation by overexpressing NQO1, which fed back to the diselenide bond and BDOX, thus promoting the substantial release of DOX. Moreover, the generated ROS and DOX synergistically induced tumor apoptosis. ROS levels in healthy cells were largely unchanged by β-Lapachone due to low expression of NQO1, ensuring that BDOX remained inert in these cells.
Materials and Methods
Materials
Pullulan (MW 0.2 MDa) was obtained from Hayashibara Biochemical Laboratory (Okayama, Japan). Selenium powder, 4-Bromobenzyl alcohol, hydrazine hydrate, 1-ethyl-3-[3-(dimethylaminopropyl)]carbodiimide hydrochloride (EDCI), 4-Dimethylaminopyridine (DMAP), p-nitrobenzoyl chloride, 4-(hydroxymethyl) phenylboronic acid pinacol ester, fluorescein isothiocyanate (FITC-dextran), and doxorubicin hydrochloride (DOX•HCl) were obtained from Sigma-Aldrich (Milwaukee, USA). β-Lapachone were purchased from Sinopharm Chemical Reagent Co., Ltd (China). Also, 2,7-dichlorodihydrofluorescein diacetate (DCFH-DA) was procured from Beyotime Biotechnology Co., Ltd (China). All chemicals were of analytical grade and were used as received without further purification. Solvents were dried and distilled before use.
Synthesis and Characterization of Pullulan-Di-(4,1-Hydroxybenzylene) Diselenide (Pu-HBSe) Conjugates
Hydrazine monohydrate (100%; 1 mL, 25 mmol) was added dropwise to a mixture of sodium hydroxide (1.52 g, 38 mmol) and selenium powder (1.98 g, 25 mmol) in anhydrous dimethylformamide (DMF, 100 mL). Then, the mixture was stirred for 2 h at room temperature. Next, 4-Bromobenzyl alcohol (4.675 g, 25 mmol) was added to this mixture, stirred, and refluxed at 120°C for 4 h. The mixture was cooled to room temperature, diluted using water, and extracted using ethyl acetate. Na2SO4 was used to dry the organic phase, followed by solvent removal using a rotary evaporator. The purified di-(4,1-hydroxybenzylene) diselenide (HBSe, 2 (Scheme S1)) was isolated by drying in vacuo.
Carboxymethyl pullulan (CMP, 1 (Scheme S1)) was prepared following a previously described method.42 CMP and EDCI were dissolved using deionized water and stirred for 30 min. Next, HBSe in DMF was added to this mixture, followed by stirring for 5 h. Ethyl acetate was used to extract the reaction mixture, followed by the collection of the aqueous phase. The mixture was dialyzed against deionized water for 24 h, followed by lyophilization to collect pullulan-di-(4,1-hydroxybenzylene) diselenide (Pu-HBSe, 3 (Scheme S1)) conjugates, which were analyzed using proton nuclear magnetic resonance (1H NMR; Varian, Palo Alto, CA, 400MHz) and Fourier transform infrared spectroscopy (FTIR; Nicolet iS50 FT-IR, Thermo SCIENTIFIC, USA).
Synthesis of BDOX
The prodrug BDOX was synthesized according to the procedure described in Scheme S2.
Compound (1) was synthesized following the previously described methods.43,44 First, 4-(hydroxymethyl) phenylboronic acid pinacol ester (0.50 g, 2.2 mmol) and DMAP (0.40 g, 3.3 mmol) was dissolved in 15 mL of anhydrous dichloromethane (DCM) and cooled to 0°C. Then, a solution of p-nitrobenzoyl chloride (0.62 g, 3.1 mmol) in DCM was added dropwise to this mixture, which was stirred for 12 h at room temperature. The insoluble salt was removed by filtration, and the product was concentrated by rotary evaporation to get compound (1). The 1H NMR spectrum of this product was obtained on Varian. 1H NMR in DMSO, δ ppm: 7.78, 7.28, 6.95, 6.77, 5.13, 1.20.
Doxorubicin hydrochloride (0.20 g, 0.34 mmol), compound (1) (0.20 g, 0.52 mmol), and triethylamine (TEA, 143 μL, 1.03 mmol) were dissolved in 5 mL of anhydrous DMF and reacted at room temperature in the dark for 10 h. After completion of the reaction, the product was precipitated using diethyl ether to obtain BDOX as a red powder. 1H NMR in DMSO, δ ppm: 7.86, 7.78, 7.67, 7.36, 7.28, 5.37, 5.30, 5.05, 4.91, 4.87, 4.69, 4.65, 4.58, 3.90,3.70, 3.24, 2.12, 1.20, 1.0.
Preparation of Drug-Loaded Non-Cross-Linked and Drug-Loaded Core Cross-Linked Nanosystems
Pu-HBSe Conjugates (20 mg) were dissolved in deionized water (2 mL). Simultaneously, a mixture of BDOX (8 mg, 9.92 mmol) and β-Lapachone (2 mg, 8.25 mmol) was dissolved in 2 mL DMSO. Then, these two solutions were mixed and stirred for 12 h. The β-Lapachone/BDOX-loaded non-cross-linked nanosystem (β-Lapachone/BDOX-NCS) was obtained. Next, the aqueous dispersion of β-Lapachone/BDOX-NCS was irradiated using a 25 W incandescent light bulb without filter at room temperature at 182–184 Lux for 3 h to fabricate β-Lapachone/BDOX-loaded core cross-linked nanosystem (β-Lapachone/BDOX-CCS). Following the same procedure, BDOX-NCS and BDOX-CCS were polymerized by changing the loaded-drug. The amount of incorporated BDOX was measured using a UV spectrophotometer (NanoDrop 2000, USA) at an excitation wavelength of 485nm. Loading capacity (LC) was determined as follows: LC (wt.%) = [weight of loaded drug/weight of drug-loaded nanosized drug delivery system] ×100. The size and morphology of the NDDSs were separately characterized using a DLS spectrometer (Nano series ZEN3600, Malvern Instruments Ltd., UK), and a TEM (JEM-1200EX, Japan) after staining with water-soluble uranyl acetate.
Assessment of the Critical Micelle Concentration (CMC)
Pyrene fluorescent probe was used to measure the CMC values, by preparing a range of polymer solutions (10 mg mL−1, 5mg mL −1, 2 mg mL−1, 1 mg mL−1 –10−4 mg mL−1), after which 5 × 10−7 M pyrene in acetone was added. Acetone was removed under reduced pressure, and the emission spectra were recorded between 0–750 nm. The CMC values were determined based on the 373 to 384 nm ratio (I1/I3) as a function of polymer concentrations.
Stability Test of β-Lapachone/BDOX-NCS/CCS in FBS
The β-Lapachone/BDOX-NCS/CCS (15 mg mL−1) was prepared and mixed with phosphate-buffered saline (PBS) with or without fetal bovine serum (FBS) at a ratio of 9:1 (v/v). The mixture was incubated at 37°C and tested at intervals using DLS. The Z-average diameters from every test were recorded and plotted as a function of time.
In vitro Release from β-Lapachone/BDOX-CCS
The β-Lapachone/BDOX-CCS (5.2 mg mL−1) was transferred into dialysis bags (MWCO 8000–14,000 Da) that were submerged into 27 mL deionized water solution containing 2 mL DMSO containing glutathione (GSH; concentrations: 0 mM, 2 mM, 5 mM, and 10 mM) and H2O2 (1 μM, 10 μM, and 100 μM) and kept in a horizontal shaker maintained at 120 rpm. The amount of BDOX released was determined using a UV spectrophotometer at the specific time points.
Cell Culture
Human hepatocellular carcinoma cells (HepG2) from the Chinese Academy of Science cells Bank (Shanghai, China) were cultured in DMEM containing 10% FBS and penicillin/streptomycin at 37°C in a 5% CO2 humidified incubator. Cells in the logarithmic stage of growth were used in further experiments.
In vitro Cytotoxicity Assays
Cell Counter Kit-8 (CCK-8, Dojindo Laboratories, Kumamoto, Japan) assays were used to assess the cytotoxicity of different formulations. Briefly, HepG2 cells were added to 96 well plates (4×104 cells well−1) and allowed to adhere. Then, they were treated with fresh media containing free drugs (different formulations, DOX: β-Lapachone = 1:0, 8:2, 6:4, 5:5, 4:6, 2:8, and 0:1) along with free DOX, BDOX-NCS, BDOX-CCS, β-Lapachone/BDOX-NCS, and β-Lapachone/BDOX-CCS. The equivalent DOX/BDOX concentrations were 0.01, 0.1, 1.0, 10.0, and 100.0 mg L−1, respectively. Subsequently, the cells were incubated for 24 h. After washing with PBS, the CCK-8 solution was added for 4 h. Finally, the absorbance was measured at 450 nm using a microplate reader (ELX 800, Bio Tec, USA). Percent viability was normalized based on untreated cells.
