

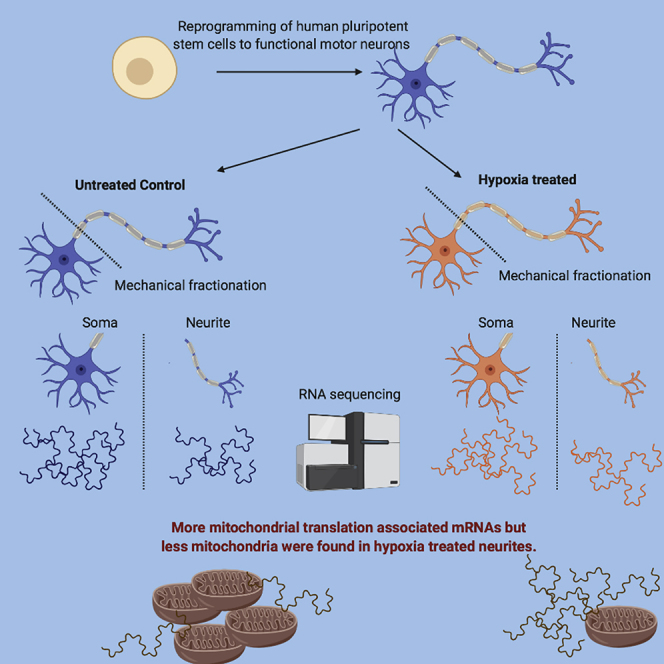
Summary
Human motor neuron (MN) diseases encompass a spectrum of disorders. A critical barrier to dissecting disease mechanisms is the lack of appropriate human MN models. Here, we describe a scalable, suspension-based differentiation system to generate functional human MN diseases in 3 weeks. Using this model, we translated recent findings that mRNA mis-localization plays a role in disease development to the human context by establishing a membrane-based system that allows efficient fractionation of MN cell soma and neurites. In response to hypoxia, used to mimic diabetic neuropathies, MNs upregulated mitochondrial transcripts in neurites; however, mitochondria were decreased. These data suggest that hypoxia may disrupt translation of mitochondrial mRNA, potentially leading to neurite damage and development of neuropathies. We report the development of a novel human MN model system to investigate mechanisms of disease affecting soma and/or neurites that facilitates the rapid generation and testing of patient-specific MN diseases.
Keywords: stem cell-derived human motor neurons, diabetic neuropathies, cell compartments, fractionation, neurites, soma, hypoxia
Graphical Abstract
Highlights
-
•
Differentiation of human pluripotent stem cells into motor neurons in 3 weeks
-
•
Global transcriptomes of motor neuron soma and neurite fractionations
-
•
Modeling of diabetic neuropathies by hypoxia reveals neurite-specific defects
-
•
Study suggests mRNA mis-localization as novel disease mechanism
Russ and colleagues present here a rapid, scalable, suspension-culture system for the efficient generation of functional human motor neurons from pluripotent stem cells. Using a soma and neurite fractionation approach, they reveal that mitochondrial transcripts, but not proteins, are neurite enriched under conditions modeling diabetic neuropathies. These findings suggest mRNA mis-localizations as a potential novel disease mechanism in motor neurons.
Introduction
Human motor neuron (MN) diseases encompass a wide range of progressive disorders, including amyotrophic lateral sclerosis (ALS) and diabetic motor neuropathies that lead to severe disabilities and/or death. MNs are unique in that their cell bodies are located within the motor cortex, brainstem, and spinal cord, and extend long axonal projections to innervate peripheral tissues. In addition, RNA transcripts are transported to these axons where they are translated to provide highly regulated localization of specific proteins. Recently, several studies using animal models have suggested that mis-localization of transcripts is a critical contributor to MN degeneration in several diseases, including fragile X syndrome, ALS, and myotonic dystrophy (Fallini et al., 2012, Wang et al., 2016). A recent study using human induced pluripotent stem cells (iPSCs) generated from ALS patients or by mutation of the FUS gene also showed mis-localization of the Fos-B mRNA in ALS samples compared with controls (Akiyama et al., 2019). Based on these findings, we speculate that transcript mis-localization could be a common mechanism of MN dysfunction in other diseases, including ischemic diabetic neuropathy, the mechanism of which is currently unknown.
Human pluripotent stem cells (hPSCs) provide unique opportunities to investigate previously inaccessible aspects of MN disease and can provide functional cell types for disease modeling and cell therapy approaches. hPSCs can be subclassified into human embryonic stem cells (ESCs) that are derived from the inner cell mass of the blastocyst and iPSCs that are generated by introducing dominant reprogramming factors into somatic cells that reverse their differentiated phenotype into a pluripotent state; iPSCs are comparable with ESCs but have the advantage of being patient specific. hPSCs are characterized by two key features: (1) rapid and indefinite cell division, thus having the potential to produce large numbers of cells, and (2) their ability to give rise to any cell type found in the human body when appropriate differentiation signals are provided. There has been substantial progress in directed differentiation of hPSCs into diverse functional cell types using stepwise differentiation approaches that employ small molecules, growth factors, and extracellular signals to recapitulate key aspects of organogenesis in vivo. MNs have been generated in vitro from hPSCs, but the current protocols require long maturation times and/or generate heterogeneous cell populations that contain non-MN cell types, hence complicating experimental setup and data interpretation (Hester et al., 2011, Li et al., 2011, Reinhardt et al., 2013). In addition, most differentiation protocols are conducted in conventional attachment cultures that are not amendable to the upscaling necessary for large experiments, including unbiased genetic screens or cell replacement therapy approaches in patients.
