

Abstract
Motivation
Accurate prediction of binding between a major histocompatibility complex (MHC) allele and a peptide plays a major role in the synthesis of personalized cancer vaccines. The immune system struggles to distinguish between a cancerous and a healthy cell. In a patient suffering from cancer who has a particular MHC allele, only those peptides that bind with the MHC allele with high affinity, help the immune system recognize the cancerous cells.
Results
MHCAttnNet is a deep neural model that uses an attention mechanism to capture the relevant subsequences of the amino acid sequences of peptides and MHC alleles. It then uses this to accurately predict the MHC-peptide binding. MHCAttnNet achieves an AUC-PRC score of 94.18% with 161 class I MHC alleles, which outperforms the state-of-the-art models for this task. MHCAttnNet also achieves a better F1-score in comparison to the state-of-the-art models while covering a larger number of class II MHC alleles. The attention mechanism used by MHCAttnNet provides a heatmap over the amino acids thus indicating the important subsequences present in the amino acid sequence. This approach also allows us to focus on a much smaller number of relevant trigrams corresponding to the amino acid sequence of an MHC allele, from 9251 possible trigrams to about 258. This significantly reduces the number of amino acid subsequences that need to be clinically tested.
Availability and implementation
The data and source code are available at https://github.com/gopuvenkat/MHCAttnNet.
1 Introduction
Major histocompatibility complex (MHC) classes I and II play a significant role in identifying the cancerous cells in one’s body. These sets of genes, when bonded with peptides, help the immune system by bringing the bonded complex to the surface of a cancerous cell, making it visible to T-cells which eventually destroy it. This antigen presentation to the T-cells is a crucial part of the training and development of the adaptive immune response. The formation of a peptide–MHC allele complex depends on several factors, including proteasome cleavage preference of a peptide and the MHC-peptide binding affinity, thereby making it a difficult task (Zeng and Gifford, 2019a).
There have been several works in the past that use computational methods to estimate the binding affinity between an MHC allele and a peptide. MHCflurry (O’Donnell et al., 2018) is considered to be one of the state-of-the-art methods for this task. MHCflurry is trained on 130 MHC alleles and works well only on MHC alleles on which it is trained. MHCflurry also limits the length of a peptide to 8–15 amino acids. Other widely used state-of-the-art models are NetMHC (Andreatta and Nielsen, 2016; Nielsen et al., 2003) and NetMHCpan (Hoof et al., 2009; Jurtz et al., 2017; Nielsen and Andreatta, 2016). NetMHC is allele-specific and thereby has a different predictive model for each allele. NetMHC-4.0 is trained on 118 MHC alleles. NetMHCpan is pan-specific and makes binding affinity prediction even for unseen alleles, as long as the allele’s amino acid sequence is known. NetMHCpan-4.0, on the other hand, is trained on 169 MHC alleles. NetMHC and NetMHCpan tools are not open-sourced and hence, the workflow of training their models are not clear. In convolutional neural network-based models, like PUFFIN (Zeng and Gifford, 2019b), the architecture needs to work with a fixed-length input amino acid sequence. In these models, the variable-length sequences have to be made equal length by padding. The PUFFIN model predicts the expected affinity of an MHC-peptide binding, for both classes I and II alleles, as well as the uncertainty of its prediction. The MHCSeqNet (Phloyphisut et al., 2019) model considers the input as an equally-weighted linear amino acid chain and can handle 92 alleles of class I. It looks at all amino acid subsequences equally which is not necessary, as only certain subsequences contribute to determining the binding affinity. An artificial neural network, NN-Align (Nielsen and Lund, 2009), focuses on class II alleles. It, however, handles only 14 class II alleles. A newer tool from NetMHCpan series, NetMHCII and NetMHCIIpan (Jensen et al., 2018), computes binding affinities on 36 different HLA-DR alleles.
Despite the progress made so far, there is still a need for a more robust method for predicting binding affinity. There is a need to generalize the confidence of the predicted interactions between peptides and MHC alleles without having a significant drop in precision. MHCAttnNet overcomes the shortcomings of earlier methods by: (i) releasing the source code as an open-source package; (ii) enabling prediction even for variable-length peptides; (iii) focusing only on relevant subsequences of amino acids; and (iv) training and testing on a larger number of alleles.
MHCAttnNet uses a bidirectional long short-term memory (Bi-LSTM) styled encoder to deal with variable-length peptide sequences. This permits the model to handle a large variety of peptides, and hence makes it more general. MHCAttnNet is trained and tested on the Immune Epitope Database (IEDB) (Sahin et al., 2017) (as of 2019) and is capable of working well with both class I and class II MHC alleles separately. This has been made possible with the help of the attention mechanism used in the neural network. The attention mechanism is used to identify relevant subsequences responsible for determining the binding affinity and thereby increase the weights of these relevant subsequences. This permits the model to focus on these important subsequences of the amino acid sequence, making it more targeted and informative. MHCAttnNet is trained on 161 different class I MHC alleles and 49 different class II alleles as only these many MHC alleles are presently available in the dataset. MHCAttnNet can compute binding predictions on other MHC alleles as well as long as their amino acid sequences are available. Even while handling a large variety of MHC alleles, our model outperforms the current state-of-the-art models for predicting binding between peptide and class I MHC alleles while at the same time, is competitive in case of class II MHC alleles.
The structure of rest of this article is as follows: Section 2 presents the background about what personalized cancer vaccines are and how they work. The dataset, pre-processing steps, and the MHCAttnNet model are explained in Section 3. Section 4 describes the analysis of our results with respect to the state-of-the-art models. We summarize our approach and discuss some relevant aspects in Section 5.