Cellular Uptake, Intracellular Distribution, and in vitro ROS Assessments
HepG2 cells (initial density of 4 × 105 cells well−1) were cultured in six-well plates until adherent. After replacing the culture media with fresh media containing free DOX, BDOX-NCS, BDOX-CCS, β-Lapachone/BDOX-NCS, and β-Lapachone/BDOX-CCS (DOX/BDOX content: 10 mg L−1), cells were cultured for another 4 h. Then, cells were gently washed with PBS thrice and stained with DCFH-DA in a serum-free medium for 30 min. Subsequently, cells were harvested with trypsin, collected by centrifugation (1000 rpm, 5 min), resuspended in PBS, and measured by flow cytometry (CytoFLEX, Beckman Coulter, USA) at an excitation wavelength of 495nm.
HepG2 cells (4 × 104 cells well−1) were cultured in glass-bottom dishes until adherent. After replacing the culture media with fresh media containing free DOX, BDOX-NCS, BDOX-CCS, β-Lapachone/BDOX-NCS, β-Lapachone/BDOX-CCS, and β-Lapachone/BDOX-CCS (different formulations, DOX/BDOX content: 10 mg L−1), cells were grown for another 4 h. Then, cells were washed thrice using PBS. Subsequently, fixed cells were stained with DCFH-DA for 30 min and Hoechst 33342 for 7 min, and visualized using Confocal Laser Scanning Microscopy (CLSM; Nikon, Tokyo, Japan) at excitation wavelengths of 485 nm and 495 nm, respectively.
Animal Experiments
Female BALB/c nude mice (4–6 weeks old, 20–25g) and female Sprague-Dawley (SD) rats (8–10 weeks old, 200–220g) were obtained from the Laboratory Animal Center, Chongqing Medical University, China. All animal experiments were carried out according to the guidelines of the Experimental Animal Center of Chongqing Medical University and approved by the Ethics Committee of Chongqing Medical University.
Ultrasound Apparatus
The experimental ultrasonic devices were obtained from Chongqing Haifu Medical Technology Co., Ltd (Chongqing, China). While performing the in vivo experiments, a focused ultrasound transducer was used. The load power (LP) indicated as 1.3 (us 1), 3.3 (us 2), and 4.1 w (us 3) with a frequency of 1.0 MHz was adopted in this study.
Ultrasound Enhanced Tumor Vessel Permeability Assays
The ultrasound-enhanced tumor vessel permeability was evaluated in vivo by using FITC-dextran to monitor their variation in tumor-bearing female BALB/c mice. Female BALB/c nude mice were subcutaneously inoculated with HepG2 cells (5 × 106) into the flanks of their back. When the tumor size reached 100 mm3, we tested the impact of ultrasound treatment time on tumor vessel permeability. First, the tumor was pre-treated with ultrasound irradiation (4.1 w) for 1, 3, 5, or 7 min. Next, the tumor-bearing mice were treated with FITC-dextran formulations at a concentration of 100 mg kg−1 (FITC-dextran/body weight) through an intravenous tail vein injection. After 20 min, the mice were sacrificed, 0.1 g tumor tissue was homogenized and diluted with 1 mL PBS. Then, this mixture was centrifuged, and FITC-dextran in the supernatant was measured via UV-Vis Spectrometer (NanoDrop 2000, Thermo SCIENTIFIC, USA). Second, we tested the impact of the dose of ultrasound on tumor vessel permeability. The tumor was pre-treated with ultrasound irradiation (1.3, 3.3, or 4.3 w) for 5 min. The remaining procedure was the same as described above.
Hemolysis Assessment
Blood samples from female BALB/c mice were combined with a range of concentrations of β-Lapachone/BDOX-CCS (5, 25, 50, 100, and 200 µg mL−1) at 37°C for 4 h. Next, the samples were spun for 3 min at 1200 rpm, and supernatants were isolated to quantify the rates of hemolysis. For this classification, 100 µL supernatant samples were added to 96-well plates (n = 6/condition), and absorbance was measured at 541 nm via a microplate reader. RBCs in water were used as the positive control, and RBCs in PBS were used as the negative control. The rate of hemolysis was determined using the following equation: 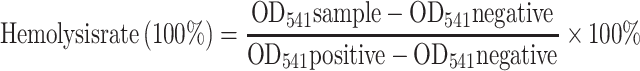
Study of Plasma Pharmacokinetics
Female SD rats were randomly assigned to 3 groups (n = 3) and were intravenously administered free DOX, β-Lapachone/BDOX-NCS, or β-Lapachone/BDOX-CCS at a concentration of 5 mg kg−1 DOX-equivalent dose. Blood was collected into heparinized tubes at different time intervals and spun for 10 minutes at 3000 rpm and 4°C. The supernatant plasma (100 μL) was combined with acetonitrile (900 μL) for protein precipitation. Following centrifugation, the organic layer was collected, concentrated, and subjected to a UV-Vis scan to determine the DOX/BDOX levels. Previously constructed standard curves for DOX and BDOX were used to determine the exact plasma concentration.
In vivo Biodistribution
In vivo fluorescence imaging was performed to monitor the biodistribution of free DOX, β-Lapachone/BDOX-NCS, and US + β-Lapachone/BDOX-CCS in HepG2 tumor-bearing mice. Female BALB/c mice were subcutaneously inoculated with HepG2 cells (4 × 106 cells per animal in 100 μL) in their back. After cellular inoculation, when the tumor size reached 100 mm3, they were pre-treated with ultrasound irradiation (4.1 w, 5 min, 1.0 MHz), followed by intravenous injections of 200 μL of β-Lapachone/BDOX-CCS formulations at a concentration of 10 mg kg−1 (DOX/body weight). At specific time intervals, animals were anesthetized and evaluated using an IVIS imaging device (PerkinElmer, Germany) at an excitation wavelength of 485 nm. In the end, DCFH-DA (50 μL, 25 μM) was injected into the tumors, and then the mice were euthanized after 30 minutes. Next, 10 μm tumor sections were prepared for CLSM, with DCF fluorescence was measured at 495 nm. Nuclei were stained with DAPI.
Determination of DOX Distribution
HepG2 tumors were implanted in the left flanks of female BALB/c mice by injecting 1 × 106 cells subcutaneously. Once tumor size reached 100 mm3, animals were randomly assigned into two groups (n = 3) and were treated with free DOX or US + β-Lapachone/BDOX-CCS at a DOX-equivalent dose of 10 mg kg−1. The mice were sacrificed 24 h after the treatment. Major organs such as the heart, liver, spleen, lung, kidneys, and tumor were collected, washed in saline solution, and weighed. Next, they were minced, homogenized, and the obtained mixture was centrifuged, and DOX in the supernatant was extracted using chloroform/isopropanol solution (3:1 v/v) and measured using UV spectrophotometer, and the corresponding DOX tissue levels were determined.
Ultrasound-Guided β-Lapachone/BDOX-CCS Penetration into Tumors
Mice with 100 mm3 tumors were used to evaluate the tumor penetration capacity of the β-Lapachone/BDOX-CCS after ultrasound irradiation. Tumors in the treatment group were sonicated for 5 min (4.1w, 1.0 MHz) to enhance the vascular permeability, with the tumors in the control group did not receive ultrasound irradiation. Subsequently, at 24 h or 48 h post-injection, the tumors were sectioned to analyze the distribution of DOX fluorescence via CLSM. Nuclei were stained with DAPI.
The ability of β-Lapachone/BDOX-CCS along with ultrasound irradiation to penetrate tumors was quantitatively assessed by injecting β-Lapachone/BDOX-NCS, β-Lapachone/BDOX-CCS, or US + β-Lapachone/BDOX-CCS containing 5 mg kg−1 DOX equivalent dose intravenously into mice when the tumor size reached 700 mm3 (with ultrasound, 4.1 w, 5 min, 1.0 MHz), and then sacrificed at 24 or 48 h after a single intravenous injection. Central tumor interstitial regions (100 mm3) or whole tumors (700 mm3) were isolated for DOX measurement via UV-Vis spectrometer.
In vivo Antitumor Study
The antitumor efficacy study was done when the tumor size reached 70–100 mm3. The HepG2 tumor-bearing mice were treated with saline, free DOX, BDOX-CCS, β-Lapachone/BDOX-NCS, β-Lapachone/BDOX-CCS, or US + β-Lapachone/BDOX-CCS (ultrasound: 4.1 w, 5 min, 1.0 MHz) five times every 4 days. DOX/BDOX was administered at a dose of 5 mg kg−1. Tumor volumes and body weight were measured using a caliper and an electronic balance every alternate day, respectively. Also, the tumor change was visually assessed and photographed. Tumor volumes were calculated using the following equation: V = (L × W2)/2.
