

Abstract
Computationally driven engineering of proteins aims to allow them to withstand an extended range of conditions and to mediate modified or novel functions. Therefore, it is crucial to the biotechnological industry, to biomedicine and to afford new challenges in environmental sciences, such as biocatalysis for green chemistry and bioremediation. In order to achieve these goals, it is important to clarify molecular mechanisms underlying proteins stability and modulating their interactions. So far, much attention has been given to hydrophobic and polar packing interactions and stability of the protein core. In contrast, the role of electrostatics and, in particular, of surface interactions has received less attention. However, electrostatics plays a pivotal role along the whole life cycle of a protein, since early folding steps to maturation, and it is involved in the regulation of protein localization and interactions with other cellular or artificial molecules. Short- and long-range electrostatic interactions, together with other forces, provide essential guidance cues in molecular and macromolecular assembly. We report here on methods for computing protein electrostatics and for individual or comparative analysis able to sort proteins by electrostatic similarity. Then, we provide examples of electrostatic analysis and fingerprints in natural protein evolution and in biotechnological design, in fields as diverse as biocatalysis, antibody and nanobody engineering, drug design and delivery, molecular virology, nanotechnology and regenerative medicine.
Prologue: scope and contents of this mini-review
Several excellent reviews and exhaustive chapters can be found in literature about electrostatics theoretical background and its influence on protein properties [1], [2], [3], [4], [5], therefore, the need for one more may seem futile. However, the aim of such articles was “to present a comprehensive coverage on the uses of electrostatic interactions by proteins in tuning their basic biophysical properties and in performing their biological functions” [1]. However, according to the scope of this Journal, we aim to highlight how structural computational studies focusing on protein electrostatics may boost applied science and biotechnology projects. In this mini-review, we provide examples of bioinformatic tools used in prediction-driven projects that led, via the computational analysis of electrostatics, to the successful engineering of proteins or the identification of functional fingerprints. Indeed, apart from its crucial role in mediating basic functions, protein electrostatics can also be studied for biotechnological purposes, i.e. to provide information on its role in modulating enzyme or antibody/nanobody features of industrial interest (e.g. solubility, catalytic efficacy, binding strength and specificity), protein-nanoparticle interactions exploited in regenerative medicine and drug delivery, mechanisms underlying subcellular sorting to membranes and organelles and viral antigenic drift and host jump, which are highly important to human and animal health and vaccine design.
Section 1 and its subsections depict the overall role of electrostatics in modulating protein stability and function; computational methods and tools that could be used to infer functional insights are reviewed in section 2, while section 3 focuses on aforementioned examples of applied science/biotechnology projects in which protein electrostatics investigation played a central role.
1. Electrostatics: a pivotal player in protein structure and interactions
Each protein consists of a peculiar combination of amino acids, which, in turn, can be classified according to shared features, e.g hydrophobic/polar, small/large, presence/absence of a positive/negative charge. Hydrophobic and electrostatic interactions influence the fate of any protein since its synthesis and folding to its degradation. Hereafter, we shortly summarize the main processes in which protein electrostatics play a role. In subsections 1.1 and 1.2, intra-chain electrostatics and interactions between protein residues and the solvent are considered as determinants for proper folding and stability, as well as a pivotal player in enzyme catalysis; then, we report about the role of electrostatics in regulating protein interactions with other proteins (subsection 1.3) or with molecules other than proteins, either cellular/natural or not (subsection 1.4). An overall picture of protein electrostatics is presented in Fig. 1.
Fig. 1.

Examples of interactions mediated by electrostatic forces. (a) Intramolecular salt bridges are involved in protein folding and stability; (b) Cotranslational interaction with ribosomes may influence secondary structure formation; (c) Long-range interaction between charged residues is involved in tertiary structure acquisition; (d) Surface charge can act as a direct proteasome signal and trigger protein degradation ; (e) Electrostatic can facilitate complex formation; (f) Conformational exchange; (g) Positive charge is required for rycin binding to rRNA (h) Protein binding to DNA could hamper binding of other molecules modulating DNA accessibility
1.1. Protein folding and stability
Proper protein folding and stability are maintained by many interactions between inner and outer residues. Traditionally, hydrophobic effect, hydrogen bonding, and packing interactions between buried protein residues have been considered as the dominant ones, while the role of surface features – particularly electrostatic ones – has often been neglected [3]. Although electrostatic interactions are long in range, and thus weakly specific, they can contribute to proper folding and stability e.g. via salt bridges or by further modulating the folding landscape.
The relevance of electrostatics in the interactions of proteins with other polypeptides or non-proteinaceous molecules is widely acknowledged (see next subsections). In contrast, the role it can play in establishing specifically funneled landscapes and their relevance in determining the final protein structure (and its conformational changes) are still unclear [2]. The influence of electrostatic forces on proteins arises as early as during translational events. The ribosomal exit tunnel is negatively charged, but the local potential through its length could be influenced by charged amino acids of nascent peptides as they move along the tunnel, generating a wave of electrostatic potential; this, in turn, regulates secondary structure acquisition and protein elongation rates and pauses, drastically contributing to nascent peptide folding or misfolding [6]. Furthermore, the ribosome outer surface is also characterized by a diffused negative electrostatic potential (mainly due to rRNA), and there is some evidence pointing out at its interaction with charged residues of nascent polypeptides, in turn affecting their conformational sampling ability, cotranslational events including interactions, aggregation and degradation [7] and ultimately folding kinetics [8].
In addition to synthesis, folding, and maturation, electrostatics is involved in protein degradation. Although the majority of proteins is degraded through a ubiquitin dependent mechanism, several direct proteasome signals (DPSs) exist that can mediate proteasome-directed protein degradation in a ubiquitin independent manner.
Recently, Kudriaeva and co-workers [9] characterized the myelin basic protein (MBP) DPS, showing that it is enriched in basic and flexible amino acids and its signaling function is charge-mediated. Notably, MBP mutants with negative or less positive surface charge showed a decrease in proteasomal degradation rate in vitro. It seems possible that MBP high cationic charge may function as a built-in ubiquitin-mimetic signal that can be attracted by the proteasome. The resulting interaction could be mediated by ionic complexes between basic amino acids in MBP and anionic residues of the proteasome.
The role played by electrostatic forces in governing non-specific protein-protein interactions and stability has been investigated by “supercharging” mutation analysis [10]. Even though such an approach can be of interest to biotechnology projects (see section 3.1), the relationship between net charge and refolding ability still needs complete elucidation, as diverse sets of supercharging mutations have different outcomes, likely because of the specific structural contributions given by each residue [11].
The impact of electrostatics on protein stability is very complex because of the competition between several phenomena, including local dipolar interactions, attraction and repulsion between charged residues and desolvation penalties of their ionized groups (which depend on the solvent dielectric properties, in turn affected by the temperature [4], [12]). Given the ionizable nature of such residues, pH also influences protein stability [13]. Furthermore, the balance between electrostatic and hydrophobic contributions should be taken into account when dealing with protein stability. As a result of this complexity, many studies seem to point out conflicting results on the stabilization/destabilization potency of electrostatic interactions. For example, the aforementioned supercharging strategy for improving protein refolding goes along with the observation of an increased amount of charged residues and a high number of salt bridges and hydrogen bonds in thermophilic proteins [12]. On the other hand, this is in contrast to the abundance of highly charged proteins in aggregates found in aging organisms and to their proposed pronounced susceptibility to destabilization by age-related oxidation and charge modification [14]. Furthermore, some works calculated a destabilizing effect of ionizable groups due to charged residues desolvation during protein folding [5] and to like charge repulsion, especially in proteins with a high net charge [14]. In this context, amino acids position and surrounding environment are critical: as a rule of thumb to avoid a destabilizing effect, hydrophobic residues are usually buried into the protein core whereas polar and charged amino acids are more often solvent-exposed [1], unless charge burial into protein core entails the establishment of favorable interactions (not necessarily ion-pairing [13]). Analogously, nonpolar and charged residues are usually positioned in the core and at the rim of interacting protein interfaces, respectively [1]. Interestingly, Kumar and Nussinov [12] proposed that the notable amount of buried salt bridges found in hyperthermophilic proteins maintain a stabilizing effect since many cooperative electrostatic interactions are established both between the salt-bridging side chains and between these residues and others inside the protein, without suffering from solvent screening.
1.2. Enzyme catalysis
Understanding how enzymes work remains one of the challenges of modern biophysics [15]. Even though electrostatic effects are the best candidates to explain enzyme catalysis, early studies in this field did not consider this possibility, likely because studies on reactions in solution focused on rate constants rather than on rate-limiting activation barriers. In the absence of a proper computational model, it is almost impossible to assess the dielectric effects in the protein and thus to estimate the strength of electrostatic effects. However, pioneering work [16] provided the first quantitative hint that electrostatic effects may have a major role in enzyme catalysis, suggesting that the catalytic power of enzymes is almost exclusively due to electrostatic effects and specifically to the preorganized electrostatic environment of their active sites [15], [17]. Since all quantum chemistry is electrostatic ,“electrostatic catalysis” [16] takes into account the effects of the protein charges, permanent/induced dipoles, metal ions and the solvation by bound water molecules [15]. Studies that identified electrostatic effects as a key factor in enzyme catalysis concerned several enzymes and enzyme classes, as reviewed by Warshel and co-workers [15]. In addition to the direct effect on catalysis mediated by transition-state stabilization, electrostatics also influences the allosteric control of catalytic efficiency. For instance, in the activation of Ras, GAP binding results in a major electrostatic stabilization favoring GTP hydrolysis by an arginine finger and by stabilization of the catalytic configuration [15]. The importance for catalysis of electrostatic preorganization over conformational motions could be depicted by accurate computational analyses. In particular, a work on dihydrofolate reductase and other enzymes demonstrated that the role of flexibility and conformational dynamics in catalysis is negligible, while the largest contribution arises from electrostatic preorganization, as changes in motion depend in turn on changes in the reaction potential surface, modifying the reorganization free energy [18], [19].
Krzemińska and co-workers investigated the dynamic and electrostatic effects governing the catalytic power of HIV-1 PR protease, by computing the free energy surfaces for all possible mechanisms, in terms of potentials of mean force. Such calculations showed that the contribution of dynamic effects to the activation free energy is relatively low, while the electric field created in the active site is critical for the electronic reorganization required during the reaction [20].
Computational combined quantum mechanics and molecular mechanics (QM/MM) methods are currently considered as the most appropriate to simulate dynamics and to dissect steps of chemical catalysis as it proceeds in very large and flexible systems such as enzymes. These methods have been applied to the reaction catalyzed by a rat-liver glycine N-methyltransferase (GNMT), that is the transfer of a methyl group from the S atom of the donor molecule S-adenosyl-L-methionine (SAM) to the acceptor N atom of a glycine. The wild-type enzyme and three mutants, in which a single active-site tyrosine had been changed either in alanine, glycine or phenylalanine, were compared to check whether mechanical compression by the protein structure could destabilize the reactant state more than the transition state (resulting in a lower free-energy barrier). Slight differences were observed, in accordance with similar calculated values of activation free energy, but mutants showed a diminished ability to achieve a determined donor-acceptor distance. Mechanical constrains were consequently ruled out and results were interpreted by suggesting the WT enzyme provided the most favorable electrostatic environment to stabilize the charge of the methyl group all along its transfer (i.e., as the reaction proceeds, in the stabilization of the transition state with respect to the reactant state, in the accommodation of the donor-acceptor distance, in each conformational fluctuation till the release of the product) [21].
1.3. Protein-protein interactions
Electrostatic forces play an established role in protein-protein interactions (PPIs). The electrostatic interaction energy between two molecules carrying only one unit of net charge and positioned 10 Å away from each other is much higher (at such distances) than any other energy component contributing to binding [22]. Of course, in addition to electrostatic forces depending on residues of protein partners, salt concentration and pH also play an important role in modulating electrostatics guidance of PPIs, as reviewed by Jensen [23]. The intracellular environment is populated by a huge crowd of molecules, including proteins, each needing to find the right partner among hundreds of thousands of candidates from the cellular proteomic complement; this notwithstanding, a fast recognition process is needed in most instances [24]. Indeed, the recognition process can be enhanced by long-range electrostatic guidance, a force that selects and brings the interacting partners together. A few examples, among many others, of PPIs favored by electrostatic steering, are interactions between (i) fasciculin and acetylcholinesterase [25], (ii) formation of α-lactalbumin–lysozyme heterodimers [26], (iii) antibody Fc heterodimer formation in bispecific antibodies [27], and Cdc42 recognition by Wiskott-Aldrich syndrome proteins [28]. Formation of the protein complex KIX-pKID is favored by electrostatic interactions providing charge-charge stabilization of the native state and contributing to the funneling of the binding landscape [2].