2 Background
Personalized cancer vaccines have shown promising results in their early stages. To synthesize a personalized cancer vaccine, first the genomes of cancerous cells are collected, which helps in the identification of tumor-specific peptides called neoepitopes. Neoepitopes, when combined with adjuvants or other immune-stimulatory agents and injected into the patient, helps the immune system identify cancerous cells and kill them using the body’s own T-cells. This way, the human body learns to kill the cancerous cells on its own, without having the risk of autoimmune diseases (Hu et al., 2018b; Ott et al., 2017; Reddy et al., 2006; Sahin et al., 2017).
For the identification of neoepitopes, next-generation sequencing data from tumor and healthy cells are compared with that of the human reference genome. RNA sequencing narrows the focus to mutations of expressed genes. The potential sequences are validated by using computational models that predict the binding affinity of neoepitopes with the individual’s MHC proteins that would present the neoepitopes to the surface. This filters the candidate neoepitopes for personalized vaccines, as shown in Figure 1.
Fig. 1.
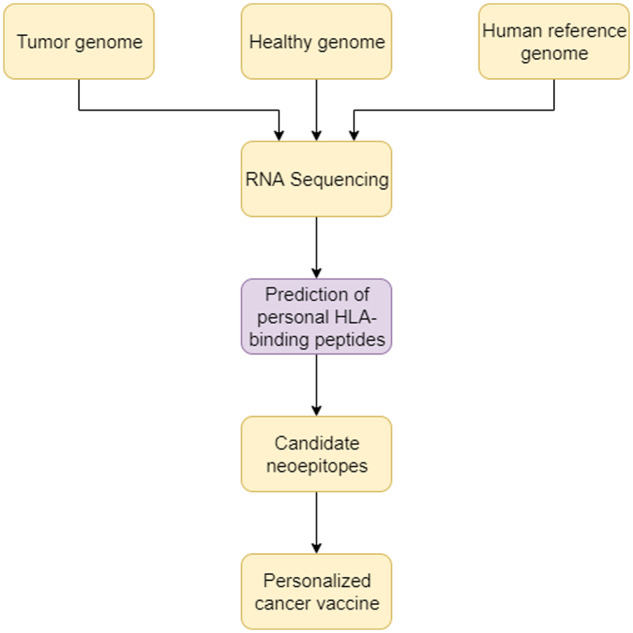
Steps to synthesize personalized cancer vaccine
The MHC allele is referred to as HLA complex in humans. There are three types of MHC genes in humans, classes I, II and III. We only focus on class I and class II MHC alleles (there is no publicly available data for class III alleles). Class I MHC alleles present endogenous antigens that originate from the cytoplasm to cytotoxic T-cells (CD8+ T-cells). Class II MHC alleles present exogenous antigens that originate extra-cellularly from foreign bodies, such as bacteria to helper T-cells (CD4+ T-cells) (Abbas et al., 2014; Delves et al., 2017). Previous research (Comber and Philip, 2014; Garrido et al., 1993) has shown that class I MHC alleles play a major role in identification of cancerous cells. Recent work (Pyke et al., 2018) however suggests that patient-specific variations in class II MHC alleles have as significant an effect on the mutations that arise in tumors, as that of class I MHC alleles.
3 Implementation
In this section, we discuss the dataset used and explain the MHCAttnNet model in detail.
3.1 Dataset
We use the IEDB (Sahin et al., 2017) (as of 2019) to prepare our training and testing data as done in previous works (Hu et al., 2018a; Jurtz et al., 2017; Nielsen and Lund, 2009; Phloyphisut et al., 2019; Zeng and Gifford, 2019b). We filter the data-points that correspond to ‘human’ or ‘homo-sapiens’ and the data-points that have MHC alleles belonging to classes I or II. We take into consideration the qualitative affinity measurements, which are labeled in the IEDB as ‘Positive’, ‘Positive High’, ‘Positive Intermediate’, ‘Positive Low’ and ‘Negative’. Based on Zhao and Sher (2018) and our analysis of quantitative affinity measurements, the classes ‘Positive’, ‘Positive High’ and ‘Positive Intermediate’ have IC50 values of <500 nM for class I and 1000 nM for class II alleles. Hence, all the data-points with these three classes are labeled as binding and the remaining two classes are labeled as non-binding.
Our final dataset comprises of 491 018 class I MHC-allele data-points covering 161 different HLAs and 64 954 class II MHC-allele data-points covering 49 different HLAs. The peptide lengths range from 3 to 43 for both classes, while the lengths of amino acid sequences of HLAs range from 180 to 347 for class I and from 85 to 232 for class II.
We get 451 484 HLA-A*, 39 424 HLA-B* and 110 HLA-C* class I alleles, out of which 379 783 are binding and 111 235 are non-binding. Similarly, we get 64 926 HLA-DRB1*, 22 HLA-DRB3*, 4 HLA-DRB4* and 2 HLA-DRB5* class II alleles out of which 36 035 are binding and 28 919 are non-binding.
3.2 Model
As shown in Figure 2, the embedding layers are used to encode the input amino acids into a low-dimensional vector. The Bi-LSTM encoder is used to extract abstract token-level features from these embeddings. The attention mechanism produces a weight vector, which is multiplied with the token-level features to build a sentence-level feature vector. The obtained weighted sentence-level feature vector is passed through the fully connected layers to perform the classification task.
Fig. 2.
