

Abstract
Motivation
The computational prediction of gene function is a key step in making full use of newly sequenced genomes. Function is generally predicted by transferring annotations from homologous genes or proteins for which experimental evidence exists. The ‘ortholog conjecture’ proposes that orthologous genes should be preferred when making such predictions, as they evolve functions more slowly than paralogous genes. Previous research has provided little support for the ortholog conjecture, though the incomplete nature of the data cast doubt on the conclusions.
Results
We use experimental annotations from over 40 000 proteins, drawn from over 80 000 publications, to revisit the ortholog conjecture in two pairs of species: (i) Homo sapiens and Mus musculus and (ii) Saccharomyces cerevisiae and Schizosaccharomyces pombe. By making a distinction between questions about the evolution of function versus questions about the prediction of function, we find strong evidence against the ortholog conjecture in the context of function prediction, though questions about the evolution of function remain difficult to address. In both pairs of species, we quantify the amount of information that would be ignored if paralogs are discarded, as well as the resulting loss in prediction accuracy. Taken as a whole, our results support the view that the types of homologs used for function transfer are largely irrelevant to the task of function prediction. Maximizing the amount of data used for this task, regardless of whether it comes from orthologs or paralogs, is most likely to lead to higher prediction accuracy.
Availability and implementation
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Whole-genome sequencing of new species continues to outpace the experiments needed to annotate the function of every gene in these genomes. As a result, computational prediction of gene function is an essential tool for researchers in a range of biomedical fields. The prediction of gene function generally proceeds by the transfer of function from genes with experimental evidence to unannotated, or less-annotated, genes that are similar by some measure (Radivojac et al., 2013). While several methods use multiple data types to carry out predictions (Cozzetto et al., 2016; Lan et al., 2013; Sokolov et al., 2013), many solely rely on evolutionary relationships (Clark and Radivojac, 2011; Cozzetto and Jones, 2017; Engelhardt et al., 2005; Hawkins et al., 2006) and are the focus of this study.
One of the most important distinctions in evolutionary relationships among genes is between orthologs and paralogs (Fitch, 1970). Orthologous genes originate via a speciation event, whereas paralogous genes arise through a duplication event. By definition, orthologs are always found in different species, though paralogs can be found in either the same or different species (e.g. when duplication precedes a speciation event; Fig. 1). Identifying orthologous genes across species is important for many tasks, including the inference of species relationships (but see Du et al., 2019; Legried et al., 2019; Zhang et al., 2019). In the context of function prediction, orthologs have traditionally taken a privileged role based on the belief that they are more functionally similar to one another than are paralogs (Dolinski and Botstein, 2007; Fang et al., 2010; Gabaldón and Koonin, 2013; Tatusov et al., 1997). Indeed, prediction of function often proceeds by first identifying orthologs, and sometimes only single-copy orthologs, discarding all genes with other relationship; e.g. as in Wang et al. (2018).
Fig. 1.
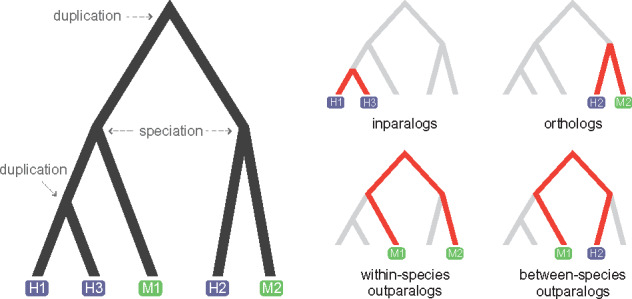
Four different types of homology relations. A family of five genes sampled from human (in blue) and mouse (in green) evolves through speciation and duplication events (left-hand tree). The relationships among genes are highlighted in the four trees on the right. Human genes H1 and H3 are inparalogs, arising from a duplication event after the most recent speciation event in the tree. Genes H2 and M2 are orthologs, as they are related through a speciation event. Gene pairs M1–M2 and M1–H2 are outparalogs because they arose from a duplication that predates the reference speciation event. Pair M1–M2 is within-species outparalogs, while M1–H2 is between-species outparalogs
The idea that orthologous genes share greater functional similarity than do paralogous genes has been termed the ‘ortholog conjecture’ (Nehrt et al., 2011). Historically, this conjecture has rarely been questioned in either evolutionary biology (but see Gibson and Goldberg, 2009; Koonin, 2005; Studer and Robinson-Rechavi, 2009) or function prediction studies (but see Engelhardt et al., 2005; Mika and Rost, 2006; Nadimpalli et al., 2015). In a previous study, we tested the ortholog conjecture using experimental evidence gleaned from the Gene Ontology (GO) database and gene expression data from 25 different tissues in human and mouse (Nehrt et al., 2011). We found no evidence that orthologs were more functionally similar than paralogs of equivalent levels of protein divergence, and in fact showed that the highest functional similarity was shared by within-species homologs. A simple model was proposed to explain these data: functional differences evolve over time, so that pairs of genes that have been diverged for a smaller amount of time are more functionally similar, and that genes found in the same species share a cellular context, making their functions more similar (Nehrt et al., 2011). Because within-species paralogs can have a common ancestor much more recently than any particular speciation event (which will delimit the age of orthologs)—and are obviously found within the same species—such genes will share higher levels of functional similarity.