Chaperones recognition of unfolded proteins is commonly believed to rely mostly on hydrophobic interactions [29]; without diminishing the importance of hydrophobic effects, long-range electrostatic interactions have been recently found to play a crucial role in the first binding stage of some molecular chaperones (e.g. Escherichia coli Spy) to their client proteins [29]. In contrast, other chaperones (i.e. DnaK of E. coli) not only benefit from electrostatic steering effects but specifically recognize patterns including charged residues amongst nonpolar ones [30].
Methods for driving alteration of the electrostatic properties are considered for designing proteins with optimized binding and activity [31]; for instance, electrostatic optimization was used for modulating protein-protein association rates [32]. As reported by Ritchie and Webb [33], structural and electrostatic factors are crucial in the affinity and specificity of macromolecular interactions and chemical reactivity. For example, specific electrostatic interactions between charged amino acid residues regulate the binding of (i) von Willebrand factor to blood platelets [34] and (ii) of GTP binding proteins of the RAS superfamily with downstream effector proteins [33]. It is well-known that clusters of charged and polar residues that are located at protein-protein interfaces may enhance complex stability [35].
1.4. Protein binding to other molecules
Electrostatics plays a central role in the interaction of proteins with highly charged biological molecules such as DNA, RNA and membrane phospholipids. For instance, it is widely known that most nucleic-acid binding proteins are rich in positively charged residues (especially in their binding regions), which mediate attraction with the negatively charged backbone of DNA/RNA. Intriguingly, several nucleic acid-binding proteins may cause frustration in the DNA-binding region, which favors their binding to DNA but hampers binding of other protein partners. These interactions are mainly driven by electrostatic forces that, therefore, contribute to modulating nucleic acid accessibility [2].
Long- and short-range interactions play a pivotal role in the assembly of the spliceosomal U1 small nuclear ribonucleoprotein. In particular, long-range electrostatic interactions mediate high-affinity binding between the positive charge of U1A protein and the negatively charged stem loop 2 RNA of the U1 snRNA, while short-range interactions (hydrogen bonds among RNA bases and amino acid side chains) favor a specific binding [36].
DCL-1 is an intrinsically disordered protein involved in miRNA biogenesis that recognizes the dsRNA by acquiring a well-folded structure after engagement with its interaction partner; electrostatics was found to play a key role in both the protein-RNA recognition and in folding of the complex [37].
3dRPC, an algorithm for prediction of 3D RNA-protein complex structures, has been used for sampling possible RNA-protein complex conformations by calculating the geometric and electrostatic complementarities and stacking interactions at the RNA-protein interface [38].
The long-range character of electrostatic forces has been exploited by molecular recognition processes that need fast rates. As an example, very rapid degradation of some molecules may represent an adaptive advantage, e.g. high rate in neurotransmitters clearance is crucial to fast resetting of neuromuscular junctions and to mediate quick escape from predators and dangerous agents [39]. Indeed, electrostatic steering enhances by few hundred fold the degradation of the positively charged acetylcholine molecule by the negatively charged active-site region of synaptic acetylcholinesterase [40].
Another example of electrostatics involvement in fast biological processes is represented by ricin poisoning. The catalytic A chain of ricin acts as a glycosidase able to remove a specific adenine residue from an exposed loop of the 28S rRNA, leading to rRNA breakage, ribosome inactivation, and final block of protein synthesis. The high toxicity of ricin depends on the capacity of its A chain to inactivate a few thousand ribosomes per minute, i.e. faster than the cell can make new ones (https://www.uniprot.org/uniprot/P02879). The rate constant for this reaction at low ionic strength is increased by roughly a 3 million factor because of electrostatic attraction between ricin and the ribosome [41].
1.5. Electrostatic interactions in protein binding to plasma membrane and organelles
The role of electrostatic interactions as mediator of protein localization at different cell compartments has been studied by several research groups. It has been proposed that peripheral proteins attach to biological membranes either due to the structural and geometric arrangement of the latter, that is curvature and lipid packing, or to electrostatic forces exerted by negatively charged lipids [42]. Coordination of these two “protein adsorption strategies” fundamentally impacts on protein localization at well-defined microdomains and consequently, on the initiation of processes that require biological membranes, like autophagosomes formation and vesicle budding or fusion [42], [43]. The three main classes of lipids present in eukaryotic membranes are glycerophospholipids (GPLs), sphingolipids and sterols. The GPL class is the most enriched one, accounting for phosphatidylcholine (PtdCho), phosphatidylethanolamine (PtdEtn), phosphatidylserine (PtdSer) and phosphatidylinositol (PtdIns). The most prominent lipid involved in membrane electrostatics is the negatively charged PtdSer. PtdSer is produced through phospholipid exchange between the Endoplasmic Reticulum and mitochondria at sites where the two organelles lie in close proximity [44], [45]. The membranes that define these points of juxtaposition are known as mitochondria associated membranes (MAMs): despite being produced there, PtdSer is not a main component of MAMs, being retrieved at high percentages (up to 10%) on the Plasma Membrane (PM). Along with the phosphoinositide phosphatidylinositol(4,5) bisphosphate (PIP2), PtdSer mediates the association of proteins at membranes of the late secretory pathway, which is directly proportional to their electrostatic potential (the extent of their charge) without any dependence on the exact aminoacidic sequence [46]. An interesting example of electrostatically-driven subcellular localization concerns the tumor suppressor PTEN, whose attachment to PM is mediated by polycationic stretches within its sequence. Phosphorylation at its C-terminus hides this positive charge, thus controlling its localization and function [47]. Another example regards the G proteins Rac1/2, which associates to the highly charged surfaces of endosomes and cytosolic side of the PM proportionally to the number of basic residues included at their C-termini [46], [48], [49]. Bacterial toxins also exploit electrostatics to strive small G proteins, thus exerting their cytotoxic effects [50].
Phagocytic membranes facing the cytosol are enriched of PtdSer and PIP2, playing a role in pathogen engulfment [51]. The negative charge however decreases along the endocytic pathway, due to the gradual decrease of PIP2 abundance from the initial phagosome to lysosomes [48], [49], [52]. The importance of electrostatics in the phagocytic process is corroborated by two findings. First, some bacteria (e.g. Salmonella typhimurium) have developed as a pro-survival mechanism the expression of a phosphatase able to reduce PIP2 and PtdSer levels, hence impairing the endocytic path and hampering their lysosomal-mediated degradation [53]. Second, electrostatic dynamics associated with phagocytosis underlie a recent drug delivery approach based on the use of Cell Penetrating Peptides (CPPs). Some CPPs are enriched of positively charged aminoacids which favour the phagocytic internalization of their cargo. While drug internalization with this approach is effective, the success of CCPs-fused therapeutics is blunted because drugs are degraded during the final steps of the phagocytic process. To avoid this issue, secondary/tertiary amine groups having a pKa close the pH of the endosome have been introduced. Their proton buffering ability reduces endosomal vesicle acidification, in turn blocking the extrusion of chloride through endosomal pumps and enhancing water uptake into the vescicle, thus leading to osmotic swelling and drugs release into the cytosol [54], [55].
Protein linkage to anionic lipids is pivotal not only for protein localization and related intracellular processes, but also for regulatory feedback loops on the membrane composition itself. Negative surface potential indeed attracts cationic sphingolipid hydrolases/transferases and phospholipases that, through their activity, lower or increase the abundance of their substrates thus contributing to membrane remodelling [56], [57]. Electrostatic interactions at the surface of organelles and cytosolic side of PM are also regulated by scramblases, floppases and flippases, enzymes responsible for the flip-flop of GPLs and hence asymmetry of biological membranes. Reversing the distribution of GPLs between the two sides of the lipid bilayer is yet another signal transduction mechanism. Exposure of the negatively charged PtdSer moiety to the external side of the PM results in cell signaling cascades both intracellularly and at extracellular level and cell death. PtdSer depletion of the PM cytosolic leaflet would induce the release of electrostatically-associated proteins into the cytosol, while its exposure on the surrounding environment would represent a cue for phagocytes recruitment and clearance of dead cell [58], [59].
Changes in electrostatic interactions during apoptosis occur not only at PM: negative charges on the surface of mitochondria contribute to the recruitment of cationic proteins, such as K-Ras, Bid and caspase 8. A dianionic molecule, cardiolipin, is exposed on the surface of these organelles during early steps of the apoptotic process: this seems pivotal for the progression of the cell death cascade as dampening of the negative membrane charge through cardiolipin binding proteins blocks the process [60], [61].
On the contrary, electrostatics play minor roles in the association of proteins to lipid rafts, which are membrane portions enriched of cholesterol, sphingomyelin, phospholipids with saturated fatty acid chains and gangliosides at the expense of phospholipids with unsaturated acyl chains [62].
Basic electrostatic features (and organellar association) of proteins are further modulated by post-translational modifications, like phosphorylation and lipidation (introducing negative charges), proteolysis that removes charged aminoacidic sequences [63], [64], [65].
Overall this evidence highlight that electrostatic interactions are key mediators of protein localization and that keeping the balance between charged and non-charged membranes domains is critical for cell physiology.
2. Computational analysis and prediction tools
Solvent plays a pivotal role in mediating biochemical processes such as the interaction between proteins or between proteins and ligands. The environment in which these events take place, is often made up of water and ions. While modeling proteins in such a complex setting, water molecules and ions can be treated in explicit or implicit (continuum) way. The former one allows for more accurate simulation but it is more expensive from a computational point of view. Conversely, implicit solvent methods are affected by drawbacks related to lower accuracy, but are faster and more suitable for studies focusing on the solute (the protein) behavior. Therefore, we choose to discuss only the continuum solvent treatment. Several methods are available in the field and this section is not a comprehensive list; hereafter, instead, we provide an overview of popular methods used in the protein electrostatic computations field, which are user-friendly and suitable for biotechnologists (see Table 1).
Table1.
Selected tools for PBE computation.
| Name | URL | Features | References |
|---|---|---|---|
| APBS | http://nbcr-222.ucsd.edu/pdb2pqr_2.0.0/ | Electrostatic focusing, pKa calculations | [72], [77], [78] |
| WebPIPSA | http://pipsa.eml.org | Electrostatic potential comparison | [73], [79] |
| DelPhi Suite | http://compbio.clemson.edu/sapp/delphi_webserver/ | Object-oriented, Gaussian-based smooth dielectric function, pKa calculations, DelPhiForce | [75] |
| PBEQ-Solver | http://www.charmm-gui.org/input/pbeqsolver | Electrostatic focusing, pKa calculations, protein-protein/DNA/RNA electrostatic interaction energy calculation | [76] |
| Bluues | http://protein.bio.unipd.it/bluues/ | Born radii calculations, solvation electrostatic free energy calculations, electrostatic forces calculation, pKa calculations, electrostatic potential calculation | [71] |
2.1. Basis of continuum electrostatic calculations
Electrostatic properties depend on the distribution of whole and partial charge on the 3D protein structure [31]. Coulomb’s law expresses the electrostatic potential V(r) as follows:
where: q = charge; r = distance; ε = dielectric constant relative to vacuum permittivity and ε0= dielectric medium. However, Coulomb’s law is not suitable for describing electrostatics in proteins, as it describes a system with a single dielectric medium, whereas proteins show a hydrophobic core enveloped by the solvent. Therefore, electrostatic calculations for proteins are carried out using the Poisson-Boltzmann Equation (PBE):
Here, the solvent is treated as implicit: in this way, dynamic effects of water are not directly internalized, leading to a better analysis of electrostatics [31]. Protein dielectric constants express the effect of the protein environment, reflecting proteins structure and sequence properties [66]. Moreover, continuum electrostatic studies and molecular dynamic simulations revealed that different structural motifs located in the protein may present different dielectric constant values [67]. Implicit solvent calculations (continuum electrostatics) provide a water phase atomic detail reduction and intrinsically an equilibrium solution [68]. The medium dielectric constant is 80 (ε = 80, water), whereas a dielectric constant of 4 (ε = 4), used for protein, should account for electronic polarization and small backbone fluctuations. Amin and Küpper, in a recent paper calculated the mean dielectric constant of more than 150000 proteins in the PDB, presenting a value of 3.23. Once calculations have been performed (e.g. by software tools reported below), an electrostatic map is computed and can be visualized onto the molecule, as reported in Fig. 2.
Fig. 2.
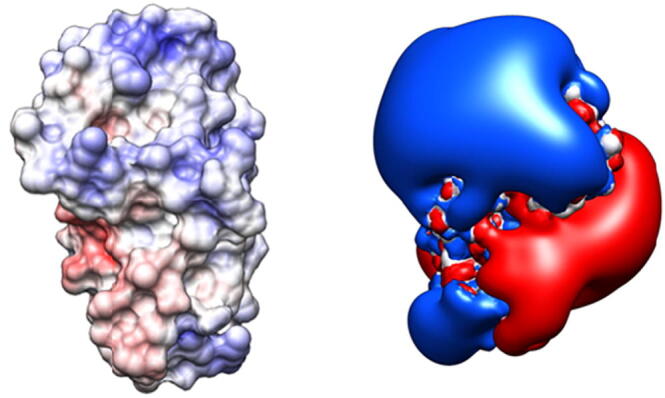
Electrostatic representations: surface projection (left) and isopotential contours (right). Positive potential is highlighted in blue, negative one in red. Images obtained via UCSF Chimera [69].