Relative tumor volumes were normalized based on the starting volume. The tumor inhibition rate (IRT) was calculated as follows: IRT = 100% × (mean relative tumor volumes of the control group-mean relative tumor volumes of the experimental group)/mean relative tumor volumes of the control group.
The antitumor efficacy was further evaluated by preparing 10 µm sections of tumors and major organs for H&E staining, 25 days after treatment. For proliferating cell nuclear antigen (PCNA) and terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assays, stained tumor sections were assessed via an optical microscope (OLYMPUS BX50). The survival of the mice was also tracked and recorded. Animals were euthanized when tumors size reached > 1000 mm3.
In vivo Toxicity Analyses
Female BALB/c mice were intravenously administered equal doses of free DOX and β-Lapachone/BDOX-CCS (200 μL, 5 mg kg−1 per animal) five times every 4 days, with saline as the control. After 25 days, blood was collected from anesthetized animals for routine blood biochemistry, including the measurement of white blood cells (WBC), red blood cells (RBC), hemoglobin (HGB), hematocrit (HCT), and platelet (PLT). Blood aspartate aminotransferase (AST), alanine aminotransferase (ALT), blood urea nitrogen (BUN) and serum creatinine (CREA) were also determined.
Statistical Analysis
Data were compared via one-way ANOVAs and Student’s t-tests as appropriate using SPSS v24.0. Data means ± SD unless otherwise noted. *P<0.05; **P<0.01.
Results and Discussion
Preparation and Characterization of Drug-Loaded Core Cross-Linked Nanosized System (β-Lapachone/BDOX-CCS)
Pullulan-di-(4,1-hydroxybenzylene) diselenide (Pu-HBSe) conjugates were synthesized by the esterification of diselenide (Se-Se) bond in HBSe with the carboxyl-modified pullulan polysaccharide (CMP) backbone according to Scheme S1. Next, visible light exposure was used to induce diselenide bond exchange within the core to yield a core cross-linked nanosized system. The chemical structures of Pu-HBSe conjugates were assessed via 1H NMR spectra and FTIR spectra by comparing the characteristic peaks of CMP, HBSe, and Pu-HBSe. The 1H NMR spectrum of CMP showed the characteristic peaks at δH 3.6 and δH 4.51 (Figure S1A). The spectrum of HBSe showed characteristic peaks at δH 5.27 and δH 7.28, δH 7.36 (Figure S1A), while all the characteristic peaks mentioned above were present in the spectrum of Pu-HBSe (Figure S1A), which confirmed efficient synthesis. Successful esterification was confirmed based on the FTIR spectrum, which showed the peak for the ester group C=O at 1736 cm−1 (Figure S1B). These results demonstrated the successful preparation of the Pu-HBSe conjugates.
Scheme S2 shows the strategy for the synthesis of ROS-responsive prodrug (BDOX). This drug was derived from doxorubicin and contained an additional boronate moiety to decrease toxicity before ROS-mediated activation. This prodrug had high hydrophobicity, and its structure was confirmed via 1H NMR (Figure S2).
The nanoprecipitation method was used to prepare the β-Lapachone/BDOX-loaded non-cross-linked nanosized systems (β-Lapachone/BDOX-NCS). Then, the β-Lapachone/BDOX-NCS was irradiated with a 25 W incandescent light bulb at room temperature at 182–184 Lux without any reagent for 3 h to fabricate the core cross-linked nanosized systems (β-Lapachone/BDOX-CCS). Additionally, BDOX-NCS or BDOX-CCS was also synthesized to compare and demonstrate the advantages of β-Lapachone. The particle size and morphology of the NDDSs were characterized via dynamic light scattering (DLS) and transmission electron microscopy (TEM). As shown in Figure S3A–C and Figure 2A, the average size of BDOX-NCS, BDOX-CCS, β-Lapachone/BDOX-NCS, and β-Lapachone/BDOX-CCS were 181.4±5.1, 140.1±2.2, 193.0±4.3, and 134.4±5.6 nm. The average particle size of cross-linked nanosized drug delivery system was smaller than that of non-cross-linked ones, suggesting that the core was cross-linked effectively and that cross-linking resulted in a more compact structure to ensure the stability of the NDDSs. Representative TEM images in Figure 2B show that β-Lapachone/BDOX-CCS was individually dispersed with a spheroidal shape.
Figure 2.

Characterization of the NDDSs. (A) The size distribution. (B) TEM images of β-Lapachone/BDOX-CCS. (C) The average size change of β-Lapachone/BDOX-NCS and β-Lapachone/BDOX-CCS when incubated with PBS (pH = 7.4) and 10% fetal bovine serum (FBS). (D) In vitro β-Lapachone/BDOX-CCS drug release profiles in PBS at different concentrations of glutathione (GSH) and H2O2. Data are presented as mean ± SD (n = 3).
The LC and stability of the NDDSs are closely correlated to CMC. The CMC of the NDDSs in aqueous solution was determined using a pyrene fluorescent probe based on the concentration gradient of the NDDSs and pyrene intensity ratio of I373/I384 (Figure S4). The CMC test results showed that the core cross-linked nanosized drug delivery system had a low CMC value (0.7 mg mL−1) than the non-cross-linked one’s (1.3 mg mL−1), which ensured the stability of the cross-linked nanosized drug delivery system, and that the structure was not damaged even after the dilution of the injected substances by blood.
The LC of BDOX in β-Lapachone/BDOX-NCS and β-Lapachone/BDOX-CCS was 13.4%, and 19.3%, respectively. The high drug LC of the β-Lapachone/BDOX-CCS was due to the strong π–π stacking interactions between phenyl groups in the hydrophobic core, making the core of the NDDSs more compact and enhancing the effectiveness of the cross-linking reaction.
Protein adsorption can cause agglomeration or dispersion of the NDDSs, in turn resulting in its rapid clearance from the blood. Thus, the stability of NDDSs in the blood is a key pharmacokinetic parameter.45–47 When the β-Lapachone/BDOX-NCS and β-Lapachone/BDOX-CCS were incubated in PBS with or without 10% FBS at 37°C for 24 h, the β-Lapachone/BDOX-CCS incubated with 10% FBS displayed remarkable physiological stability with no discernible size variation. However, the size of β-Lapachone/BDOX-NCS incubated with 10% FBS fluctuated, rising to over 82 nm (Figure 2C). This result confirmed the core cross-linking of NDDSs, which endowed unique physiological stability for extended blood circulation and improved the EPR effect.
The diselenide bonds are highly sensitive to GSH and ROS in tumor cells,48–50 which can trigger the rapid release of payload drugs of NDDSs. The release of β-Lapachone/BDOX-CCS (Figure 2D) was further investigated in PBS (pH = 7.4) in the presence of different concentrations of GSH and H2O2 (mimic the ROS environment). β-Lapachone/BDOX-CCS exhibited significant sensitivity towards GSH and H2O2. Also, β-Lapachone/BDOX-CCS was highly stable in PBS (pH = 7.4) without GSH or in the presence of H2O2 (1 μM) since less than 21.2% or 24.8% of BDOX was released over 48 h, which indicated that the visible-light-induced core cross-linked nanosized drug delivery system had a stable structure with minimal untimed drug release. However, BDOX was released relatively rapidly in the same solution in the presence of 2 mM GSH, with more than 39.3% released after 4 h and 73.1% released after 48 h. In the presence of 5 or 10 mM GSH, which is identical to the amount of GSH present in the tumor cells,51,52 approximately 43.6% or 47.5% of BDOX was released after 4 h, and more than 82.1% or 90.1% was released within 48 h. Additionally, for H2O2, the data showed that BDOX was rapidly released from β-Lapachone/BDOX-CCS, with 69.1% or 89.4% of BDOX being released within 48 h, implying that β-Lapachone/BDOX-CCS disassembled rapidly to release the payload drug following the rupture of the diselenide bond. These results inferred that the core cross-linking enhanced the stability of the NDDSs and effectively prevented premature drug release following the intravenous injection in response to the tumor environment. This triggered faster payload drug release for tumor chemotherapy, while effectively limiting the organ damage and payload drug-induced side effects.
In vitro Cytotoxicity Assays
In vitro cytotoxic analysis of free drugs (different formulations) and NDDSs (different formulations) against HepG2 cells were evaluated using the CCK-8 assays (Figure 3A and B). Following a 24 h incubation, dose-dependent inhibition of cellular proliferation was detected for all formulations.
Figure 3.