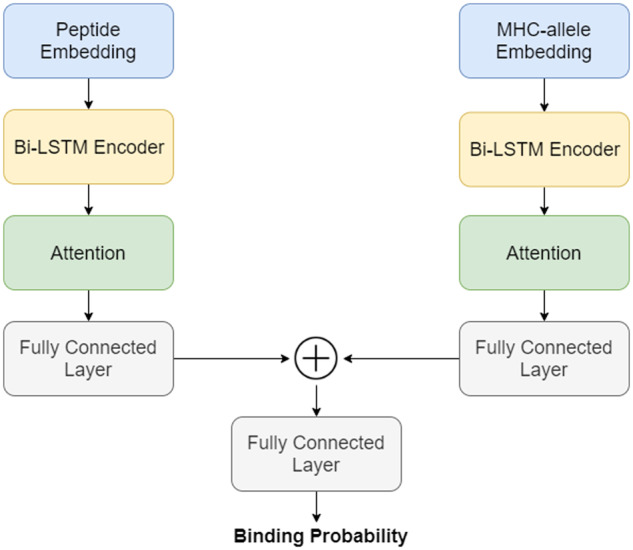
An overview of the MHCAttnNet architecture
This architecture is capable of handling both class I and class II MHC alleles. The model weights are shared across all MHC alleles as opposed to building one model per MHC allele [as done elsewhere (O’Donnell et al., 2018)]. The Adam optimization algorithm (Kingma and Ba, 2015) is used for the training with binary cross-entropy loss function. Our model accepts both the peptide and the MHC allele in the form of a sequence of amino acids.
3.2.1. Embedding
The inputs to the model are peptides and MHC alleles, which are represented as sequences of amino acids. To encode these amino acid sequences, we use a continuous vector representation called embedding (Collobert et al., 2011). Embeddings, when used as the underlying input representations, have been shown to boost the performance of various Natural Language Processing (NLP) tasks as they capture the semantic meanings of words in sentences. The input, a sequence of amino acids, can be treated as a sentence where the individual words are the amino acids. The embedding representation for the tokens can be learned from a large corpus in an unsupervised manner, and can be later fine-tuned for the required upstream task. Given a sentence consisting of T words , every word ei is converted into a real-valued vector xi. The sentence S, represented through the real-valued vectors of words, is passed to the Bi-LSTM Encoder (Section 3.2.2).
To pre-train the continuous vector embedding for the tokens, 1 or 3 consecutive amino acids (non-overlapping 1-g or 3-g) in an amino acid sequence, we use the Skip-Gram model (Mikolov et al., 2013). The lengths of amino acid sequences of peptides are much shorter than those of MHC alleles. Therefore, we train a 1-gram embedding for peptides, but use a 3-gram model called ProtVec for MHC alleles as suggested by an earlier study (Asgari and Mofrad, 2015). We fix the embedding dimension at 100, which was reported as the optimal parameter (Asgari and Mofrad, 2015).
3.2.2 Bi-LSTM encoder
A Bi-LSTM (Schuster and Paliwal, 1997) was chosen to encode the sequence of amino acids as it is capable of processing sequences with variable lengths, unlike the fixed n-mer peptides, and generalizing the relationship in the amino acid sequence.
The following notations and equations have been borrowed from Zhou et al. (2016). An LSTM cell consists of four components: one input gate it with the trainable weight matrices Wxi, Whi, Wci and bias bi; one forget gate ft with the trainable weight matrices Wxf, Whf, Wcf and bias bf and one output gate ot with the trainable weight matrices Wxo, Who, Wco and bias bo. The current cell state, ct, is generated by calculating the weighted sum using both the previous cell state and the current information generated by the cell.
| (1) |
| (2) |
| (3) |
| (4) |
| (5) |
| (6) |
To learn the relationship within the amino acids sequences, it would be beneficial to have access to future as well as past context. Bidirectional LSTM (1997) networks permit this by extending the unidirectional LSTM networks by introducing a second layer. In this second layer, the hidden connections are aligned in the opposite direction. The model is therefore able to exploit information from both the past and the future context. The output of the every word is the concatenation of the forward and backward hidden states (6) for that word.
3.2.3. Attention
Attentive neural networks (Vaswani et al., 2017) have recently demonstrated success over a wide range of fields ranging from NLP (Bahdanau et al., 2015) to Computer Vision (Xu et al., 2015). Here, we discuss the attention mechanism used for our classification task.
| (7) |
| (8) |
| (9) |
| (10) |
The u vector is randomly initialized, which is learned during the training process, to weight the amino acid subsequences. ut can be thought of as a non-linearity applied over the Bi-LSTM hidden state output, as in (6). vt is the exponential of the dot-product of ut with the u vector. Intuitively, the value of vt is high if u and ut are similar. We compute the weight of each token, αt, by normalizing over the vt vectors and thereby compute the final sentence pair-representation used for classification by taking the weighted sum of the Bi-LSTM hidden vectors ht (6).
3.2.4. Fully connected layers
The neurons are fully pairwise connected across adjacent linear layers, but the neurons within a single layer do not share connections. In the MHCAttnNet model, a non-linear activation function is applied in every fully connected layer. The Rectified Linear Unit (ReLU) (Nair and Hinton, 2010) activation function is used which computes the function . This forces the activation to be thresholded at zero.
The attended vectors, (10), from the peptide and MHC-allele branch of the network are concatenated before being passed onto another fully connected layer. The output of the model is the probability of the input being in a particular class which is obtained from the softmax function (Goodfellow et al., 2016), defined as in (11).
| (11) |
The softmax function squeezes the outputs for each class between 0 and 1, by dividing each class output by the sum of the outputs of all classes.