In this study, we describe a suspension culture-based differentiation protocol that produces a highly pure MN population within 3 weeks and expresses markers consistent with the phenotype of MNs located at the thoracic/lumbar intersection. In addition, we established an effective method to investigate RNA transcript localization in MN soma and neurites in an unbiased high-throughput manner. We then applied these technologies to compare the localization of RNA transcripts in MN soma and neurites in normal versus hypoxic conditions that mimic diabetic microvascular disease in control hESCs and iPSCs and a new onset type 1 diabetic (T1D) iPSC sample.
Results
To generate MN in a high-throughput and rapid fashion from hPSCs, we employed a scalable three-dimensional suspension-culture system previously described (Russ et al., 2015). Independent of culture method (feeder-dependent or defined, feeder-free medium) or hPSC type (ESC or iPSC), single-cell suspensions were aggregated overnight to form individual clusters containing approximately 5,000 cells, followed by a stepwise differentiation scheme (Figure 1A). During the first 6 days (Figure 1B), cultures were differentiated into neuroepithelial progenitors by dual SMAD inhibition, as described previously (Chambers et al., 2009). At day 7 (d7), clusters exhibited obvious morphological changes that included visible cell layers, indicating distinct cell organization and potential polarization of the developing epithelium (Figure 1B′). Thereafter, efficient patterning into motor neuron progenitor cells (MNPs) was achieved by activating the sonic hedgehog (SHH) and retinoic acid pathways. Following completion of MNP induction at d10, clusters were dissociated into single cells, yielding ~10 × 106 MNPs per seeded 1 × 106 hPSCs (~10-fold cell expansion). At this stage, MNPs were frozen or plated immediately on Matrigel-coated dishes for further maturation into MN in medium containing gamma-secretase inhibitors, brain-derived neurotrophic factor (BDNF), and glia cell-derived neurotrophic factor (GDNF). Comparison of fresh or frozen MNPs at d10 showed that fresh MNPs can be plated at a density of 53,000 cells per cm2, whereas frozen MNPs need to be thawed and plated at a higher density of 106,000 cells per cm2 to account for cell loss. Importantly, freezing did not impair subsequent differentiation into MNs. Plated MNPs gave rise to neurite outgrowths after 3 days in culture (Figure 1B″), indicating differentiation into mature neurons. These cells further self-organized into discrete structures of dense MN cell body clusters that were highly interconnected through extending neurites (Figure 1B‴).
Figure 1.

Suspension Culture-Based Direct Differentiation Approach of Pluripotent Stem Cells into Human Motor Neurons
(A) Schematic outlining the direct differentiation approach.
(B) Representative images taken at the indicated magnification at key stages of the differentiation protocol. Scale bar, 200 μm.
(C and D) Immunofluorescence analysis for neuronal markers ISL (C), SMI-32 (C'), merge (C”) as well as HB9 (D), βIII-TUBULIN (D') and merge (D”) of d20 MNs. Scale bar, 20 μm.
(E) Quantification of HB9 and ISL1 positive cells over DAPI at d20, n = 3–4 independent experiments.
Immunohistochemical analysis indicated that the hPSC-derived MNs expressed several mature MN markers, including insulin gene enhancer protein 1 (ISL1), motor neuron and pancreas homeobox 1 (MNX1/HB9), the neurofilament marker SMI-32, and βIII-TUBULIN (Figures 1C–1D″). Quantification of either ISL1 or HB9 positive nuclei over total nuclei (DAPI positive) revealed that approximately 80% of cells expressed ISL1 and 63% expressed HB9 (Figure 1E). Global gene expression profiling of d20 MNs showed that they resemble functionally mature MNs at the molecular level when compared with recently published datasets from in vitro-derived MNs (Ziller et al., 2018) (Figure 2A). Furthermore, quantitative PCR (qPCR) analysis at intermediate stages of the differentiation shows rapid loss of pluripotent stem cell markers NOGGIN and POU class 5 homeobox 1 (POU5F1, also known as OCT4) (Figure 2B) and upregulation of the neuronal progenitor marker Oligodendrocyte transcription factor 2 (OLIG2) at days 8–14 (Imayoshi and Kageyama, 2014). We also validated expression of the mature neuronal markers ISL1 and HB9 in the final d20 MNs (Figure 2C). Live Ca2+ imaging using Fluo-4 imaging dye (Grienberger and Konnerth, 2012) further revealed that d20 MNs exhibited robust baseline activities, with many active connections between unstimulated cells under basal conditions (Figure 2D and Video S1). Upon depolarization by 60 mM KCl, MNs displayed a strong and immediate Ca2+ influx (Figure 2D′ and Video S1), demonstrating the ability to potentially mount action potentials. Consistent with the functional MN phenotype, whole-cell patch clamping analysis of iPSC-derived MNs at d20 revealed single and multiple action potentials in response to 25–50 mV (Figures 2E and E″). To determine if d20 MNs represent a distinct regional subpopulation, we analyzed mRNA expression of segmentation marker genes, HOX A1, B3, C4, C5, C6, C8, B8, and D8 by qPCR. HOXC8 expression was significantly higher than all the other HOX genes analyzed (Figure 2F), suggesting that we generated a very specific subset of lateral motor column MNs located in the thoracic/lumbar region of the spinal cord. Paralogs HOXB8 and HOXD8 were expressed at lower levels in MNs, which supports previously published data showing that different cells within a specific tissue express typically one, but not all, region-specific HOX paralogs (Fabre et al., 2018, Mucenski et al., 2019). Taken together, these data provide compelling evidence that our scalable differentiation approach generates relatively pure populations of functional MNs within 3 weeks, allowing us to move forward with important questions related to altered MN responses in disease.
Figure 2.

