

Abstract
Motivation
Extra-long tandem repeats (ETRs) are widespread in eukaryotic genomes and play an important role in fundamental cellular processes, such as chromosome segregation. Although emerging long-read technologies have enabled ETR assemblies, the accuracy of such assemblies is difficult to evaluate since there are no tools for their quality assessment. Moreover, since the mapping of error-prone reads to ETRs remains an open problem, it is not clear how to polish draft ETR assemblies.
Results
To address these problems, we developed the TandemTools software that includes the TandemMapper tool for mapping reads to ETRs and the TandemQUAST tool for polishing ETR assemblies and their quality assessment. We demonstrate that TandemTools not only reveals errors in ETR assemblies but also improves the recently generated assemblies of human centromeres.
Availability and implementation
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Tandem repeats are formed by multiple consecutive nearly identical sequences that are often generated by unequal crossover (Smith, 1976). The early DNA sequencing projects revealed that tandem repeats are abundant in eukaryotic genomes (Bacolla et al., 2008; Yunis and Yasmineh, 1971). Recent studies of tandem repeats revealed their role in various cellular processes and demonstrated that mutations in tandem repeats may lead to genetic disorders (Black and Giunta, 2018; Giunta and Funabiki, 2017; McFarland et al., 2015; Song et al., 2018).
We distinguish between extensively studied short tandem repeats (Gymrek et al., 2016; Saini et al., 2018; Willems et al., 2014) and extra-long tandem repeats (ETRs) that range in length from tens of thousands to millions of nucleotides. Centromeric and pericentromeric regions contain some of the longest ETRs that account for ∼3% of the human genome and span megabase-long regions (Miga, 2019). Centromeres and pericentromeres represent the ‘dark matter’ of the human genome that evaded all attempts to sequence until recently and are the largest gaps in the reference human genome (Hayden et al., 2013; Miga et al., 2019). The goal of the telomere-to-telomere (T2T) consortium is to generate a complete assembly of the human genome, including all centromeres and pericentromeres (Miga et al., 2019). This effort recently resulted in assemblies of chromosomes X and Y (Jain et al., 2018b; Miga et al., 2019) but centromeres in other chromosomes are waiting to be assembled.
Human and primate centromeres are comprised of retrotransposon repeats and alpha-satellites, a DNA repeat based on a 171 bp monomer (Manuelidis and Wu, 1978). In humans and many primates, consecutive monomers are arranged tandemly into higher-order repeat (HOR) units (Willard and Waye, 1987a). The number of monomers and their order in a HOR are chromosome-specific. For example, the chromosome X HOR, referred to as DXZ1, consists of 12 monomers (Willard and Waye, 1987b). The monomer sequences are divided into five distinct monomer subtypes, denoted as A, B, C, D and E, where monomers from the same subtype are more closely related to each other than to monomers of other subtypes (Willard and Waye, 1987b). According to this classification, DXZ1 can be represented as C1D1E1 A1B1C2D2E2A2B2C3D3. For consistency with Bzikadze and Pevzner (2019), we took the liberty to refer to the chromosome X HOR as ABCDEFGHIKL.
Emergence of long-read technologies, such as Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT), have greatly altered the landscape of whole-genome sequencing. The development of long-read assemblers (Chin et al., 2016; Kolmogorov et al., 2019; Koren et al., 2017; Li, 2016; Lin et al., 2016; Ruan and Li, 2020) and hybrid assemblers that combine long and short reads (Antipov et al., 2016; Wick et al., 2017; Zimin et al., 2017) significantly increased the contiguity of assembled genomes compared to short-read assemblies. In addition, long reads contributed to successful semi-manual approaches for reconstructing human centromeres (Jain et al., 2018b; Miga et al., 2019). The Flye assembler successfully resolves bridged tandem repeats that are spanned by long reads and even some unbridged tandem repeats that are not spanned by long reads (Kolmogorov et al., 2019). The centroFlye assembler (Bzikadze and Pevzner, 2019) was designed to automatically assemble unbridged ETRs, such as centromeres.
Various alternative strategies for ETR assembly and absence of the ground truth for benchmarking these assemblies raise the problem of their quality evaluation. Similar problems have been addressed by the short-read quality assessment tools for genome assemblies, such as GAGE (Salzberg et al., 2012) and QUAST (Gurevich et al., 2013; Mikheenko et al., 2018) as well as specialized quality assessment tools metaQUAST (Mikheenko et al., 2016) and rnaQUAST (Bushmanova et al., 2016). However, these tools are based on known references and thus are not applicable to analyzing ETRs since their analysis requires reference-free approaches to evaluating assembly quality. At the same time, existing reference-free tools are based on analyzing gene content or mapping reads to the assembled sequences (Clark et al., 2013; Ghodsi et al., 2013; Hunt et al., 2013; Simão et al., 2015) and are not applicable to ETRs either.
Existing reference-free assembly quality assessment approaches rely on sequence alignment tools (Langmead et al., 2009; Li, 2013, 2016, 2018; Li and Durbin, 2009) to accurately map reads to assemblies. However, our benchmarking revealed that these tools often fail in ETRs. The BWA-MEM tool (Li, 2013), primarily designed for short-read mapping, incorrectly maps many long reads to ETRs. Minimap2 (Li, 2018) incorrectly maps some long reads to ETRs (especially in regions with assembly errors) and thus is not well suited for ETR assembly quality evaluation. The recently developed Winnowmap tool (Jain et al., 2020) was specifically designed for mapping reads to repetitive genomic regions. However, our benchmarking demonstrated that Winnowmap is limited with respect to detecting assembly errors: while it works well in the case of error-free assemblies, its accuracy deteriorates in the case of assembly errors (Table 1). We thus developed the TandemMapper tool that efficiently maps long error-prone reads to ETRs. TandemMapper not only enabled TandemQUAST development but also led to an improvement in ETR assemblies due to more accurate read mapping and subsequent polishing.
Table 1.