4 Results
MHCAttnNet produces competitive results in terms of predicting MHC-peptide bindings for both MHC classes I and II. The metrics used to compare the models are described in Section 4.1. Comparison with the state-of-the-art models is done in Sections 4.2 and 4.3. Attention in MHCAttnNet provides some interesting insights into which subsequences of amino acids of peptides and MHC alleles play major roles in determining the MHC-peptide bindings. These insights are explained in detail in Section 4.4. In Section 4.5, we define sequence reduction, a list of trigrams of amino acid sequences of MHC alleles, which captures all the information, relevant for predicting binding, corresponding to a particular amino acid. This list consists of just about 2.8% of the total possible trigrams, and would greatly reduce the number of possible clinical trials needed for vaccine development without major loss of information. This means that only 2.8% of the total trigrams have any significance in predicting the binding between a peptide amino acid and an MHC allele.
4.1 Model performance
To test the performance of MHCAttnNet, we look at the following metrics:
Precision or positive predictive value (PPV) (Alpaydin, 2014), which denotes the fraction of results which are relevant (12).
Recall or sensitivity (Trevethan, 2017), which denotes the fraction of relevant instances that the model was able to retrieve (13).
Accuracy (Alpaydin, 2014) is the percentage of correctly classified instances (14).
F1-score (Alpaydin, 2014), which gives a good balance between precision and recall (15).
Area under receiver operating characteristics (AUC-ROC) (Fawcett, 2006), which is the area under the curve plotted when true positive rate is plotted against false positive rate.
Area under precision-recall curve (AUC-PRC) (Saito and Rehmsmeier, 2015), which is defined as the area under the curve plotted when Precision is plotted against Sensitivity.
Pearson correlation coefficient (PCC) (Kirch, 2008), which is a linear correlation between two normally distributed continuous variables.
AUC-PRC is an appropriate metric to compare the results of models as there is a large class imbalance in the dataset, especially for class I MHC alleles. Precision-recall plots provide a good estimate of future classification performance as they evaluate the fraction of true positives among the positive predictions (Saito and Rehmsmeier, 2015).
In the following expressions, TP, TN, FP and FN denote true positive, true negative, false positive and false negative, respectively.
| (12) |
| (13) |
| (14) |
| (15) |
The performance of MHCAttnNet in class I MHC alleles is measured using AUC-ROC, AUC-PRC, PCC, PPV, F1-score and Sensitivity. In class II alleles, the class imbalance is not significant, and hence to evaluate the performance of MHCAttnNet on class II MHC alleles, accuracy, AUC-ROC, PCC, Sensitivity, PPV and F1-score are used.
These metrics are computed by us on a 5-fold cross-validation data (Refaeilzadeh et al., 2009), which is derived from IEDB (Vita et al., 2015) and processed as mentioned in Section 3.1. The 5-fold cross-validation data are different for class I and class II MHC alleles. All experiments are run on these 5-fold cross-validation data. Each of the 5 test files for class I MHC alleles consists of 97 000 data-points, while for class II MHC alleles, there are around 13 000 data-points each.
4.2 Performance on class I MHC alleles
MHCAttnNet is the first model that takes into consideration 161 different class I MHC alleles and yet achieves a 5-fold cross-validation AUC-PRC score of 94.18%. The model also attains a high cross-validation F1-score of 94.22%. Four different hyper-parameter configurations score higher than 94% for AUC-PRC, as seen in Table 1. MHCAttnNet, therefore, has high accuracy with low variance. The scores for the best model configuration are shown in bold in Table 1. This best model configuration was used for all the experiments.
Table 1.
The 5-fold cross-validation performance of MHCAttnNet with different hyper-parameters on class I MHC alleles
| Bi-LSTM | Number of layers | Number of layers | Context | AUC-PRC | AUC-ROC | F1- |
|---|---|---|---|---|---|---|
| hidden dimension | in peptide Bi-LSTM | in MHC Bi-LSTM | dimension | score | ||
| 64 | 3 | 3 | 16 | 0.9418 | 0.8893 | 0.9422 |
| 64 | 3 | 1 | 16 | 0.9418 | 0.8893 | 0.9416 |
| 64 | 3 | 3 | 32 | 0.9409 | 0.8874 | 0.9398 |
| 64 | 3 | 1 | 32 | 0.9416 | 0.8887 | 0.9404 |
Note: The best performance is indicated in bold. The hyper-parameter setting, corresponding to this, was used to run all the experiments.
MHCAttnNet is compared with the widely used state-of-the-art models, MHCflurry (O’Donnell et al., 2018), NetMHC-4.0 (Andreatta and Nielsen, 2016) and PUFFIN (Zeng and Gifford, 2019b).
We tested MHCflurry on our 5-fold cross-validation data using mhctools (https://github.com/openvax/mhctools). MHCflurry takes into consideration 130 class I MHC alleles as compared to 161 for MHCAttnNet. The predictions were computed for all the data-points in the test data, except for those data-points that had MHC alleles, which are not supported by MHCflurry. The performance of MHCflurry is computed without those data-points. MHCflurry outperforms MHCAttnNet on (PPV) by only 0.1% while MHCAttnNet achieves better results on the remaining metrics as seen in Table 2. MHCAttnNet achieves AUC-ROC score of 88.93% as compared to 82.34% of MHCflurry, which is an improvement of about 6.5%. The AUC-PRC score of MHCAttnNet is 94.18% while it is 90.74% for MHCflurry, an improvement of 3.5%. Moreover, MHCAttnNet shows a significant improvement in terms of Sensitivity (around 21.5%), PCC (around 19.5%) and F1-score (around 12%).
Table 2.