Direct Differentiation Results in Functional, Spatially Defined Human Motor Neurons
(A) Global gene expression analysis of differentiated MNs in comparison with previously published datasets of hESC-derived motor neurons (Ziller et al., 2018). Gene expression values from each sample were correlated. Values represent Spearman rho correlation coefficients.
(B) Quantitative PCR analysis of pluripotency markers NANOG and OCT4 normalized to TBP at subsequent stages of differentiation. n = 3–4 independent experiments; data are presented as standard error of the mean (SEM).
(C) Quantitative PCR reveals expression of mature MN markers OLIG2 and MNX1 at d14, which is maintained at d23, at which point ISL1 expression also becomes highly enriched. n = 3–4 independent experiments; data are presented as SEM.
(D–D″) Representative images of calcium imaging before, during, and after depolarization. Scale bar, 50 μm.
(E) Whole-cell patch clamp recordings of cultured MNs. An example of evoked action potentials with a 50 pA step. Four sweeps overlaid. Y axis, millivolts; X axis, milliseconds. (E′ and E″) Additional example of evoked single or multiple action potentials from MNs. Action potentials were evoked by 25–50 pA steps. Y axis, millivolts; X axis, milliseconds.
(F) Quantitative PCR analysis for HOX gene expression of mature MNs reveals a strong enrichment of HOXC8. Schematic showing the developmental distribution of HOX genes in humans. n = 4–8 independent experiments; data are presented as SEM.
The subcellular localization of RNA transcripts within neurons allows MNs to spatially regulate gene expression (Sahoo et al., 2018), and many studies have identified specific transcripts that are trafficked to neurites (Cajigas et al., 2012, Taliaferro et al., 2016, Zappulo et al., 2017, Zivraj et al., 2010). Recently, mis-regulation of this process has been linked to several neurological diseases including fragile X syndrome, spinal muscular atrophy, myotonic dystrophy (Fallini et al., 2012, Wang et al., 2016), and ALS (Akiyama et al., 2019). To investigate whether the in vitro-derived MNs could be employed for the study of RNA localization in the human context, we applied a mechanical fractionation method that has been used previously for mouse neuronal cells to separate neurites from soma (Figure 3A, Taliaferro et al., 2016). Cells were plated on porous membranes that allow growth of neurites through the membrane, but restrict soma to the top of the membrane, allowing the mechanical separation of cell neurites from soma by simply scraping the membrane for sample collection. Protein and RNA samples from both fractions were collected and analyzed. We validated the efficiency of the fractionation by assaying for beta-actin, which is present in both soma and neurites, and histone H3 protein, which is restricted to the nucleus (Figure 3B). High-throughput RNA sequencing was performed on the fractionated samples from three independent ESC experiments, two control iPSCs and four T1D iPSCs, and the data were analyzed by hierarchical clustering. Soma samples exhibited distinct transcriptomes when compared with neurite samples as demonstrated by clustering of samples based on cell fraction (Figures 3C and 3D). We then compared gene expression using the ratio of a gene's expression in the neurite versus its expression in the soma, a metric we have previously termed the localization ratio (LR) (Taliaferro et al., 2016). Neurite-enriched genes have positive log(LR) values, whereas soma-enriched genes have negative log(LR) values. Comparison of soma and neurite expression data using a standard statistical framework (Love et al., 2014) identified 1,380 genes significantly enriched in neurites and 1,020 genes significantly enriched in soma (p < 0.01, log2(fold change) > 1.5) in the ESCs, 914 neurite enriched, 823 soma enriched in iPSC controls and 1,335 neurite enriched, 1,059 soma enriched in T1D iPSC-derived MNs. Importantly, these datasets included three genes known to be enriched in neurites, ACTB, NGRN, and RANBP1, further supporting efficient fractionation. NGRN and RANBP1 were significantly neurite enriched and ACTB transcripts were more abundant in neurites than soma (Figures 3E and E″). We also observed strong enrichment of transcripts encoding ribosomal protein genes and nuclear-encoded members of the electron transport chain, which have been shown to be hallmarks of neurite-associated RNAs (Taliaferro et al., 2016, Gumy et al., 2011) (Figures 3F, 3F″, S3A, and S3B). As a further validation, we compared the observed LR values for all genes in ESC-derived MNs with those previously reported from a fractionation of mouse cortical neurons (Taliaferro et al., 2016) to demonstrate a highly significant correlation of LR values for genes between the two groups (Figure 3G).
Figure 3.

Global Analysis of Motor Neuron Soma and Neurite-Enriched RNAs
(A) Fractionation schematic. Neurons are plated on porous membranes that allow neurite growth through the membrane but restrict soma to the top of the membrane. Cells are then mechanically fractionated, and RNA and protein from each fraction are isolated and analyzed.
(B) Dot blot of soma and neurite protein fractions from three independent fractionation experiments. Beta-actin is present in both fractions while histone H3 is restricted to the soma fraction.
(C and D) (C) Hierarchical clustering of gene expression values from motor neuron RNA fractionations from ESC-derived MNs and (D) control iPSCs and T1D iPSC MNs. Values represent Spearman rho correlation coefficients.
(E) Comparison of gene expression values in soma and neurite samples for ESCs, iPSC controls (E′), and T1D iPSCs (E″). Genes significantly enriched (p < 0.01, log2(fold change) >1.5) in either fraction are represented in purple. Three known neurite-enriched genes (NGRN, ACTB, and RANBP1) are highlighted and are neurite enriched in the data.
(F) LR values for genes from two gene ontology categories: electron transport chain and structural constituent of ribosome in all three cell types (F, F″,F″). These ontology categories have been previously observed to contain many neurite-enriched genes.
(G) Comparison of LR values from motor neuron data with previously published LR values from E15.5 mouse cortical neurons. Only one-to-one human-to-mouse orthologs are considered.