Results questioning the ortholog conjecture have been received with a range of reactions. Some researchers were ‘baffled’ that such an obvious assumption would be tested (Sonnhammer et al., 2014), whereas others pointed out important limitations in using GO similarity for answering questions about the evolution of function (Altenhoff et al., 2012; Chen and Zhang, 2012; Thomas et al., 2012). In the rush to cement the primacy of orthologs, several analyses were reported in which orthologs appeared significantly more similar than paralogs (Adipietro et al., 2012; Altenhoff et al., 2012; Chen and Zhang, 2012; Kryuchkova-Mostacci and Robinson-Rechavi, 2016; Rogozin et al., 2014). While orthologs may be more similar than paralogs for some traits (Soria et al., 2014) and for some types of genes (Adipietro et al., 2012), the higher similarity of orthologs in some newer datasets was accompanied by either no change in functional similarity over time (Altenhoff et al., 2012; Chen and Zhang, 2012) or the increase in functional similarity over time (Rogozin et al., 2014). As a decrease in structural (Peterson et al., 2009; Mohan et al., 2009) and functional (Cao and Cowen, 2017; Coolon et al., 2014; Gu et al., 2002; Laurent et al., 2019; Liao and Chang, 2014; Makova and Li, 2003) similarity with divergence is a widely expected and observed pattern for both paralogs and orthologs, the patterns of evolution in these studies are indeed baffling. Further examination of several of these studies has uncovered problems with the analyses such that there is either no longer support for the ortholog conjecture (Dunn et al., 2018) or that there was no statistical support for the ortholog conjecture in the first place, as in the case of human–mouse comparisons by Altenhoff et al. (2012).
Further testing of the ortholog conjecture using experimental data must deal with several issues, mostly concerned with the nonrandom nature of experiments done by individual researchers and in individual species (Altenhoff et al., 2012; Nehrt et al., 2011; Thomas et al., 2012). An important distinction that can help to overcome these issues can be drawn between two different interpretations of the ortholog conjecture, one evolutionary and one predictive. Evolutionarily, questions about the tempo and mode of functional evolution require that the same traits and experimental methods be used in all species considered. If this is not done, then differences in the types of traits studied can bias results in favor of within-species paralogs (Nehrt et al., 2011; Thomas et al., 2012). For instance, if tails are only studied in mice, then obviously more genes in mice will be predictive of tail-related functions. But a second interpretation of the ortholog conjecture is only concerned with the prediction of function, regardless of the evolutionary history of these functions. For this interpretation of the ortholog conjecture, there is no bias associated with the nonrandom collection of experimental data, as long as we assume that all types of annotations are equally accurate on average. Following from the example given above, if more genes in mice are useful for predicting tail-specific functions, then these genes should be preferred when predicting function.
Regardless of the validity of the ortholog conjecture, its importance to the task of function prediction remains unclear. That is, even if orthologs are slightly better for predicting functions than paralogs, this should not imply that paralogs should be ignored, and vice versa. In fact, including paralogs in prediction results in higher accuracy than orthologs alone (e.g. Škunca et al., 2012) and methods that include both orthologs and paralogs (Engelhardt et al., 2005, 2011) are some of the most successful in the Critical Assessment of Functional Annotation (CAFA) challenge (Jiang et al., 2016; Radivojac et al., 2013; Zhou et al., 2019). Therefore, quantifying the increase in the number and accuracy of functional predictions made possible by including paralogs represents an equally valuable task in the context of the ortholog conjecture. Given a large enough benefit of inclusion, it may be that there is no need to distinguish between orthologs and at least some types of paralogs in the first place.
In this article, we revisit the ortholog conjecture and related questions using experimentally verified functional annotations from almost 43 000 genes. We attempt to control for some of the factors that can bias evolutionary tests of the ortholog conjecture, finding that within-species paralogs are overwhelmingly favored in comparisons between two mammalian species, Homo sapiens and Mus musculus, and between two yeast species, Saccharomyces cerevisiae and Schizosaccharomyces pombe. We also quantify the enormous gain in number and accuracy of predictions that is manifested when all types of homologs, and not just orthologs, are included. Our results reaffirm the lack of support for the ortholog conjecture, and further suggest that its accuracy is irrelevant to the task of predicting function.
2 Materials and methods
2.1 Sequence data and evolutionary relationships
We collected protein-coding genes from H.sapiens (Hs), M.musculus (Mm), Sa.cerevisiae (Sc) and Sc.pombe (Sp). Ensembl Biomart (release 91, December 2017) and Ensembl Fungimart (release 38, January 2018) gene trees were used to specify different homologous relationships for human–mouse and cerevisiae–pombe comparisons, respectively (Zerbino et al., 2018). A total of 8606 gene trees contained human and mouse genes, whereas 3059 gene trees contained cerevisiae and pombe genes.
Homologous relationships between proteins were divided into four main categories, two for homologs found in different species (orthologs and between-species outparalogs) and two for homologs found in the same species (inparalogs and within-species outparalogs). Orthologs were further classified as one-to-one, one-to-many and many-to-many. We used duplication events inferred from gene trees to distinguish between inparalogs and within-species outparalogs: if the duplication event occurred more recently than the speciation event, the protein pairs were identified as inparalogs; otherwise, they were identified as within-species outparalogs (Fig. 1). All genes included in the final dataset had at least one type of homologous relationship with another gene. This dataset was composed of 19 514 human genes, 21 398 mouse genes, 4205 cerevisiae genes and 3487 pombe genes.