In vitro cytotoxicity evaluation and intracellular trafficking. (A) In vitro cytotoxicity assays at seven molar ratios (1:0, 8:2, 6:4, 5:5, 4:6, 2:8, 0:1) of free DOX and free β-Lapachone formulations against HepG2 cells for 24 h. (B) Cell viability of HepG2 cells treated with free DOX, BDOX-NCS, BDOX-CCS, β-Lapachone/BDOX-NCS, and β-Lapachone/BDOX-CCS at various DOX concentrations for 24 h; Results were expressed as mean ± SD (n = 6). (C) Flow cytometric analysis of intracellular ROS levels (DCF Fluorescence intensity: ×104) in HepG2 cells treated with free DOX, BDOX-NCS, BDOX-CCS, β-Lapachone/BDOX-NCS, β-Lapachone/BDOX-CCS, and control (culture medium). (D) Confocal images of intracellular DOX distribution and ROS generation in HepG2 cells after treatment with free DOX, BDOX-NCS, BDOX-CCS, β-Lapachone/BDOX-NCS, and β-Lapachone/BDOX-CCS at 37°C for 4 h. Cells were exposed to a DOX-equivalent dose of 10 mg L−1. (E) Average DOX fluorescence in the nucleus and ROS in whole cells was calculated from the confocal images. DOX is red, nuclei (Hoechst 33342-stained) are blue, and ROS are shown in green (DCFH-DA stained). Merged images of all three are also shown. Data are expressed as mean ± SD (n = 3). *p<0.05, **p<0.01.
The combined effect could be attributed to the dose ratios,53,54 as some forms of combinatorial drug delivery can facilitate synergistic efficacy.55,56 HepG2 cells were treated with free drugs (different formulations) and NDDSs (different formulations) to explore the combined effects of this system. Table S1 shows the IC50 values of free drugs (different formulations). The lowest IC50 value of 0.097 was discovered in the free drug (8:2) group, which exhibited optimal cytotoxicity and maximal synergy for all formulations. Therefore, BDOX/β-Lapachone (8:2) was adopted as the optimal ratio for the following experiments.
In vitro cytotoxicity of the β-Lapachone/BDOX-CCS revealed high cytotoxicity compared with free DOX, BDOX-NCS, BDOX-CCS, and β-Lapachone/BDOX-NCS (Figure 3B). Specifically, IC50 values were determined to be 1.85 µg mL−1 (free DOX), 2.04 µg mL−1 (BDOX-NCS), 1.92 µg mL−1 (BDOX-CCS), 0.237 µg mL−1 (β-Lapachone/BDOX-NCS), and 0.229 µg mL−1 (β-Lapachone/BDOX-NCS) (Table S2). These results support the hypothesis that BDOX-NCS and BDOX-CCS were significantly less cytotoxic than free DOX, which was vital for their antitumor activity, was blocked by the phenylboronic ester moiety. However, the β-Lapachone/BDOX-CCS exhibited strong cytotoxicity against NQO1-overexpressing tumor cells. This was because, after the release of β-Lapachone from β-Lapachone/BDOX-CCS, it promoted ROS generation, and these ROS interacted with β-Lapachone/BDOX-CCS to transform BDOX into DOX, resulting to a more rapid release of DOX. These ROS also blocked the efflux of DOX, facilitating nuclear transportation of DOX, thus, synergically inducing cellular apoptosis.
Cellular Uptake and ROS Generation of the β-Lapachone/BDOX-CCS on HepG2 Cells
We assessed the efficiency of β-Lapachone-induced production of ROS in HepG2 cells using a green fluorescent DCFH-DA ROS-sensitive probe.57 We used this probe to stain cells after treatment with free DOX, BDOX-NCS, BDOX-CCS, β-Lapachone/BDOX-NCS, or β-Lapachone/BDOX-CCS for 4 h, followed by flow cytometric assessment. It was observed that the ROS levels were markedly elevated after treatment with β-Lapachone/BDOX-CCS and the value of DCF fluorescence became two-fold relative to BDOX-CCS. On the contrary, free DOX had little impact on levels of ROS in cells (Figure 3C), indicating β-Lapachone selectively enhanced ROS generation via NQO1 due to its overexpression within the HepG2 cells.
Next, the intracellular uptake, distribution, and ROS generation of β-Lapachone/BDOX-CCS were investigated using CLSM in HepG2 cells. These cells were treated with free DOX, BDOX-NCS, BDOX-CCS, β-Lapachone/BDOX-NCS, β-Lapachone/BDOX-CCS, BDOX and β-Lapachone different ratios of β-Lapachone/BDOX-CCS for 4 h and stained with DCFH-DA. It was observed that NDDSs could be internalized effectively by the HepG2 cells, and DCF fluorescence was barely measurable in the free DOX, BDOX-NCS, and BDOX-CCS groups. On the contrary, treatment with β-Lapachone/BDOX-NCS and β-Lapachone/BDOX-CCS enhanced this fluorescence by approximately two-fold and three-fold, respectively, consistent with a marked increase in the intracellular ROS levels (Figure 3D). The β-Lapachone/BDOX-CCS group exhibited brighter fluorescence compared with the β-Lapachone/BDOX-NCS group, which was ascribed to its higher stability.
DOX interacts with DNA and topoisomerase II in cellular nuclei; thus, nuclear DOX accumulation is essential to its ability to induce tumor cell apoptosis.58 After 4 h treatment with β-Lapachone/BDOX-CCS, we observed that the red fluorescence of DOX was colocalized throughout the cells and accumulated mostly in the nuclei (Figure 3D). NIS-Elements AR 4.20.00 software was used to quantify nuclear fluorescence (Figure 3E). Strong nuclear fluorescence was detected following treatment with free DOX due to its non-selectivity, while this fluorescence was weak in HepG2 cells treated with BDOX-NCS and BDOX-CCS as these compounds were not sufficiently activated. On the contrary, the HepG2 cells treated with β-Lapachone/BDOX-CCS showed high nuclear fluorescence, indicating that the β-Lapachone in the β-Lapachone/BDOX-CCS could radically raise ROS levels by elevating the NQO1 levels in the tumor cells, mediating the transformation of BDOX to DOX. Therefore, the elevated ROS levels in tumor cells could facilitate the rapid release of DOX. Figure 4A and B show that after 4 h incubation under different ratios of β-Lapachone/BDOX-CCS, a relatively efficient release of DOX was observed at the nuclear of β-Lapachone/BDOX-CCS (DOX: β-Lapachone = 8:2). These in vitro cellular uptake results were consistent with the cytotoxicity studies and suggested that β-Lapachone/BDOX-CCS that was prepared in this study could be internalized by the HepG2 cells, displaying great potential as a platform for antitumor drug delivery.
Figure 4.

Confocal images of DOX and ROS within the cells following treatment with (A) BDOX and β-Lapachone in β-Lapachone/BDOX-CCS in the following ratios 8:2, 6:4, 2:8 in molar ratio (n=6). (B) Average DOX fluorescence in the nucleus and ROS in whole cells was calculated based on the confocal images. (C–F) Analysis of ultrasound-enhanced tumor vascular permeability, biocompatibility, and pharmacokinetics of β-Lapachone/BDOX-CCS. In vivo Quantification of the variable degree of permeability performed at different (C) times (4.1w) and doses (5 min) (D). The FD 40 fluorescence in tumor tissues was evaluated using a UV-Vis spectrometer. (E) The hemolysis test of β-Lapachone/BDOX-CCS at different concentrations (5, 25, 50, 100, and 200 µg mL−1) in PBS. (F) Blood drug concentrations against time following one intravenous dosage with free DOX, β-Lapachone/BDOX-NCS, and β-Lapachone/BDOX-CCS at a DOX-equivalent dose of 5 mg kg−1. All results were calculated based on three independent experiments (*p<0.05, **p<0.01).
Ultrasound Enhanced Tumor Vascular Permeability Assays
FD40 is a macromolecule that combines FITC with dextran. Due to its high molecular weight (40,000 Da), it can enter the cell only when the vascular endothelial cellular permeability is increased. By injecting FD40 into the tail vein of rodents, the content of fluorescein in the tumor tissues could be detected to estimate the vascular endothelial cellular permeability. Figure 4C and D show the FITC content in tumor tissues irradiated by ultrasound under different irradiation times and powers. The results showed that the FITC content in tumor tissues increased with the increase of ultrasound irradiation time and power. The intravascular ultrasound effect caused the FITC to accumulate more readily within the tumor due to an increase in vascular permeability, and the acoustic radiation, microstreaming, and shock wave generation induced deeper penetration of FITC into the tumor.
The use of ultrasound radiation at the frequency of 1.0 MHz, power of 4.1w, and time of 5 min could safely and effectively increase the tumor vascular permeability, providing an experimental basis for the further study of ultrasound combined with NDDSs to penetrate deeply into the tumor.