Hypoxia is believed to play important roles in the development of neuropathies (Gupta and Agarwal, 2006, Mayer et al., 1999.), particularly those related to diabetes, due to vascularization defects that occur during disease progression (Malik et al., 1990, Hendriksen et al., 1992). To determine the effects of hypoxia on MNs, we cultured d20 MNs under normoxic (atmospheric) and hypoxic (5% O2) conditions for 24 h, followed by cell fractionation into soma and neurite samples. After verifying efficient fractionation (Figures 3B–3D), global RNA gene expression was analyzed by high-throughput sequencing as described above. We observed a high degree of reproducibility across treatments and fractionations as the samples clustered according to hypoxia treatment and subcellular compartment regardless of their ESC or iPSC origin (Figures 4A and 4A″). Because soma samples contain approximately 99% of the total cellular RNA, we compared soma samples between normoxia and hypoxia conditions as a proxy for general gene expression changes in response to hypoxia. We observed significant (p < 0.01, log2(fold change) > 1.5) expression changes in 2,737 genes of the ESC MNs (Tables S1, S2, and S3), many of which were known to be involved in the hypoxic response (Figures S4A–S4C). In addition, many genes involved in glycolysis were highly upregulated (Figures S4A and S4D), presumably in response to the increased need for glycolytic energy production due to the decreased efficiency of oxidative phosphorylation. To determine how RNA localization to neurites was regulated in response to hypoxic conditions, we compared LR values observed in normoxia and hypoxia in ESCs, iPSC controls, and T1D iPSCs (Figures S4B and S4C). Using a software package designed to identify changes in the ratios of gene expression values across conditions (Xiao et al., 2016), we identified 1,511 genes that became significantly (p < 0.05) more neurite localized upon hypoxia treatment and 718 genes that became less neurite localized in ESC MNs (Figure 4B); in T1D cells, 1,226 genes were more neurite enriched in hypoxia and 582 genes were less neurite enriched (Figure S4H). In control iPSCs, we only detected one gene that was significantly more neurite enriched in hypoxia and two that were less neurite enriched (Figure S4G); this was due to the smaller sample size, thus reducing the power to detect significant changes considerably. Interestingly, we observed that nuclear-encoded genes involved in mitochondrial function, including those involved in mitochondrial translation and the electron transport chain, were strongly more neurite localized upon hypoxia treatment in all cell types (Figures 4C–4E, S4I, and S4J). Genes involved in cytoplasmic translation were unchanged in their localization following hypoxia, suggesting that this effect is specific to mitochondria-related genes and not a general stress response (the data are accessible under accession number GEO: GSE134737).
Figure 4.

Hypoxia Induces Significant Changes in Gene Expression and RNA Localization in Motor Neurons
(A) Principal component analysis of gene expression values from soma and neurite samples of normoxia and hypoxia-treated ESC MNs, (A′) iPSC controls, and (A″) iPSC T1D MNs.
(B) LR values from normoxia and hypoxia-treated MNs. Genes with significant (p < 0.05) changes in LR between conditions are colored in purple.
(C–E) Nuclear-encoded genes related to mitochondrial function, including those involved in the electron transport chain and the mitochondrial translation machinery are more localized to neurites upon hypoxia treatment, while those involved in cytosolic translation show no change in localization. In ESC-derived MNs (C), mitochondrially encoded genes are significantly less localized to neurites but show no significant changes in control iPSCs (D) and T1D iPSC MNs (E). p values are Wilcoxon rank sum values.
(F and G) (F) Representative images (scale bar, 20 μm; inset, 50 μm) and (G) quantification of mitochondrial staining in normoxic and hypoxic conditions show a significant reduction of mitochondria stain intensity in the neurites cultured under hypoxic conditions n = 2 independent experiments, n = 8 technical replicates.
To assess whether the observed transcript enrichment translates into an increase of functional mitochondria within neurites, we used MitoTracker to visualize mitochondria. We found that the number of mitochondria decreased significantly in neurites of hypoxic MNs compared with controls (Figures 4F and 4G). Consistently, neurite localization of genes encoded by the mitochondrial genome was also significantly reduced upon hypoxia treatment (Figure 4C). Taken together, these data suggest that mitochondria loss in MN neurites occurs in response to hypoxia, causing the cells to upregulate nuclear-encoded mitochondrial genes and their transport to neurites in an attempt to compensate for this loss. These data provide novel insight into critical neuronal responses to hypoxia and provide a framework for future investigations into the development and progression of neuropathies.
Discussion
Human MN diseases encompass a wide range of disorders, including ALS, viral poliomyelitis, Guillain-Barré syndrome, and diabetic motor neuropathies. These progressive diseases are debilitating and often irreversible; thus, increasing our understanding of mechanisms of MN disease is essential to reduce patient mortality and morbidity. In this study, we demonstrate the ability to reliably and quickly generate highly pure populations of spatially defined, functional human MNs that can be used to assess disease mechanisms and therapies in a patient-specific manner. These efforts will include the emerging concept of RNA transcript mis-localization, which is not well characterized in the MN field.
In the present study, we sought to recapitulate and improve upon previously published protocols to produce large numbers of MNs from hPSCs. We generated large numbers of MN progenitors, which can be frozen and thawed using a suspension-culture system in 10 days. We were able to generate functional MNs in just 18 days. In addition, the protocol produced a spatially defined subpopulation of MNs expressing high levels of HOXC8, placing them in the thoracic/lumbar region of the spinal cord, which specifically innervates muscles of the legs, therefore providing an ideal model system to study neuropathies of the lower extremities. Future studies will attempt to determine the culture conditions necessary to generate alternative populations of MNs.