Isopotential contours are plotted at levels of different ±nkBT/e which means that the electrostatic component of the potential energy of interaction between the protein field and an elementary charge of +1e located somewhere on the +nkBT/e isopotential surface, would be equal to:
Another formalism involved in continuum electrostatics is Generalized Born (GB) method. This approach also takes part in protein and nucleic acids Molecular Dynamics (MD) simulations in order to model hydration effects and obtain solvent-dependent forces [70]. GB model is an approximation for the linear PBE:
Where
where is the permittivity of free space, is the dielectric constant of the solvent, is the charge on particle i, is the charge on particle j, is the distance between the particles I and j and is the Born radius, expressing the distance between the atom and the molecular surface. The Born equation describes the transfer free energy of a single spherical ion having a single charge at its center from the gas phase to an environment characterized by . The main goal of GB model relies on the estimation of polarization charge contributions to the self-energy of each charge and the interaction energy of each pair of charges [71].
2.2. Prediction tools
2.2.1. APBS
Adaptive Poisson-Boltzmann Solver (APBS) software package can solve PBE using electrostatic “focusing”. This is a popular finite difference technique for generating accurate solutions to the PBE in subsets of the problem domain, such as a binding or titrable sites within a protein. This approach implies that charges and dielectric constants are discretized over a grid. Protein molecular surface (MS) is mapped onto a user-defined density 3D grid, then used to obtain the finite difference solutions of the PBE. The product of the electrical potential and charge at each voxel (grid point), where a real charge has been mapped, provides the electrostatic free energies. APBS performs calculations by using initially a coarser grid and then a finer one for the refinement. APBS carries out calculations with a grid spacing of 0.5 Å. APBS first defines the solvent accessible regions of the protein and calculates the electrostatic potential for each of the grid points. Finally, the electrostatic potential is mapped onto the MS (.dx file). Electrostatic calculations can be applied on molecules of a wide size range. APBS can be used through PDB2PQR server [72] at http://nbcr-222.ucsd.edu/pdb2pqr_2.0.0/
2.2.2. WebPIPSA
WebPIPSA [73] (http://pipsa.eml.org) allows computing and comparing electrostatic potential among a large number of proteins. This pipeline launches a workflow combining several algorithms thus it is not based on a single calculation method. Specifically, after structures upload, WebPIPSA workflow can be summarized as follows:
- Structures superimposition: with “sup2pdb” option selected, the sequence of one structure, referred to as the template, undergoes a pairwise sequence alignment with the remaining coordinate files. Alignments are then used to perform structures superimpositions;
- Polar hydrogens addiction: WHAT IF [74] adds polar hydrogen atoms to the structures. Protonation is executed at pH 7 for all residues except for His, which is treated as singly or doubly protonated;
- Electrostatic potentials calculations: this step can be performed using APBS or UHBD. Electrostatic potentials are automatically calculated. The user must choose ionic strength and temperature. The solvent is treated as implicit;
- Electrostatic potentials comparison: a probe of radius 2Å defines the protein surface. PIPSA compares potentials in the complete protein surface skins. The skin extends out from the protein surface with a thickness of 3Å. Electrostatic protein comparison is possible due to the implementation of the Hodgkin or Carbo similarity indexes. The similarity indexes range from -1 (anti-correlated potentials) through 0 (uncorrelated) to +1 (identical potentials). These values are converted into distances given by where SI stands for similarity index. Distance values are comprised between 0 (identical) and 2 (anti-correlated potentials);
- Clustering analysis and epograms generation: in this last step WebPIPSA generates an output consisting of a heat map (representing the distance matrix) and an epogram, allowing for the fast identification of inter-protein relations.
2.2.3. DelPhi Suite
DelPhi Suite [75] is available at http://compbio.clemson.edu/sapp/delphi_webserver/ or as a standalone software. This popular platform has been redesigned, utilizing the object-oriented programming technique and other unique features provided by C++ to ensure various levels of multiprocessing and memory distribution. This leads to a significant improvement in computational time needed to solve the linear and nonlinear PBEs and large-scale modeling. In the case of relatively small systems (less than 3003 grid points), the best choice is the regular single-CPU implementation. For medium-size problems (3003 to 6003 grid points) the OpenMP implementation is the most suitable choice. Finally, extremely large problems (>6003 grid points) can be handled by the MPI DelPhi implementation. Both standalone and webserver versions are characterized by the following features:
- Gaussian-based smooth dielectric function: this approach takes into account the inhomogeneity of both the macromolecule and the space between the solute and the solvent. Here, there is a smooth transition between macromolecule and water phase: densities are represented as Gaussian density functions and the dielectric constant as a function of space. This method assigns higher dielectric constant at the protein surface with respect to the hydrophobic core; moreover, the cavities inside the protein are described by a dielectric constant higher than in the rest of the macromolecule but smaller than for bulk water. The Gaussian-based smooth dielectric function shows that surface water molecules are not much freer to move than the bulk ones.
- Mobile ions treatment: the treatment of mobile ions relies on a desolvation penalty term in the PBE in order to avoid the ions to enter the protein.
- Zeta-potential: it reflects the aggregation tendency. High values of Zeta-potential mean electrostatic repulsion and therefore, no particles/macromolecules aggregation.
- pKa computational approach: it is located into a Gaussian-based approach protocol. Here macromolecules ionizable groups pKas are calculated without defining molecular surface (surface-free pKa approach). This method is able to predict the pKa of polar groups and it takes into account salt effects.
- DelPhiForce: the electrostatic force between two molecules (proteins, DNAs, lipids, small molecules, etc.) is computed via the FRC module. Electrostatic forces are pivotal in both molecules docking and mutual orientation prior to this physical event. The electrostatic force between two objects can be highlighted by vector representations.
- SAAFEC method: SAAFEC stands for “Single Amino Acid Folding Free Energy Changes”. DelPhi Suite is able to model the effect of nonsynonymous variants on the folding free energy, particularly important for personalized medicine applications due to the correlation between the magnitude of folding/binding free energy change and the propensity of a given mutation to be pathogenic. SAAFEC method is based on Molecular Mechanics Poisson-Boltzmann (MM/PBSA) approach.
- SAAMBE method: using sequence- and structure-based approaches, the SAAMBE (Single Amino Acid Mutation related change of Binding Energy) method computes binding free energy changes due to amino acid substitutions. The method is characterized by a MM/PBSA-based component and a set of statistical terms.
- SAMPDI method: it computes the effects of amino acids substitutions on the binding free energy of protein-DNA/RNA complexes. This is a MM/PBSA-based feature.
- MSSM method: the MultiScale Sampling Method is able to model the docking between large and small molecules via a Monte Carlo procedure. First, MSSM calculates the electrostatic energy of the entire system at a coarse-grained resolution. Then, this information is shifted to a region of interest, calculating the electrostatic energy at a significantly finer resolution.
2.2.4. PBEQ-Solver
PBEQ-Solver software [76] (http://www.charmm-gui.org/input/pbeqsolver) carries out PBE calculations using coarse-grid spacing (1.5 Å before and 1 Å after focusing). This program is available as a web GUI, implementing the CHARMM PBEQ module to solve the PB equation. The electrostatic potential of the solvent accessible surface and the isopotential contours are visualized by a Java applet (MarvinSpace molecular visualization software). PBEQ-Solver is able to compute protein-protein/DNA/RNA electrostatic interaction energy and pKa of selected titrable residues in both aqueous and membrane environments. Calculations are provided in two steps:
1. Conversion of a PDB file into CHARMM readable files: the user can select part of a protein chain or a model in the case of NMR structures, modify engineered residues, select terminal group, protonation, disulfide bonds, phosphorylation state, or generate a biologically functional unit or a crystal packing. In this first step, PDB Reader is able to detect and display disulfide bonds and they can be removed. Moreover, the software can spot engineered residues, converting them to the corresponding natural ones. Finally, PDB Reader is able to manage undetermined coordinates by building them using a predetermined internal coordinate table.
2. PB calculations: the PBEQ-Solver module can perform three types of PB calculations for:
- Electrostatic potential and solvation free energy:
where is the electrostatic solvation energy and is the reaction field potential.
- Protein-protein/DNA/RNA interaction energy:
where , and are the electrostatic potentials respectively of the complex and its components A and B in solvent or membrane environments.
- pKa of a selected titrable residue:
where
2.2.5. Bluues
Bluues software [71], available at http://protein.bio.unipd.it/bluues/, performs electrostatic calculations using the GB model. At first, the program computes generalized Born radii from numerical surface integrals. Then, Born radii are used to carry out electrostatic analyses. Depending on user’s requirement, the output will show:
- the generalized Born radius of each atom;
- the solvation electrostatic free energy;
- the electrostatic forces on each atom;
- the pH-dependent properties;
- the pKa of all ionizable groups;
- the electrostatic potential at the surface of the molecule;
- the electrostatic potential in a volume surrounding the molecule.
3. From functional fingerprints to biotech applications
Biological activity requires proper interaction between proteins and their interactors. Since electrostatics plays a pivotal role in defining molecular recognition, the assessment of electrostatic complementarity of protein-ligand complexes provides insights on why ligands bind and on how binding can be modulated. These issues have been addressed by Bauer and Mackey [80], who developed a system to predict electrostatic complementarity (EC) between enzymes and substrates. Particularly, the authors suggest that displaying EC onto molecule surfaces might be a straightforward method to determine which mutations may improve binding efficiency and, therefore, opening the road for next steps in protein engineering.
Hereafter we provide several examples of biotechnological applications where electrostatics modulation acts as the main player.
3.1. Electrostatic fingerprints in enzyme stability and solubility
Covalent disulfide bridges, representing the strongest intra-chain bonds in proteins, are often crucial to protein thermal stability and thus of special interest in the stabilization of biotechnologically relevant mesophilic enzymes by rational engineering. However, covalent bridges could be too rigid to allow proper conformational changes required for protein function and stabilization can be obtained by modifying the number and positioning of salt bridges, as electrostatic forces are strong enough to boost stabilization, while still allowing for local conformational plasticity needed to mediate biochemical functions.
Stability and solubility are essential characteristics for enzymes used as biocatalysts. In the presently imperative research of new tools and methods for green chemistry applications, attention has mainly been focused on (i) accumulating genomic and metagenomic data [81]; (ii) the engineering of enzymes by rational and/or directed evolution [82]; and, only more recently, on (iii) the development of new compatible ionic liquids (ILs) to be employed in substitution of harmful organic solvents. Advanced ILs are sought in biocatalysis as potential ‘greener’ solvents, being non-volatile, non-flammable, biodegradable, less expensive and generally well-tolerated by proteins [83], [84]. In any case, engineering approaches aiming to increase enzyme stability and solubility also have to safeguard (or possibly improve) their enzymatic activity. So far, when computational analysis has been employed as a preliminary or synergic tool to genetic engineering, it has been preferentially focused on electronic interactions occurring in internal shells rather than on the surface of protein structures. Indeed, charges in or nearby the catalytic site (coordinating e.g. metal ions, cofactors, substrate molecules, reaction intermediates), defining its entrance or, also, circumscribing tunnels for access to or exit from it, are the most obvious determinants of activity and stability. In spite of this, charged amino acid distribution on the surface of an enzyme has been almost exclusively considered as a mere determinant of protein solubility. However, surface charges have a key role in mediating the conformational response of enzymes to the chemical-physical behavior of the medium in which the reaction occurs. This response, even though not always evident, in turn, has an unavoidable impact on the catalytic activity, in terms of both kinetic parameters and regio- or stereo-selectivity. A recent example of this is provided by a work studying the effects of mutating charged amino acids exclusively located on the surface of Bacillus subtilis lipase A (BsLA) [85], an enzyme widely utilized in industrial biocatalysis and thoroughly studied by site-directed mutagenesis. The authors planned to merge eight different mutations on the surface of the protein, all introducing negatively charged amino acids (glutamic and aspartic acids): four of them had been previously identified in a set of twelve substitutions capable of increasing BsLA thermostability [86]; the other four substitutions had been associated to an IL-tolerant phenotype [87]. The obtained octuple variant of BsLA demonstrated a surprisingly incremental effect on both thermal resistance and solubility in ILs. Noteworthy, its specific activity was shown to be comparable to the wild-type enzyme and intermediate between the two control quadruple variants maintaining the four thermo-stabilizing and the four IL-solubilizing mutations, separately (with the former control variant having an almost double activity with respect to the wild-type enzyme).
Efforts in engineering proteins surface charges to produce more useful and applicable biocatalysts have been addressed to different classes of enzymes and have been reviewed elsewhere [88]. Besides the aforementioned example regarding a lipase, we can also mention (i) a research producing negatively supercharged cellulases resistant to lignin and better performing on simulated pretreated lignocellulosic biomass [89] and (ii) a work introducing the profitable properties of halophilic enzymes to the mesophilic bovine carbonic anhydrase II, so producing an extreme halotolerant biocatalyst [90].