In vivo Biocompatibility and Plasma Pharmacokinetics of β-Lapachone/BDOX-CCS
Biocompatibility is essential for NDDSs since it can facilitate prolonged circulation in vivo and enhance intratumoral drug bioavailability.59,60 Hemolysis is a method to gauge the potential biocompatibility of NDDSs, as it provides insights into their impact on cellular membranes.61 Thus, we conducted a hemolysis test, where RBCs collected via retro-orbital puncture from a healthy female BALB/c nude mice were exposed to a range of concentrations of β-Lapachone/BDOX-CCS for 4 h, which revealed minimal hemolysis (<5%) up to concentrations of 200 µg mL−1 (Figure 4E). Thus, β-Lapachone/BDOX-CCS was assumed to be biocompatible, increasing intratumoral bioavailability.
Accumulation of NDDSs in the tumor vasculature depends on in vivo stability. We evaluated the in vivo pharmacokinetics of free DOX, β-Lapachone/BDOX-NCS, and β-Lapachone/BDOX-CCS by collecting retro-orbital blood samples at different time points following the intravenous injection of different formulations (DOX-equivalent dose of 5 mg kg−1, n = 3), and DOX/BDOX plasma concentrations were assessed via UV-Vis spectrometry. β-Lapachone/BDOX-CCS exhibited a prolonged half-life in circulation compared with free DOX or β-Lapachone/BDOX-NCS (Figure 4F). Approximately 24 h after injection, about 16% of the injected β-Lapachone/BDOX-CCS remained in the plasma compared with 10.9% of the β-Lapachone/BDOX-NCS, suggesting that the structure of the core was typically tight after cross-linking with excellent stability and long circulation. Therefore, core cross-linked nanosized drug delivery systems were used to improve the EPR effect.
In vivo Biodistribution of β-Lapachone/BDOX-CCS in Tumors
Next, we assessed the biodistribution of different formulations of free DOX, β-Lapachone/BDOX-NCS, US + β-Lapachone/BDOX-CCS (Figure 5A). The tumor site was irradiated with ultrasound (4.1w, 5 min, 1.0 MHz) after injection of β-Lapachone/BDOX-CCS in the tail vein of mice. The IVIS imaging system was used to monitor the biodistribution of the DOX signals in vivo. At 4 h post-injection of US + β-Lapachone/BDOX-CCS, strong DOX fluorescence was seen at the tumor site, and the fluorescence intensity reached the maxima after 12 h and was consistent up to 24 h. However, β-Lapachone/BDOX-NCS exhibited less tumor accumulation, underscoring the key roles of the core cross-linked diselenide bond and ultrasound, which helped in achieving better circulation and the opening of the tumor vascular endothelial cell gaps, thus helping β-Lapachone/BDOX-CCS achieve excellent penetration and accumulation at the tumor site. Free DOX treatment led to weak fluorescence that disappeared over 24 h owing to the poor accumulation and short half-life of this DOX in vivo. The average radiant efficiency in the images was quantified using IVIS Spectrum software and plotted in Figure S5A. The average radiant efficiency in the tumor tissues treated with US + β-Lapachone/BDOX-CCS was much higher than in the tumor tissues treated with free DOX or β-Lapachone/BDOX-NCS. After β-Lapachone/BDOX-CCS was internalized by the tumor cells, β-Lapachone enhanced ROS production. Then, 24 h after injecting animals with different formulations, we injected DCHF-DA into the tumor, and then ROS levels in tumors were assessed based on fluorescence in the tumor sections (Figure 5B). The DOX and ROS fluorescence distribution were monitored by CLSM in tumors. Unlike the low ROS levels in free DOX or β-Lapachone/BDOX-NCS-treated groups, the US + β-Lapachone/BDOX-CCS group exhibited robust fluorescence consistent with enhanced intratumoral ROS levels. In the US + β-Lapachone/BDOX-CCS-treated group, there was even distribution in the cell nucleus, while there were some bright DOX spots located in the tumor tissue in the free DOX-treated group. Notably, compared with β-Lapachone/BDOX-NCS, the fluorescence intensity of the US + β-Lapachone/BDOX-CCS group was significantly higher, indicating that the elevated intratumoral ROS levels accelerated apoptosis and DOX release from β-Lapachone/BDOX-CCS. The bioavailability of β-Lapachone and DOX was enhanced by the combinatorial treatment, including ultrasound and β-Lapachone/BDOX-CCS, which improved the EPR effect by promoting the permeability of blood vessels and tumor tissue to increase the penetration of β-Lapachone/BDOX-CCS into the tumor.
Figure 5.

The biodistribution of β-Lapachone/BDOX-CCS. (A) images of in vivo fluorescence of different formulations injected into the mice at different time points (n = 3). (B) Fluorescent images of HepG2 tumors, 24 h post-treatment with the indicated formulations, collected using CLSM. ROS (green) was measured by injecting DCFH-DA into tumors. Nuclei were stained blue with DAPI.
The other treated mice were euthanized 24 h after treatment, and their tumors and major organs were collected to quantitate the DOX distribution by UV-Vis spectrometry (Figure S5B, Supplementary Material). The US + β-Lapachone/BDOX-CCS group revealed more robust intratumoral drug accumulation compared with the free DOX group, with a two-fold increase from 8.9% to 17.7% injected dose g−1, which was probably be attributed to deep tumor penetration and efficient cellular uptake. Additionally, in the β-Lapachone/BDOX-CCS group, substantial renal accumulation of DOX was observed to be three-fold higher than the free DOX group treatment, indicating that the payload drug in β-Lapachone/BDOX-CCS group could undergo renal excretion without significant toxicity, demonstrating that it had excellent biocompatibility and safety. These results further verify that core cross-linking improved the physiological stability and extended the blood circulation, and ultrasound increased the permeability of blood vessels, which markedly enhanced the penetration of β-Lapachone/BDOX-CCS into the tumor and improved tumor accumulation.
Ultrasound-Guided Penetration of β-Lapachone/BDOX-CCS into the Tumor Interstitium
We used the tumor-bearing mice to evaluate the intratumoral penetration capacity of the β-Lapachone/BDOX-CCS after ultrasound irradiation. When tumor size reached 100 mm3 in volume, they were sonicated for 5 min (4.1 w, 1.0 MHz) and injected to enhance tumor vascular permeability, with control group tumors not being subjected to US irradiation. Subsequently, 24 h or 48 h post-injection, tumors were sectioned to assess DOX fluorescence using CLSM. Figure 6A shows that β-Lapachone/BDOX-CCS-treated group had the best penetration profile with bright red fluorescence spread throughout the tumor tissues, indicating that β-Lapachone/BDOX-CCS could penetrate deeply into the tumor tissue after ultrasound irradiation. On the contrary, in the non-ultrasound-treated group, there was minimal DOX fluorescence observed in the outer edge of tumor tissues. Moreover, the free DOX-treated or β-Lapachone/BDOX-NCS-treated tumors showed a limited distribution of DOX spots at the peripheral regions with undetectable signals in the core of the tumor tissue (Figure S6). Two factors contributed to the significant accumulation of β-Lapachone/BDOX-CCS inside the tumor. First, the β-Lapachone/BDOX-CCS could passively accumulate in tumor sites due to the EPR effect owning to its excellent stability. Second, the ultrasound irradiation enhanced the tumor vascular permeability and improved the ability of β-Lapachone/BDOX-CCS to extravasate out of the blood vessels and penetrate the tumor interstitium.
Figure 6.

Tumor penetration behavior of β-Lapachone/BDOX-CCS. (A) tumor sections were collected, and DOX fluorescence was assessed via CLSM, 24h and 48 h post-treatment. (B and C) UV-Vis spectrometry was used to quantify DOX levels in (B) central tumor interstitial regions (100 mm3) and (C) throughout the HepG2 tumors after one intravenous injection of β-Lapachone/BDOX-NCS, β-Lapachone/BDOX-CCS, and US + β-Lapachone/BDOX-CCS. (D) Penetration indexes (P-index) of β-Lapachone/BDOX-CCS were obtained by dividing central interstitial DOX levels by the total tumor DOX levels. All results were calculated based on three independent experiments (**p<0.01).
The quantitative ability of β-Lapachone/BDOX-CCS combined ultrasound irradiation to penetrate the tumors, animals with tumors (700 mm3 in size) were intravenously injected with β-Lapachone/BDOX-NCS, β-Lapachone/BDOX-CCS, or β-Lapachone/BDOX-CCS (with ultrasound, 4.1w, 5 min, 1.0 MHz) containing 5 mg kg−1 equivalent dose of DOX, and then sacrificed 24 or 48 h post-injection. Next, we assessed DOX levels via UV-Vis spectrometry in the central interstitial regions (100 mm3) or whole tumors to calculate the penetration index (P-index),62 allowing for objective evaluation of the ability of NDDSs to penetrate the tumor tissues. P-indexes of β-Lapachone/BDOX-NCS, β-Lapachone/BDOX-CCS, or US + β-Lapachone/BDOX-CCS were determined by dividing the interstitial DOX levels (Figure 6B, 100 mm3) by total tumor DOX levels (Figure 6C). The P-index in Figure 6D clearly shows that the strongest tumor penetration was exhibited by US + β-Lapachone/BDOX-CCS. The superb tumor penetration of β-Lapachone/BDOX-CCS was probably due to its stable structure and ultrasound-assisted opening of the tumor vascular endothelial cell gap, which increased the number of β-Lapachone/BDOX-CCSs entering the tumor tissue.
