

Abstract
We carried out chymotryptic digestion of multimeric ATP-dependent Lon protease from Escherichia coli. Four regions sensitive to proteolytic digestion were located in the enzyme and several fragments corresponding to the individual structural domains of the enzyme or their combinations were isolated. It was shown that (i) unlike the known AAA+ proteins, the ATPase fragment (A) of Lon has no ATPase activity in spite of its ability to bind nucleotides, and it is monomeric in solution regardless of the presence of any effectors; (ii) the monomeric proteolytic domain (P) does not display proteolytic activity; (iii) in contrast to the inactive counterparts, the AP fragment is an oligomer and exhibits both the ATPase and proteolytic activities. However, unlike the full-length Lon, its AP fragment oligomerizes into a dimer or a tetramer only, exhibits the properties of a non-processive protease, and undergoes self-degradation upon ATP hydrolysis. These results reveal the crucial role played by the non-catalytic N fragment of Lon (including its coiled-coil region), as well as the contribution of individual domains to creation of the quaternary structure of the full-length enzyme, empowering its function as a processive protease.
Keywords: AAA+ protein, ATP-dependent proteases, Lon, Lon domains, Limited proteolysis
INTRODUCTION
ATP-dependent Lon proteases (EC 3.4.21.53; MEROPS: clan SJ, ID S16), as well as other enzymes which are involved in selective, energy-dependent degradation of intracellular proteins (FtsH, ClpAP, ClpXP, HslUV, and 26S proteasome), belong to the AAA+ protein superfamily (Neuwald et al., 1999; Maurizi & Li, 2001; Lupas & Martin, 2002; Gottes-man, 2003; Frickey & Lupas, 2004). Members of this superfamily are generally organized in hexameric or heptameric ring-shaped structures formed by their ATPase modules (Vale, 2000). The diverse functions of the AAA+ proteins are principally mediated by their ability to hydrolyze ATP. The binding of ATP and its hydrolysis to ADP in the presence of Mg2+ ions leads to conformational changes of the protein, expressed in vivo through a variety of functional relationships (Ogura & Wilkinson, 2001). For example, substantial nucleotide-dependent structural alterations within the hexamers were clearly demonstrated by electron AAA+ protein p97 (Rouiller et al., 2002).
The main biological function of the proteolytic enzymes that couple ATP hydrolysis to degradation of protein substrates is control of the cellular level of regulatory proteins and digestion of damaged ones (Goldberg, 1992; Gottesman et al., 1995; Wickner et al., 1999; Tsilibaris et al., 2006). The overall structural organization of heterooligomeric ATP-dependent proteases (ClpAP, ClpXP, and HslUV) for which crystal structures have been already reported is quite well understood. The ATPase and proteolytic sites of these enzymes are located in different subunits that form “sandwich-type” oligomers (Schmidt et al., 1999; Sauer et al., 2004). In such oligomers, the double hexa- or heptameric rings of the proteolytic subunits are flanked from both sides by the hexameric rings of the ATPase subunits. The latter act as molecular machines that unfold and denature protein substrates and subsequently translocate them to the inner cavity of the enzyme, where the substrates are further degraded by the proteolytic sites (Weber-Ban et al., 1999; Reid et al., 2001; Ramachandran et al., 2002; Joshi et al., 2004; Burton et al., 2005; Kenniston et al., 2005). The activity of each ATPase subunit is strictly dependent on the state of the other subunits of the oligomer (Rouiller et al., 2002; Ogura et al., 2004; Martin et al., 2005).