Indeed, enzyme stability can be influenced by surface electrostatic forces, especially when considering multimeric enzymes, since surface features modulate PPIs and thus are crucial to either proper dimerization/oligomerization, docking to other proteins, or undesired formation of insoluble aggregates.
To provide hints into protein solubilization strategies, Lawrence et al. investigated the effect of protein net charge on aggregation and precipitation [10]. The introduction of many surface mutations that led to both negative or positive “supercharging” caused a very strong improvement in protein solubility upon thermal and chemical denaturation and function restoring upon refolding. Interestingly, the formation of multimers was not significantly reduced and the physiological function was retained (albeit with protein dependent alterations) [10].
In addition to structure-based mutational attempts to increase protein solubility and purification yields by improved electrostatic features, helpful insights can derive from comparative computational analyses of homologous soluble and insoluble enzymes. A recent example of such an approach is provided by a study in which a dataset of Baeyer-Villiger Mono Oxygenases (BVMOs), enzymes of interest to biocatalysis hence to biotechnology, was investigated by comparative electrostatic distance (ED) analysis. In this study, enzyme clustering by ED was found to be independent of phylogenetic classification, as insoluble enzymes from different phylogenetic BVMO classes clustered altogether rather than with soluble members from the same class, opening the route to in silico predictive protocols for the selection of multiple mutants in rational design projects [91].
Indeed, electrostatic forces are crucial to catalysis itself and proper calculation may provide the basis for engineering. For instance, findings about the electrostatic properties of the active site could be used as a mold for future drug design [20]. Since their role in enzyme catalysis is now recognized, computation of long-ranged electric fields could become a primary design parameter in the discovery of new catalytic materials. In their perspective paper [92], Welborn and co-workers discuss how to use electric fields for computational optimization of the biocatalytic performance of a synthetic enzyme. At the same time, the authors suggest that such computation may represent a unifying descriptor for catalytic design when considering a range of catalysts [92]. An accurate analysis of the electrostatic environmental effects may open new routes toward the rational design and optimization of efficient catalysts; however, when using electric field alignments in the reactive centers of complex catalytic systems, much more predictive capacity is needed for computational design providing results transferable to the experimental field [92]. Electrostatic preorganization has been brought into focus by works studying artificial enzymes to be used in biocatalysis and, in particular, diverse variants of Kemp eliminase, an unnatural enzyme used as benchmark for testing protocols for de novo design [92], [93], [94]. Kemp elimination, that is the conversion of benzisoxazoles into salicylonitriles, takes place in solution via a delocalized transition state in which a proton transfer and a bond breaking in a 5-membered ring occur simultaneously; no natural enzyme capable of sustaining such conversion as primary and specific reaction has been identified so far. These studies report on progresses of computational ground plans toward the achievement of enzymes having efficiencies (only) close to those of natural enzymes. Indeed, the best Kemp eliminase obtained so far, a variant named HG-3.17, arose from 17 rounds of mutagenesis (including directed evolution) and screening from the previous, totally designed variant HG-3 [95]. Hybrid QM/MM molecular dynamics simulations have been used to explain reasons for the limited efficiency of the computationally designed enzyme(s) compared to the one evolved in laboratory. Similarly to the truly natural GNMT enzyme (see sub-section 1.2), the analysis revealed the fundamental contribute of electrostatic topography not only in providing an optimal conformational complementarity between the synthetic protein and the substrate but, noticeably, between its active site and the reaction process itself [94].
3.2. Antibodies and nanobodies
The interactions of antibodies (Ab) with their target antigens (Ag) are of particular interest because of their high affinity and specificity and their notable medical and biotechnological relevance. Charged residues directly affect antigen-antibody binding by interacting with other local charges on the protein interactor. Moreover, they are involved in intramolecular interactions that determine secondary structures, in turn indirectly modulating the biophysical properties of an antigen-antibody complex. Such a relevant role in determining binding abilities of proteins justify the frequent targeting of charged amino acids in protein engineering works aiming to modulate antibody affinity and selectivity, often driven by energy calculations and electrostatic potential predictive tools. In a computational design study that led to improving the affinity of different antibodies for their protein targets, the electrostatic term turned out to be a better predictor for enhanced binding than the total calculated free energy [96]. According to the authors, the designed mutations improved binding by either increasing electrostatic interactions following the introduction of a charged residue or by substituting a polar residue with unfavorable desolvation with a hydrophobic one.
The antibody 11K2 underwent a 4.7 fold affinity enhancement for its target (MCP-1) following the introduction of a single mutation involving a charged residue in an in silico engineering work and the enthalpic contribution in the transition state (probably as early as in long-range electrostatic attraction) was suggested as the main driving force of the improvement [97]. It is worth noting that 5 out of 12 proposed mutations (all involving charged residues) had an enhancing effect on affinity. Yoshida et al. [98] investigated the effect of surface charge manipulations at the interface between IFNγR domain D1 and a neutralizing antibody, the complexing of which was abolished or notably reduced by singularly disrupting three electrostatic interactions between the antigen and the antibody and restored by charge-exchanged Ag–Ab pairs. These results showed that electrostatic forces have a pivotal role in PPIs (especially by long-range attraction of partner residues). However, the failure of binding restoration in one case, together with proteins biophysical characterization and molecular dynamics of the different complexes analyzed, confirmed electrostatics crucial impact on protein conformation, in turn further affecting binding events. The authors proposed surface charge conversion as a new strategy for boosting the production of antibodies against weakly immunogenic antigens [98], as depicted in Fig. 3a.
Fig. 3.

. Protein electrostatic manipulation for immunology applications. (a) As proposed by Yoshida et al. (2019), a weakly immunogenic antigen (left) could be charge-modified to elicit the production of antibodies (center), which could be further engineered to make the surface of their Ag-binding site complementary to the native antigen (right). (b) Homodimerization competes with heterodimerization in bispecific antibodies production (left). The formation of heterodimers can be enhanced by engineering surface charges on the Fc portion so that two identical monomers repel each other while different monomers attract (right).
While complementarity determining regions (CDRs) on antibody heavy and light chains variable domains (VH and VL) mostly determine antibody affinity for its target, the sequences of framework regions (FRs) on variable domains and constant domains are main determinants of antibody stability [99]. Although these considerations should be taken into account in protein engineering, some case studies have shown that such a distinction is not exhaustive, especially in electrostatic manipulation.
A single charged mutation (D32H) mapping on the L-CDR1 region of hu3F8 antibody was able to significantly increase its affinity for GD2 ganglioside by altering the surface charge distribution and improved its ADCC, CDC and in vivo anti-tumor activities [100]. Even though a second charge mutation (E1K) mapping on L-FR1 further strengthened hu3F8-GD2 affinity, the double mutant did not show better effector functions than the single D32H mutant [100].
Fukunaga and Tsumoto [101] have demonstrated that besides commonly investigated CDRs, antibodies framework regions could also represent the target for mutations aimed at enhancing antibody binding to its target. Following the introduction of cationic and anionic residues in the FRs of single-chain variable fragments (scFvs) directed toward an acidic and a basic epitope, respectively, they observed higher association rates (kon) values by SPR and thus hypothesized that long-range electrostatic attraction might improve antibody-antigen affinity. Some concerns about possible scFv stability reduction upon FR engineering were raised according to the high degree of sequence conservation in this region and experimentally confirmed [101].
The particular influence of long-range electrostatic interactions on the association constant (rather than on the dissociation constant) has also been demonstrated for other protein-protein complexes, e.g. the kon and the affinity of the protein inhibitor BLIP for its target, the β-lactamase TEM1, were improved more than 200-fold (while leaving koff rather unaffected) by modulating BLIP surface charges near the binding interface to increase the electrostatic attraction between the two proteins [102].
Antibody-based therapeutics could benefit from electrostatic manipulation by mechanisms other than increasing affinity for the recognized epitope. The manipulation of few charged residues on Fc fragments has been reported as a feasible way to modulate their interchain interaction, in particular enhancing the formation of desired heterodimers and suppressing side Fc homodimerization in bispecific antibodies production [27], [103]. Analogously, the introduction of complementary charged protein tags in the constant domains of a Fab heavy and light chains – in proximity to the interchain disulfide bond – led to electrostatic steering-mediated increase in Fab assembly, thus improving secretion from insect cells [104].
The 10E8 antibody, targeting the membrane-proximal external region (MPER) of the gp41 subunit of the HIV-1 envelope glycoprotein (Env), shows a positively-charged patch on the Fab surface which is suggested to favor the binding to its target by engaging electrostatic interactions with negatively-charged membrane phospholipids [105]. 10E8 neutralizing activity against many HIV-1 isolates was increased by broadening its positive surface patch with the introduction of three solvent-exposed arginine residues [105].
Nanobodies are emerging as potent tools in therapeutics development, diagnosis, and research because of their limited dimensions, easy recombinant production and peculiar binding abilities to difficult-to-target antigens [106]. Electrostatic interactions are gaining a notable interest also in nanobodies engineering.
Recently a structure-based computational method was used to design a single point mutation involving a charged amino acid on the VHNAC1 nanobody, which consequently showed an increase by more than an order of magnitude in affinity for nonamyloid component (NAC) region of human α-synuclein: this improved binding was predicted to stem from electrostatic complementarity optimization at the binding interface between the nanobody and its target [107]. Analogously, Cheng et al. [108] applied an in silico affinity maturation workflow to an anti-CD47 nanobody, obtaining a set of four mutations that improved both target binding and thermostability, combining the introduction of a salt bridge with other different interactions. The last observation suggests that electrostatic engineering could reach better protein optimization if applied together with other design strategies.
The strong binding of a recently developed nanobody to HIV-1 p24 capsid protein was revealed to be due to several interactions, including hydrogen bonds, hydrophobic interactions, and most notably, a salt bridge and the alignment of electrostatic complementary regions [109]. In particular, the same nanobody was able to bind several p24 subtypes that conserved electrostatic potential homogeneity, while if the broad electrostatic complementarity between antigen and nanobody surfaces was altered by mutations in either of the two proteins, then their binding was diminished [109]. According to this observation, antigenic types classification based on their surface electrostatic potential could be useful in predicting antibodies cross-reactivity against them.
3.3. Electrostatic binding to small molecules and nanomaterials
Electrostatic interactions are particularly relevant in small molecules binding to proteins, the former being a substrate, an effector/modulator or an inhibitor. Electrostatic interactions, either transient or stable, together with shape complementarity and hydrophobic interactions, play a key role in defining the enthalpic contribution in ligand recognition. Since electrostatics impact slowly decreases with distance, it allows the establishment of long-range interactions.
Medicinal chemistry studies devoted to developing and optimizing hits and leads that could target the activity of enzymes or receptors rely on the availability of structural data as well as on our proficiency in predicting such interactions where direct experimental evidence is missing [110], [111]. Although with different degree of success, a large number of strategies have been recently proposed to evaluate and simulate the impact of electrostatic interactions on this process [80], [112], [113], [114]
For example, Waldner and co-workers developed an algorithm that was able to predict the correlation between electrostatic features and substrate specificity of nine representative members of the chymotrypsin family of serine proteases [113]. In serine protease, target recognition occurs through a pattern of sub-pockets that build up the active site cleft features and define upstream and downstream amino acids selectivity within the peptide substrate [113].
Through the building and usage of electrostatic molecular interaction fields (by the software GRID; [115]), their method was able to infer electrostatic substrate preferences both at subpockets close to the cleavage site and far from it, correctly predicting similarities and differences within evaluated proteases. The success of this approach, where Van der Waals interactions contribution was minimized on purpose, demonstrated the relevance of electrostatic interactions in the activity and selectivity of chymotrypsin family members.
Another example of a powerful methodology for estimating the electrostatic contribution in view of drug design purposes is offered by the work of Bauer and collaborators [80]. They developed an electrostatic complementarity (EC) calculation and visualization tool that allows evaluating ligand-protein matching at the interface between the two partners and could guide binding and selectivity improvement. Examples from protein families as diverse as kinase inhibitors, glutamate receptors, G protein-coupled receptors and the leukemia drug target Mcl-1, member of bcl-2 family, have been used by the authors to prove the efficacy of the proposed method as well as good matching between predicted electrostatic complementarity and small molecules bioactivity. Rapid identification of suboptimal electrostatic interactions can be used to guide the introduction of changes able to improve binding.
Activities targeted by such approaches could be highly specific, as well as quite promiscuous. The level of promiscuity in substrate recognition by the macromolecule of interest should be taken into account since off-target binding events as well as elusive results might be related to the selectivity of the targeted interactions.
On the other hand, the necessity of broad-spectrum activity inhibitors, as in the case of ß-lactamases inhibitors, definitely pose a demand for tackling with families of enzymes with few common features and large variability in their subclasses [116]. A successful example is offered by boronic-acid based inhibitor taniborbactam, a molecule currently under clinical development active against both serine and metallo ß-lactamases (sBLIs and mBLIs [117], [118]). Pan-inhibition by boron-based molecules is achieved by mimicking the transition state structure of both metal and serine catalyzed reactions, responsible for hydrolyzing even last resort carbapenems. Both cyclic and acyclic boronic acid derivatives have been proved to establish a covalent complex with catalytic serine in sBLIs, while react and coordinate the bridging hydroxide between Zn metal ions and the metals themselves by boron–oxygens [119], [120].