In vivo Antitumor Effects
Based on the excellent therapeutic efficiency of the β-Lapachone/BDOX-CCS in vitro and its ability to penetrate and accumulate in tumors in vivo, we investigated the in vivo antitumor effects of the saline, free DOX, BDOX-CCS, β-Lapachone/BDOX-NCS, β-Lapachone/BDOX-CCS, and US + β-Lapachone/BDOX-CCS in HepG2 tumor-bearing mice. Different formulations with equal DOX concentrations (5 mg kg−1) were injected into the tail vein, and then the tumor area was exposed to local ultrasound irradiation (4.1 w, 5 min, 1.0 MHz). Next, same administrations (intravenous injection of NDDSs and local tumor ultrasound irradiation) were repeated five times. We assessed the antitumor properties of these formulations by measuring and observing the tumor volume (Figure 7A and E). Relative to saline, all formulations delayed tumor growth. Figure 7B shows the tumor inhibition rate. The tumor inhibition rate (98.44%) of the US + β-Lapachone/BDOX-CCS group was much higher than that of the β-Lapachone/BDOX-CCS group (87.04%), free DOX group (51.15%), BDOX-CCS (60.54%), and β-Lapachone/BDOX-NCS group (84.32%). The antitumor efficacy of the free DOX group was limited because of multidrug resistance (MDR) in the tumor cells. BDOX-CCS was significantly less cytotoxic than free DOX, which was key to its antitumor activity, which was disrupted due to the presence of a phenylboronic ester moiety. β-Lapachone/BDOX-CCS exhibited superior tumor suppression relative to the β-Lapachone/BDOX-NCS group due to the extended circulation and improved passive accumulation of the NDDSs. US + β-Lapachone/BDOX-CCS remarkably inhibited the tumor growth due to ultrasound-enhanced deep tumor penetration and improved tumor accumulation of β-Lapachone/BDOX-CCS.
Figure 7.

In vivo antitumor efficacy in a xenograft HepG2 mice model. (A) Xenograft HepG2 tumor growth profiles following treatment using saline, free DOX, BDOX-CCS, β-Lapachone/BDOX-NCS, β-Lapachone/BDOX-CCS, and US + β-Lapachone/BDOX-CCS (n = 6). (B) Tumor volume inhibition ratios (IRT), 1: Saline, 2: free DOX, 3: BDOX-CCS, 4: β-Lapachone/BDOX-NCS, 5: β-Lapachone/BDOX-CCS, 6: US + β-Lapachone/BDOX-CCS. (C) Body weight changes over time. (D) Survival rates. (E) Representative photographs of mice treated with different formulations. Tumors are represented by circles in red. Data are means ± SD; **p < 0.01.
DOX can effectively treat many tumor types, but it exhibits high organ toxicity restricts its utility. Mice intravenously injected with free DOX exhibited a substantial reduction in body weight, which is consistent with substantial systemic toxicity (Figure 7C). On the contrary, mice in other treatment groups showed healthy body weight over time. The survival rate of US + β-Lapachone/BDOX-CCS treatment was 100% at 33 days (Figure 7D). Since cardiotoxicity and renal toxicity are the main adverse events associated with free DOX, we assessed the morphology of H&E stained major organs. DOX induced the cytoplasmic segmental aggregation of myocardial cells with uneven distribution, while no apparent histopathological abnormality was observed in the myocardium of the β-Lapachone/BDOX-CCS, β-Lapachone/BDOX-NCS, and BDOX-CCS groups (Figure 8A). Treatment with free DOX also induced clear renal damage, particularly in the renal cortex (Figure 8A). Glomeruli were degenerated in the kidneys of free DOX-treated mice, with some exhibiting atrophy and the dilation of Bowman’s space. On the contrary, other treatment groups showed minimal congestion within the parietal layers of Bowman’s capsule. No inflammation or organ damage was observed from the results of H&E staining of other major tissues of other treatment groups (Figure S7), indicating that the core cross-linked nanosized system loaded with BDOX and β-Lapachone could minimize/avoid the severe side effects of DOX.
Figure 8.

Pathological analysis in xenograft nude mice with HepG2 tumor. (A) Histological sections of tumor tissues and main organs stained with H&E. (B) PCNA and TUNEL staining of tumors isolated on day 25 for observing proliferation and apoptosis. (C–K) Biochemical studies, including liver functions (ALT, AST), renal functions (CREA, BUN), and hematology data (WBC, RBC, HGB, HCT, PLT) in healthy BALB/c mice treated with saline, free DOX, and β-Lapachone/BDOX-CCS. Data are represented as mean ± SD, n = 3; *p < 0.05, **p < 0.01.
Further, this combined therapeutic effect on tumors was assessed via histological analyses such as H&E, PCNA, and TUNEL assays, which revealed that treatment with US + β-Lapachone/BDOX-CCS resulted in substantial cellular necrosis (H&E, Figure 8A), lower proliferative activity (PCNA staining, Figure 8B; Figure S8A) but with a large area of cancer cell remission in the tumor tissue with a noticeable level of cellular apoptosis and DNA damage (TUNEL assay, Figure 8B; Figure S8B) compared with other treatments. β-Lapachone/BDOX-CCS, which has a stable cross-linked diselenide bond structure, after undergoing ultrasound irradiation at the tumor site, could leak through the tumor vasculature via the gaps between the endothelial cells and penetrate deeply into the tumor tissue, which significantly enhanced its therapeutic efficacy. This enhanced antitumor efficacy was linked to enhanced tumor penetration and accumulation, efficient cellular uptake, and subsequent rapid drug release.
These results were consistent with the tumor accumulation and intratumoral distribution of the payload drug. After ultrasound irradiation on the tumor site, which was enough to improve its tumoral and vascular permeability, BDOX and β-Lapachone were effectively co-delivered deep within the tumors, which was vital for synergistic anticancer efficacy. This was probably responsible for the best survival rates and tumor growth inhibition by US + β-Lapachone/BDOX-CCS group.
Next, we treated healthy BALB/c mice (n = 3) with saline, free DOX, and β-Lapachone/BDOX-CCS at 5 mg kg−1 for a thorough assessment of in vivo toxicity. After 25 days, the kidney and liver function were evaluated based on the serum biochemistry and blood panels of these animals. Serum biochemistry showed that DOX significantly increased the levels of serum CREA (Figure 8E) and BUN (Figure 8F), consistent with kidney damage, while β-Lapachone/BDOX-CCS prevented DOX-induced renal damage. Also, ALT (Figure 8C) and AST (Figure 8D) levels showed that hepatotoxicity was markedly decreased in the β-Lapachone/BDOX-CCS treatment group. Compared with the saline and β-Lapachone/BDOX-CCS treated group, free DOX reduced the levels of WBC (Figure 8G), RBC (Figure 8H), HGB (Figure 8I), HCT (Figure 8J), and PLT (Figure 8K). Blood chemistry results showed that β-Lapachone/BDOX-CCS caused hepatic or systemic toxicity, even 25 days post-administration. This underscores the safety and suitability of the use of β-Lapachone/BDOX-CCS as an NDDSs for tumor therapy.
Conclusion
In this study, we developed a core cross-linked nanosized system for transport β-Lapachone and BDOX into the tumor. β-Lapachone/BDOX-CCS possesses unique structural stability under physiological conditions, extended blood circulation, and enhanced passive accumulation in blood vessels by the EPR effect with minimal cytotoxicity. Also, the β-Lapachone/BDOX-CCS, combined with ultrasound irradiation, showed deeper tumor penetration and high accumulation in central tumor regions. GSH and ROS, after cellular uptake and entering redox-sensitive cytosolic milieu, triggered the cleavage of diselenide bonds in β-Lapachone/BDOX-CCS to release β-Lapachone and DOX. Co-delivery of these two agents eliminated DOX-associated chemotherapeutic drug resistance, leading to near-total tumor growth inhibition with no adverse effects in xenograft mice. Our findings underscore the potential of this specific strategy to enhance the penetration ability and synergistic anti-tumor efficacy of NDDSs.
Funding Statement
This project was financially supported by Chongqing Research Program of Basic Research and Frontier Technology (no. cstc2019jcyj-msxmX0334).
Disclosure
The authors report no conflicts of interest in this work.