Benchmarking of TandemMapper, minimap2 and Winnowmap on the simulated dataset
| Correctly mapped reads | Incorrectly mapped reads | # alignments extended through the deletion breakpoint | Running time (s) | Memory footprint (GB) | |
|---|---|---|---|---|---|
| TandemMapper (unique k-mers) | 97.9% (1155) | 0.01% (1) | 0 | 511 | 5.2 |
| TandemMapper (solid k-mers) | 98.3% (1160) | 0.01% (1) | 0 | 590 | 5.6 |
| minimap2 | 96.0% (1133) | 2.7% (32) | 58 | 357 | 5.8 |
| Winnowmap | 95.8% (1130) | 2.8% (33) | 58 | 84 | 1.2 |
Note: Minimap2 and Winnowmap were run using recommended parameters for mapping ONT reads (-cx map-ont). The best value for each column is indicated in bold. A read is considered correctly mapped if its starting position is within 100 bp from the read simulated position calculated for the longest read alignment (an alignment is elongated to both ends of a read). Only reads longer than 5 kb with alignments longer than 3 kb were considered. The total number of such reads in this read-set is 1180. Although minimap2 mapped 4 more reads than TandemMapper (1165 versus 1161), 3 out of these 4 reads came from the region of the deletion and 1 read was mapped incorrectly. The benchmarking was done on a server with Intel Xeon X7560 2.27 GHz CPUs using 16 threads.
The initial attempt to evaluate the quality of ETR assemblies was centromere-specific (Bzikadze and Pevzner, 2019) and has not resulted in a general quality assessment tool for ETR assemblies. Species- and chromosome-specific nature of centromeres prevents applications of the same approach to other ETRs. However, the common principles of centromere organization can be utilized for developing a universal assembly evaluation tool for ETRs.
Here, we present the TandemTools package that includes the TandemMapper tool for mapping reads to ETRs, and the TandemQUAST tool for evaluating and improving ETR assemblies. We used TandemTools and subsequent polishing to improve assemblies of the human centromere X (cenX) generated by both centroFlye (Bzikadze and Pevzner, 2019) and the curated semi-manual approach (Miga et al., 2019). These improvements suggest that TandemTools will become a useful tool for evaluating the quality and polishing of many assemblies since nearly all genomes have ETRs. We also applied TandemTools to the GAGE gene cluster at the human chromosome X (Miga et al., 2019) and to the assembly of the human centromere 8 generated by the recently developed HiCanu assembler (Nurk et al., 2020) and demonstrated that it reveals assembly errors in these ETRs. The results are presented in Supplementary Appendices ‘Analyzing ETRs in the GAGE locus at the human X chromosome’ and ‘TandemTools results on cen8 assembly’.
TandemTools is open-source software that is freely available as a command-line utility on GitHub at https://github.com/ablab/TandemTools.
2 Materials and methods
2.1 TandemTools input
As an input, TandemTools requires one or several ETR assemblies and the set of long reads (PacBio continuous long reads or ONT) that contributed to these assemblies. Additionally, error-prone long reads can be complemented by accurate long reads, such as PacBio high-fidelity (HiFi) reads. We do not consider short Illumina reads since it is nearly impossible to unambiguously map them to ETRs.
2.2 TandemTools modules
TandemTools consists of the read-mapping module that aligns reads to the assembly (TandemMapper), the polishing module for improving the assembly quality based on the identified read alignments and the quality assessment module (TandemQUAST). TandemQUAST uses general metrics for evaluating ETRs of any kind and centromeric metrics designed specially to account for the HOR structure of centromeric ETR.
2.3 Selection of k-mers
2.3.1. Selecting solid k-mers in ETRs
Most long-read mapping algorithms are based on minimizers (Jain et al., 2018a; Li, 2016, 2018), k-mers that are chosen as the anchors for the read mapping. However, mapping a long read to an ETR is a non-trivial problem since minimizers are expected to be reduced in numbers and irregularly arranged due to local expansions of identical tandem repeats. Bzikadze and Pevzner (2019) used unique k-mers (that appear just once in the assembly) to improve read mapping to ETRs.
The density of unique k-mers may significantly vary along an assembly (Fig. 1), leading to drops in coverage or incorrect mappings in some regions. To address this problem, TandemMapper uses rare k-mers that appear less than MaxOccurrences times in the assembly. To obtain uniform k-mer density, we compute MaxOccurrences as the assembly length divided by 100 kb.Figure 1 illustrates that the density of rare k-mers is significantly larger than the density of unique k-mers, thus providing more ‘signposts’ for read mapping.
Fig. 1.
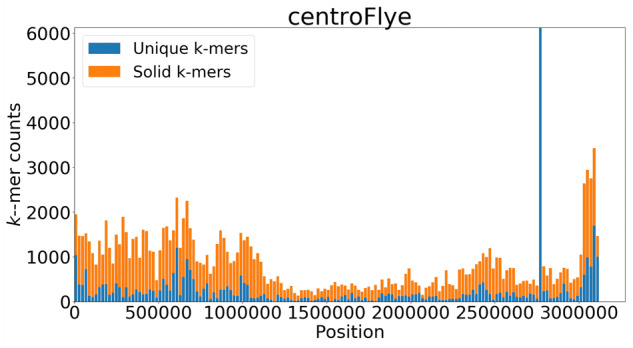
Distribution of unique and solid 19-mers along the cenX assembly of the CHM13 cell line constructed by centroFlye. Each bar shows the number of unique (solid) 19-mers in a bin of length 20 kb. The total number of unique (solid) 19-mers is 39 530 (57 318). The peak at ∼2750 kb corresponds to a LINE element and contains 6128 unique 19-mers and only 1499 solid 19-mers after filtration
Since ETR assemblies can be error-prone, some rare k-mers may represent assembly errors rather than low-frequency k-mers in the genome. To filter out such rare k-mers, we analyze their frequencies in the read-set. We assume that a k-mer from an assembly was erroneously classified as rare if it has an unusually low frequency (lower than MinFrequency) or an unusually high frequency (higher than MaxFrequency) in reads. The MinFrequency (MaxFrequency) threshold is defined as a fifth (95th) quantile of k-mer frequencies in the read-set. We, thus classify a rare k-mer as solid if it occurs in reads at least MinFrequency and at most MaxFrequency times.