2.2 Function data
We used Biological Process, Molecular Function and Cellular Component ontologies released by the Gene Ontology (GO) consortium in March 2018 (Ashburner et al., 2000; Consortium, 2016). Functional annotations were obtained from the UniProt-GOA database (release 176, March 2018) (Huntley et al., 2015). Only annotations supported with evidence codes EXP, IDA, IMP, IPI, IGI, IEP, TAS or IC were considered. The comparative genomics data obtained from Ensembl uses Ensembl gene IDs, whereas protein functional annotation data obtained from the UniProt-GOA database uses UniProt accession numbers. In some cases, especially for human and mouse, when there were one-to-many mappings from Ensembl gene to UniProt accession numbers, we only kept annotations for the protein with the longest sequence. This gave 22 280 human proteins, 12 859 mouse proteins, 5135 cerevisiae proteins and 2669 pombe proteins annotated with at least one functional term from any ontology. A total of 81 332 unique publications were used to assign experimental annotations to these proteins.
Our analysis requires pairs of homologs that both have functional annotations to quantify their similarity. Therefore, our final dataset consisted of 8637 ortholog pairs, 1917 inparalog pairs, 33 741 between-species outparalog pairs and 40 021 within-species outparalog pairs for human–mouse comparisons. For cerevisiae and pombe, there were 1724 ortholog pairs, 2072 inparalog pairs, 193 between-species outparalog pairs and 892 within-species outparalog pairs.
We also identified proteins that had been annotated in the same publication or by the same researchers. To do so, we retrieved PubMed identifiers for each protein-term assignment and, ultimately, associated a list of authors with each protein. This list was then used to narrow the analysis only to pairs of proteins that were studied by nonoverlapping groups of investigators.
2.3 Similarity calculations
2.3.1 Sequence identity
Pairs of protein sequences were aligned using the Needleman–Wunsch algorithm (Needleman and Wunsch, 1970), with the BLOSUM62 matrix (Henikoff and Henikoff, 1992), a gap opening penalty of 11 and a gap extension penalty of 1. Sequence identity was obtained by dividing the number of matches in the alignment by the length of the longer protein sequence.
2.3.2 Functional similarity
To calculate functional similarity between pairs of annotated proteins, we used two groups of similarity measures. The first group uses topological measures of the structure of the GO graph to measure similarities of its subgraphs, whereas the second group uses information-theoretic (probabilistic) measures that further incorporate the database of all characterized proteins with their respective annotations. Each measure considered in this work returns values between , with 1 indicating identical annotations. As our main functional similarity measure is Yang–Clark similarity, we introduce it below. Two alternative measures, i.e. the Maryland bridge similarity (Glazko et al., 2005) and Schlicker’s similarity (Schlicker et al., 2006), are defined in the Supplementary Materials.
Yang–Clark semantic similarity. The Yang–Clark distance metric is based on the previously introduced concept of information content of a subgraph within an ontology (Clark and Radivojac, 2013). We used the normalized version of this semantic distance, as proposed by Yang et al. (2019), to calculate functional similarities between protein pairs. This model first assumes that protein annotations are generated by a probabilistic model; i.e. a Bayesian network that has the same structure as GO, with each node in the ontology treated as a binary random variable. The marginal probability for a consistent subgraph T associated with a protein is then defined as
| (1) |
where v is a node in the annotation graph and is the set of parent nodes of v. defines the probability that a node v belongs to the functional annotation of a protein in the database given that all of its parents are present in the annotation. The information content of a subgraph T annotating a protein is then defined as
| (2) |
where stands for information accretion of a node (Clark and Radivojac, 2013). We estimated ia(v) using the maximum likelihood approach over the entire UniProt-GOA database as the negative binary logarithm of the relative frequency that the term v is present in a protein’s annotation given that all its parent terms are also present. We also considered species-specific term frequencies to understand the impact of averaging over the entire UniProt-GOA.
The semantic distance between annotations of two proteins can now be calculated as follows. Suppose that A and B are two sets of nodes presenting propagated annotations for two proteins a and b, respectively, and that A is used as a prediction of B. Misinformation (mi) is defined as the total information content of the nodes that are present in annotation A but not in B, whereas remaining uncertainty (ru) is defined as the total information content of the nodes that are present in annotation B but not in A (Clark and Radivojac, 2013). More formally,
| (3) |
We define the total normalized Yang–Clark distance of the order between the two annotations A and B as
| (4) |
The Yang–Clark semantic similarity is then defined as
| (5) |
Based on previous works (Clark and Radivojac, 2013; Jiang et al., 2014; Yang et al., 2019), we used p = 2 throughout this study.
2.3.3 Background similarity
Background functional similarity is defined as the expected functional similarity for a pair of randomly selected genes. We calculate such similarity for different groups of genes with the same labels, such as orthologs or inparalogs. To calculate background similarity, we randomly selected with replacement 1000 protein pairs from a pool of all proteins forming a certain group; e.g. the pool of all proteins forming human–mouse orthologous pairs. The average functional similarity over these pairs can then be subtracted from the functional similarity of the actual homologous pairs to form the so-called excess similarity (Altenhoff et al., 2012).
We calculated background similarities separately for each homology type. Orthologs were further split between one-to-one and other orthologs (one-to-many, many-to-many). Background similarities for nonhomologous genes both within and between species were also calculated.