References
- 1.Li C, Wang J, Wang Y, et al. Recent progress in drug delivery. Acta Pharm Sin B. 2019;9(6):1145–1162. doi: 10.1016/j.apsb.2019.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Wang C, Chen S, Wang Y, et al. Lipase-triggered water-responsive “pandora’s box” for cancer therapy: toward Induced neighboring effect and enhanced drug penetration. Adv Mater. 2018;30(14):e1706407. doi: 10.1002/adma.201706407 [DOI] [PubMed] [Google Scholar]
- 3.Zhu W, Lee SJ, Castro NJ, Yan D, Keidar M, Zhang LG. Synergistic effect of cold atmospheric plasma and drug loaded core-shell nanoparticles on inhibiting breast cancer cell growth. Sci Rep. 2016;6(1):21974. doi: 10.1038/srep21974 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mukherjee S, Kotcherlakota R, Haque S, et al. Improved delivery of doxorubicin using rationally designed PEGylated platinum nanoparticles for the treatment of melanoma. Mater Sci Eng C. 2020;108:110375. doi: 10.1016/j.msec.2019.110375 [DOI] [PubMed] [Google Scholar]
- 5.Petersen GH, Alzghari SK, Chee W, Sankari SS, La-Beck NM. Meta-analysis of clinical and preclinical studies comparing the anticancer efficacy of liposomal versus conventional non-liposomal doxorubicin. J Control Release. 2016;232:255–264. doi: 10.1016/j.jconrel.2016.04.028 [DOI] [PubMed] [Google Scholar]
- 6.Stirland DL, Nichols JW, Miura S, Bae YH. Mind the gap: a survey of how cancer drug carriers are susceptible to the gap between research and practice. J Control Release. 2013;172(3):1045–1064. doi: 10.1016/j.jconrel.2013.09.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Barenholz YC. Doxil®—the first FDA-approved nano-drug: lessons learned. J Control Release. 2012;160(2):117–134. doi: 10.1016/j.jconrel.2012.03.020 [DOI] [PubMed] [Google Scholar]
- 8.Moghimi SM, Hunter AC, Murray JC. Long-circulating and target-specific nanoparticles: theory to practice. Pharmacol Rev. 2001;53(2):283–318. doi: 10.0000/PMID11356986 [DOI] [PubMed] [Google Scholar]
- 9.Li S-D, Huang L. Pharmacokinetics and biodistribution of nanoparticles. Mol Pharm. 2008;5(4):496–504. doi: 10.1021/mp800049w [DOI] [PubMed] [Google Scholar]
- 10.Perrault SD, Walkey C, Jennings T, Fischer HC, Chan WC. Mediating tumor targeting efficiency of nanoparticles through design. Nano Lett. 2009;9(5):1909–1915. doi: 10.1021/nl900031y [DOI] [PubMed] [Google Scholar]
- 11.Liang H, Ren X, Qian J, et al. Size-shifting micelle nanoclusters based on a cross-linked and pH-sensitive framework for enhanced tumor targeting and deep penetration features. ACS Appl Mater Interfaces. 2016;8(16):10136–10146. doi: 10.1021/acsami.6b00668 [DOI] [PubMed] [Google Scholar]
- 12.Wong C, Stylianopoulos T, Cui J, et al. Multistage nanoparticle delivery system for deep penetration into tumor tissue. Proc Natl Acad Sci. 2011;108(6):2426–2431. doi: 10.1073/pnas.1018382108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Yuan F, Leunig M, Huang SK, Berk DA, Papahadjopoulos D, Jain RK. Mirovascular permeability and interstitial penetration of sterically stabilized (stealth) liposomes in a human tumor xenograft. Cancer Res. 1994;54(13):3352–3356. doi: 10.1007/BF01517183 [DOI] [PubMed] [Google Scholar]
- 14.Caliceti P, Veronese FM. Pharmacokinetic and biodistribution properties of poly (ethylene glycol)–protein conjugates. Adv Drug Deliv Rev. 2003;55(10):1261–1277. doi: 10.1016/s0169-409x(03)00108-x [DOI] [PubMed] [Google Scholar]
- 15.Owen SC, Chan DP, Shoichet MS. Polymeric micelle stability. Nano Today. 2012;7(1):53–65. doi: 10.1016/j.nantod.2012.01.002 [DOI] [Google Scholar]
- 16.Mitragotri S, Burke PA, Langer R. Overcoming the challenges in administering biopharmaceuticals: formulation and delivery strategies. Nat Rev Drug Discov. 2014;13(9):655. doi: 10.1038/nrd4363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ekladious I, Colson YL, Grinstaff MW. Polymer–drug conjugate therapeutics: advances, insights and prospects. Nat Rev Drug Discov. 2019;18(4):273–294. doi: 10.1038/s41573-018-0005-0 [DOI] [PubMed] [Google Scholar]
- 18.Cho K, Wang X, Nie S, Shin DM. Therapeutic nanoparticles for drug delivery in cancer. Clin Cancer Res. 2008;14(5):1310–1316. doi: 10.1158/1078-0432.CCR-07-1441 [DOI] [PubMed] [Google Scholar]
- 19.Moghimi SM, Hunter C. Capture of stealth nanoparticles by the body’s defences. Crit Rev Ther Drug. 2001;18(6). doi: 10.1615/CritRevTherDrugCarrierSyst.v18.i6.30 [DOI] [PubMed] [Google Scholar]
- 20.Ruoslahti E. Drug targeting to specific vascular sites. Drug Discov Today. 2002;7(22):1138–1143. doi: 10.1016/s1359-6446(02)02501-1 [DOI] [PubMed] [Google Scholar]
- 21.Wei R, Cheng L, Zheng M, et al. Reduction-responsive disassemblable core-cross-linked micelles based on poly (ethylene glycol)-b-poly (N-2-hydroxypropyl methacrylamide)–lipoic acid conjugates for triggered intracellular anticancer drug release. Biomacromolecules. 2012;13(8):2429–2438. doi: 10.1021/bm3006819 [DOI] [PubMed] [Google Scholar]
- 22.Jackson AW, Fulton DA. Making polymeric nanoparticles stimuli-responsive with dynamic covalent bonds. Polym Chem. 2013;4(1):31–45. doi: 10.1039/C2PY20727C [DOI] [Google Scholar]
- 23.Samarajeewa S, Ibricevic A, Gunsten SP, et al. Degradable cationic shell cross-linked knedel-like nanoparticles: synthesis, degradation, nucleic acid binding, and in vitro evaluation. Biomacromolecules. 2013;14(4):1018–1027. doi: 10.1021/bm3018774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ren J, Zhang Y, Zhang J, et al. pH/sugar dual responsive core-cross-linked PIC micelles for enhanced intracellular protein delivery. Biomacromolecules. 2013;14(10):3434–3443. doi: 10.1021/bm4007387 [DOI] [PubMed] [Google Scholar]
- 25.Zhao Z, Yao X, Zhang Z, Chen L, He C, Chen X. Boronic acid shell‐crosslinked dextran‐b‐PLA micelles for acid‐responsive drug delivery. Macromol Biosci. 2014;14(11):1609–1618. doi: 10.1002/mabi.201400251 [DOI] [PubMed] [Google Scholar]
- 26.Read ES, Armes SP. Recent advances in shell cross-linked micelles. Chem Commun (Camb). 2007;(29):3021–3035. doi: 10.1039/b701217a [DOI] [PubMed] [Google Scholar]
- 27.Maiti C, Parida S, Kayal S, Maiti S, Mandal M, Dhara D. Redox-responsive core-cross-linked block copolymer micelles for overcoming multidrug resistance in cancer cells. ACS Appl Mater Interfaces. 2018;10(6):5318–5330. doi: 10.1021/acsami.7b18245 [DOI] [PubMed] [Google Scholar]
- 28.Pretto C, van Hest JC. Versatile reversible cross-linking strategy to stabilize CCMV virus like particles for efficient siRNA delivery. Bioconjug Chem. 2019;30(12):3069–3077. doi: 10.1021/acs.bioconjchem.9b00731 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tanaka J, Moriceau G, Cook A, et al. Tuning the structure, stability, and responsivity of polymeric arsenical nanoparticles using polythiol cross-linkers. Macromolecules. 2019;52(3):992–1003. doi: 10.1021/acs.macromol.8b02459 [DOI] [Google Scholar]
- 30.Partlow BP, Applegate MB, Omenetto FG, Kaplan DL. Dityrosine cross-linking in designing biomaterials. ACS Biomater Sci Eng. 2016;2(12):2108–2121. doi: 10.1021/acsbiomaterials.6b00454 [DOI] [PubMed] [Google Scholar]