To directly address RNA localization in human MNs, we adopted a methodology that we previously reported using animal cells (Taliaferro et al., 2016). In this approach, MNs are grown at high density on filters that allow passage of neurites but restrict cell bodies from migrating through. We successfully separated the two cell compartments and performed RNA sequencing on these samples, thus providing a novel and comprehensive dataset of transcripts of human MN soma and neurites (the data are accessible under accession number GEO: GSE134737).
A widely accepted common cause for peripheral neuropathies is the development of a hypoxic environment that damages MNs due to the loss of surrounding blood vessels. While still poorly understood, this mechanism is believed to be a key feature of many neuropathies, including ones caused by long-term diabetes. Approximately 50% of patients with long-standing T1D develop neuropathies of the extremities independent of glucose control. This suggests a potential genetic component and urged us to perform fractionation experiments with T1D iPSCs. To investigate potential skewing of transcript localization in human MNs under diabetic neuropathic conditions, we exposed MNs derived from ESCs, controls, and iPSCs from T1D patients to hypoxia and performed cell fractionation and global RNA analysis. Reassuringly, RNA samples from hypoxic cell soma were significantly enriched for genes involved in the hypoxic response. In addition, we found an upregulation of genes in the glycolytic pathway, likely a compensatory mechanism to counteract the decrease in energy production through oxidative phosphorylation. Interestingly, we observed neurite enrichment of mitochondrial translation and electron transport associated genes while cytoplasmic translation genes were not enriched, indicating an active stress response of the genes localized to neurites, the cell compartment believed to be most affected by hypoxia. However, quantitative analysis showed that the compensatory stress response observed on the transcriptomic level did not result in an increase but rather a decrease of neurite-localized mitochondria. These results provide first insights into the underlying molecular mechanisms that result in the loss of functional MNs, leading to the development of neuropathies. While we did not observe obvious developmental phenotypes or different hypoxia responses in T1D-derived MNs, the experiments provide an important proof of principle of our approach using patient-specific cells and will be followed up with more detailed analyses. Mitochondrial dysfunction has been proposed to be a central mediator in the development and progression of diabetes and diabetic neuropathies (Fernyhough, 2015, Fernyhough et al., 2003) (Pinti et al., 2019), and we provide here strong evidence that mitochondrial dysfunction occurs specifically in MN neurites. Future studies will need to employ patient-specific iPSCs from patients with long-standing T1D with neuropathies in comparison with healthy and/or patients with long-standing T1D without complications to illuminate unknown (genetic) disease contributors. In addition, the approach provided here can be effectively employed to test the effects of current drugs used to treat neuropathies and to assay novel drug candidates. Taken together, we present here a rapid and efficient approach to generate human MNs from PSCs and provide a reliable assay system to investigate RNA mis-localization in MNs derived from multiple hPSC sources. We anticipate that skewed subcellular localization or RNAs will prove to be an important contributor to MN diseases. Indeed, studies performed in our laboratories using patient-specific MNs from fragile X patients and healthy controls show differential enrichment of RNAs containing a G-quadruplex sequence (Goering et al., 2019), thereby corroborating our notion that RNA localization exhibits a critical role in MN biology and disease development.
Experimental Procedures
Generation of Patient-Specific Induced Pluripotent Stem Cells
Isolated human peripheral blood mononuclear cells were expanded for 6 days in StemSpan SFEM II medium with Erythroid Expansion Supplement (Stem Cell Technologies). Deidentified PBMCs constitute not human subject research as per Colorado Multiple Institutional Review Board. Erythroid progenitor cells were electroporated with plasmids containing Okita factors (Okita et al., 2013) using a P3 Primary Cell 4D Nucleofector X Kit L (Lonza) according to the manufacturer’s instructions. Electroporated cells were plated on human embryonic-qualified Matrigel (Corning)-coated six-well plates and cultured in ReproTeSR medium (Stem Cell Technology). The medium was changed every other day for 14 days. Cultures were then transitioned to mTeSR1 (Stem Cell Technology) medium with the medium changed daily. Between days 14 and 18, clonal colonies with pluripotent morphology were hand picked, expanded, and phenotypically assessed. G-Band karyotyping was performed by WiCell.
hPSC Culture
Human ESCs were grown as previously described (Russ et al., 2015) or in Nutristem (Corning) or mTESR1 or mTESR plus (Stem Cell Technology) medium on hESC-qualified Matrigel-coated cell culture plates. iPSCs were grown in mTESR1 or mTESR plus (Stem Cell Technology) medium on hESC-qualified Matrigel-coated cell culture plates. Clusters were formed using Aggrewells800. To do so, 70%–100% confluent PSC cultures were washed once with PBS (GIBCO) followed by incubation for 6 min at 37°C in TrypLE (GIBCO) to generate a single-cell suspension. Digested cultures were quenched with growth medium, live cells were counted using an MOXI Z cell counter (Orflow Technologies), and 3,000 cells per microwell were added to a 24-well Aggrewell800 plate (Stem Cell Technology), spun down for 3 min at 1,200 rpm and incubated overnight in the presence of 10 μM Rock Inhibitor Y-27632 (RI, Tocris) to allow cluster formation before proceeding with direct differentiation. hPSC cultures were regularly tested for mycoplasma contamination and found negative.
Motor Neuron Differentiation
Base Media
KO DMEM (Gibco), 1× N2-A Supplement (Stem Cell Technologies), 1× SM1 (Stem Cell Technologies), 1× Glutamax (Gibco), and 50 mM ascorbic acid (Sigma).