The k-mer selection procedure can be affected by the fact that ETRs may harbor various transposable elements (TEs), such as LINE repeats, Alu repeats, etc. Even a single copy of a TE within an ETR is likely to contain many solid k-mers that may affect the mapping accuracy and complicate further analysis. To minimize the influence of TEs on the choice of solid k-mers, we set the MaxKmers limit on the maximum number of solid k-mers that can be selected in each window of a fixed length L (default value L = 1000 bp). Given an array KmerDensity of the number of k-mers in each window of length L in the assembly, MaxKmers is calculated as median(KmerDensity) + 2·σ(KmerDensity), where σ is the SD within KmerDensity. Thus, if the number of solid k-mers in a window exceeds the threshold, we randomly select MaxKmers of them.
2.3.2. Compatible k-mers
The TandemMapper algorithm is inspired by the minimap2 (Li, 2018) and Flye mappers (Kolmogorov et al., 2019; Lin et al., 2016). As solid k-mers are not necessarily unique in the assembly, we consider each occurrence of each solid k-mer separately.
Let aR and bR (aA and bA) be occurrences of solid k-mers a and b in the read R (assembly A). To make aR and bR uniquely defined for each read, we limit attention to solid k-mers that appear exactly once in this read. Note that, while aR and bR are uniquely defined, there may be multiple choices for aA and bA. We define d(aR, bR) and d(aA, bA) as distances between a and b in R and A, respectively.
We refer to the pair of aA and aR (bA and bR) as a match aM (bM) and define:
To assess the distribution of differences between distances in reads and the assembly, we collect all penalties taken over all consecutive non-overlapping unique k-mers a and b in all reads where these k-mers appear once into the Penalties array. We define distortion C as median(Penalties) + IQR(Penalties), where IQR stands for the interquartile range.
In addition, we define MissedKmers(aM, bM) as the number of solid k-mers in assembly A between aA and bA. We call aM and bM compatible if distance(aM, bM) < maxDistance (maxDistance is defined as the largest distance between two consecutive unique k-mers in the assembly), MissedKmers(aM, bM) < maxMissed (the default value maxMissed = 500) and diff(aM, bM) < C · distance(aM, bM), where C is the distortion.
2.4 Tandemmapper module
Given a read, we define a directed weighted compatibility graph with a vertex-set equal to the set of all matches of solid k-mers between R and A. We connect vertices aM and bM by an edge if (i) a precedes b in R and (ii) aM and bM are compatible. We further define the weight of this edge as premium—penalty(aM, bM), where premium is a constant selected to optimize the number of correctly mapped reads (default value premium =0.1). A chain between a read R and an assembly A is defined as the longest path in the compatibility graph. Note, that since all considered solid k-mers appear just once in R, no solid k-mer can be present in the chain more than once.
A chain for a given read can be used to map this read to the assembly. TandemMapper finds a chain for each read using dynamic programing, filters out short chains (shorter than 3 kb in length or containing less than 20 solid k-mers) and constructs the corresponding nucleotide-level alignments within the derived chain boundaries for each remaining chain. Table 1 in Section 3 illustrates that TandemMapper improves on other long-read mapping tools in ETRs.
2.5 Polishing module
Due to the high error rate in reads, most long-read assemblers have a polishing step to improve base-calling accuracy of the assembly (Chin et al., 2013; Lin et al., 2016; Loman et al., 2015; Vaser et al., 2017). However, Miga et al. (2019) demonstrated that standard polishing tools may even decrease the assembly quality in ETRs due to incorrect and ambiguous read alignments against the assembly. On the other hand, Miga et al. (2019) demonstrated that the marker-assisted read mapping (based on unique k-mers) significantly improves accuracy of ETR assemblies. TandemQUAST uses read alignments generated by TandemMapper as an input for a modified Flye polishing module (Kolmogorov et al., 2019; Lin et al., 2016). Section 3 demonstrates that this polishing procedure fixes erroneous deletions and base-calling errors.
2.6 Quality assessment module (TandemQUAST)
To evaluate the assembly quality and reveal possible errors, we developed two general metrics (indel-based and k-mer-based) and a centromeric metric (monomer-based) that we describe below. Former metrics are applicable to any ETRs and the latter metric is applicable to centromeric ETRs only.
2.6.1. Indel-based metrics
ETR assemblies are prone to large-scale deletions and duplications that lead to misassembly breakpoints. QUAST (Gurevich et al., 2013) defines a misassembly breakpoint based on differences between an assembly and a reference genome. In contrast, since the reference is not available, TandemQUAST detects breakpoints based on abnormalities in the read coverage. Below we describe the coverage metric and the breakpoint metric and use them to reveal putative breakpoints.
Coverage metric. Assembly errors may affect the coverage near the assembly breakpoints. TandemQUAST uses the read alignments (truncated with respect to their longest chains) to construct the coverage plot and reveal regions with abnormal coverage that may point to assembly errors (Fig. 3).
Fig. 3.

Coverage (top) and breakpoint (bottom) metrics for simulated (left) and simulateddel (right) assemblies. The coverage plot does not show a significant drop at the point of the deletion but the breakpoint plot reveals peak at the position of the deletion (400 kb). The red plot is based on the breakpointRatio(Kmer) values, the gray plot is based on the breakpointRatio+(Kmer) values
Breakpoint metric. Since long-read assemblers often fail to distinguish various repeat copies and erroneously collapse repetitive regions, indels represent the most frequent assembly errors in ETRs. The breakpoint metric was designed specifically to detect indels based on the analysis of mapped reads. In case, an assembly contains a breakpoint caused by a long indel, longest chains for the majority of reads spanning this indel breakpoint cannot be extended through this indel due to a substantial discrepancy in distances between solid k-mers in reads spanning this breakpoint and the assembly. Thus, if longest chains for many reads start or end in a certain region, this region may contain an assembly breakpoint. However, stochastic differences in coverage and various biases may also result in drops or peaks in read coverage. Our goal is to distinguish these cases and reveal assembly breakpoints.