Electrostatic interactions stabilize the oxyanion intermediate that is formed in the acylation step of the catalyzed hydrolysis in sBLIs and developed inhibitors take advantage of such interactions by mimicking the oxyanion tetrahedral intermediate through a reversible covalent complex embedded in a network of non-covalent interactions. On the other side, mBLIs activity is based on one or two zinc atoms located in the active site. All the inhibitors that target such subfamily explore electrostatic interactions with both metals and bridging activated hydroxide as well as local interactions that confer high affinity toward the active site [121]. A further beneficial contribution is often introduced in BLIs inhibitors by the presence of a carboxylate side group, still mimicking ß-lactams, since it establishes electrostatic interactions with Ser/Thr/Asn residues highly conserved in sBLIs and usually binding carboxylate moiety of antibiotics.
Protein electrostatic interactions also play a pivotal role in nanomedicine and nanotechnology, where they are involved in conferring a biological identity to nanomaterials [122], [123]. It is now well known that as soon as a nanomaterial comes in contact with a biological fluid, a protein corona forms around it and that it influences its targeting efficiency, biodistribution, biocompatibility, drug release, clearance rates and stability [124]. Despite the forces involved in protein corona formation depend on both protein and nanoparticle physicochemical properties (as reviewed by Nel and co-wokers [125]), the main factors driving its formation are represented by surface charge [126], [127] and zeta potential (i.e. the potential at the boundary of the hydrodynamic shear plane of a charged particle [128]). Proteins are able to bind different nanoparticles based on their isoelectric point. In general, proteins with an isoelectric point below 5.5 show a tendency to bind positively charged nanoparticles while the ones with a pI above 5.5 adsorb to negatively charged nanoparticles [129]. Furthermore, serum proteins are more likely to bind negatively charged nanoparticles, reducing nonspecific cellular uptake [130]. Similarly, when composite materials are used as biocompatible scaffold for cell growth [131], [132], protein absorption influences cell adhesion, proliferation, differentiation, cell signaling and tissue regeneration contributing to scaffold final effect [133].
3.4. Electrostatic fingerprints in the evolution of pandemic viruses
Widespread outbreaks (pandemics) of viruses able to mediate severe respiratory diseases, depending on sporadic “host jump” events [134], resulted since 1918 in the death of tens of million people worldwide [135]. Especially in the case of influenza, this picture is further complicated by the emergence of novel reassortant viruses, especially where multiple strains and clades co-circulate [136]. These considerations highlight the importance of shedding light on mechanisms underlying viruses evolution and spreading, in order to boost a coordinated global surveillance network by monitoring genetic changes and predicting ‘evolutionary trends’ among emerging viruses for which animal/human host switching has been reported or is likely to occur [137]. Despite an extensive body of literature concerning evolutionary aspects of viral pandemics linked, for example, to antigenic drift, immune escape, interspecies transmission, host specificity shift and low-to-high pathogenicity shift, any “functional” overall explanation and identification of major protein players in viral clade evolution and spreading is still largely missing. Therefore, improving strategies in functional studies about virus variation is dramatically relevant to human health and vaccine efficacy, as well as to animal husbandry.
In Avian influenza (AI) viruses, haemagglutinin (HA) is the central player in infection and sensitivity to vaccines, as it is the major capsidic protein and main viral surface antigen, which mediates attachment and penetration into the host cell; mutations at the HA surface may result in antigenic drift and allow the virus to escape anti-HA Ab neutralization [138]. Mature HA monomers - which form trimers at the viral surface - show a globular head or Receptor-Binding Domain (RBD) that mediates docking to the host cell by binding sialic acids (SA) as cell entry receptors [138]. This mechanism explains why HA is the pivotal player in host specificity [139] and, when mutations result in increased binding to α 2-6 SA, affinity to the human host is improved [140], [141], [142]. In recent years, a comparative functional analysis of AI virus HA was performed by computational prediction of the haemagglutinin RBD structure and surface electrostatics. Finding that electrostatic closeness can group HAs and their RBDs from different H5N1 virus phylogenetic groups suggested that this kind of analysis could provide function-related, rather than taxonomy-related, classification and thus unveil functional fingerprints [143]. This was confirmed by a deeper analysis on H5N1 clades and subclades, which unveiled electrostatic fingerprints related to clades evolution and spreading. In particular, charge redistribution at the RBD surface was found to relate to the branching of still-circulating clades relative to no longer circulating ones, hence being likely involved in antigenic drift events [143]. Further comparative analyses on a different AI virus subtype, H9N2, allowed to confirm that the electrostatic variation by surface charge redistribution is a general fingerprint in AI virus evolution, and a hallmark for AI viruses [144]. Evidence that surface electrostatics is a major player in the evolution and antigenic drift of AI viruses prompted further studies. Specifically, they focused on electrostatic distance (ED)-based grouping of model AI subtypes, either having “pandemic history” (H5 and H7) or a poor story of infections in mammals (H4 and H6), and aimed to infer possible difference in electrostatic clustering and/or in shared fingerprints, among viruses isolated from avian and human/mammalian host. Such a first systematic analysis of the surface electrostatics versus host specificity relationship also provided a deeper analysis of different HA subregions [145]. In particular, clustering by electrostatic closeness of the RBD resulted in groups including mixed avian- and human/mammalian-host viruses and, when the relative distribution among electrostatic groups was considered, H5 and H7 subtypes showed preferential (even if not 100% specific) clustering, as one electrostatic group was fully populated by viruses from human or mammalian host. In contrast, in other groups, viruses from avian hosts represented the major population [145]. Even though the predictive power of electrostatic isocontours for host jump was found to be lower than fingerprints for antigenic drift and clades evolution, the observed preference in clustering suggested electrostatic changes could somehow be involved (likely together with other surface features) in the modulation of host specificity. This discovery elicited deeper analyses by progressively zooming in relevant antigenic epitopes of the RBD. Preferential clustering by electrostatic distance was confirmed by next “zoom in” analyses with progressively smaller HA fragments and focused around the most antigenic 130-loop to 220-loop fragment of the RBD [145]. This evidence is in agreement with another recent work, highlighting the relevance for host specificity of changes at the 130-loop [146]. Furthermore, it is known that decreasing the positive electrostatic charge in the vicinity of RBD epitopes involved in immune escape could also lead to a lowering of the affinity to sialic acid analogs of cell receptors [147]. The aforementioned works suggest that surface electrostatics can modulate, rather than drive, host specificity anyway representing a determinant more important than hydropathy [145]. Next investigations on changes in surface electrostatics should be combined to the analysis of local changes in e.g. solvent accessible surface area and/or specific linear and conformational motifs, and integrated by docking simulations, for predicting changes in relative affinities to the different types of sialic acid, and thus trends in host jump and pandemic events.
Indeed, electrostatics studies may be of further help to virologists. For instance, diverse electrostatic characteristics at host-pathogen interfaces influence virus pathogenesis [148], and electrostatics is the major determinant in keeping the influenza virus Matrix Protein M1 conformation stable at different pH values [149]. Electrostatic analysis and clustering could have a positive impact on the pharmacological level: as an example, interfering with electrostatics was of help to develop drugs inhibiting HIV-1 fusion [150] and inhibitors for the influenza virus neuraminidase [151].
4. Concluding remarks
Even though a large computational toolbox is currently available for studying role(s) played by electrostatics in regulation of protein life cycle and its interaction, electrostatic features are still a neglected factor in both basic science and applied, biotechnological projects. However, protein electrostatics can deeply influence protein stability, from capacity to properly dimerize/oligomerize or form complexes to undesired aggregation and precipitation. Furthermore, it is crucial to selection of cofactors, substrates, binding to other macromolecules, subcellular compartments, nanomaterials.
Last but not least, an important indication is emerging, not only from electrostatics studies, which concerns the overall structural and protein surface features. In many fields, “functional similarity” is still inferred as directly related to sequence closeness. This is often true, but not always, as different substitutions may dramatically diverge in functional outcome. For instance, several substitutions and thus high divergence depending on the very long, separate evolutionary history of two proteins derived from a common ancestor might suggest a weak functional relationship, especially when compared with another protein showing only a few substitutions. This can be misleading when a high number of substitutions concerns non conserved regions, or anyway all functional motifs are not changed or keep their relevant features. Conversely, two proteins with highly similar sequences might strongly diverge from a functional point of view, when a limited number of substitutions affect residues (even only one) crucial to e.g. catalysis, or binding of a cofactor, a molecular partner etc. In a few words, sequence-based comparison can fail in properly sorting proteins together based on shared functions, and this task can be better achieved by taking advantage of structure based, computational comparison.
In addition to sharing functions based on structural closeness rather than sequence similarity, the analysis of protein surface features is quite relevant to biotechnological project design. Indeed, this review illustrates some examples in which electrostatic features are clearly able to sort proteins in phylogeny-independent clusters where the shared feature is a function, rather than taxonomic classification. Indeed, most protein interactions occur at the protein surface and accessible cavities, i.e. where motifs are exposed and where electrostatics can act as driving force in guidance, specificity and strength in binding events, i.e. ultimately functional events.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This work was supported by a “DOR” grant from Padua University to F.F., grant Progetto PRIN 2017JL8SRX and Cariparo Visiting programme 2018 to L.C. and grant Unipd Stars Consolidator FIRMESs and PRIN 2017 FS5SHL “RADIUS” to M.Gi. F.V. is supported by a PhD fellowship from the Padua University. The authors have no conflict of interests to disclose.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.csbj.2020.06.029.
Contributor Information
Francesco Filippini, Email: francesco.filippini@unipd.it.