The homooligomeric proteases of the Lon and FtsH families are unique among the ATP-dependent proteases and other proteins of the AAA+ superfamily in that their ATPase and proteolytic domains are part of a single polypeptide chain. Lon proteases are peptide hydrolases that contain a Ser-Lys catalytic dyad in their active sites (Rotanova, 2002; Rotanova et al., 2003). They are represented by two subfamilies, designated A and B (Rotanova et al., 2004). Division into these subfamilies is based on significant differences in the sequences around the strictly conserved Ser and Lys residues in their proteolytic centers, that coincide with the differences in their overall molecular architecture (Iyer et al., 2004; Rotanova et al., 2004; Maupin-Furlow et al., 2005). Lon protease from Escherichia coli (here called EcLon, or simply Lon) is a typical member of the LonA subfamily (Rotanova et al., 2004) and the first ATP-dependent protease to be identified (Charette et al., 1981; Chung & Goldberg, 1981; Swamy & Goldberg, 1981; Zehnbauer et al., 1981). Comparative sequence analysis (Amerik et al., 1990; Gottesman et al., 1995; Rotanova, 1999) indicated that each subunit of EcLon (or any other LonA protease) is composed of three principal structural domains. The N-terminal (N) domain does not contain any identifiable catalytic site, and its function is not yet completely clear. The amino-acid sequences of the N domains are not highly conserved, however structure prediction indicates the presence of coiled-coil regions (Lupas et al., 1991). The N domain of EcLon has been shown to consist of at least two structurally distinct subdomains; crystal structure of one of them (residues 1–119) was recently reported (Li et al., 2005). The central domain of LonAs is an ATPase that includes a typical AAA+ module containing two structural domains, α/β and α (Neuwald et al., 1999; Lupas & Martin, 2002; Iyer et al., 2004), whereas the proteolytic activity resides in the C-terminal (P) domain (Amerik et al., 1991).
The lack of crystal structures of full-length Lon proteases makes it difficult to detail the structure-function relationships for this family of homooligomeric ATP-dependent enzymes. However, it is evident that elucidation of the interactions between the domains within a Lon oligomer is a necessary step leading to understanding the organization and function of that enzyme. Limited proteolysis is one of the possible approaches to such investigations. This method allows one not only to obtain information on the organization of complex enzymes and their domain-domain interactions, but could also be used to estimate the contribution of each domain to the quaternary structure. Furthermore, limited proteolysis is of special value for the selection of fragments that might be more amenable to crystallization.
We have utilized chymotryptic digestion for fragmentation of EcLon. Two fragments resulting from such experiments, the α domain of the AAA+ module and the proteolytic domain, have already been successfully used for solving crystal structures (Botos et al., 2004a; 2004b). Here we present, for the first time, a common scheme of chymotryptic fragmentation of EcLon and preparative isolation of the enzyme fragments which are likely to correspond to the structural domains of Lon or their combinations. They include the N-terminal (N) and ATPase (A) fragments, α-helical (α) and proteolytic (P) domains, as well as the combination of the A fragment and P domain (AP). The oligomeric states of these fragments in solution and the impact of the individual domains on creation of the quaternary structure of full-length enzyme are discussed. The enzymatic properties of the fragments containing their respective catalytic centers are also characterized.
MATERIALS AND METHODS
Expression and purification of full-length Lon protease.
The intact wild-type EcLon and its proteolytically inactive mutant EcLon-S679A were expressed in the Lon-deficient E. coli strain BL21 (Novagen, Madison, WI, USA) using plasmid constructs based on pBR327 (Amerik et al., 1990; 1991). Transformed cells were grown overnight at 37°C in LB media containing 100 μg/ml ampicillin, harvested by centrifugation, and stored at −80°C. Thawed cells (40 g of cell paste collected from 9 1 of culture) were suspended in 140 ml of 20 mM Tris/HCl, pH 7.5, 5 mM EDTA buffer. The cell suspension was divided into six equal portions of approx. 30 ml each, mixed with lysozyme (final concentration 0.1 mg/ml), and incubated for an hour at 4°C. The partially lysed cells were disrupted by sonication and the homogenate was centrifuged at 40 000 × g for 2 h. Supernatant (cell-free extract) was filtered through a 0.45 μm pore-size cellulose acetate membrane and subjected to chromatography on P-11 cellulose. All pre-packed columns were from GE Healthcare (Pis-cataway, NJ, USA).