2.4 Protein function prediction and its evaluation
To understand and quantify the influence of particular types of homologs in protein function prediction, we selected one of the most intuitive predictors used in this field. This predictor transfers protein annotations from a database of experimentally annotated ‘target’ proteins to an unannotated ‘query’ protein as follows: (i) the query protein is first aligned to all target proteins; (ii) each annotation term is transferred from the target database to the query with a score equaling the largest global sequence identity between the query and any of the target proteins containing that term. Eventually, each term in the query protein is associated with a score between 0 and 1. This model is equivalent to the BLAST baseline algorithm that has been used in the CAFA experiments (Jiang et al., 2016; Radivojac et al., 2013; Zhou et al., 2019), except that we used global sequence identity instead of local identity. We selected this algorithm because of the ability to easily track the influence of target proteins from which the annotations were transferred.
The performance of protein function prediction was evaluated using a leave-one-out strategy based on both topological and information-theoretic accuracy measures. As in pairwise functional similarity between proteins, the Yang–Clark semantic similarity was used as the main evaluation metric and is presented below. The topological metric, a criterion regularly seen in CAFA (Jiang et al., 2016; Radivojac et al., 2013; Zhou et al., 2019), was used as an alternative measure and is presented in Supplementary Materials.
To summarize this performance evaluation, we consider a prediction algorithm on a set of n proteins, where each protein i is assigned a score (say, between 0 and 1) for each functional term v in the ontology. The normalized remaining uncertainty (nru) and misinformation (nmi) are defined as
| (6) |
| (7) |
where contains predicted terms with a score greater than or equal to τ for the ith protein, Ti is the experimental annotation for the ith protein, and is an indicator function. The term ia(v), as before, is the information accretion corresponding to the ontology term v (Clark and Radivojac, 2013). The maximum semantic similarity, , is now defined as
| (8) |
for . takes values between 0 and 1. Higher values correspond to better predictions, with the value of 1 corresponding to a perfect prediction for each protein in the dataset. As before, we used p = 2.
It is important to mention that all functional similarity measures between proteins are susceptible to problems caused by incomplete (Dessimoz et al., 2013) and noisy (Schnoes et al., 2009) experimental annotations. There is a small effect of annotation incompleteness on topological measures and a somewhat larger effect on unnormalized semantic distance (Jiang et al., 2014). However, compared with topological measures, semantic similarity avoids a form of double-counting of nodes caused by the directed acyclic graph structure of GO, and thus properly treats hierarchical dependencies in the ontology.
3 Results
3.1 Higher functional similarity in within-species homologs
We analyzed patterns of functional similarity versus sequence identity for pairs of proteins separated by their type of homology: orthologs, inparalogs, within-species outparalogs and between-species outparalogs (Fig. 2). We observe that for both pairs of species and across all three functional ontologies, within-species homologs—especially inparalogs—generally exhibit higher average functional similarity than between-species homologs (orthologs and between-species outparalogs). Inparalogs by definition share a more recent common ancestor with each other than do orthologs, and it therefore may not be surprising that they are more functionally similar than pairs of orthologs at essentially all levels of divergence (Nehrt et al., 2011). Outparalogs are not constrained by such relationships, but our results show that within-species pairs are consistently more functionally similar than are between-species pairs. This result is also consistent with the previously proposed effect of cellular and organismal context on measured protein function (Nehrt et al., 2011). The patterns, shown for the Yang–Clark similarity measure in Figure 2, also hold when using Maryland bridge and Schlicker’s similarity measures (Supplementary Figs S2 and S3).
Fig. 2.

Left: The relationship between Yang–Clark functional similarity and sequence identity for human and mouse over three different ontologies in GO. The breakdown is presented for four types of homologous relationships between pairs of proteins (orthologs, between-species outparalogs, within-species outparalogs and inparalogs). The dashed line in each panel shows the estimated functional similarity for a randomly selected pair of proteins, obtained by averaging 1000 randomly selected proteins from the available pool. The data are presented for each sequence identity bin in which at least three pairs of proteins could be used to calculate functional similarity. Right: The relationship between Yang–Clark functional similarity and sequence identity for Sa.cerevisiae and Sc.pombe over three different ontologies in GO. The breakdown is presented for four types of homologous relationships between pairs of proteins (orthologs, between-species outparalogs, within-species outparalogs and inparalogs). The dashed line in each panel shows the estimated functional similarity for a randomly selected pair of proteins, obtained by averaging 1000 randomly selected proteins from the available pool. The data are presented for each sequence identity bin in which at least three pairs of proteins could be used to calculate functional similarity. The results in which term frequencies were separately computed over each pair of species are very similar, as presented in Supplementary Figure S1
3.2 Controlling for potential annotation bias
While the results presented in Figure 2 clearly show that within-species homologs have more annotated functional terms in common than do between-species homologs, it is not clear whether this is due to underlying biological differences. We studied two sources of bias that could inflate functional similarities of within-species homologs, in an attempt to control for them. The first factor examined was ‘authorship bias’, which proposes that pairs of proteins experimentally annotated by the same authors will be annotated more similarly (Altenhoff et al., 2012; Nehrt et al., 2011). This effect could be due either to a limited range of GO terms known by individual authors or to experiments preferentially testing similar functions. In either case, proteins studied by the same authors could have higher functional similarity compared to pairs annotated by different authors. The second potential factor is referred to as ‘background similarity’ between pairs of homologs (Altenhoff et al., 2012). Such similarity arises because certain functions are studied more or less in different organisms, leading to similarities even between nonhomologous proteins found in the same species.