Differentiation Protocol
Clusters were washed once with base medium and subsequently placed in progenitor medium containing Compound C, SB431542, CHIR99021, and RI and placed on a shaker for 6 days. Afterward, clusters were placed in induction medium containing Compound C, SB431542, RI, SAG, and TTNPB for 3 days. At d10, the clusters were dissociated using Accumax for 12 min at 37°C. Live cells were counted, and 53,000 or 106,000 cells per cm2 of fresh or frozen MNPs, respectively, were plated on Matrigel-coated dishes in maturation medium containing glucose, non-essential amino acids (NEAA), RI, SAG, TTNPB, BDNF, and GDNF for 4 days. At day 14, cells were switched into base medium containing gamma-secretase inhibitor, BDNF/GDNF, and RI for 3 days, after which point cells were cultured in base medium with BDNF/GDNF alone until d18–21 when they were analyzed. See Table S4 for company and concentration details for reagents.
Immunohistochemistry
Cells were cultured on coverslips starting on d10 and harvested at various time points. Cells were fixed in 4% paraformaldehyde for 30 min at room temperature, washed three times for 5 min in PBS, and blocked in CAS block (Invitrogen) for 1 h at room temperature. Cells were incubated with primary antibodies: SMI-32 mouse 1:500 (Calbiochem, catalog no. NE1023), HB9 mouse 1:50 (DSHB 81.5C10-c), ISL-1 polyclonal 1:100 (Millipore, catalog no. AB4326), βIII-Tubulin Rabbit 1:100 (Cell Signaling, catalog no. 5568) in CAS block (Invitrogen) overnight at 4°C. Cells were then washed three times for 5 min with PBS and incubated with Alexa secondary antibodies (1:500, Invitrogen) for 1 h at room temperature, washed three times with PBS, and incubated with DAPI for 5 min at room temperature. The coverslips were mounted on slides using Vectashield Mounting medium (Vectorlabs, catalog no. H-1000).
To visualize mitochondria, cells were placed on Mattek dishes and incubated in BMHH buffer plus MitoTracker Green FM (Thermo Fisher, catalog no. M7514) for 30 min. MNs were imaged using a LSM800 Zeiss confocal microscope with a 40× or 63× objective.
Quantification was performed using Fiji software by analyzing three to five images from two independent experiments. Soma and neurite intensity/area was calculated followed by neurite/soma ratios shown in the graph. p values were calculated using the standard Student t test.
Ca2+ Imaging
Cells were incubated in BMHH buffer plus Fluo-4 dye (Thermo Fisher, catalog no. F14201) for 30–45 min at 37°C and imaged on a Zeiss LSM 800 confocal microscope. Cells were imaged for 1 min at baseline after which they were subjected to depolarization with 30 mM KCl. Calcium dynamics were analyzed using Fiji (ImageJ).
Patch Clamping
MNPs were plated on glass coverslips that were coated with 2% Matrigel and matured for 10 days as described above. Glass coverslips were then placed in a recording chamber and perfused with recording solution which contained 120 mM NaCl, 2.5 mM KCl, 26 mM NaHCO3, 1 mM NaH2PO4, 10 mM glucose, 1 mM MgCl2, 2 mM CaCl2, adjusted to 285−290 mosmol/kg H2O. All solutions were bubbled in 95% O2, 5% CO2. Recording pipettes ranged from 4 to 7 MΩ. Internal solutions contained 123 mM K-gluconate, 2 mM KCl, 2 EGTA, 2 mM Na-ATP, 0.5 mM Na-GTP. Recordings were acquired using Axograph X and a MultiClamp 700B amplifier (Molecular Devices). Data were low-pass filtered at 2 kHz and acquired at 10 kHz.
Quantitative PCR
RNA was isolated using the QIAGEN RNA MINI extraction kit. cDNA was synthesized using the iScript (Bio-Rad) kit. Four nanograms of cDNA was used per reaction, and reactions were performed in duplicate. Data were normalized to control housekeeping genes ACTIN, TBP, or GAPDH using the delta Ct method. Primer and probe information can be found in Table S4.
RNA Sequencing
Cell Fractionation
To fractionate neuronal cells, medium was removed and both sides of the membrane were rinsed with PBS. One milliliter of PBS was placed on top of the membrane. Cell bodies were scraped in the PBS from the top of the membrane using a cell scraper. The membrane, still containing neurites, was then cut out of its plastic housing and incubated with RNA lysis buffer (Zymo Research) at room temperature for 15 min. Between 6 and 12 membranes were combined for a single preparation, depending on the cell density on the membranes. RNA was purified from both the soma and neurite fractions using a Quick RNA Microprep kit (Zymo Research). Typically, between 200 and 500 ng of total RNA was collected from neurite fractions in a single preparation.
RNA-Sequencing Library Construction
Stranded, polyA-selected RNA-sequencing libraries were prepared from 100 ng of total RNA using the Kapa mRNA Hyperprep kit. The final PCR amplification of the libraries consisted of 15 cycles. The libraries were sequenced to a depth of 20–35 million 150-bp paired end read pairs per sample on a NovaSeq sequencer (Illumina).
Analysis of High-Throughput Sequencing Data
Adapter sequences (AGA TCG GAA GAG CAC ACG TCT GAA CTC CAG TCA and AGA TCG GAA GAG CGT CGT GTA GGG AAA GAG TGT) were removed using cutadapt (Martin, 2011). Reads were then processed to remove the final 75 nt of each read, essentially converting the sequencing data from 2 × 150 to 2 × 75. Transcript expression was quantified using Salmon v0.8 (Patro et al., 2017) against the human transcriptome (hg38, Gencode 28). Gene-level expression data were calculated from the transcript expression data using txImport (Soneson et al., 2015). The extent of neurite localization of individual genes was calculated by taking the ratio of expression (TPM) of the gene in neurites to its expression in soma. Significantly localized genes were identified using DESeq2 (Love et al., 2014). When comparing soma and neurite samples, significantly localized genes had adjusted p values of less than 0.05 and log2(fold change) values of at least 1.5.