A chain for a read R defines its partitioning into prefix(R), middle(R) and suffix(R), where middle(R) is the mapped part of a read that starts (ends) at the first (last) k-mer in the chain. The region in the assembly corresponding to middle(R) is referred to as a chain-segment. We also define an elongated chain-segment as a chain-segment extended by |prefix(R)| and |suffix(R)| nucleotides in the beginning and the end, respectively.
Given a solid k-mer Kmer, we define breaks(Kmer) [breaks+(Kmer)] as the number of chain-segments (elongated chain-segments) starting or ending in this k-mer (over all reads). We also define number(Kmer) [number+(Kmer)] as the number of chain-segments (elongated chain-segments) containing this k-mer. Finally, we define breakpointRatio(Kmer) as breaks(Kmer)/number(Kmer) and breakpointRatio+(Kmer) as breaks+(Kmer)/number+(Kmer).
While drops in values of breakpointRatio usually correspond to poorly covered regions, peaks in values may reveal breakpoints in the assembly. We expect that regions, where breakpointRatio(Kmer) has significantly higher values than breakpointRatio+(Kmer), contain assembly breakpoints because the longest chains for many reads were not extended through this region (Fig. 3).
2.6.2 K-mer-based metrics
In contrast to the TandemMapper tool (that considers k-mers that appear more than once in the assembly), the k-mer-based metrics need a reliable set of k-mers that appear just once in the assembly. We, thus filter out solid k-mers that occur more than once in the assembly or more than once in a single read and refer to the rest as unique solid k-mers.
After constructing read alignments, TandemQUAST finds where a unique solid k-mer in a read maps to the assembly and calculates coordinates of all found alignments across all reads containing this k-mer. Afterward, it clusters these coordinates (for a given unique solid k-mer) if they are located within MaxClumpDistance from each other (default value MaxClumpDistance =1 kb). After single linkage clustering, we define a cluster as a clump if it contains more than MinClumpSize elements (default value MinClumpSize =2). Ideally, all occurrences of a unique solid k-mer should form a single clump. We divide all k-mers having at least MinClumpSize occurrences in reads into three groups: a single clump, multiple clumps and spurious k-mers that do not form clumps (Fig. 2).
Fig. 2.
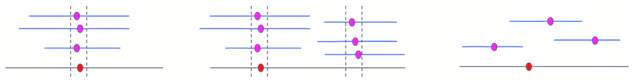
Coordinates of unique solid k-mers in the assembly and reads. Purple and red dots represent k-mer position in reads (shown as blue lines) and in the assembly (shown as a gray line), respectively. Clumps are flanked by vertical lines. (left) k-mers forming a single clump, (middle) k-mers forming multiple clumps in different parts of the assembly and (right) k-mers that do not form clumps (spurious k-mers)
TandemQUAST reports absolute and relative abundance of such k-mers and generates a plot showing their distribution (Table 2 and Fig. 4 in Section 3). Multiple clumps or spurious k-mers appearing along the entire assembly may point to poor base-calling quality of this assembly. Multiple clumps or spurious k-mers appearing in a certain region of an assembly reflect either a poor base-calling quality in these regions or collapsed duplications with subsequent ‘consensus’ polishing with reads from both copies.
Table 2.
Distribution of different types of unique solid k-mers in the T2T4, T2T4polish, T2T7, centroFlye and centroFlyepolish assemblies
| T2T4 | T2T4polish | T2T7 | centroFlye | centroFlyepolish | |
|---|---|---|---|---|---|
| Single clump | 13 130 (75%) | 15 158 (96%) | 16 114 (97%) | 16 550 (96%) | 15 858 (97%) |
| Multiple clumps | 1058 (6%) | 524 (3%) | 294 (2%) | 422 (2%) | 396 (2%) |
| No clumps | 3217 (17%) | 197 (1%) | 237 (1%) | 302 (2%) | 180 (1%) |
Note: Assemblies do not utilize information derived from accurate PacBio HiFi reads.
Fig. 4.

Distribution of different types of unique solid k-mers in the simulated (left) and simulatedmut (right) assemblies. Each bar shows the number of different types of k-mers in a bin of length 5 kb
In the case when a complementary set of accurate PacBio HiFi reads is available, TandemQUAST compares k-mer frequencies in the assembly and the HiFi reads. If the assembly contains k-mers that do not occur in HiFi reads or frequent k-mers from reads have a low frequency or are even absent in the assembly, it is likely that the assembly requires additional polishing (Supplementary Fig. S8).
2.6.3. Centromeric metrics
The additional set of metrics takes into account the centromere organization into monomers and HOR units. When a set of specific monomer sequences is known, TandemQUAST can analyze the assembly using the monomer-based metric described below and the unit-based statistic described in Supplementary Appendix ‘Unit-based statistic’.
Centromere assemblies may include difficult-to-detect indels of multiple monomers. In case monomer sequences are known, TandemQUAST attempts to detect discrepancies between reads and the assembly at the monomer level. The assembled centromere and all reads are aligned to the provided monomer sequences and are subsequently translated into the monomer alphabet using the StringDecomposer tool (Dvorkina et al., 2020), resulting in a monocentromere and monoreads.
For each monomer ReadMonomer in each monoread, TandemQUAST uses nucleotide-based read alignments to identify the starting nucleotide position of ReadMonomer in the monocentromere [referred to as Start(ReadMonomer)]. In case, ReadMonomer is aligned against a deletion in the monocentromere, Start(ReadMonomer) is recursively defined as Start(NextReadMonomer), where NextReadMonomer is the next monomer in the monoread. For each monomer CenMonomer in the monocentromere, we define Start(CenMonomer) as the starting position of this monomer in the centromere. We say that a monomer in a read (ReadMonomer) and a monomer in a centromere (CenMonomer) are co-located if |Start(ReadMonomer) - Start(CenMonomer)| is below MaxStartDistance (the default value MaxStartDistance =50 bp).