To examine the effects of these factors on our results, we first calculated functional similarity between pairs of proteins annotated by different authors. We did this by removing any homologous pair for which experimental annotations were derived from either the same paper or different papers sharing one or more of their authors (Section 2). Across all ontologies, and for both species pairs, functional similarity for inparalogs remained higher than for orthologs for high sequence similarity, whereas the results were mixed in the lower sequence identity groups (Supplementary Fig. S4). The effect of filtering of functional annotations was more noticeable for within-species outparalogs: these become comparable in functional similarity to orthologs across the entire sequence identity range, with fluctuations likely influenced by smaller dataset sizes (Supplementary Fig. S4). Finally, between-species outparalogs remained the least predictive of functional annotations (Supplementary Fig. S4). These results were consistent regardless of the similarity measure used (Supplementary Figs S4–S6). To control for background similarity, we calculated this measure separately for the different types of homologous relationships (Supplementary Figs S7–S9). We then subtracted the background similarity from the total functional similarity between homologous pairs of the same type; these values were subtracted from the similarities calculated above using annotations only from different authors. Inparalog pairs remained functionally slightly more similar to each other than orthologs after accounting for both sources of biases (using all three measures and over all three ontologies; Supplementary Figs S10–S12). While there remain several unavoidable problems with experimental data collected from different species (Section 4), these results suggest that orthologs do not evolve functions more slowly than paralogs.
3.3 The impact of ignoring paralogs: number of genes
Regardless of whether paralogs are more or less functionally similar than orthologs of the same sequence identity, using only orthologs for function prediction means that we are discarding some amount of functional information. We therefore attempted to characterize the impact of discarding paralogs both in terms of the number of proteins that are ignored and the loss of predictive ability. As before, these results are shown for human versus mouse and cerevisiae versus pombe. Using Ensembl gene trees (Zerbino et al., 2018), we extracted 8606 gene families for human–mouse and 3059 for cerevisiae–pombe that contained at least two proteins within them of some homology type. We were interested in quantifying the number of families and the number of proteins in the two pairs of species where functional transfer is possible given: (i) only orthologs, (ii) only paralogs and (iii) both orthologs and paralogs.
Figure 3 shows a significant overlap between protein families where functional transfer is possible using both orthologs and any type of paralogs. In human and mouse, a total of 4744 families with 9488 proteins contain only orthologous assignments, whereas 260 families with 1396 proteins contain only paralogous assignments, mostly from the same species (i.e. either human–human or mouse–mouse). In contrast, 3602 gene families containing 30 000 proteins contain both types of homologs and thus could potentially benefit from both groups in function transfer. For Sa.cerevisiae versus Sc.pombe, we find that 1969 gene families (3968 proteins) only have orthologous assignments, 571 families (1590 proteins) only have paralogous assignments, whereas 519 families (1772 proteins) contain both types of homologous assignments. Additional breakdowns are provided in Supplementary Materials.
Fig. 3.
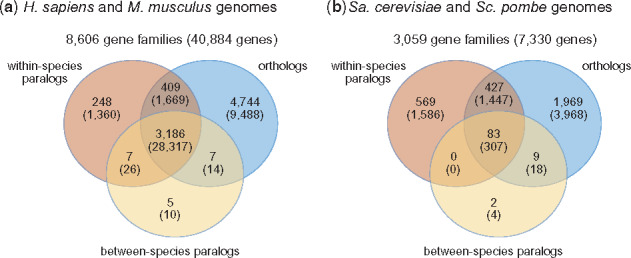
The numbers of gene families with different types of homologous relationships in them. The numbers in parentheses represent the counts of genes in the respective gene families. (a) Gene families containing H.sapiens and M.musculus proteins and (b) gene families containing Sa.cerevisiae and Sc.pombe proteins
These results point to the potentially large impact paralogs could have on function transfer. Although orthologs are the only source of function transfer in 55% of human and mouse families and 64% of yeast families, 73% of proteins in human and mouse and 24% of proteins in the yeast species are in families containing genes with both orthologous and paralogous relationships. In addition, 3% of families in human and mouse (3% of proteins) contain only paralogous relationships and 19% of families (22% of proteins) in the yeast species contain only paralogous proteins. Together, these numbers suggest that a large amount of functional information is potentially ignored if paralogs are not included in function transfer.
3.4 Impact of ignoring paralogs: prediction performance
We estimated the performance accuracy of protein function prediction when different groups of targets (i.e. different types of homologs) were used to transfer GO terms. We first estimated the prediction accuracy when orthologs and all paralogs were used for the prediction. We then removed orthologs and paralogs one at a time to gauge the impact on prediction performance. Figure 4a–c shows the prediction performance in each ontology for the proteins that have an experimentally characterized ortholog and at least one paralog, regardless of the type (gray circles). Removing orthologs (blue circles) resulted in significantly reduced performance across all ontologies and across both pairs of species (10.2% reduction on average in human–mouse and 13.4% reduction on average in cerevisiae–pombe). Similarly, removing paralogs from function transfer (yellow circles) resulted in reduced performance by an equivalent margin to that for the case of orthologs (10.0% reduction on average in human–mouse and 16.8% on average in cerevisiae–pombe). These results suggest that the two groups of homologs provide approximately equivalent contributions to accurate function transfer.
Fig. 4.