Comparison of performance of MHCAttnNet with the state-of-the-art methods for class I MHC alleles
| Model | AUC- | AUC- | PCC | PPV | F1- | Sensitivity |
|---|---|---|---|---|---|---|
| ROC | PRC | score | ||||
| NetMHC4.0 | 0.8237 | 0.9048 | 0.5738 | 0.9486 | 0.8531 | 0.7757 |
| MHCflurry | 0.8234 | 0.9074 | 0.5624 | 0.9695 | 0.8230 | 0.7149 |
| PUFFIN | 0.8185 | 0.9129 | 0.5398 | 0.9668 | 0.8264 | 0.7215 |
| MHCAttnNet (no attention) | 0.8822 | 0.9380 | 0.7463 | 0.9643 | 0.9402 | 0.9303 |
| MHCAttnNet | 0.8893 | 0.9418 | 0.7570 | 0.9683 | 0.9422 | 0.9304 |
Note: The best performances are indicated in bold.
We tested NetMHC-4.0 using the publicly available prediction software (2016). NetMHC-4.0 is trained on 118 class I MHC alleles and hence, there are some class I MHC alleles in our test data, which are not covered by NetMHC-4.0. Those data-points were removed from the test data on which the performance of NetMHC-4.0 is computed. Table 2 shows that MHCAttnNet outperforms NetMHC-4.0 on all the metrics. MHCAttnNet achieves an AUC-ROC score of 88.93% and shows a gain of more than 6% over NetMHC-4.0. Similarly MHCAttnNet is able to achieve AUC-PRC and PPV scores of 94.18 and 96.83%, respectively, while NetMHC4.0 achieves 90.48 and 94.86%, respectively. MHCAttnNet shows a significant gain of around 15% in both, Sensitivity and PCC.
We tested the pre-trained PUFFIN model using the provided official implementation (2019) on our test data. Table 2 also shows that MHCAttnNet scores better than PUFFIN on all the metrics for class I MHC alleles. MHCAttnNet outperforms PUFFIN by about 7% on AUC-ROC metric and about 3% on AUC-PRC metric. PUFFIN achieves the PPV score of 96.68%, which is just about 0.15% shy of MHCAttnNet’s score. MHCAttnNet shows improved performances on PCC, F1-score and Sensitivity than PUFFIN by about 22, 11.5 and 21%, respectively.
We also tested the performance of MHCAttnNet without the Attention Module to show the importance of attention. The comparison of performances with and without attention is shown in Table 2. It is clear that having attention produces better results. The obtained attention weights are also useful to understand how the model is weighting the amino acids while predicting.
4.3 Performance on class II MHC alleles
For class II MHC alleles, MHCAttnNet reaches as high as 75.79% on the AUC-ROC metric with 5-fold cross-validation on 49 different class II MHC alleles. NN-Align (Nielsen and Lund, 2009) is considered as one of the state-of-the-art methods to compute binding predictions between peptides and class II MHC alleles. NN-Align handles only 14 alleles as compared to 49 alleles for MHCAttnNet. NN-Align is also allele-specific and hence, has different predictive models for each allele. On the contrary, MHCAttnNet is pan-allele and hence, has only one model for the task of peptide and class II MHC-allele binding. We were unable to do a class-wise analysis on the benchmark Wang et al. (2008) dataset, which is used for analysis of the NN-Align model, as the number of data–points, there are too low for our model to learn. We however compared MHCAttnNet to a newer model for class II, NetMHCIIpan-3.2 (Jensen et al., 2018). We tested NetMHCIIpan using the software that has been made publicly available. The performance is computed on our 5-fold cross-validation data after removing the data-points that correspond to MHC alleles which are not handled by NetMHCIIpan. The results are compared on different metrics in Table 3. MHCAttnNet outperforms NetMHCIIpan on all metrics by around 7%–9%.
Table 3.
Comparison of performance of MHCAttnNet with the state-of-the-art methods for class II MHC alleles
| Model | Accuracy | AUC-ROC | PCC | Sensitivity | PPV | F1-score |
|---|---|---|---|---|---|---|
| NetMHCIIpan | 0.6822 | 0.6751 | 0.4297 | 0.3888 | 0.9039 | 0.5435 |
| PUFFIN | 0.7657 | 0.7756 | 0.5520 | 0.6853 | 0.8644 | 0.7645 |
| MHCAttnNet (no attention) | 0.7503 | 0.7530 | 0.5032 | 0.7275 | 0.8732 | 0.7636 |
| MHCAttnNet | 0.7549 | 0.7579 | 0.5130 | 0.7298 | 0.8769 | 0.7675 |
Note: The best performances are indicated in bold.
We also compare MHCAttnNet’s performance with PUFFIN. We run the pre-trained PUFFIN model, as given by Zeng and Gifford (2019b), on our test data for class II MHC alleles. PUFFIN scores better than MHCAttnNet on three metrics–AUC-ROC, Accuracy and PCC by ∼1.5, 1 and 4%, respectively. On the other hand, MHCAttnNet scores better on Sensitivity, PPV and F1-score. It achieves 72.98% on Sensitivity, 87.69% on PPV and 76.75% on F1–score, which is more than PUFFIN by ∼4.5, 1.3 and 0.3%, respectively. Overall, MHCAttnNet’s performance on class II MHC alleles is comparable to the PUFFIN model as seen in Table 3. Although the task of prediction is difficult on class II MHC alleles (Dimitrov et al., 2010), MHCAttnNet outperforms or does as well as the state-of-the-art models for class II MHC alleles.