To identify genes whose localization changed across conditions, the R package Xtail was used (Xiao et al., 2016). This software package is designed for the analysis of ribosome profiling across conditions. Ribosome profiling experiments monitor the ratio of expression data in ribosome footprint and RNA-sequencing datasets (otherwise known as translational efficiency or ribosome occupancy). Xtail asks if this ratio changes for a gene across conditions. We adapted this package to ask if LRs (neurite/soma) changed across conditions. Gene ontology enrichments were calculated using GOrilla (Eden et al., 2009), and differential expression data are available in Tables S1, S2, and S3.
Author Contributions
L.I.H., L.S., J.M.T., and H.A.R. conceived the experimental study. L.H., A.B., T.T., C.S.N., J.M.T., and H.A.R. performed experiments and analyzed data. L.I.H., A.B., L.S., M.N., J.M.T., and H.A.R. interpreted data. L.I.H., J.M.T., and H.A.R. wrote the manuscript with help from L.S. and M.N.. All authors read and agreed on the manuscript.
Acknowledgments
This work was supported by a Novel Methods Development grant from the Colorado Clinical and Translational Science institute (CCTSI) to M.N. and H.A.R. CCTSI is supported by NIH/NCATS Colorado CTSA grant number UL1 TR002535. L.I.H. is supported by F32 DK118803-01A1 and a Guild ACORN Postdoctoral Fellowship. T.T. is supported by NIDDK K12DK094712. Work in the laboratory of H.A.R. is supported by the Children’s Diabetes Foundation, NIH/NIDDK grants DK120444 and AI140044, a new investigator award from the NIH/HIRN consortium, the Culshaw Junior Investigator Award in Diabetes and the JDRF. H.A.R. is a consultant to Sigilon Therapeutics and SAB member at Prellis Biologics. L.I.H., L.S., and H.A.R. are inventors on a patent application filed based on the results presented in this work. Contents are the authors' sole responsibilities and do not necessarily represent the official NIH views.
Published: May 7, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.stemcr.2020.04.003.
Accession Numbers
The sequencing data have been deposited under accession number GEO: GSE134737.
Supplemental Information
References
- Akiyama T., Suzuki N., Ishikawa M., Fujimori K., Sone T., Kawada J. Aberrant axon branching via Fos-B dysregulation in FUS-ALS motor neurons. EBioMedicine. 2019;45:362–378. doi: 10.1016/j.ebiom.2019.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cajigas I.J., Tushev G., Will T.J., tom Dieck S., Fuerst N., Schuman E.M. The local transcriptome in the synaptic neuropil revealed by deep sequencing and high-resolution imaging. Neuron. 2012;74:453–466. doi: 10.1016/j.neuron.2012.02.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers S.M., Fasano C.A., Papapetrou E.P., Tomishima M., Sadelain M., Studer L. Highly efficient neural conversion of human ES and iPS cells by dual inhibition of SMAD signaling. Nat. Biotechnol. 2009;27:275–280. doi: 10.1038/nbt.1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eden E., Navon R., Steinfeld I., Lipson D., Yakhini Z. GOrilla: a tool for discovery and visualization of enriched GO terms in ranked gene lists. BMC Bioinformatics. 2009;10:48–57. doi: 10.1186/1471-2105-10-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabre P.J., Leleu M., Mascrez B., Giudice, Lo Q., Cobb J., Duboule D. Heterogeneous combinatorial expression of Hoxd genes in single cells during limb development. BMC Biol. 2018;16:101–115. doi: 10.1186/s12915-018-0570-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fallini C., Bassell G.J., Rossoll W. The ALS disease protein TDP-43 is actively transported in motor neuron axons and regulates axon outgrowth. Hum. Mol. Genet. 2012;21:3703–3718. doi: 10.1093/hmg/dds205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernyhough P. Mitochondrial dysfunction in diabetic neuropathy: a series of unfortunate metabolic events. Curr. Diab. Rep. 2015;15:89. doi: 10.1007/s11892-015-0671-9. [DOI] [PubMed] [Google Scholar]
- Fernyhough P., Huang T.-J., Verkhratsky A. Mechanism of mitochondrial dysfunction in diabetic sensory neuropathy. J. Peripher. Nervous Syst. 2003;8:227–235. doi: 10.1111/j.1085-9489.2003.03028.x. [DOI] [PubMed] [Google Scholar]
- Goering R., Hudish L.I., Guzman B.B., Raj N., Bassell G.J., Russ H.A., Dominguez D., Taliaferro M.J. FMRP promotes RNA localization to neuronal projections through interactions between its RGG domain and G-quadruplex RNA sequences. bioRxiv. 2019:784728. doi: 10.1101/784728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grienberger C., Konnerth A. Imaging calcium in neurons. Neuron. 2012;73:862–885. doi: 10.1016/j.neuron.2012.02.011. [DOI] [PubMed] [Google Scholar]
- Gumy L.F., Yeo G.S.H., Tung Y.C.L., Zivraj K.H., Willis D., Coppola G. Transcriptome analysis of embryonic and adult sensory axons reveals changes in mRNA repertoire localization. RNA. 2011;17:85–98. doi: 10.1261/rna.2386111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta P.P., Agarwal D. Chronic obstructive pulmonary disease and peripheral neuropathy. Lung India. 2006;23:25–33. [Google Scholar]
- Hendriksen P.H., Oey P.L., Wieneke G.H., van Huffelen A.C., Gispen W.H. Hypoxic neuropathy versus diabetic neuropathy. An electrophysiological study in rats. J. Neurol. Sci. 1992;110:99–106. doi: 10.1016/0022-510x(92)90015-d. [DOI] [PubMed] [Google Scholar]