For each monomer CenMonomer in the monocentromere, TandemQUAST constructs the set ReadMonomers(CenMonomer) of all monomers in reads that are co-located with this monomer. For an error-free assembly, we expect that the vast majority of monomers in ReadMonomers(CenMonomer) coincide with CenMonomer, i.e. the ratio of CenMonomer in ReadMonomers(CenMonomer) is high. If this ratio [denoted as Ratio(CenMonomer)] is below a threshold MinRatio (the default value MinRatio =0.8), the assembly is likely to have an error (Supplementary Fig. S1). However, in the case of heterozygous monomers, this ratio is close to 0.5 as roughly half of the reads support (do not support) the monomer.
Although individual monomers may significantly vary in sequence, their length is fairly conserved within species that have alpha-satellites (Haaf and Willard, 1998; Hall et al., 2003). Thus, variations in monomer length across the centromere in such species may point to flaws in the assembly. Using StringDecomposer output, TandemQUAST generates an interactive HTML-page that provides a general monomer-level overview of the assembly and demonstrates the distribution of monomer lengths (Fig. 5).
Fig. 5.
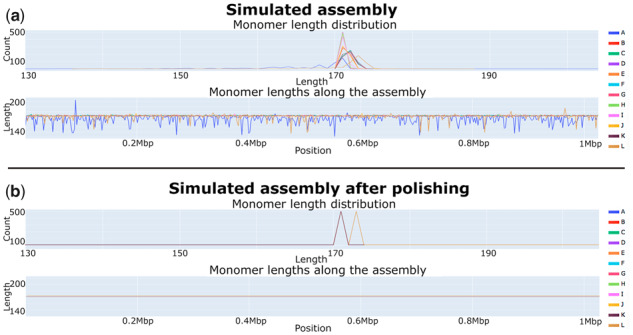
Monomer length distribution for the simulated (a) and simulatedpolish (b) assemblies. Monomer sequences forming a consensus DXZ1* sequence, derived in Bzikadze and Pevzner (2019), were used for analysis. In the simulated assembly, the length of the A-monomers varies from 131 to 203 bp (mean 165 bp) and the length of the L-monomers varies from 137 to 187 bp (mean 171 bp). In the simulatedpolish assembly, the length of all A-monomers (L-monomers) is equal to 171 (173) bp. Since all monomers, except for L, have lengths 171 bp after polishing, they all are represented by the color corresponding to the K-monomer
2.6.4. Comparison of various ETR assemblies
TandemQUAST performs pairwise comparison for each pair of analyzed assemblies using the bi-mapping plot and the discordance test.
A bi-mapping plot (Supplementary Fig. S2) provides an overview of read alignments from the perspective of both assemblies. Each read aligned to both assemblies represents a dot with its starting mapping positions in two assemblies as the x- and y-coordinates. Positions of read alignments for two assemblies can be compared to reveal structural discrepancies between them.
The discordance test was introduced in Bzikadze and Pevzner (2019) for comparing two assemblies. Supplementary Appendix ‘Discordance test’ describes its implementation in TandemQUAST.
3 Results
3.1 Simulated assembly
To benchmark TandemTools, we simulated an ETR of length ∼1.03 Mbp, which is a concatenation of 500 randomly mutated copies of the consensus HOR sequence on chromosome X (DXZ1) that diverge from the consensus sequence by 1% (substitutions only). Afterward, we simulated 1200 reads from this ETR using NanoSim (Yang et al., 2017) trained on the real ONT dataset enriched for ultra-long reads (longer than 50 kb) generated by the T2T consortium (Miga et al., 2019). We refer to the centroFlye assembly of these reads as simulated. We further introduced various artificial errors (described below) into the simulated assembly and ran TandemTools. An additional example of TandemTools performance on a centromere with more complex structure is presented in Supplementary Appendix ‘TandemTools results on the simulated datasets (D6Z1)’.
3.1.1. Benchmarking TandemMapper, minimap2 and Winnowmap
We compared TandemMapper with minimap2, the widely used long-read mapper that achieves excellent results outside repeated regions, and Winnowmap (Jain et al., 2020) that is designed specifically for mapping reads to repetitive genomic regions. To analyze how these tools handle assembly errors, we generated simulateddel assembly by introducing an artificial deletion of length 10 kb in the simulated assembly at position 400 kb.
We benchmarked mapping tools by aligning simulated reads to the simulateddel assembly and comparing their known exact positions in the assembly to the inferred positions (Table 1). TandemMapper correctly stopped all read alignments at the breakpoint of this deletion, while minimap2 and Winnowmap erroneously extended alignments through this breakpoint due to the highly repetitive sequence of the ETR. Using solid k-mers instead of unique k-mers slightly increased the number of correctly mapped reads even in an easy case of the simulated assembly with the uniform density of unique k-mers.
3.1.2. Indel-based metrics
To analyze how these metrics capture breakpoints, we used the simulateddel assembly (Fig. 3). Although the coverage plot does not show a significant drop at the point of the deletion, the breakpoint plot reveals a peak at the position of the deletion (400 kb).
3.1.3. k-mer-based metrics
To benchmark metrics evaluating the base-calling accuracy of an assembly, we introduced 10 000 (∼1% of the sequence length) random single-nucleotide substitutions in the simulated assembly (we refer to this assembly as simulatedmut). TandemQUAST reports the number of each group of unique solid k-mers and their distribution in the assembly (Fig. 4). The percent of unique solid k-mers forming a single clump decreased from 91% in the simulated assembly to 74% in the simulatedmut assembly, mostly due to the increased number of spurious k-mers.
3.1.4. Centromeric metrics
In order to illustrate the monomer-based metric and the unit-based statistic, we generated the simulateddel_monomer assembly by introducing a deletion of three consecutive monomers in the simulated assembly at position 226 kb. The results are presented in Supplementary Appendices ‘TandemTools results on the simulated datasets (DXZ1)’ and ‘Unit-based statistic’.
In addition, we demonstrated how these metrics might be affected by the assembly quality. Figure 5 shows that most monomers have conserved length across the assembly. However, the first monomer A and the last monomer L show surprising variability in length, suggesting that the accuracy of the simulated assembly deteriorates at the ends of HOR units due to imperfect polishing. This imperfect polishing is caused by limitations of the existing read-mapping tools in ETRs, forcing centroFlye to perform separate polishing for each HOR. Since the polishing procedure (Lin et al., 2016) is known to have limitations in the very beginning/end of each segment subjected to polishing, the beginning of the first (A) and the end of the last (L) monomers in each HOR can be cut off in a polished assembly. Just a single round of polishing with TandemQUAST resulted in the simulatedpolish assembly with an increased assembly length (by ∼4 kb) and corrected sequences of the first and the last monomers along the entire assembly (Fig. 5).