Irene Righetto, Email: irene.righetto@bio.unipd.it.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Zhou H.-X., Pang X. Electrostatic Interactions in Protein Structure, Folding, Binding, and Condensation. Chem Rev. 2018;118:1691–1741. doi: 10.1021/acs.chemrev.7b00305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsai M.-Y., Zheng W., Balamurugan D., Schafer N.P., Kim B.L., Cheung M.S. Electrostatics, structure prediction, and the energy landscapes for protein folding and binding. Protein Sci. 2016;25:255–269. doi: 10.1002/pro.2751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Strickler S.S., Gribenko A.V., Gribenko A.V., Keiffer T.R., Tomlinson J., Reihle T. Protein Stability and Surface Electrostatics: A Charged Relationship. Biochemistry. 2006;45:2761–2766. doi: 10.1021/bi0600143. [DOI] [PubMed] [Google Scholar]
- 4.Roca M., Messer B., Warshel A. Electrostatic contributions to protein stability and folding energy. FEBS Lett. 2007;581:2065–2071. doi: 10.1016/j.febslet.2007.04.025. [DOI] [PubMed] [Google Scholar]
- 5.Hendsch Z.S., Tidor B. Do salt bridges stabilize proteins? A continuum electrostatic analysis. Protein Sci. 1994;3:211–226. doi: 10.1002/pro.5560030206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lu J., Deutsch C. Electrostatics in the Ribosomal Tunnel Modulate Chain Elongation Rates. J Mol Biol. 2008;384:73–86. doi: 10.1016/j.jmb.2008.08.089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Knight A.M., Culviner P.H., Kurt-Yilmaz N., Zou T., Ozkan S.B., Cavagnero S. Electrostatic effect of the ribosomal surface on nascent polypeptide dynamics. ACS Chem Biol. 2013;8:1195–1204. doi: 10.1021/cb400030n. [DOI] [PubMed] [Google Scholar]
- 8.Kaiser C.M., Goldman D.H., Chodera J.D., Tinoco I., Bustamante C. The ribosome modulates nascent protein folding. Science. 2011;334:1723–1727. doi: 10.1126/science.1209740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kudriaeva A., Kuzina E.S., Zubenko O., Smirnov I.V., Belogurov A. Charge-mediated proteasome targeting. FASEB J. 2019;33:6852–6866. doi: 10.1096/fj.201802237R. [DOI] [PubMed] [Google Scholar]
- 10.Lawrence M.S., Phillips K.J., Liu D.R. Supercharging Proteins Can Impart Unusual Resilience. J Am Chem Soc. 2007;129:10110–10112. doi: 10.1021/ja071641y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Der B.S., Kluwe C., Miklos A.E., Jacak R., Lyskov S., Gray J.J. Alternative Computational Protocols for Supercharging Protein Surfaces for Reversible Unfolding and Retention of Stability. PLoS One. 2013;8 doi: 10.1371/journal.pone.0064363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kumar S., Nussinov R. How do thermophilic proteins deal with heat? Cell Mol Life Sci. 2001;58:1216–1233. doi: 10.1007/PL00000935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Giletto A., Pace C.N. Buried, charged, non-ion-paired aspartic acid 76 contributes favorably to the conformational stability of ribonuclease T1. Biochemistry. 1999;38:13379–13384. doi: 10.1021/bi991422s. [DOI] [PubMed] [Google Scholar]
- 14.De Graff A.M.R., Hazoglou M.J., Dill K.A. Highly Charged Proteins: The Achilles’ Heel of Aging Proteomes. Structure. 2016;24:329–336. doi: 10.1016/j.str.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 15.Warshel A., Sharma P.K., Kato M., Xiang Y., Liu H., Olsson M.H.M. Electrostatic basis for enzyme catalysis. Chem Rev. 2006;106:3210–3235. doi: 10.1021/cr0503106. [DOI] [PubMed] [Google Scholar]
- 16.Warshel A., Levitt M. Theoretical studies of enzymic reactions: Dielectric, electrostatic and steric stabilization of the carbonium ion in the reaction of lysozyme. J Mol Biol. 1976;103:227–249. doi: 10.1016/0022-2836(76)90311-9. [DOI] [PubMed] [Google Scholar]
- 17.Mhashal A.R., Pshetitsky Y., Eitan R., Cheatum C.M., Kohen A., Major D.T. Effect of Asp122 Mutation on the Hydride Transfer in E. coli DHFR Demonstrates the Goldilocks of Enzyme Flexibility. J Phys Chem B. 2018;122:8006–8017. doi: 10.1021/acs.jpcb.8b05556. [DOI] [PubMed] [Google Scholar]
- 18.Moliner V. “Eppur si muove” (yet it moves) Proc Natl Acad Sci U S A. 2011;108:15013–15014. doi: 10.1073/pnas.1112014108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Adamczyk A.J., Cao J., Kamerlin S.C.L., Warshel A. Catalysis by dihydrofolate reductase and other enzymes arises from electrostatic preorganization, not conformational motions. Proc Natl Acad Sci U S A. 2011;108:14115–14120. doi: 10.1073/pnas.1111252108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krzemińska A., Moliner V., Świderek K. Dynamic and Electrostatic Effects on the Reaction Catalyzed by HIV-1 Protease. J Am Chem Soc. 2016;138:16283–16298. doi: 10.1021/jacs.6b06856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Świderek K., Tuñón I., Williams I.H., Moliner V. Insights on the Origin of Catalysis on Glycine N -Methyltransferase from Computational Modeling. J Am Chem Soc. 2018;140:4327–4334. doi: 10.1021/jacs.7b13655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang Z., Witham S., Alexov E. On the role of electrostatics in protein-protein interactions. Phys Biol. 2011;8 doi: 10.1088/1478-3975/8/3/035001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jensen J. Calculating pH and Salt Dependence of Protein-Protein Binding. Curr Pharm Biotechnol. 2008;9:96–102. doi: 10.2174/138920108783955146. [DOI] [PubMed] [Google Scholar]
- 24.Carbonell P., Nussinov R., Del Sol A. Energetic determinants of protein binding specificity: Insights into protein interaction networks. Proteomics. 2009;9:1744–1753. doi: 10.1002/pmic.200800425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elcock A.H., Gabdoulline R.R., Wade R.C., McCammon J.A. Computer simulation of protein-protein association kinetics: Acetylcholinesterase-fasciculin. J Mol Biol. 1999;291:149–162. doi: 10.1006/jmbi.1999.2919. [DOI] [PubMed] [Google Scholar]
- 26.Persson B.A., Lund M. Association and electrostatic steering of α-lactalbumin–lysozyme heterodimers. Phys Chem Chem Phys. 2009;11:8879. doi: 10.1039/b909179c. [DOI] [PubMed] [Google Scholar]
- 27.Gunasekaran K., Pentony M., Shen M., Garrett L., Forte C., Woodward A. Enhancing antibody Fc heterodimer formation through electrostatic steering effects: Applications to bispecific molecules and monovalent IgG. J Biol Chem. 2010;285:19637–19646. doi: 10.1074/jbc.M110.117382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hemsath L., Dvorsky R., Fiegen D., Carlier M.F., Ahmadian M.R. An electrostatic steering mechanism of Cdc42 recognition by Wiskott-Aldrich syndrome proteins. Mol Cell. 2005;20:313–324. doi: 10.1016/j.molcel.2005.08.036. [DOI] [PubMed] [Google Scholar]
- 29.Koldewey P., Stull F., Horowitz S., Martin R., Bardwell J.C.A. Forces Driving Chaperone Action. Cell. 2016;166:369–379. doi: 10.1016/j.cell.2016.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu W., Bratko D., Prausnitz J.M., Blanch H.W. Electrostatic Interactions between Peptides and the Molecular Chaperone DnaK. J Phys Chem B. 2003;107:11563–11569. doi: 10.1021/jp035872c. [DOI] [Google Scholar]
- 31.Gorham R.D., Kieslich C.A., Morikis D. Electrostatic clustering and free energy calculations provide a foundation for protein design and optimization. Ann Biomed Eng. 2011;39:1252–1263. doi: 10.1007/s10439-010-0226-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schreiber G, Shaul Y, Gottschalk KE. Electrostatic Design of Protein–Protein Association Rates. Protein Des., vol. 340, New Jersey: Humana Press; 2006, p. 235–50. https://doi.org/10.1385/1-59745-116-9:235. [DOI] [PubMed]
- 33.Ritchie A.W., Webb L.J. Understanding and Manipulating Electrostatic Fields at the Protein-Protein Interface Using Vibrational Spectroscopy and Continuum Electrostatics Calculations. J Phys Chem B. 2015;119:13945–13957. doi: 10.1021/acs.jpcb.5b06888. [DOI] [PubMed] [Google Scholar]
- 34.Interlandi G., Yakovenko O., Tu A.Y., Harris J., Le J., Chen J. Specific electrostatic interactions between charged amino acid residues regulate binding of von Willebrand factor to blood platelets. J Biol Chem. 2017;292:18608–18617. doi: 10.1074/jbc.M117.797456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sheinerman F. Electrostatic aspects of protein–protein interactions. Curr Opin Struct Biol. 2000;10:153–159. doi: 10.1016/S0959-440X(00)00065-8. [DOI] [PubMed] [Google Scholar]
- 36.Ghaemi Z., Guzman I., Gnutt D., Luthey-Schulten Z., Gruebele M. Role of Electrostatics in Protein–RNA Binding: The Global vs the Local Energy Landscape. J Phys Chem B. 2017;121:8437–8446. doi: 10.1021/acs.jpcb.7b04318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhao L., Suarez I.P., Gauto D.F., Rasia R.M., Wang J. The key role of electrostatic interactions in the induced folding in RNA recognition by DCL1-A. Phys Chem Chem Phys. 2018;20:9376–9388. doi: 10.1039/C7CP07889G. [DOI] [PubMed] [Google Scholar]
- 38.Huang Y., Li H., Xiao Y. 3dRPC: a web server for 3D RNA-protein structure prediction. Bioinformatics. 2018;34:1238–1240. doi: 10.1093/bioinformatics/btx742. [DOI] [PubMed] [Google Scholar]
- 39.McCammon J.A. Darwinian biophysics: Electrostatics and evolution in the kinetics of molecular binding. Proc Natl Acad Sci. 2009;106:7683–7684. doi: 10.1073/pnas.0902767106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Radić Z., Kirchhoff P.D., Quinn D.M., McCammon J.A., Taylor P. Electrostatic Influence on the Kinetics of Ligand Binding to Acetylcholinesterase. J Biol Chem. 1997;272:23265–23277. doi: 10.1074/jbc.272.37.23265. [DOI] [PubMed] [Google Scholar]
- 41.Qin S., Zhou H.-X. Dissection of the high rate constant for the binding of a ribotoxin to the ribosome. Proc Natl Acad Sci. 2009;106:6974–6979. doi: 10.1073/pnas.0900291106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bigay J., Antonny B. Curvature, Lipid Packing, and Electrostatics of Membrane Organelles: Defining Cellular Territories in Determining Specificity. Dev Cell. 2012;23:886–895. doi: 10.1016/j.devcel.2012.10.009. [DOI] [PubMed] [Google Scholar]
- 43.Oshima A., Sumitomo K. Vesicle fusion with bilayer lipid membrane controlled by electrostatic interaction. Biochem Biophys Reports. 2017;11:58–63. doi: 10.1016/j.bbrep.2017.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rusiñol A.E., Cui Z., Chen M.H., Vance J.E. A unique mitochondria-associated membrane fraction from rat liver has a high capacity for lipid synthesis and contains pre-Golgi secretory proteins including nascent lipoproteins. J Biol Chem. 1994;269:27494–27502. [PubMed] [Google Scholar]
- 45.Giacomello M., Pellegrini L. The coming of age of the mitochondria–ER contact: a matter of thickness. Cell Death Differ. 2016;23:1417–1427. doi: 10.1038/cdd.2016.52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yeung T., Terebiznik M., Yu L., Silvius J., Abidi W.M., Philips M. Receptor Activation Alters Inner Surface Potential During Phagocytosis. Science. 2006;313:347–351. doi: 10.1126/science.1129551. [DOI] [PubMed] [Google Scholar]
- 47.Das S., Dixon J.E., Cho W. Membrane-binding and activation mechanism of PTEN. Proc Natl Acad Sci. 2003;100:7491–7496. doi: 10.1073/pnas.0932835100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Magalhaes M.A.O., Glogauer M. Pivotal Advance: Phospholipids determine net membrane surface charge resulting in differential localization of active Rac1 and Rac2. J Leukoc Biol. 2010;87:545–555. doi: 10.1189/jlb.0609390. [DOI] [PubMed] [Google Scholar]
- 49.Ueyama T., Eto M., Kami K., Tatsuno T., Kobayashi T., Shirai Y. Isoform-Specific Membrane Targeting Mechanism of Rac during FcγR-Mediated Phagocytosis: Positive Charge-Dependent and Independent Targeting Mechanism of Rac to the Phagosome. J Immunol. 2005;175:2381–2390. doi: 10.4049/jimmunol.175.4.2381. [DOI] [PubMed] [Google Scholar]
- 50.Mesmin B., Robbe K., Geny B., Luton F., Brandolin G., Popoff M.R. A Phosphatidylserine-binding Site in the Cytosolic Fragment of Clostridium sordellii Lethal Toxin Facilitates Glucosylation of Membrane-bound Rac and Is Required for Cytotoxicity. J Biol Chem. 2004;279:49876–49882. doi: 10.1074/jbc.M406903200. [DOI] [PubMed] [Google Scholar]
- 51.Yeung T., Gilbert G.E., Shi J., Silvius J., Kapus A., Grinstein S. Membrane Phosphatidylserine Regulates Surface Charge and Protein Localization. Science. 2008;319:210–213. doi: 10.1126/science.1152066. [DOI] [PubMed] [Google Scholar]
- 52.Yeung T., Heit B., Dubuisson J.F., Fairn G.D., Chiu B., Inman R. Contribution of phosphatidylserine to membrane surface charge and protein targeting during phagosome maturation. J Cell Biol. 2009;185:917–928. doi: 10.1083/jcb.200903020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bakowski M.A., Braun V., Lam G.Y., Yeung T., Do Heo W., Meyer T. The Phosphoinositide Phosphatase SopB Manipulates Membrane Surface Charge and Trafficking of the Salmonella-Containing Vacuole. Cell Host Microbe. 2010;7:453–462. doi: 10.1016/j.chom.2010.05.011. [DOI] [PubMed] [Google Scholar]
- 54.Parodi A., Corbo C., Cevenini A., Molinaro R., Palomba R., Pandolfi L. Enabling cytoplasmic delivery and organelle targeting by surface modification of nanocarriers. Nanomedicine. 2015;10:1923–1940. doi: 10.2217/nnm.15.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yuan Y., Zhang C.-J., Liu B. A Photoactivatable AIE Polymer for Light-Controlled Gene Delivery: Concurrent Endo/Lysosomal Escape and DNA Unpacking. Angew Chemie Int Ed. 2015;54:11419–11423. doi: 10.1002/anie.201503640. [DOI] [PubMed] [Google Scholar]
- 56.Abe A., Shayman J.A. The role of negatively charged lipids in lysosomal phospholipase A2 function. J Lipid Res. 2009;50:2027–2035. doi: 10.1194/jlr.M900008-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Sonnino S., Aureli M., Loberto N., Chigorno V., Prinetti A. Fine tuning of cell functions through remodeling of glycosphingolipids by plasma membrane-associated glycohydrolases. FEBS Lett. 2010;584:1914–1922. doi: 10.1016/j.febslet.2009.11.020. [DOI] [PubMed] [Google Scholar]