P-11 cellulose phosphate resin was prepared according to the manufacturer’s protocol (Whatman Inc., Clifton, NJ, USA). The cell-free extract was diluted 5-fold with buffer A (50 mM potassium phosphate, pH 6.8, 10% (v/v) glycerol and 1 mM EDTA) and applied at 0.3 ml/min to a 150 ml P-11 column pre-equilibrated with buffer A. The column was washed with 1 1 of buffer A, and the protein was eluted with 600 ml of buffer B (300 mM potassium phosphate, pH 6.8, 10% (v/v) glycerol and 1 mM EDTA) at 3 ml/min. Fractions containing the target protein were pooled and used for Q-Sepharose chromatography.
The protein solution was diluted 4-fold with buffer C (50 mM Tris/HCl, pH 7.5, 10% glycerol, 1 mM EDTA), filtered through a 0.2 μm pore-size cellulose acetate membrane and loaded at 1 ml/min onto three pre-equilibrated 5 ml HiTrap Q-Sepharose HP columns connected in series. The columns were washed with 150 ml of buffer C and the protein was eluted with a 225-ml linear gradient of NaCl from 0 to 1 M. Fractions eluted within 0.2–0.5 M NaCl were pooled and loaded at 1 ml/min onto a 5 ml HiTrap Heparin-Sepharose HP column equilibrated in a buffer containing 50 mM Tris/HCl, pH 7.5, 300 mM NaCl, 10% glycerol and 1 mM EDTA. The column was washed with 50 ml of buffer C and the protein was eluted with a 100 ml linear gradient of NaCl from 0.3 to 1 M. Fractions eluted within 0.4–0.7 M NaCl were pooled, concentrated on Centriprep 50 (Millipore, Bedford, MA, USA) in the presence or absence of 1 mM ADP, and used for size-exclusion chromatography on a HiPrep 26/60 Sephacryl S-300 column equilibrated with buffer D containing 50 mM Tris/HCl, pH 7.5, and 0.2 M NaCl. These procedures yielded approx. 100 mg of EcLon from 40 g of cell paste, with more than 90% purity.
Limited proteolysis of full-length Lon and purification of the fragments.
The procedures used to obtain the C-terminal α domain of the AAA+ module and the P domain have been described previously (Botos et al., 2004a; 2004b). Both the wild-type Lon and the proteolytically inactive mutant Lon-S679A were subjected to chymotryptic digestion.
All reactions of limited proteolysis that yielded the N, A, and AP fragments of EcLon were performed in the presence of 1 mM ADP. In addition, 20 mM MgCl2 was used in the reaction that led to obtaining the AP fragment. Purified enzyme (100 mg) was digested with α-chymotrypsin (0.5 mg, Sigma, St. Louis, MO, USA) in 50 ml of buffer D at 30°C. After 2 h incubation, the reaction was stopped by adding PMSF to 1 mM final concentration. The solution was cooled to 4°C, filtered through a 0.2 μm pore-size cellulose acetate membrane and loaded onto a 5 ml HiTrap Heparin-Sepharose HP column equilibrated with buffer D. The flow-through containing the unbound N fragment was collected and used as described below. The column (with bound intact Lon, A, and AP fragments) was washed with 50 ml of buffer D and eluted with a 100-ml linear gradient of NaCl from 0.2 to 1 M in the same buffer.
Fractions that eluted with 0.4–0.8 M NaCl were pooled, diluted 3-fold with 50 mM Tris/HCl, pH 7.5, and loaded onto 5 ml HiTrap Q-Sepharose column pre-equilibrated in the dilution buffer. The flow-through (containing mainly the A fragment) was concentrated for size-exclusion chromatography on a HiLoad 16/60 Superdex 75 column. The Q-Sepharose column was washed with 25 ml of the equilibration buffer, the bound protein (containing AP) was eluted with 20 ml 50 mM Tris/HCl, pH 7.5, 0.4 M NaCl buffer, concentrated, and loaded on a HiPrep 26/60 Sephacryl S-300 column.