3.2 Analysis of cenX assemblies
We analyzed the following cenX assemblies: the T2T consortium assembly v0.4 (T2T4), v0.7 (T2T7) (Miga et al., 2019) and centroFlye v0.8.3 assembly (centroFlye) (Bzikadze and Pevzner, 2019). Note that, the T2T4 assembly is an interim version that was not polished with the marker-assisted methods described in Miga et al. (2019). We added it to the comparison to show how TandemQUAST analyzes unpolished assemblies. The T2T7 version was first semi-manually assembled and further improved based on centroFlye assembly as described in Miga et al. (2019). The T2T7 and centroFlye assemblies were additionally polished using ONT reads.
We also applied our polishing method to the T2T4 and centroFlye assemblies (resulting in T2T4polish and centroFlyepolish assemblies) to demonstrate how TandemQUAST improves assemblies.
3.2.1. Selecting solid k-mers in ETRs
The centroFlye assembly of the cenX has 39 530 unique 19-mers distributed across the 3.1 Mbp of the cenX length, with the largest distance between consecutive unique 19-mers =30 kb (Bzikadze and Pevzner, 2019). The number of rare 19-mers using MaxOccurrences =30 is 66 785 (Fig. 1).
Applying the filtration of k-mers by MinFrequency and MaxFrequency removes 5801 out of 66 785 rare k-mers, leaving 60 984 solid 19-mers. Comparison with PacBio HiFi reads generated from the same cell line (Vollger et al., 2019) revealed that 4844 of 5801 filtered out 19-mers are absent in the HiFi read-set or, on the contrary, have a very high frequency (higher than a frequency of 95% of 19-mers in the read-set). Applying the additional filtration by MaxKmers further reduces the number of solid 19-mers in the assembly from 60 984 to 55 173.
3.2.2. Indel-based metrics
Figure 6 illustrates that all assemblies have slightly lower read coverage at the center of the centromere at ∼1300–1600 kb that has a low concentration of unique k-mers (Supplementary Fig. S6).
Fig. 6.

The breakpoint metric for the T2T4, T2T4polish, T2T7, centroFlye and centroFlyepolish assemblies. The red and the gray plot are based on the breakpointRatio(Kmer) and breakpointRatio+(Kmer) values, respectively. The vertical light gray bands represent regions with low coverage (<10×). Discrepancies in these regions do not necessarily reflect flaws in an assembly
Low base-calling accuracy of an assembly can prevent chain extension in TandemMapper. As a result, the longest chains for many reads may end in a poorly polished region, causing an increase in breakpointRatio values. Thus, to verify breakpoints found in the T2T4 assembly, we compared them to the T2T4polish assembly. Both assemblies have peaks in breakpointRatio values at ∼270, 800, 1500, 2000 and 2500 kb that correlate with the bi-mapping plot (Supplementary Fig. S7). The breakpoint metric for centroFlye and T2T7 assemblies are generally consistent between breakpointRatio(Kmer) and breakpointRatio+(Kmer) values, suggesting that these assemblies do not have large indels and rearrangements.
3.2.3. k-mer-based metrics
Supplementary Figure S6 and Table 2 show the distribution of different types of unique solid k-mers across the assemblies. The T2T4 assembly has a high number of spurious k-mers as expected for an unpolished assembly, while T2T4polish demonstrates significant improvement in base-calling accuracy across the assembly. The high percentage (92–96%) of k-mers forming a single clump in the T2T7 and centroFlye assemblies suggest a high base-level quality in these assemblies.
In addition, we compared k-mer frequencies in assemblies and in accurate PacBio HiFi reads generated from the same cell line CHM13 (Vollger et al., 2019). The number of k-mers that do not occur in the HiFi read-set was the highest in the unpolished T2T4 assembly (223 579) and the lowest (842) in the T2T7 assembly (Supplementary Fig. S4).
3.2.4. Monomer metrics
Figure 7 presents the monomer length distribution across various assemblies. The T2T7 and centroFlye assemblies have a few unusually short (145–146 bp) A-monomers at ∼1 Mbp. We checked these monomers further and confirmed that they are supported by reads. Besides that, the T2T7 assembly has very conserved monomer lengths except for a few monomers at ∼2.15 Mbp.
Fig. 7.
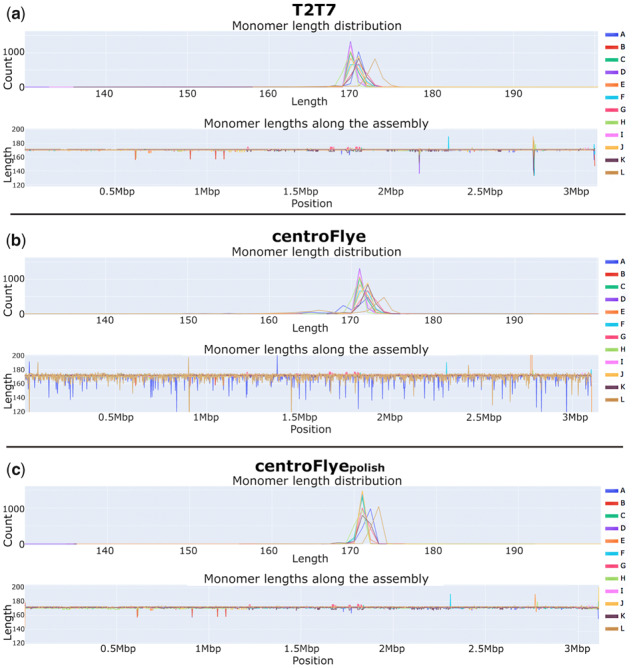
Monomer length distribution along the assembly in the T2T7 (a), centroFlye (b) and centroFlyepolish (c) assemblies
In the centroFlye assembly, L-monomers significantly vary in length as in the simulated assembly (Fig. 5), suggesting that centroFlye assembly requires additional polishing of HOR unit ends. The centroFlyepolish assembly has significantly more uniform monomer lengths as compared to the centroFlye assembly.