- 31.Thanneeru S, Li W, He J. Controllable self-assembly of amphiphilic tadpole-shaped polymer single-chain nanoparticles prepared through intrachain photo-cross-linking. Langmuir. 2019;35(7):2619–2629. doi: 10.1021/acs.langmuir.8b03095 [DOI] [PubMed] [Google Scholar]
- 32.Mane S, Ponrathnam S, Chavan N. Effect of chemical cross-linking on properties of polymer microbeads: a review. Can Chem Trans. 2015;3(4):473–485. doi: 10.13179/canchemtrans [DOI] [Google Scholar]
- 33.Ji S, Cao W, Yu Y, Xu H. Visible‐light‐induced self‐healing diselenide‐containing polyurethane elastomer. Adv Mater. 2015;27(47):7740–7745. doi: 10.1002/adma.201503661 [DOI] [PubMed] [Google Scholar]
- 34.Ji S, Cao W, Yu Y, Xu H. Dynamic diselenide bonds: exchange reaction induced by visible light without catalysis. Angew Chem Int Ed. 2014;53(26):6781–6785. doi: 10.1002/anie.201403442 [DOI] [PubMed] [Google Scholar]
- 35.Cheng Z, Al Zaki A, Hui JZ, Muzykantov VR, Tsourkas A. Multifunctional nanoparticles: cost versus benefit of adding targeting and imaging capabilities. Science. 2012;338(6109):903–910. doi: 10.1126/science.1226338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chauhan VP, Stylianopoulos T, Martin JD, et al. Normalization of tumour blood vessels improves the delivery of nanomedicines in a size-dependent manner. Nat Nanotechnol. 2012;7(6):383. doi: 10.1038/nnano.2012.45 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Netti PA, Berk DA, Swartz MA, Grodzinsky AJ, Jain RK. Role of extracellular matrix assembly in interstitial transport in solid tumors. Cancer Res. 2000;60(9):2497–2503. doi: 10.1046/j.1523-5394.2000.83010.x [DOI] [PubMed] [Google Scholar]
- 38.McKee TD, Grandi P, Mok W, et al. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res. 2006;66(5):2509–2513. doi: 10.1158/0008-5472.CAN-05-2242 [DOI] [PubMed] [Google Scholar]
- 39.Krasovitski B, Frenkel V, Shoham S, Kimmel E. Intramembrane cavitation as a unifying mechanism for ultrasound-induced bioeffects. Proc Natl Acad Sci. 2011;108(8):3258–3263. doi: 10.1073/pnas.1015771108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sirsi SR, Borden MA. State-of-the-art materials for ultrasound-triggered drug delivery. Adv Drug Deliv Rev. 2014;72:3–14. doi: 10.1016/j.addr.2013.12.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu P, Jia Y, Qu F, et al. Ultrasound-responsive polymeric micelles for sonoporation-assisted site-specific therapeutic action. ACS Appl Mater Interfaces. 2017;9(31):25706–25716. doi: 10.1021/acsami.7b05469 [DOI] [PubMed] [Google Scholar]
- 42.Li H, Cui Y, Sui J, et al. Efficient delivery of DOX to nuclei of hepatic carcinoma cells in the subcutaneous tumor model using pH-sensitive pullulan–DOX conjugates. ACS Appl Mater Interfaces. 2015;7(29):15855–15865. doi: 10.1021/acsami.5b03150 [DOI] [PubMed] [Google Scholar]
- 43.Ye M, Han Y, Tang J, et al. A tumor-specific cascade amplification drug release nanoparticle for overcoming multidrug resistance in cancers. Adv Mater. 2017;29(38):1702342. doi: 10.1002/adma.201702342 [DOI] [PubMed] [Google Scholar]
- 44.Caroline DGL, Joshi-Barr S, Nguyen T, et al. Biocompatible polymeric nanoparticles degrade and release cargo in response to biologically relevant levels of hydrogen peroxide. J Am Chem Soc. 2012;134(38):15758–15764. doi: 10.1021/ja303372u [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ferrari M. Cancer nanotechnology: opportunities and challenges. Nat Rev Cancer. 2005;5(3):161. doi: 10.1038/nrc1566 [DOI] [PubMed] [Google Scholar]
- 46.Hubbell JA, Chilkoti A. Nanomaterials for drug delivery. Science. 2012;337(6092):303–305. doi: 10.1126/science.1219657 [DOI] [PubMed] [Google Scholar]
- 47.Butcher NJ, Mortimer GM, Minchin RF. Drug delivery: unravelling the stealth effect. Nat Nanotechnol. 2016;11(4):310. doi: 10.1038/nnano.2016.6 [DOI] [PubMed] [Google Scholar]
- 48.Xu H, Cao W, Zhang X. Selenium-containing polymers: promising biomaterials for controlled release and enzyme mimics. Acc Chem Res. 2013;46(7):1647–1658. doi: 10.1021/ar4000339 [DOI] [PubMed] [Google Scholar]
- 49.Cao W, Wang L, Xu H. Selenium/tellurium containing polymer materials in nanobiotechnology. Nano Today. 2015;10(6):717–736. doi: 10.1016/j.nantod.2015.11.004 [DOI] [Google Scholar]
- 50.Zhang W, Lin W, Zheng X, He S, Xie Z. Comparing effects of redox sensitivity of organic nanoparticles to photodynamic activity. Chem Mater. 2017;29(4):1856–1863. doi: 10.1021/acs.chemmater.7b00207 [DOI] [Google Scholar]
- 51.Kuppusamy P, Li H, Ilangovan G, et al. Noninvasive imaging of tumor redox status and its modification by tissue glutathione levels. Cancer Res. 2002;62(1):307–312. doi: 10.1046/j.1523-5394.2002.101008.x [DOI] [PubMed] [Google Scholar]
- 52.Balendiran GK, Dabur R, Fraser D. The role of glutathione in cancer. Cell Biochem Funct. 2004;22(6):343–352. doi: 10.1002/cbf.1149 [DOI] [PubMed] [Google Scholar]
- 53.Chou T-C. Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev. 2006;58(3):621–681. doi: 10.1124/pr.58.3.10 [DOI] [PubMed] [Google Scholar]
- 54.Lee S-M, O’Halloran TV, Nguyen ST. Polymer-caged nanobins for synergistic cisplatin− doxorubicin combination chemotherapy. J Am Chem Soc. 2010;132(48):17130–17138. doi: 10.1021/ja107333g [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mayer LD, Harasym TO, Tardi PG, et al. Ratiometric dosing of anticancer drug combinations: controlling drug ratios after systemic administration regulates therapeutic activity in tumor-bearing mice. Mol Cancer Ther. 2006;5(7):1854–1863. doi: 10.1158/1535-7163.MCT-06-0118 [DOI] [PubMed] [Google Scholar]
- 56.Luo S, Gu Y, Zhang Y, et al. Precise ratiometric control of dual drugs through a single macromolecule for combination therapy. Mol Pharm. 2015;12(7):2318–2327. doi: 10.1021/mp500867g [DOI] [PubMed] [Google Scholar]
- 57.Wang H, Joseph JA. Quantifying cellular oxidative stress by dichlorofluorescein assay using microplate reader. Free Radic Biol Med. 1999;27(5–6):612–616. doi: 10.1016/s0891-5849(99)00107-0 [DOI] [PubMed] [Google Scholar]
- 58.Wang F, Wang Y-C, Dou S, Xiong M-H, Sun T-M, Wang J. Doxorubicin-tethered responsive gold nanoparticles facilitate intracellular drug delivery for overcoming multidrug resistance in cancer cells. ACS Nano. 2011;5(5):3679–3692. doi: 10.1021/nn200007z [DOI] [PubMed] [Google Scholar]
- 59.Nagayasu A, Uchiyama K, Kiwada H. The size of liposomes: a factor which affects their targeting efficiency to tumors and therapeutic activity of liposomal antitumor drugs. Adv Drug Deliv Rev. 1999;40(1–2):75–87. doi: 10.1016/S0169-409X(99)00041-1 [DOI] [PubMed] [Google Scholar]
- 60.Larsen EK, Nielsen T, Wittenborn T, et al. Size-dependent accumulation of PEGylated silane-coated magnetic iron oxide nanoparticles in murine tumors. ACS Nano. 2009;3(7):1947–1951. doi: 10.1021/nn900330m [DOI] [PubMed] [Google Scholar]
- 61.Lin Y-S, Haynes CL. Impacts of mesoporous silica nanoparticle size, pore ordering, and pore integrity on hemolytic activity. J Am Chem Soc. 2010;132(13):4834–4842. doi: 10.1021/ja910846q [DOI] [PubMed] [Google Scholar]
- 62.Zhang P, Wang J, Chen H, et al. Tumor microenvironment-responsive ultrasmall nanodrug generators with enhanced tumor delivery and penetration. J Am Chem Soc. 2018;140(44):14980–14989. doi: 10.1021/jacs.8b09396 [DOI] [PubMed] [Google Scholar]