This shows that the overall performance of MHCAttnNet is better than the previous state-of-the-art methods. It may be noted that MHCAttnNet has a single architecture that works for both class I and class II MHC alleles, and that even without making any changes to the layers (Fig. 2) of MHCAttnNet, it predicts the binding of peptides with both class I and class II MHC alleles accurately.
4.4 Analysis of attention weights
Attention mechanisms play a crucial role in NLP (Bahdanau et al., 2015), where they focus on the sensitive parts of the input during output generation. Such mechanisms are most important in long sentences as it is not required to encode the full source sentence into a fixed-length vector. The encoded vector, produced by the attention layer, depends on the weighted combination of all the hidden states of the Bi-LSTM layer and not just the last state. Hence, such mechanisms can boost the contributions of the key features. The placed attention mechanism is effective as it results in higher scores, over the model without attention, as seen in Tables 2 and 3.
One of the most widely used approaches is interpretation by visualizing the weights of the model. In long sequence domains, like amino acid sequences, attention is visualized using text heatmaps (Yang and Zhang, 2018). A heatmap is an efficient representation that uses a system of color codes to graphically show the importance of different values of the input. As shown in Figures 3 and 4, the 1D heatmaps are superimposed on input text, showing the aggregate attention, which provides an overview of consequential portions of input. The input MHC allele and peptide, as sequences of amino acids, are tokenized as indicated in Section 3. The intensity of the background color is the magnitude of the attention weight placed at that token.
Fig. 3.

Attention weights (2018) are highlighted on class I MHC allele (HLA-A*02:01) and peptide. respectively
Fig. 4.

Attention weights (2018) are highlighted on class II MHC allele (HLA-DRB1*04:01) and peptide, respectively
A big advantage of attention mechanism is that it gives us the ability to reason and visualize what the model is doing. The attention mechanism makes it easier to interpret the results by giving us better insights about which subsequences of the amino acid sequence are more relevant (for the upstream classification task)—the visualized attention weights give insights into how the model made the prediction. The understanding of actual binding process between an MHC allele and a peptide is still not very clear (Rajapakse et al., 2007) and hence, the attention weights can help the researchers to focus their studies on the particular subsequences of amino acids to get a better understanding of the binding mechanism. The significant amino acids, based on the attention values placed over them, may serve as a good starting point for the clinicians but the reliability of this is subject to clinical validation.
4.5 Sequence reduction
We analyze the cumulative information coverage obtained by going through the important trigrams corresponding to a particular amino acid, with attention weights as given by (10) being >0.001, from most important to least. Out of the 9261 possible trigrams of amino acid sequences of MHC alleles, we find that only about 258 of these trigrams contain all the information about a particular amino acid present in a peptide, as seen in Figure 5. This reduced list of trigrams we call sequence reduction.
Fig. 5.
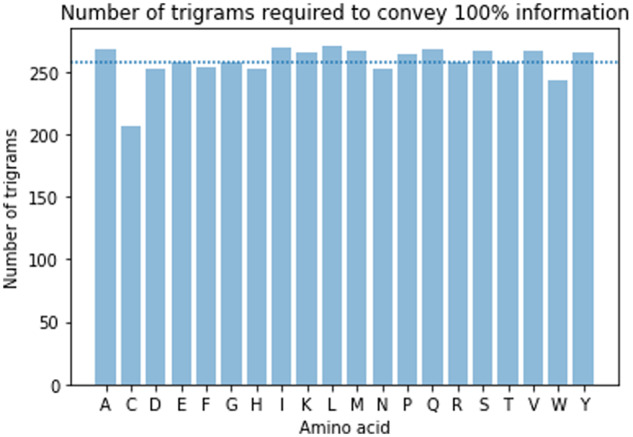
Number of trigrams needed to collectively convey 100% of information. The average number of trigrams needed to convey the complete information is 258 and is indicated with the dotted line
The threshold of 0.001 was chosen because we found it to be the most appropriate value without any significant loss of information. We ran the experiments for different thresholds. We noticed that when we selected a higher threshold value (say 0.01), our reduced list had around 211 trigrams, hence lost on some significantly informative trigrams whereas when we selected a lower threshold value (say 0.0001), our sequence reduction list had 301 trigrams, which contained a large number of low-contributing trigrams. In Figure 6, which has been plotted with a threshold of 0.001, we can see that for almost all the amino acids, the number of trigrams that cover most of the information is around 220. A similar curve profile is noticed for other thresholds. The biological significance of this threshold can be empirically tested by conducting clinical trials. The sequence reduction gives us a correlation between an amino acid of a peptide with a trigram of an MHC allele, while helping in significantly reducing the number of amino acid sequences that need to be clinically tested.
Fig. 6.

Relevance of particular trigrams of MHC alleles for some amino acids in peptides
The Pareto Principle (Lipovetsky, 2009) suggests that a small fraction (typically given as 20%) of items in a set has a disproportionate impact (typically given as 80%). While analyzing sequence reduction values, we found that the Pareto Principle holds for the amino acid E. As shown in Figure 6, 20% of the top relevant trigrams contribute to 77.27% of the information provided by all the important trigrams for the amino acid E. On the other hand, for other amino acids, the top 20% of the attended trigrams contribute for an average 60% of the relevant information, with a standard deviation of <10% of the mean. This shows that even in the sequence reduction list, it is only a small percentage of trigrams that play a significant role in the prediction of binding. While conducting clinical trials, this reduced list can help determine which peptides are likely to have a higher impact on a particular allele.