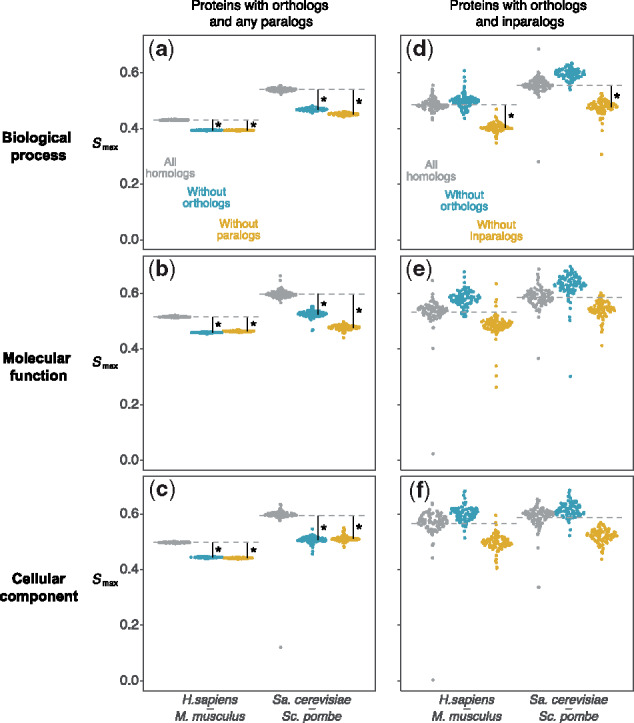
The impact of excluding orthologs or paralogs from prediction. values for proteins having orthologs and one other homolog (panels a-c) and proteins having orthologs and inparalogs (panels d-f), in human–mouse (Hs–Mm) and Sa.cerevisiae–Sc.pombe (Sc–Sp), are lower when ignoring either homolog. Points are 100 bootstrap samples of each protein set, and asterisks indicate a significant decrease in (bootstrap P < 0.05)
Figure 4d–f summarizes a similar experiment, but where we only considered proteins with both orthologs and inparalogs having experimentally determined functions. Once again, the removal of inparalogs resulted in a significant decrease in prediction performance across all ontologies and both pairs of species (12.6% reduction on average in human–mouse and 11.1% reduction on average in cerevisiae–pombe). Interestingly, however, the exclusion of orthologs from function transfer resulted in slight performance increases in all six experiments, although this result was not statistically significantly (6.2% increase on average in human–mouse and 6.5% increase on average in cerevisiae–pombe). These results suggest that the annotated inparalogs, when available, constitute the most reliable source of functional annotation and can be readily used in function transfer. The results show that accuracy actually decreased when orthologs were included alongside the inparalogs. Further experiments with the removal of other types of homologs and different performance measures are summarized in the Supplementary Materials (Supplementary Figs S13–S24).
4 Discussion
We have revisited the problem of the ‘ortholog conjecture,’ with a focus on assessing the value of orthologs and paralogs in the task of sequence-based protein function prediction. Exploiting significantly larger datasets of experimentally characterized proteins than have been used before, we repeated the analysis of Nehrt et al. (2011), finding that those original results still hold for two different pairs of species. We then moved beyond this analysis to quantify the value of different types of homologs on both the number of possible predictions that can be made and their accuracy. Several conclusions and implications of our results deserve additional discussion.
We believe that the ortholog conjecture as originally operationalized in our previous work conflated two different ideas about the value of orthologs: their role in automated function prediction versus their rate of evolution of function (Nehrt et al., 2011). Although these are obviously related issues, different types of experiments—and more importantly, different types of experimental biases—can affect the conclusions drawn about the roles of orthologs in each. Questions about the role of orthologs in functional annotation have been satisfactorily answered, with no problems due to experimental bias: the cumulative evidence suggests that paralogous genes are highly important for functional transfer, and in certain cases, even more useful than orthologs in transferring function from one gene and/or species to another. The analyses presented here continue to show that paralogs can offer both more and better predictions of protein function; that is, given a query protein and experimentally annotated orthologs and paralogs, the combination of orthologs and paralogs consistently improves functional annotation compared to prediction based on orthologs alone. These results hold across both a comparison of human and mouse and a comparison of two yeast species. Nehrt et al. (2011) and Altenhoff et al. (2012) have previously shown the same patterns in human and mouse, though Altenhoff et al. (2012) did not find the same pattern in their analysis of the two yeasts. We do not know exactly why the two results are different, but our work is based on six more years of accumulated data.
In contrast, questions regarding the evolution of function in different types of homologs are still unsettled. The results presented here are consistent with a model in which protein functions evolve over time, with no difference between orthologs and paralogs except with respect to whether two genes are found within the same species (Nehrt et al., 2011). In previous work, there was also no evidence for a greater conservation of function in orthologs between human and mouse when using GO terms, even after correcting for experimental evidence coming from the same paper (Nehrt et al., 2011) or from the same authors (Altenhoff et al., 2012). However, issues remain in the experimental data reported in the GO database that are hard to overcome in downstream analyses of protein function evolution. One such issue is that current measures of functional similarity treat a lack of overlap in GO terms as evidence for a difference in function, without properly accounting for the possibility that the relevant experiments have simply not been carried out (Thomas et al., 2012). Such problems can only be addressed as more experiments are added to the GO database, especially experiments reporting negative results. Indeed, by tracking the accumulation of data in GO, Chen and Zhang (2012) predicted that the functional similarity of ortholog pairs would be higher than for outparalog pairs by the year 2018 for all three ontologies. We used experimental data from more than six times as many publications in this study than in our previous work (Nehrt et al., 2011) and did not find evidence for this (Fig. 2). Moreover, Chen and Zhang (2012) predicted that functional similarity between orthologs would exceed those of inparalogs for the biological process and cellular context ontologies in 2013 and 2015, respectively; these predictions are also not supported by our results. In addition, a previous analysis of gene expression divergence between orthologs and paralogs, which avoids any problems of incomplete annotation, also found higher similarity between paralogs (Nehrt et al., 2011). Nevertheless, further work is clearly necessary to directly assess the rate of evolution of protein function among different classes of homologs.