3.2.5. Pairwise comparison of assemblies
Supplementary Figure S7 shows bi-mapping plots for each pair of assemblies. As expected from the analysis of the breakpoint metric (Fig. 6), the centroFlye and T2T7 assemblies are nearly identical. The T2T4polish assembly differs from the T2T7 assembly around ∼350, 1600, 2100 and 2800 kb (coordinates are given for the T2T7 assembly).
4 Discussion
We presented the TandemMapper and TandemQUAST tools and applied them to various cenX assemblies. Although these tools detect flaws in ETR assemblies and provide a possibility to assess their quality, they have certain limitations discussed below.
4.1 False assembly errors
TandemQUAST is based on mapping reads to the assembly and subsequent analysis. Such an approach implies that inherent errors or systematic biases in the sequencing platforms may affect evaluation of the assembly and bring in some discrepancies that could be considered as false assembly errors. To reduce this effect, TandemQUAST has an option of using accurate PacBio HiFi reads.
4.2 Analysis of arbitrary ETRs in human and other genomes
Sequence and structural organization of ETRs, and particularly centromeres, varies widely across species. Since assembly of arbitrary ETRs remains an open problem, there is currently only one tool (centroFlye) for an automatic assembly of some ETRs and few examples of ETR assemblies. We thus limited the scope of our study to the recently completed assemblies of the human centromeres and the GAGE locus (Supplementary Appendix ‘Analyzing ETRs in the GAGE locus at the human X chromosome’). Since the T2T consortium aims to generate a gap-free assembly of the entire human genome (Miga et al., 2019), we anticipate that more high-quality ETR assemblies will soon be generated. These new assemblies will help us to improve the TandemMapper and TandemQUAST tools.
4.3 Analysis of diploid assemblies
Since centroFlye is now limited to haploid assemblies, the current version of TandemQUAST also focuses on haploid assemblies. Extending TandemQUAST functionality to diploid assemblies presents a complex algorithmic challenge. However, even effectively haploid cell lines may contain somatic heterogeneity due to clonal genomic instability in the cell culture. In this case, TandemQUAST can report heterozygous sites based on the discrepancies in mapped reads.
4.4 Using additional data types for assessing quality of ETR assemblies
We used accurate HiFi PacBio reads to analyze various centromere assemblies but not bacterial artificial chromosomes (BACs) and other alternative technologies that represent valuable resources for analyzing tandem repeats (see Supplementary Appendix ‘Alternative technologies for ETR assembly quality assessment’).
For example, a BAC from an ETR is often easier to assemble than an entire long ETR, such as a centromere. For example, centromere Y was recently sequenced using ONT reads to generate assemblies of BACs spanning this centromere (Jain et al., 2018b). However, certain limitations of the BAC technology make BACs a non-ideal option for ETRs sequence classification (Miga et al., 2019). In particular, BACs (i) do not represent a high-throughput approach and thus limit the scope of studies, (ii) have severe differences in coverage that complicate the analysis, (iii) require partial restriction digests that introduce biases in cloning, (iv) may have secondary structures making them incompatible with a bacterial host and (v) since existing short-read assemblers are unable to assemble highly repetitive centromeric BAC from short reads (or even Sanger reads), it is not clear how to reproduce the semi-manual assemblies of such BACs (some of them assembled two decades ago) with current state-of-the-art assemblers like SPAdes (Bankevich et al., 2012). It is also difficult to accurately assemble BACs from centromeres using long error-prone reads, e.g. recent large BAC sequencing effort has not resulted in assembling such BACs (Dennis et al., 2017). Thus, if a BAC sequence and a centromere assembly disagree, it is not clear whether this disagreement is caused by an error in the BAC assembly or an error in the centromere assembly. A possible way to address this challenge is a hybrid BAC assembly that combines short and long reads like in Jain et al. (2018b).
Supplementary Material
Acknowledgements
We are grateful to Ivan Alexandrov for many insightful comments and Andrey Prjibelski for helpful discussions and suggestions.
Funding
This work was supported by St. Petersburg State University, St. Petersburg, Russia [ID PURE 51555639].
Conflict of Interest: none declared.