5 Conclusion
MHCAttnNet shows improved results on a larger variety of peptides and MHC alleles, including both class I and class II MHC alleles. The model handles variable-length peptides because of the use of Bi-LSTMs. It is important to note that unlike many of the previous approaches, MHCAttnNet is a pan-allele method. MHCAttnNet has only one model architecture that works well with both class I and class II MHC alleles. The model weights are different for class I and class II MHC alleles but the architecture remains the same. The improvement in results for MHCAttnNet is mainly due to the use of the ProtVec Embedding (Asgari and Mofrad, 2015) and the use of Bi-LSTMs to encode the amino acid information. The attention layers help improve the results marginally, but the most important benefit of the attention layers is to help illustrate the importance given to any subsequence of the amino acid sequence (either peptide or MHC allele). The use of an attention mechanism helps MHCAttnNet produce more focused and insightful results.
The bio-medicine community can gain from such a deep learning model that can not only predict with higher precision, but also gives an insight into relevant subsequences of amino acids of MHC alleles and their correlations with particular amino acids of peptides. Therefore, MHCAttnNet can contribute to improved processes for the design and manufacture of personalized cancer vaccines.
In MHCAttnNet, the input sequence of amino acids is encoded using the ProtVec embedding [trained using the Skip-Gram approach of the Word2Vec (Mikolov et al., 2013) algorithm]. Further work could be to use a contextualized word embedding, like ELMo (Peters et al., 2018), Flair (Akbik et al., 2018) or BERT (Devlin et al., 2019), to encode the input sequence. Such a contextualized word embedding could capture the structural aspects of amino acid sequences by looking into their contexts and hence, would further improve the binding predictions. It would also be interesting to see if the contextualized embedding can lead us to look at the amino acid sequences differently. The focused subsequences (as seen in Section 4.4) can lead to a better understanding of the mechanism of binding between alleles and peptides.
Our use of sequence reduction, a reduced relevant list of trigrams of amino acids obtained using the attention mechanism, opens the door to further research in this area. Sequence reduction reduces the search space significantly, making clinical trials easier. Understanding where the binding takes place in peptides and MHC alleles is an interesting and important problem. Further work on sequence reduction can lead to better development of cancer vaccines.
Acknowledgement
This work was supported by an Amazon AWS Machine Learning Research Award.
Conflict of Interest: none declared.
References
- Abbas A.K. et al. (2014) Cellular and Molecular Immunology E-Book. Elsevier Health Sciences; Philadelphia, USA. https://www.clinicalkey.com/dura/browse/bookChapter/3-s2.0-C20150023565. [Google Scholar]
- Akbik A. et al. (2018) Contextual string embeddings for sequence labeling. In: Proceedings of the 27th International Conference on Computational Linguistics pp. 1638–1649. Association for Computational Linguistics ACL, Santa Fe, NM, USA. https://www.aclweb.org/anthology/C18-1139.
- Alpaydin E. (2014) Introduction to Machine Learning. 3rd edn.MIT Press, Cambridge, Massachusetts, USA. [Google Scholar]
- Andreatta M., Nielsen M. (2016) Gapped sequence alignment using artificial neural networks: application to the MHC class I system. Bioinformatics, 32, 511–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asgari E., Mofrad M.R. (2015) Continuous distributed representation of biological sequences for deep proteomics and genomics. PLoS One, 10, e0141287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bahdanau D. et al. (2015) Neural machine translation by jointly learning to align and translate. In: Published as a conference paper at International Conference on Learning Representations, ICLR. San Diego, California, USA.
- Collobert R. et al. (2011) Natural language processing (almost) from scratch. J. Mach. Learn. Res., 12, 2493–2537. [Google Scholar]
- Comber J.D., Philip R. (2014) MHC class I antigen presentation and implications for developing a new generation of therapeutic vaccines. Ther. Adv. Vaccines Immunother., 2, 77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delves P.J. et al. (2017) Essential Immunology. John Wiley & Sons, Massachusetts, USA. [Google Scholar]
- Devlin J. et al. (2019) BERT: pre-training of deep bidirectional transformers for language understanding. In: Proceedings of the 2019 Conference of the North American Chapter of the Association for Computational Linguistics: Human Language Technologies, Vol. 1 (Long and Short Papers) pp. 4171–4186. Association for Computational Linguistics, Minneapolis, MN, USA.
- Dimitrov I. et al. (2010) MHC class II binding prediction’a little help from a friend. BioMed Res. Int., 2010, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fawcett T. (2006) An introduction to ROC analysis. Pattern Recognit. Lett., 27, 861–874. [Google Scholar]
- Garrido F. et al. (1993) Natural history of HLA expression during tumour development. Immunol. Today, 14, 491–499. 10.1016/0167-5699(93)90264-L. [DOI] [PubMed] [Google Scholar]
- Goodfellow I. et al. (2016) Deep Learning. MIT press, Cambridge, Massachusetts, USA. [Google Scholar]
- Hoof I. et al. (2009) NetMHCpan, a method for MHC class I binding prediction beyond humans. Immunogenetics, 61, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y. et al. (2018. a) ACME: pan-specific peptide-MHC class I binding prediction through attention-based deep neural networks. Bioinformatics, 35, 4946-4954. [DOI] [PubMed] [Google Scholar]
- Hu Z. et al. (2018. b) Towards personalized, tumour-specific, therapeutic vaccines for cancer. Nat. Rev. Immunol., 18, 168–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen K.K. et al. (2018) Improved methods for predicting peptide binding affinity to MHC class II molecules. Immunology, 154, 394–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jurtz V. et al. (2017) NetMHCpan-4.0: improved peptide–MHC class I interaction predictions integrating eluted ligand and peptide binding affinity data. J. Immunol., 199, 3360–3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingma D.P., Ba J. (2015) Adam: a method for stochastic optimization. In: Published as a conference paper at International Conference on Learning Representations, ICLR.