The results presented here suggest an important modification to approaches for assigning protein function. Regardless of the validity of the ortholog conjecture for this task—that is, no matter whether orthologs are better or worse than paralogs at predicting function—adherence to the ortholog conjecture is often accompanied by the idea that only orthologs should be used to predict function (Sonnhammer et al., 2014). This is clearly not a necessary consequence of the ortholog conjecture: including paralogs could still aid in function prediction even if orthologs had more conserved functions. As we have shown, ignoring paralogs involves throwing away a huge amount of data, and often means that no prediction can be made because there are no available orthologs (Fig. 3). Stricter schemes can also involve only using one-to-one orthologs (Supplementary Fig. S25), so as to avoid the inclusion of any duplication events in the history of the orthologs. This strategy means that even fewer genes can be used in function prediction.
As sequence and function data further accumulate, future analyses, particularly those including multiple species at a time, could reveal more refined relationships between types of homology and function. Until such a time, however, we propose that homology types should be ignored in methods for transferring protein function, with a caveat that the functions from between-species outparalogs are slightly less transferable from one species to another. Our results suggest both that we are ignoring large amounts of data and that the accuracy of prediction is lower if we do not use paralogs. Even studies written in favor of the ortholog conjecture provide only slim-at-best victory margins (Adipietro et al., 2012; Altenhoff et al., 2012; Chen and Zhang, 2012; Kryuchkova-Mostacci and Robinson-Rechavi, 2016; Rogozin et al., 2014), while still presenting data that support the value of paralogs. Furthermore, evolutionary relationships among genes (i.e. a gene tree) can still be used to predict function, even when the labeling of orthologs and paralogs is ignored (Engelhardt et al., 2011). Such approaches are some of the most accurate in function prediction (Jiang et al., 2016; Radivojac et al., 2013; Zhou et al., 2019), and support the idea that having more high-quality data will almost always improve prediction accuracy. While distinguishing between orthologs and paralogs is a necessary step for answering important biological questions, we find both groups to be predictive of protein function and therefore valuable for function transfer.
Supplementary Material
Acknowledgements
The authors acknowledge the support of NSF grants DBI-1458477 and DBI-1564611, and from the Precision Health Initiative of Indiana University.
Financial Support: none declared.
Conflict of Interest: none declared.
References
- Adipietro K.A. et al. (2012) Functional evolution of mammalian odorant receptors. PLoS Genet., 8, e1002821–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altenhoff A.M. et al. (2012) Resolving the ortholog conjecture: orthologs tend to be weakly, but significantly, more similar in function than paralogs. PLoS Comput. Biol., 8, e1002514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M. et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet., 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao M., Cowen L.J. (2017) When should we not transfer functional annotation between sequence paralogs? Pac. Symp. Biocomput., 22, 15–26. [DOI] [PubMed] [Google Scholar]
- Chen X., Zhang J. (2012) The ortholog conjecture is untestable by the current gene ontology but is supported by RNA sequencing data. PLoS Comput. Biol., 8, e1002784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark W.T., Radivojac P. (2011) Analysis of protein function and its prediction from amino acid sequence. Proteins, 79, 2086–2096. [DOI] [PubMed] [Google Scholar]
- Clark W.T., Radivojac P. (2013) Information-theoretic evaluation of predicted ontological annotations. Bioinformatics, 29, i53–i61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Consortium G.O. (2016) Expansion of the Gene Ontology knowledgebase and resources. Nucleic Acids Res., 45, D331–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coolon J.D. et al. (2014) Tempo and mode of regulatory evolution in Drosophila. Genome Res., 24, 797–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cozzetto D., Jones D.T. (2017) Computational methods for annotation transfers from sequence. Methods Mol. Biol., 1446, 55–67. [DOI] [PubMed] [Google Scholar]
- Cozzetto D. et al. (2016) FFPred 3: feature-based function prediction for all Gene Ontology domains. Sci. Rep., 6, 31865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dessimoz C. et al. (2013) CAFA and the open world of protein function predictions. Trends Genet., 29, 609–610. [DOI] [PubMed] [Google Scholar]
- Dolinski K., Botstein D. (2007) Orthology and functional conservation in eukaryotes. Annu. Rev. Genet., 41, 465–507. [DOI] [PubMed] [Google Scholar]
- Du P. et al. (2019) Species tree inference under the multispecies coalescent on data with paralogs is accurate. bioRxiv 498378. [Google Scholar]
- Dunn C.W. et al. (2018) Pairwise comparisons across species are problematic when analyzing functional genomic data. Proc. Natl. Acad. Sci. USA, 115, E409–E417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt B.E. et al. (2005) Protein molecular function prediction by Bayesian phylogenomics. PLoS Comput. Biol., 1, e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt B.E. et al. (2011) Genome-scale phylogenetic function annotation of large and diverse protein families. Genome Res., 21, 1969–1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang G. et al. (2010) Getting started in gene orthology and functional analysis. PLoS Comput. Biol., 6, e1000703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitch W.M. (1970) Distinguishing homologous from analogous proteins. Syst. Biol., 19, 99–113. [PubMed] [Google Scholar]
- Gabaldón T., Koonin E.V. (2013) Functional and evolutionary implications of gene orthology. Nat. Rev. Genet., 14, 360–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gibson T.A., Goldberg D.S. (2009) Questioning the ubiquity of neofunctionalization. PLoS Comput. Biol., 5, e1000252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glazko G. et al. (2005) The choice of optimal distance measure in genome-wide datasets. Bioinformatics, 21, ii3–ii11. [DOI] [PubMed] [Google Scholar]