- 58.Segawa K., Nagata S. An Apoptotic ‘Eat Me’ Signal: Phosphatidylserine Exposure. Trends Cell Biol. 2015;25:639–650. doi: 10.1016/j.tcb.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 59.Yang Y., Lee M., Fairn G.D. Phospholipid subcellular localization and dynamics. J Biol Chem. 2018;293:6230–6240. doi: 10.1074/jbc.R117.000582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Liu J., Weiss A., Durrant D., Chi N., -w., Lee RM. The cardiolipin-binding domain of Bid affects mitochondrial respiration and enhances cytochrome c release. Apoptosis. 2004;9:533–541. doi: 10.1023/B:APPT.0000038034.16230.ea. [DOI] [PubMed] [Google Scholar]
- 61.Lutter M., Fang M., Luo X., Nishijima M., Xie X., Wang X. Cardiolipin provides specificity for targeting of tBid to mitochondria. Nat Cell Biol. 2000;2:754–756. doi: 10.1038/35036395. [DOI] [PubMed] [Google Scholar]
- 62.Pike L.J. Lipid rafts: bringing order to chaos. J Lipid Res. 2003;44:655–667. doi: 10.1194/jlr.R200021-JLR200. [DOI] [PubMed] [Google Scholar]
- 63.McLaughlin S., Aderem A. The myristoyl-electrostatic switch: a modulator of reversible protein-membrane interactions. Trends Biochem Sci. 1995;20:272–276. doi: 10.1016/S0968-0004(00)89042-8. [DOI] [PubMed] [Google Scholar]
- 64.Myhill N., Lynes E.M., Nanji J.A., Blagoveshchenskaya A.D., Fei H., Simmen K.C. The subcellular distribution of calnexin is mediated by PACS-2. Mol Biol Cell. 2008;19:2777–2788. doi: 10.1091/mbc.E07-10-0995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Levental I., Lingwood D., Grzybek M., Coskun Ü., Simons K. Palmitoylation regulates raft affinity for the majority of integral raft proteins. Proc Natl Acad Sci U S A. 2010;107:22050–22054. doi: 10.1073/pnas.1016184107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schutz C.N., Warshel A. What are the dielectric ?constants? of proteins and how to validate electrostatic models? Proteins Struct Funct Genet. 2001;44:400–417. doi: 10.1002/prot.1106. [DOI] [PubMed] [Google Scholar]
- 67.Amin M, Küpper J. Variations in Proteins Dielectric Constants. ChemistryOpen 2020;9:691–4. https://doi.org/10.1002/open.202000108. [DOI] [PMC free article] [PubMed]
- 68.Li L., Li C., Alexov E. On the modeling of polar component of solvation energy using smooth Gaussian-based dielectric function. J Theor Comput Chem. 2014;13:1440002. doi: 10.1142/S0219633614400021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C. UCSF Chimera-A visualization system for exploratory research and analysis. J Comput Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 70.Onufriev A.V., Case D.A. Generalized Born Implicit Solvent Models for Biomolecules. Annu Rev Biophys. 2019;48:275–296. doi: 10.1146/annurev-biophys-052118-115325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Fogolari F., Corazza A., Yarra V., Jalaru A., Viglino P., Esposito G. Bluues: A program for the analysis of the electrostatic properties of proteins based on generalized Born radii. BMC Bioinformatics. 2012;13:S18. doi: 10.1186/1471-2105-13-S4-S18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dolinsky T.J., Nielsen J.E., McCammon J.A., Baker N.A. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Richter S., Wenzel A., Stein M., Gabdoulline R.R., Wade R.C. webPIPSA: a web server for the comparison of protein interaction properties. Nucleic Acids Res. 2008;36:W276–W280. doi: 10.1093/nar/gkn181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Vriend G. WHAT IF: A molecular modeling and drug design program. J Mol Graph. 1990;8:52–56. doi: 10.1016/0263-7855(90)80070-V. [DOI] [PubMed] [Google Scholar]
- 75.Li C., Jia Z., Chakravorty A., Pahari S., Peng Y., Basu S. DelPhi Suite: New Developments and Review of Functionalities. J Comput Chem. 2019;40:2502–2508. doi: 10.1002/jcc.26006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Jo S., Vargyas M., Vasko-Szedlar J., Roux B., Im W. PBEQ-Solver for online visualization of electrostatic potential of biomolecules. Nucleic Acids Res. 2008;36:W270–W275. doi: 10.1093/nar/gkn314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Unni S., Huang Y., Hanson R.M., Tobias M., Krishnan S., Li W.W. Web servers and services for electrostatics calculations with APBS and PDB2PQR. J Comput Chem. 2011;32:1488–1491. doi: 10.1002/jcc.21720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Vlachakis D., Fakourelis P., Megalooikonomou V., Makris C., Kossida S. DrugOn: a fully integrated pharmacophore modeling and structure optimization toolkit. PeerJ. 2015;3 doi: 10.7717/peerj.725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Henrich S., Richter S., Wade R.C. On the use of PIPSA to guide target-selective drug design. ChemMedChem. 2008;3:413–417. doi: 10.1002/cmdc.200700154. [DOI] [PubMed] [Google Scholar]
- 80.Bauer M.R., Mackey M.D. Electrostatic Complementarity as a Fast and Effective Tool to Optimize Binding and Selectivity of Protein-Ligand Complexes. J Med Chem. 2019;62:3036–3050. doi: 10.1021/acs.jmedchem.8b01925. [DOI] [PubMed] [Google Scholar]
- 81.Distaso M.A., Tran H., Ferrer M. Golyshin PN. Metagenomic Mining of Enzyme Diversity. Consequences Microb. Interact. with Hydrocarb. Oils, Lipids Prod. Fuels Chem., Cham: Springer International Publishing. 2016:1–25. doi: 10.1007/978-3-319-31421-1_216-1. [DOI] [Google Scholar]
- 82.Arnold F.H. Directed Evolution: Bringing New Chemistry to Life. Angew Chemie Int Ed. 2018;57:4143–4148. doi: 10.1002/anie.201708408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gorke J., Srienc F., Kazlauskas R. Toward advanced ionic liquids. Polar, enzyme-friendly solvents for biocatalysis. Biotechnol. Bioprocess Eng. 2010;15:40–53. doi: 10.1007/s12257-009-3079-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Zhao H. Methods for stabilizing and activating enzymes in ionic liquids - a review. J Chem Technol Biotechnol. 2010;85:891–907. doi: 10.1002/jctb.2375. [DOI] [Google Scholar]
- 85.Zhou Y., Pérez B., Hao W., Lv J., Gao R., Guo Z. The additive mutational effects from surface charge engineering: A compromise between enzyme activity, thermostability and ionic liquid tolerance. Biochem Eng J. 2019;148:195–204. doi: 10.1016/j.bej.2018.07.020. [DOI] [Google Scholar]
- 86.Kamal M.Z., Ahmad S., Molugu T.R., Vijayalakshmi A., Deshmukh M.V., Sankaranarayanan R. In vitro evolved non-aggregating and thermostable lipase: Structural and thermodynamic investigation. J Mol Biol. 2011;413:726–741. doi: 10.1016/j.jmb.2011.09.002. [DOI] [PubMed] [Google Scholar]
- 87.Nordwald E.M., Armstrong G.S., Kaar J.L. NMR-Guided Rational Engineering of an Ionic-Liquid-Tolerant Lipase. ACS Catal. 2014;4:4057–4064. doi: 10.1021/cs500978x. [DOI] [Google Scholar]
- 88.Pedersen J.N., Zhou Y., Guo Z., Pérez B. Genetic and chemical approaches for surface charge engineering of enzymes and their applicability in biocatalysis: A review. Biotechnol Bioeng. 2019;116:1795–1812. doi: 10.1002/bit.26979. [DOI] [PubMed] [Google Scholar]
- 89.Whitehead T.A., Bandi C.K., Berger M., Park J., Chundawat S.P.S. Negatively Supercharging Cellulases Render Them Lignin-Resistant. ACS Sustain Chem Eng. 2017;5:6247–6252. doi: 10.1021/acssuschemeng.7b01202. [DOI] [Google Scholar]
- 90.Warden A.C., Williams M., Peat T.S., Seabrook S.A., Newman J., Dojchinov G. Rational engineering of a mesohalophilic carbonic anhydrase to an extreme halotolerant biocatalyst. Nat Commun. 2015;6:10278. doi: 10.1038/ncomms10278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Niero M., Righetto I., Beneventi E., Polverino de Laureto P., Fraaije M.W., Filippini F. Unique Features of a New Baeyer-Villiger Monooxygenase from a Halophilic Archaeon. Catalysts. 2020;10:128. doi: 10.3390/catal10010128. [DOI] [Google Scholar]
- 92.Welborn V.V., Ruiz Pestana L., Head-Gordon T. Computational optimization of electric fields for better catalysis design. Nat Catal. 2018;1:649–655. doi: 10.1038/s41929-018-0109-2. [DOI] [Google Scholar]
- 93.Acosta-Silva C., Bertran J., Branchadell V., Oliva A. Kemp Elimination Reaction Catalyzed by Electric Fields. ChemPhysChem. 2020;21:295–306. doi: 10.1002/cphc.201901155. [DOI] [PubMed] [Google Scholar]
- 94.Świderek K., Tuñón I., Moliner V., Bertran J. Revealing the Origin of the Efficiency of the De Novo Designed Kemp Eliminase HG-3.17 by Comparison with the Former Developed HG-3. Chem - A Eur J. 2017;23:7582–7589. doi: 10.1002/chem.201700807. [DOI] [PubMed] [Google Scholar]
- 95.Privett H.K., Kiss G., Lee T.M., Blomberg R., Chica R.A., Thomas L.M. Iterative approach to computational enzyme design. Proc Natl Acad Sci U S A. 2012;109:3790–3795. doi: 10.1073/pnas.1118082108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Lippow S.M., Wittrup K.D., Tidor B. Computational design of antibody-affinity improvement beyond in vivo maturation. Nat Biotechnol. 2007;25:1171–1176. doi: 10.1038/nbt1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Kiyoshi M., Caaveiro J.M.M., Miura E., Nagatoishi S., Nakakido M., Soga S. Affinity improvement of a therapeutic antibody by structure-based computational design: Generation of electrostatic interactions in the transition state stabilizes the antibody-antigen complex. PLoS One. 2014;9 doi: 10.1371/journal.pone.0087099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yoshida K., Kuroda D., Kiyoshi M., Nakakido M., Nagatoishi S., Soga S. Exploring designability of electrostatic complementarity at an antigen-antibody interface directed by mutagenesis, biophysical analysis, and molecular dynamics simulations. Sci Rep. 2019;9:4482. doi: 10.1038/s41598-019-40461-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Higel F., Seidl A., Sörgel F., Friess W. N-glycosylation heterogeneity and the influence on structure, function and pharmacokinetics of monoclonal antibodies and Fc fusion proteins. Eur J Pharm Biopharm. 2016;100:94–100. doi: 10.1016/j.ejpb.2016.01.005. [DOI] [PubMed] [Google Scholar]
- 100.Zhao Q., Ahmed M., Guo H., Cheung I.Y., Cheung N.-K.V. Alteration of Electrostatic Surface Potential Enhances Affinity and Tumor Killing Properties of Anti-ganglioside GD2 Monoclonal Antibody hu3F8. J Biol Chem. 2015;290:13017–13027. doi: 10.1074/jbc.M115.650903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Fukunaga A., Tsumoto K. Improving the affinity of an antibody for its antigen via long-range electrostatic interactions. Protein Eng Des Sel. 2013;26:773–780. doi: 10.1093/protein/gzt053. [DOI] [PubMed] [Google Scholar]
- 102.Selzer T., Albeck S., Schreiber G. Rational design of faster associating and tighter binding protein complexes. Nat Struct Biol. 2000;7:537–541. doi: 10.1038/76744. [DOI] [PubMed] [Google Scholar]
- 103.De Nardis C., Hendriks L.J.A., Poirier E., Arvinte T., Gros P., Bakker A.B.H. A new approach for generating bispecific antibodies based on a common light chain format and the stable architecture of human immunoglobulin G1. J Biol Chem. 2017;292:14706–14717. doi: 10.1074/jbc.M117.793497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ohmuro-Matsuyama Y., Mori K., Hamada H., Ueda H., Yamaji H. Electrostatic engineering of the interface between heavy and light chains promotes antibody Fab fragment production. Cytotechnology. 2017;69:469–475. doi: 10.1007/s10616-016-9955-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Rujas E., Leaman D.P., Insausti S., Ortigosa-Pascual L., Zhang L., Zwick M.B. Functional Optimization of Broadly Neutralizing HIV-1 Antibody 10E8 by Promotion of Membrane Interactions. J Virol. 2018;92. doi: 10.1128/JVI.02249-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ingram J.R., Schmidt F.I., Ploegh H.L. Exploiting Nanobodies’ Singular Traits. Annu Rev Immunol. 2018;36:695–715. doi: 10.1146/annurev-immunol-042617-053327. [DOI] [PubMed] [Google Scholar]
- 107.Mahajan S.P., Meksiriporn B., Waraho-Zhmayev D., Weyant K.B., Kocer I., Butler D.C. Computational affinity maturation of camelid single-domain intrabodies against the nonamyloid component of alpha-synuclein. Sci Rep. 2018;8:17611. doi: 10.1038/s41598-018-35464-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cheng X., Wang J., Kang G., Hu M., Yuan B., Zhang Y. Homology Modeling-Based in Silico Affinity Maturation Improves the Affinity of a Nanobody. Int J Mol Sci. 2019;20:4187. doi: 10.3390/ijms20174187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gray E.R., Brookes J.C., Caillat C., Turbé V., Webb B.L.J., Granger L.A. Unravelling the Molecular Basis of High Affinity Nanobodies against HIV p24. In Vitro Functional, Structural, and in Silico Insights. ACS. Infect Dis. 2017;3:479–491. doi: 10.1021/acsinfecdis.6b00189. [DOI] [PubMed] [Google Scholar]
- 110.Warshel A., Papazyan A. Electrostatic effects in macromolecules: Fundamental concepts and practical modeling. Curr Opin Struct Biol. 1998;8:211–217. doi: 10.1016/S0959-440X(98)80041-9. [DOI] [PubMed] [Google Scholar]
- 111.Warshel A., Sharma P.K., Kato M., Parson W.W. Modeling electrostatic effects in proteins. Biochim Biophys Acta - Proteins Proteomics. 2006;1764:1647–1676. doi: 10.1016/j.bbapap.2006.08.007. [DOI] [PubMed] [Google Scholar]
- 112.Siragusa L., Cross S., Baroni M., Goracci L., Cruciani G. BioGPS: Navigating biological space to predict polypharmacology, off-targeting, and selectivity. Proteins Struct Funct Bioinforma. 2015;83:517–532. doi: 10.1002/prot.24753. [DOI] [PubMed] [Google Scholar]
- 113.Waldner B.J., Kraml J., Kahler U., Spinn A., Schauperl M., Podewitz M. Electrostatic recognition in substrate binding to serine proteases. J Mol Recognit. 2018;31. doi: 10.1002/jmr.2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rathi P.C., Ludlow R.F., Verdonk M.L. Practical high-quality electrostatic potential surfaces for drug discovery using a graph-convolutional deep neural network. J Med Chem. 2019 doi: 10.1021/acs.jmedchem.9b01129. [DOI] [PubMed] [Google Scholar]
- 115.Goodford P. The Basic Principles of GRID. Molecular Interaction Fields: Applications in Drug Discovery and ADME Prediction. vol. 27. Wiley-VCH Verlag; 2006. https://doi.org/10.1002/3527607676.ch1.