- Kirch W. (2008) Pearson’s correlation coefficient In: Encyclopedia of Public Health. Springer, Dordrecht, The Netherlands, pp. 1090–2013. [Google Scholar]
- Lipovetsky S. (2009) Pareto 80/20 law: derivation via random partitioning. Int. J. Math. Educ. Sci. Technol., 40, 271–277. [Google Scholar]
- Mikolov T. et al. (2013) Distributed representations of words and phrases and their compositionality. In: Advances in Neural Information Processing Systems NIPS pp. 3111–3119. Lake Tahoe, USA. http://dl.acm.org/citation.cfm? id=2999792.2999959.
- Nair V., Hinton G.E. (2010) Rectified linear units improve restricted Boltzmann machines. In: ICML’10 Proceedings of the 27th International Conference on Machine Learning pp. 807–814. Omnipress, USA. http://dl.acm.org/citation.cfm?id=3104322.3104425.
- Nielsen M., Andreatta M. (2016) NetMHCpan-3.0; improved prediction of binding to MHC class I molecules integrating information from multiple receptor and peptide length datasets. Genome Med., 8, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M., Lund O. (2009) NN-align. An artificial neural network-based alignment algorithm for MHC class II peptide binding prediction. BMC Bioinformatics, 10, 296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen M. et al. (2003) Reliable prediction of T-cell epitopes using neural networks with novel sequence representations. Protein Sci., 12, 1007–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Donnell T.J. et al. (2018) MHCflurry: open-source class I MHC binding affinity prediction. Cell Syst., 7, 129–132. [DOI] [PubMed] [Google Scholar]
- Ott P.A. et al. (2017) An immunogenic personal neoantigen vaccine for patients with melanoma. Nature, 547, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters M. et al. (2018) Deep contextualized word representations. In: Proceedings of the 2018 Conference of the North American Chapter of the Association for Computational Linguistics (NAACL): Human Language Technologies, Vol. 1 (Long Papers). New Orleans, Louisiana, USA. doi: 10.18653/v1/n18-1202. [DOI]
- Phloyphisut P. et al. (2019) MHCSeqNet: a deep neural network model for universal MHC binding prediction. BMC Bioinformatics, 20, 270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyke R.M. et al. (2018) Evolutionary pressure against MHC class II binding cancer mutations. Cell, 175, 416–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajapakse M. et al. (2007) Predicting peptides binding to MHC class II molecules using multi-objective evolutionary algorithms. BMC Bioinformatics, 8, 459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy S.T. et al. (2006) Targeting dendritic cells with biomaterials: developing the next generation of vaccines. Trends Immunol., 27, 573–579. [DOI] [PubMed] [Google Scholar]
- Refaeilzadeh P. et al. (2009) Cross-Validation. Springer, Boston, MA, USA, pp. 532–538. [Google Scholar]
- Sahin U. et al. (2017) Personalized RNA mutanome vaccines mobilize poly-specific therapeutic immunity against cancer. Nature, 547, 222–226. [DOI] [PubMed] [Google Scholar]
- Saito T., Rehmsmeier M. (2015) The precision-recall plot is more informative than the ROC plot when evaluating binary classifiers on imbalanced datasets. PLoS One, 10, e0118432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuster M., Paliwal K. (1997) Bidirectional recurrent neural networks. Trans. Sig. Proc., 45, 2673–2681. [Google Scholar]
- Trevethan R. (2017) Sensitivity, specificity, and predictive values: foundations, pliabilities, and pitfalls in research and practice. Front. Public Health, 5, 307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaswani A. et al. (2017) Attention is all you need In: Guyon I.et al. (eds) Advances in Neural Information Processing Systems 30. Curran Associates, Inc, pp. 5998–6008. Neural Information Processing Systems (NIPS 2017), California, USA. http://papers.nips.cc/paper/7181-attention-is-all-you-need.pdf. [Google Scholar]
- Vita R. et al. (2015) The immune epitope database (IEDB) 3.0. Nucleic Acids Res., 43, D405–D412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P. et al. (2008) A systematic assessment of MHC class II peptide binding predictions and evaluation of a consensus approach. PLoS Comput. Biol., 4, e1000048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K. et al. (2015) Show, attend and tell: neural image caption generation with visual attention. In: International Conference on Machine Learning (ICML), pp. 2048–2057. Lille, France.
- Yang J., Zhang Y. (2018) NCRF++: an open-source neural sequence labeling toolkit. In: Proceedings of the 56th Annual Meeting of the Association for Computational Linguistics (ACL). Melbourne, Australia. http://aclweb.org/anthology/P18-4013.
- Zeng H., Gifford D.K. (2019. a) DeepLigand: accurate prediction of MHC class I ligands using peptide embedding. Bioinformatics, 35, i278–i283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H., Gifford D.K. (2019. b) Quantification of uncertainty in peptide-MHC binding prediction improves high-affinity peptide selection for therapeutic design. Cell Syst., 9, 159–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W., Sher X. (2018) Systematically benchmarking peptide-MHC binding predictors: from synthetic to naturally processed epitopes. PLoS Comput. Biol., 14, e1006457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou P. et al. (2016) Attention-based bidirectional long short-term memory networks for relation classification. In: Proceedings of the 54th Annual Meeting of the Association for Computational Linguistics (Vol. 2: Short Papers) pp. 207–212. Association for Computational Linguistics, Berlin, Germany.