References
- Antipov D. et al. (2016) hybridSPAdes: an algorithm for hybrid assembly of short and long reads. Bioinformatics, 32, 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bacolla A. et al. (2008) Abundance and length of simple repeats in vertebrate genomes are determined by their structural properties. Genome Res., 18, 1545–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bankevich A. et al. (2012) SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J. Comput. Biol., 19, 455–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Black E.M., Giunta S. (2018) Repetitive fragile sites: centromere satellite DNA as a source of genome instability in human diseases. Genes, 9, 615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushmanova E. et al. (2016) rnaQUAST: a quality assessment tool for de novo transcriptome assemblies. Bioinformatics, 32, 2210–2212. [DOI] [PubMed] [Google Scholar]
- Bzikadze A., Pevzner P.A. (2019) centroFlye: assembling centromeres with long error-prone reads. bioRxiv. https://www.biorxiv.org/content/10.1101/772103v1. [Google Scholar]
- Chin C.S. et al. (2013) Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat. Methods, 10, 563–569. [DOI] [PubMed] [Google Scholar]
- Chin C.S. et al. (2016) Phased diploid genome assembly with single-molecule real-time sequencing. Nat. Methods, 13, 1050–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark S.C. et al. (2013) ALE: a generic assembly likelihood evaluation framework for assessing the accuracy of genome and metagenome assemblies. Bioinformatics, 29, 435–443. [DOI] [PubMed] [Google Scholar]
- Dennis M. et al. (2017) The evolution and population diversity of human-specific segmental duplications. Nat. Ecol. Evol., 1, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dvorkina T. et al. (2020) The string decomposition problem and its applications to centromere assembly. Bioinformatics. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghodsi M. et al. (2013) De novo likelihood-based measures for comparing genome assemblies. BMC Res. Notes, 6, 334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giunta S., Funabiki H. (2017) Integrity of the human centromere DNA repeats is protected by CENP-A, CENP-C, and CENP-T. Proc. Natl. Acad. Sci. USA, 114, 1928–1933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich A. et al. (2013) QUAST: quality assessment tool for genome assemblies. Bioinformatics, 29, 1072–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gymrek M. et al. (2016) Abundant contribution of short tandem repeats to gene expression variation in humans. Nat. Genet., 48, 22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haaf T., Willard H.F. (1998) Orangutan alpha-satellite monomers are closely related to the human consensus sequence. Mamm. Genome, 9, 440–447. [DOI] [PubMed] [Google Scholar]
- Hall S.E. et al. (2003) Centromere satellites from Arabidopsis populations: maintenance of conserved and variable domains. Genome Res., 13, 195–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayden K.E. et al. (2013) Sequences associated with centromere competency in the human genome. Mol. Cell. Biol., 33, 763–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt M. et al. (2013) REAPR: a universal tool for genome assembly evaluation. Genome Biol., 14, R47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain C. et al. (2018. a) Fast approximate algorithm for mapping long reads to large reference databases. J. Comput. Biol., 25, 766–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain M. et al. (2018. b) Linear assembly of a human centromere on the Y chromosome. Nat. Biotechnol., 36, 321–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolmogorov M. et al. (2019) Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol., 37, 540–546. [DOI] [PubMed] [Google Scholar]
- Koren S. et al. (2017) Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res., 27, 722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B. et al. (2009) Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol., 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. (2009) Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2013) Aligning sequence reads, clone sequences and assembly contigs with BWA-MEM. arXiv: 1303.3997v2. [Google Scholar]
- Li H. (2016) Minimap and miniasm: fast mapping and de novo assembly for noisy long sequences. Bioinformatics, 32, 2103–2110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H. (2018) Minimap2: versatile pairwise alignment for nucleotide sequences. Bioinformatics, 34, 3094–3100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin Y. et al. (2016) Assembly of long error-prone reads using de Bruijn graphs. Proc. Natl. Acad. Sci. USA, 113, E8396–E8405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loman N.J. et al. (2015) A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat. Methods, 12, 733–735. [DOI] [PubMed] [Google Scholar]
- Manuelidis L., Wu J.C. (1978) Homology between human and simian repeated DNA. Nature, 276, 92–94. [DOI] [PubMed] [Google Scholar]
- McFarland K.N. et al. (2015) SMRT sequencing of long tandem nucleotide repeats in SCA10 reveals unique insight of repeat expansion structure. PLoS One, 10, e0135906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miga K.H. (2019) Centromeric satellite DNAs: hidden sequence variation in the human population. Genes, 10, 352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miga K.H. et al. (2019) Telomere-to-telomere assembly of a complete human X chromosome. bioRxiv. https://www.biorxiv.org/content/10.1101/735928v3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikheenko A. et al. (2016) MetaQUAST: evaluation of metagenome assemblies. Bioinformatics, 32, 1088–1090. [DOI] [PubMed] [Google Scholar]
- Mikheenko A. et al. (2018) Versatile genome assembly evaluation with QUAST-LG. Bioinformatics, 34, i142–i150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nurk S. et al. (2020) HiCanu: accurate assembly of segmental duplications, satellites, and allelic variants from high-fidelity long reads. bioRxiv. https://www.biorxiv.org/content/10.1101/2020.03.14.992248v3. [DOI] [PMC free article] [PubMed]
- Ruan J., Li H. (2020) Fast and accurate long-read assembly with wtdbg2. Nat. Methods, 17, 155–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini S. et al. (2018) Reference haplotype panel for genome-wide imputation of short tandem repeats. Nat. Commun., 9, 4397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salzberg S.L. et al. (2012) GAGE: a critical evaluation of genome assemblies and assembly algorithms. Genome Res., 22, 557–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simão F.A. et al. (2015) BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics, 31, 3210–3212. [DOI] [PubMed] [Google Scholar]
- Smith G.P. (1976) Evolution of repeated DNA sequences by unequal crossover. Science, 191, 528–535. [DOI] [PubMed] [Google Scholar]
- Song J.H.T. et al. (2018) Characterization of a human-specific tandem repeat associated with bipolar disorder and schizophrenia. Am. J. Hum. Genet., 103, 421–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaser R. et al. (2017) Fast and accurate de novo genome assembly from long uncorrected reads. Genome Res., 27, 737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollger M.R. et al. (2019) Improved assembly and variant detection of a haploid human genome using single-molecule, high-fidelity long reads. Ann. Hum. Genet., 84,125–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wick R.R. et al. (2017) Unicycler: resolving bacterial genome assemblies from short and long sequencing reads. PLoS Comput. Biol., 13, e1005595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willard H.F., Waye J.S. (1987. a) Hierarchical order in chromosome-specific human alpha satellite DNA. Trends Genet., 3, 192–198. [Google Scholar]
- Willard H.F., Waye J.S. (1987. b) Chromosome-specific subsets of human alpha satellite DNA: analysis of sequence divergence within and between chromosomal subsets and evidence for an ancestral pentameric repeat. J. Mol. Evol., 25, 207–214. [DOI] [PubMed] [Google Scholar]
- Willems T. et al. ; The 1000 Genomes Project Consortium. (2014) The landscape of human STR variation. Genome Res., 24, 1894–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang C. et al. (2017) NanoSim: nanopore sequence read simulator based on statistical characterization. Gigascience, 6, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yunis J.J., Yasmineh W.G. (1971) Heterochromatin, satellite DNA, and cell function. Structural DNA of eukaryotes may support and protect genes and aid in speciation. Science, 174, 1200–1209. [DOI] [PubMed] [Google Scholar]
- Zimin A.V. et al. (2017) Hybrid assembly of the large and highly repetitive genome of Aegilops tauschii, a progenitor of bread wheat, with the MaSuRCA mega-reads algorithm. Genome Res., 27, 787–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.