The flow-through that did not bind to Heparin-Sepharose contained the N fragment and 1 mM ADP (see above). It was diluted 6-fold with 50 mM Hepes buffer, pH 7.0, and concentrated on a YM 10 membrane (Millipore, Bedford, MA, USA) to the initial volume using a 400 ml Amicon stirred cell. In order to decrease the concentration of ADP, the dilution-concentration procedure was repeated three times. The protein solution obtained was filtered and loaded onto a 5 ml HiTrap Q-Sepharose column pre-equilibrated with the dilution buffer. The column was washed and eluted with a 50 ml linear gradient of NaCl from 0 to 0.5 M in the same buffer. Fractions eluted with 0.2–0.3 M NaCl and containing the N fragment were pooled, concentrated, and used for chromatography on a HiLoad 16/60 Superdex 75 column.
Size-exclusion chromatography was performed in buffer D for all target fragments. Protein samples were concentrated on Amicon Ultra 10 000 MWCO centrifugal filter devices. All purification procedures were performed at 4°C and fractions monitored by electrophoresis on 12% SDS/PAGE. Protein concentration was estimated with Bio-Rad Protein Assay (Bio-Rad Laboratories, GmbH, Germany) using bovine serum albumin as the standard. The purity and homogeneity of the target fragments were verified by N-terminal sequencing and electrospray ionization mass-spectrometry (Agilent 1100). The average yield of fragments obtained from about 100 mg of the full-length Lon was about 20 mg of the N fragment, 30 mg of the A fragment, 10 mg of the α domain, 40 mg of the AP fragment, and 15 mg of the P domain.
Activity assays.
The ATPase activity of the full-length EcLon and its truncated forms, A and AP, was measured by the method based on determination of the amount of inorganic phosphate resulting from ATP hydrolysis as a function of time (Bencini et al., 1983; Melnikov et al., 2000). Reactions were performed at 37°C in buffer R (50 mM Tris/HCl, pH 8.5 (at 25°C), 0.15 M NaCl, 15 mM MgCl2), in the absence or presence of 1 mg/ml β-casein. The concentration of ATP-Mg was varied between 0.3 and 2.5 mM, whereas the concentration of the enzyme (expressed as subunits) was varied within the range of 0.1−10 μM.
The thioesterase activity of the wild-type EcLon or its AP fragment and P domain was determined by the hydrolysis of substrate Suc-Phe-Leu-Phe-SBzl, as described in (Melnikov et al., 2001), at 37°C and the constant concentration of ATP (1 mM) in the buffer R containing 5% dimethylsulfoxide, and within the same range of protein concentration as for the ATPase activity assay.
The proteolytic activity of the wild-type EcLon or its AP fragment and P domain toward a protein substrate (β-casein) was analyzed by SDS/PAGE after two hours or overnight incubation at 30, 37, or 45°C. The reaction buffer was the same as for the ATPase assay. The final concentration of β-casein was 1 mg/ml, of ATP-Mg 5 mM, and of the tested protein 1 μM (EcLon) or 10 μM (AP or P domain).
The presence or absence of chymotrypsin in the final samples of EcLon fragments was verified using Suc-Ala-Ala-Phe-pNA, by detecting the absorbance of the p-nitroaniline at 410 nm, assuming ε = 8 800 M−1 cm−1 (Erlanger et al., 1961).
Sedimentation analysis of EcLon fragments.