- 116.Docquier J.D., Mangani S. An update on β-lactamase inhibitor discovery and development. Drug Resist Updat. 2018;36:13–29. doi: 10.1016/j.drup.2017.11.002. [DOI] [PubMed] [Google Scholar]
- 117.Burns CJ, Pevear DC, Lee Trout RE, Jackson RW, Hamrick J, Zulli AL, et al. Beta-lactamase inhibitors. 10464952, 2019.
- 118.Hamrick J.C., Docquier J.-D., Uehara T., Myers C.L., Six D.A., Chatwin C.L. VNRX-5133 (Taniborbactam), a broad-spectrum inhibitor of serine- and metallo-β-lactamases, restores activity of cefepime in Enterobacterales and Pseudomonas aeruginosa. Antimicrob Agents Chemother. 2019;64. doi: 10.1128/aac.01963-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Brem J., Cain R., Cahill S., McDonough M.A., Clifton I.J., Jiménez-Castellanos J.C. Structural basis of metallo-β-lactamase, serine-β-lactamase and penicillin-binding protein inhibition by cyclic boronates. Nat Commun. 2016;7:1–8. doi: 10.1038/ncomms12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Cendron L., Quotadamo A., Maso L., Bellio P., Montanari M., Celenza G. X-ray Crystallography Deciphers the Activity of Broad-Spectrum Boronic Acid β-Lactamase Inhibitors. ACS Med Chem Lett. 2019;10:650–655. doi: 10.1021/acsmedchemlett.8b00607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Linciano P., Cendron L., Gianquinto E., Spyrakis F., Tondi D. Ten Years with New Delhi Metallo-β-lactamase-1 (NDM-1): From Structural Insights to Inhibitor Design. ACS Infect Dis. 2019;5:9–34. doi: 10.1021/acsinfecdis.8b00247. [DOI] [PubMed] [Google Scholar]
- 122.Barbero F., Russo L., Vitali M., Piella J., Salvo I., Borrajo M.L. Formation of the Protein Corona: The Interface between Nanoparticles and the Immune System. Semin Immunol. 2017;34:52–60. doi: 10.1016/j.smim.2017.10.001. [DOI] [PubMed] [Google Scholar]
- 123.Lundqvist M., Augustsson C., Lilja M., Lundkvist K., Dahlbäck B., Linse S. The nanoparticle protein corona formed in human blood or human blood fractions. PLoS One. 2017;12 doi: 10.1371/journal.pone.0175871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Cai R., Chen C. The Crown and the Scepter: Roles of the Protein Corona in Nanomedicine. Adv Mater. 2019;31:1805740. doi: 10.1002/adma.201805740. [DOI] [PubMed] [Google Scholar]
- 125.Nel A.E., Mädler L., Velegol D., Xia T., Hoek E.M.V., Somasundaran P. Understanding biophysicochemical interactions at the nano–bio interface. Nat Mater. 2009;8:543–557. doi: 10.1038/nmat2442. [DOI] [PubMed] [Google Scholar]
- 126.Baimanov D., Cai R., Chen C. Understanding the Chemical Nature of Nanoparticle-Protein Interactions. Bioconjug Chem. 2019;30:1923–1937. doi: 10.1021/acs.bioconjchem.9b00348. [DOI] [PubMed] [Google Scholar]
- 127.Forest V., Pourchez J. Preferential binding of positive nanoparticles on cell membranes is due to electrostatic interactions: A too simplistic explanation that does not take into account the nanoparticle protein corona. Mater Sci Eng C. 2017;70:889–896. doi: 10.1016/j.msec.2016.09.016. [DOI] [PubMed] [Google Scholar]
- 128.Qiu Y., Liu Y., Wang L., Xu L., Bai R., Ji Y. Surface chemistry and aspect ratio mediated cellular uptake of Au nanorods. Biomaterials. 2010;31:7606–7619. doi: 10.1016/j.biomaterials.2010.06.051. [DOI] [PubMed] [Google Scholar]
- 129.Aggarwal P., Hall J.B., McLeland C.B., Dobrovolskaia M.A., McNeil S.E. Nanoparticle interaction with plasma proteins as it relates to particle biodistribution, biocompatibility and therapeutic efficacy. Adv Drug Deliv Rev. 2009;61:428–437. doi: 10.1016/j.addr.2009.03.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Alexis F., Pridgen E., Molnar L.K., Farokhzad O.C. Factors Affecting the Clearance and Biodistribution of Polymeric Nanoparticles. Mol Pharm. 2008;5:505–515. doi: 10.1021/mp800051m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Shao H., Li T., Zhu R., Xu X., Yu J., Chen S. Carbon nanotube multilayered nanocomposites as multifunctional substrates for actuating neuronal differentiation and functions of neural stem cells. Biomaterials. 2018;175:93–109. doi: 10.1016/j.biomaterials.2018.05.028. [DOI] [PubMed] [Google Scholar]
- 132.Mao A.S., Shin J.-W., Mooney D.J. Effects of substrate stiffness and cell-cell contact on mesenchymal stem cell differentiation. Biomaterials. 2016;98:184–191. doi: 10.1016/j.biomaterials.2016.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Huang B., Lou Y., Li T., Lin Z., Sun S., Yuan Y. Molecular dynamics simulations of adsorption and desorption of bone morphogenetic protein-2 on textured hydroxyapatite surfaces. Acta Biomater. 2018;80:121–130. doi: 10.1016/j.actbio.2018.09.019. [DOI] [PubMed] [Google Scholar]
- 134.Nelson M.I., Vincent A.L. Reverse zoonosis of influenza to swine: new perspectives on the human–animal interface. Trends Microbiol. 2015;23:142–153. doi: 10.1016/j.tim.2014.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kilbourne E.D. Influenza Pandemics of the 20th Century. Emerg Infect Dis. 2006;12:9–14. doi: 10.3201/eid1201.051254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Su S., Bi Y., Wong G., Gray G.C., Gao G.F., Li S. Epidemiology, Evolution, and Recent Outbreaks of Avian Influenza Virus in China. J Virol. 2015;89:8671–8676. doi: 10.1128/JVI.01034-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Al-Tawfiq J.A., Zumla A., Gautret P., Gray G.C., Hui D.S., Al-Rabeeah A.A. Surveillance for emerging respiratory viruses. Lancet Infect Dis. 2014;14:992–1000. doi: 10.1016/S1473-3099(14)70840-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Velkov T., Ong C., Baker M.A., Kim H., Li J., Nation R.L. The antigenic architecture of the hemagglutinin of influenza H5N1 viruses. Mol Immunol. 2013;56:705–719. doi: 10.1016/j.molimm.2013.07.010. [DOI] [PubMed] [Google Scholar]
- 139.Sriwilaijaroen N, Suzuki Y. Molecular Basis of a Pandemic of Avian-Type Influenza Virus. Methods Mol. Biol., vol. 1200, Humana Press Inc.; 2014, p. 447–80. https://doi.org/10.1007/978-1-4939-1292-6_38. [DOI] [PMC free article] [PubMed]
- 140.Vines A., Wells K., Matrosovich M., Castrucci M.R., Ito T., Kawaoka Y. The Role of Influenza A Virus Hemagglutinin Residues 226 and 228 in Receptor Specificity and Host Range Restriction. J Virol. 1998;72:7626–7631. doi: 10.1128/JVI.72.9.7626-7631.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Gambaryan A., Tuzikov A., Pazynina G., Bovin N., Balish A., Klimov A. Evolution of the receptor binding phenotype of influenza A (H5) viruses. Virology. 2006;344:432–438. doi: 10.1016/j.virol.2005.08.035. [DOI] [PubMed] [Google Scholar]
- 142.Chutinimitkul S., Herfst S., Steel J., Lowen A.C., Ye J., van Riel D. Virulence-Associated Substitution D222G in the Hemagglutinin of 2009 Pandemic Influenza A(H1N1) Virus Affects Receptor Binding. J Virol. 2010;84:11802–11813. doi: 10.1128/jvi.01136-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Righetto I., Milani A., Cattoli G., Filippini F. Comparative structural analysis of haemagglutinin proteins from type A influenza viruses: conserved and variable features. BMC Bioinformatics. 2014;15:363. doi: 10.1186/s12859-014-0363-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Heidari A., Righetto I., Filippini F. Electrostatic Variation of Haemagglutinin as a Hallmark of the Evolution of Avian Influenza Viruses. Sci Rep. 2018;8:1929. doi: 10.1038/s41598-018-20225-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Righetto I., Filippini F. Pandemic Avian Influenza and Intra/Interhaemagglutinin Subtype Electrostatic Variation among Viruses Isolated from Avian, Mammalian, and Human Hosts. Biomed Res Int. 2018;2018:1–10. doi: 10.1155/2018/3870508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Ni F., Kondrashkina E., Wang Q. Determinant of receptor-preference switch in influenza hemagglutinin. Virology. 2018;513:98–107. doi: 10.1016/j.virol.2017.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Timofeeva TA, Ignat’eva A V, Rudneva IA, Mochalova L V., Bovin N V., Kaverin N V. [Effect of mutations changing the antigenic specificity on the receptor-binding activity of the influenza virus hemagglutinin of H1 and H5 subtypes]. Vopr Virusol 2013;58:24–7. [PubMed]
- 148.Song H., Qi J., Haywood J., Shi Y., Gao G.F. Zika virus NS1 structure reveals diversity of electrostatic surfaces among flaviviruses. Nat Struct Mol Biol. 2016;23:456–458. doi: 10.1038/nsmb.3213. [DOI] [PubMed] [Google Scholar]
- 149.Shtykova E.V., Dadinova L.A., Fedorova N.V., Golanikov A.E., Bogacheva E.N., Ksenofontov A.L. Influenza virus Matrix Protein M1 preserves its conformation with pH, changing multimerization state at the priming stage due to electrostatics. Sci Rep. 2017;7:16793. doi: 10.1038/s41598-017-16986-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Fu S., Tong P., Tan Y., Zhu Y., Chen Y.-H. P20A inhibits HIV-1 fusion through its electrostatic interaction with the distal region of the gp41 fusion core. Microbes Infect. 2015;17:665–670. doi: 10.1016/j.micinf.2015.05.003. [DOI] [PubMed] [Google Scholar]
- 151.Dominiak P.M., Volkov A., Dominiak A.P., Jarzembska K.N., Coppens P. Combining crystallographic information and an aspherical-atom data bank in the evaluation of the electrostatic interaction energy in an enzyme-substrate complex: Influenza neuraminidase inhibition. Acta Crystallogr Sect D Biol Crystallogr. 2009;65:485–499. doi: 10.1107/S0907444909009433. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.