- Hester M.E., Murtha M.J., Song S., Rao M., Miranda C.J., Meyer K. Rapid and efficient generation of functional motor neurons from human pluripotent stem cells using gene delivered transcription factor codes. Mol. Ther. 2011;19:1905–1912. doi: 10.1038/mt.2011.135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imayoshi I., Kageyama R. bHLH factors in self-renewal, multipotency, and fate choice of neural progenitor cells. Neuron. 2014;82:9–23. doi: 10.1016/j.neuron.2014.03.018. [DOI] [PubMed] [Google Scholar]
- Li W., Sun W., Zhang Y., Wei W., Ambasudhan R., Xia P., Talantova M., Lin T., Kim J., Wang X. Rapid induction and long-term self-renewal of primitive neural precursors from human embryonic stem cells by small molecule inhibitors. Proc. Natl. Acad. Sci. U S A. 2011;108:8299–8304. doi: 10.1073/pnas.1014041108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M.I., Huber W., Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15:550–621. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malik R.A., Masson E.A., Sharma A.K., Lye R.H., Ah-See A.K., Compton A.M. Hypoxic neuropathy: relevance to human diabetic neuropathy. Diabetologia. 1990;33:311–318. doi: 10.1007/BF00403326. [DOI] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.Journal. 2011;17:10–12. [Google Scholar]
- Mayer P., Dematteis M., Pepin J.L., Wuyam B., Veale D., Vila A., Lévy P. Peripheral neuropathy in sleep apnea: a tissue marker of the severity of nocturnal desaturation. Am. J. Respir. Crit. Care Med. 1999;159:213–219. doi: 10.1164/ajrccm.159.1.9709051. [DOI] [PubMed] [Google Scholar]
- Mucenski M.L., Mahoney R., Adam M., Potter A.S., Potter S.S. Single cell RNA-seq study of wild type and Hox9,10,11 mutant developing uterus. Sci. Rep. 2019;9:4557–4616. doi: 10.1038/s41598-019-40923-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okita K., Yamakawa T., Matsumura Y., Sato Y., Amano N., Watanabe A., Goshima N., Yamanaka S. An efficient nonviral method to generate integration-free human-induced pluripotent stem cells from cord blood and peripheral blood cells. Stem Cells. 2013;31:458–466. doi: 10.1002/stem.1293. [DOI] [PubMed] [Google Scholar]
- Patro R., Duggal G., Love M.I., Irizarry R.A., Kingsford C. Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods. 2017;14:417–419. doi: 10.1038/nmeth.4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinti M.V., Fink G.K., Hathaway Q.A., Durr A.J., Kunovac A., Hollander J.M. Mitochondrial dysfunction in type 2 diabetes mellitus: an organ-based analysis. Am. J. Physiol. 2019;316:E268–E285. doi: 10.1152/ajpendo.00314.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinhardt P., Glatza M., Hemmer K., Tsytsyura Y., Thiel C.S., Höing S. Derivation and expansion using only small molecules of human neural progenitors for neurodegenerative disease modeling. PLoS One. 2013;8 doi: 10.1371/journal.pone.0059252. e59252–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russ H.A., Parent A.V., Ringler J.J., Hennings T.G., Nair G.G., Shveygert M. Controlled induction of human pancreatic progenitors produces functional beta-like cells in vitro. EMBO J. 2015;34:1759–1772. doi: 10.15252/embj.201591058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahoo P.K., Smith D.S., Perrone-Bizzozero N., Twiss J.L. Axonal mRNA transport and translation at a glance. J. Cell Sci. 2018;131 doi: 10.1242/jcs.196808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneson C., Love M.I., Robinson M.D. Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Research. 2015;4:1521. doi: 10.12688/f1000research.7563.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taliaferro J.M., Vidaki M., Oliveira R., Olson S., Zhan L., Saxena T. Distal alternative last exons localize mRNAs to neural projections. Mol. Cell. 2016;61:821–833. doi: 10.1016/j.molcel.2016.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang E.T., Taliaferro J.M., Lee J.-A., Sudhakaran I.P., Rossoll W., Gross C. Dysregulation of mRNA localization and translation in genetic disease. J. Neurosci. 2016;36:11418–11426. doi: 10.1523/JNEUROSCI.2352-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z., Zou Q., Liu Y., Yang X. Genome-wide assessment of differential translations with ribosome profiling data. Nat. Commun. 2016;7:11194. doi: 10.1038/ncomms11194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zappulo A., van den Bruck D., Ciolli Mattioli C., Franke V., Imami K., McShane E., Moreno-Estelles M., Calviello L., Filipchyk A., Peguero-Sanchez E. RNA localization is a key determinant of neurite-enriched proteome. Nat. Commun. 2017;8:583. doi: 10.1038/s41467-017-00690-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziller M.J., Ortega J.A., Quinlan K.A., Santos D.P., Gu H., Martin E.J., Galonska C., Pop R., Maidl S., Di Pardo A. Dissecting the functional consequences of de novo DNA methylation dynamics in human motor neuron differentiation and physiology. Cell Stem Cell. 2018;22:559–574.e9. doi: 10.1016/j.stem.2018.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zivraj K.H., Tung Y.C.L., Piper M., Gumy L., Fawcett J.W., Yeo G.S.H., Holt C.E. Subcellular profiling reveals distinct and developmentally regulated repertoire of growth cone mRNAs. J. Neurosci. 2010;30:15464–15478. doi: 10.1523/JNEUROSCI.1800-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.