- Gu Z. et al. (2002) Rapid divergence in expression between duplicate genes inferred from microarray data. Trends Genet., 18, 609–613. [DOI] [PubMed] [Google Scholar]
- Hawkins T. et al. (2006) Enhanced automated function prediction using distantly related sequences and contextual association by PFP. Protein Sci., 15, 1550–1556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henikoff S., Henikoff J.G. (1992) Amino acid substitution matrices from protein blocks. Proc. Natl. Acad. Sci. USA, 89, 10915–10919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntley R.P. et al. (2015) The GOA database: gene ontology annotation updates for 2015. Nucleic Acids Res., 43, D1057–D1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y. et al. (2014) The impact of incomplete knowledge on the evaluation of protein function prediction: a structured-output learning perspective. Bioinformatics, 30, i609–i616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y. et al. (2016) An expanded evaluation of protein function prediction methods shows an improvement in accuracy. Genome Biol., 17, 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koonin E.V. (2005) Orthologs, paralogs, and evolutionary genomics. Annu. Rev. Genet., 39, 309–338. [DOI] [PubMed] [Google Scholar]
- Kryuchkova-Mostacci N., Robinson-Rechavi M. (2016) Tissue-specificity of gene expression diverges slowly between orthologs, and rapidly between paralogs. PLoS Comput. Biol., 12, e1005274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lan L. et al. (2013) MS-kNN: protein function prediction by integrating multiple data sources. BMC Bioinformatics, 14, S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laurent J.M. et al. (2019) Humanization of yeast genes with multiple human orthologs reveals principles of functional divergence between paralogs. bioRxiv 668335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Legried B. et al. (2019) Polynomial-time statistical estimation of species trees under gene duplication and loss. bioRxiv 821439. [DOI] [PubMed] [Google Scholar]
- Liao B.Y., Chang A. (2014) Accumulation of CTCF-binding sites drives expression divergence between tandemly duplicated genes in humans. BMC Genomics, 15, S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makova K.D., Li W. (2003) Divergence in the spatial pattern of gene expression between human duplicate genes. Genome Res., 13, 1638–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mika S., Rost B. (2006) Protein–protein interactions more conserved within species than across species. PLoS Comput. Biol., 2, e79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohan A. et al. (2009) Influence of sequence changes and environment on intrinsically disordered proteins. PLoS Comput. Biol., 5, e1000497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadimpalli S. et al. (2015) Pervasive variation of transcription factor orthologs contributes to regulatory network evolution. PLoS Genet., 11, e1005011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Needleman S.B., Wunsch C.D. (1970) A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol., 48, 443–453. [DOI] [PubMed] [Google Scholar]
- Nehrt N.L. et al. (2011) Testing the ortholog conjecture with comparative functional genomic data from mammals. PLoS Comput. Biol., 7, e1002073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson M.E. et al. (2009) Evolutionary constraints on structural similarity in orthologs and paralogs. Protein Sci., 18, 1306–1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radivojac P. et al. (2013) A large-scale evaluation of computational protein function prediction. Nat. Methods, 10, 221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogozin I.B. et al. (2014) Gene family level comparative analysis of gene expression in mammals validates the ortholog conjecture. Genome Biol. Evol., 6, 754–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlicker A. et al. (2006) A new measure for functional similarity of gene products based on Gene Ontology. BMC Bioinformatics, 7, 302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schnoes A.M. et al. (2009) Annotation error in public databases: misannotation of molecular function in enzyme superfamilies. PLoS Comput. Biol., 5, e1000605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Škunca N. et al. (2012) Quality of computationally inferred gene ontology annotations. PLoS Comput. Biol., 8, e1002533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sokolov A. et al. (2013) Combining heterogeneous data sources for accurate functional annotation of proteins. BMC Bioinformatics, 14, S10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnhammer E.L.L. et al. (2014) Big data and other challenges in the quest for orthologs. Bioinformatics, 30, 2993–2998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soria P.S. et al. (2014) Functional divergence for every paralog. Mol. Biol. Evol., 31, 984–992. [DOI] [PubMed] [Google Scholar]
- Studer R.A., Robinson-Rechavi M. (2009) How confident can we be that orthologs are similar, but paralogs differ? Trends Genet., 25, 210–216. [DOI] [PubMed] [Google Scholar]
- Tatusov R.L. et al. (1997) A genomic perspective on protein families. Science, 278, 631–637. [DOI] [PubMed] [Google Scholar]
- Thomas P.D. et al. (2012) On the use of gene ontology annotations to assess functional similarity among orthologs and paralogs: a short report. PLoS Comput. Biol., 8, e1002386.,. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z. et al. (2018) PANDA: protein function prediction using domain architecture and affinity propagation. Sci. Rep., 8, 3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang R. et al. (2019) A new class of metrics for learning on real-valued and structured data. Data Min. Knowl. Disc, 33, 995–1016. [Google Scholar]
- Zerbino D.R. et al. (2018) Ensembl 2018. Nucleic Acids Res., 46, D754–D761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C. et al. (2019) ASTRAL-Pro: quartet-based species tree inference despite paralogy. bioRxiv 2019.12.12.874727. [Google Scholar]
- Zhou N. et al. (2019) The CAFA challenge reports improved protein function prediction and new functional annotations for hundreds of genes through experimental screens. Genome Biol., 20, 244. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.