A Beckman model E ultracentrifuge with ultraviolet scanning optics and a multiplexer was utilized for the analysis of the protein pausidispersity by velocity sedimentation. Purified EcLon or its fragments were analyzed in 50 mM Tris/HCl, pH 7.5, 0.1 M NaCl buffer, at protein concentration ranging from 0.2 to 1 mg/ml. Estimates of the average sedimentation and diffusion coefficients and of protein molecular mass were based on the results of experiments performed in capillary cells. The results of sedimentation in a charcoal-filled epon cell were used for detailed analysis of the heterogeneity of the protein samples by the Van Holde extrapolation method (Van Holde & Weischet, 1978). Estimation of the molecular mass of the components with different sedimentation coefficients was held according to the Atassi-Gandhi equation (Halsall, 1967). The relative quantity of different components making up each protein sample was calculated using light-scattering correction by the method of consecutive approximations, using the absorbance coefficient and the molecular mass of the analyzed components.
RESULTS AND DISCUSSION
The choice of protease for digestion of EcLon and the influence of effectors on fragmentation
The sensitivity of a protein substrate to cleavage by proteolytic enzymes is defined by a variety of factors. The crucial ones are peculiarities of organization of the protein target that affect the protease-accessible structural elements and the specificity of the used protease. Using enzymes of low specificity together with appropriate selection of reaction conditions, it is often possible to achieve fragmentation of target polypeptides resulting in high-molecular-mass intermediates that may represent structurally compact individual domains and/or their combinations.
In order to obtain protein fragments on a preparative scale, it is necessary to establish conditions for very rapid and highly effective inhibition of the protease that is being used for limited proteolysis. However, the inactivation procedures should not cause denaturation or chemical modification of the products, or inactivate the potential active sites if the target protein substrate is itself an active protease.
An enzyme that fulfills well the requirements outlined above is α-chymotrypsin. This serine protease cleaves peptide bonds that follow hydrophobic amino acids at the PI position (Schechter & Berger, 1967), and can be effectively suppressed by covalent modification of its active site by PMSF, which is not a very effective inhibitor of Lon. Furthermore, a chymotrypsin molecule contains five disulfide bridges, the presence of which is absolutely required for enzymatic activity, and thus this enzyme can be easily and irreversibly inactivated by reducing agents, such as dithiothreitol or β-mercaptoethanol.
We utilized α-chymotrypsin for limited proteolysis of EcLon and found that the resulting set of Lon fragments depended very much on the presence of effectors of Lon, i.e. nucleotides and magnesium ions (Fig. 1). In addition, all fragments other than the N-terminal one are only quasi-stable and undergo further degradation upon increasing concentration of the proteolytic enzyme and/or prolonged reaction time, especially at an elevated digestion temperature.
Figure 1. Fragmentation of EcLon by chymotrypsin.

A) Stabilization of the A domain by increasing concentrations of ADP (lane 1 - intact Lon); B) Influence of magnesium ions at different concentrations in the presence or absence of 1 mM ADP; C) Influence of different adenosine nucleotides and nucleotide-magnesium complexes (lane 1 - intact Lon; concentrations: nucleotides − 1 mM, MgCl2 − 20 mM). Reactions were performed at 30°C for 2 h in 50 mM Tris/HCl buffer, pH 7.5, 0.2 M NaCl, and analyzed on 15% SDS/ PAGE. Concentrations: Lon − 1 mg/ml (about 10 μM), chymotrypsin − 10 μg/ml.
In most of the experiments aimed at proteolytic digestion (Fig. 1) we used Lon preparations that were not saturated with a nucleotide before the final step of purification by size-exclusion chromatography (see Materials and Methods). Variation of pH (7–9), salt concentration (0.1–1 M) and temperature (25–37°C) did not alter the resulting set of EcLon fragments, when the time of chymotryptic digestion was limited to 2 h and the concentrations of the protein and the effectors were kept constant.
The Lon fragments shown in Fig. 1 were identified by N-terminal sequencing. The identity of the cleavage sites and the boundaries of the chymotryptic fragments of EcLon (Fig. 2) were confirmed by mass spectrometry. It is important to note that residues Q208-M234 were not present in any of the identified Lon fragments (Fig. 2B).
Figure 2. Schematic representation of EcLon subunit architecture (A) and EcLon fragments obtained by chymotryptic digestion (B).
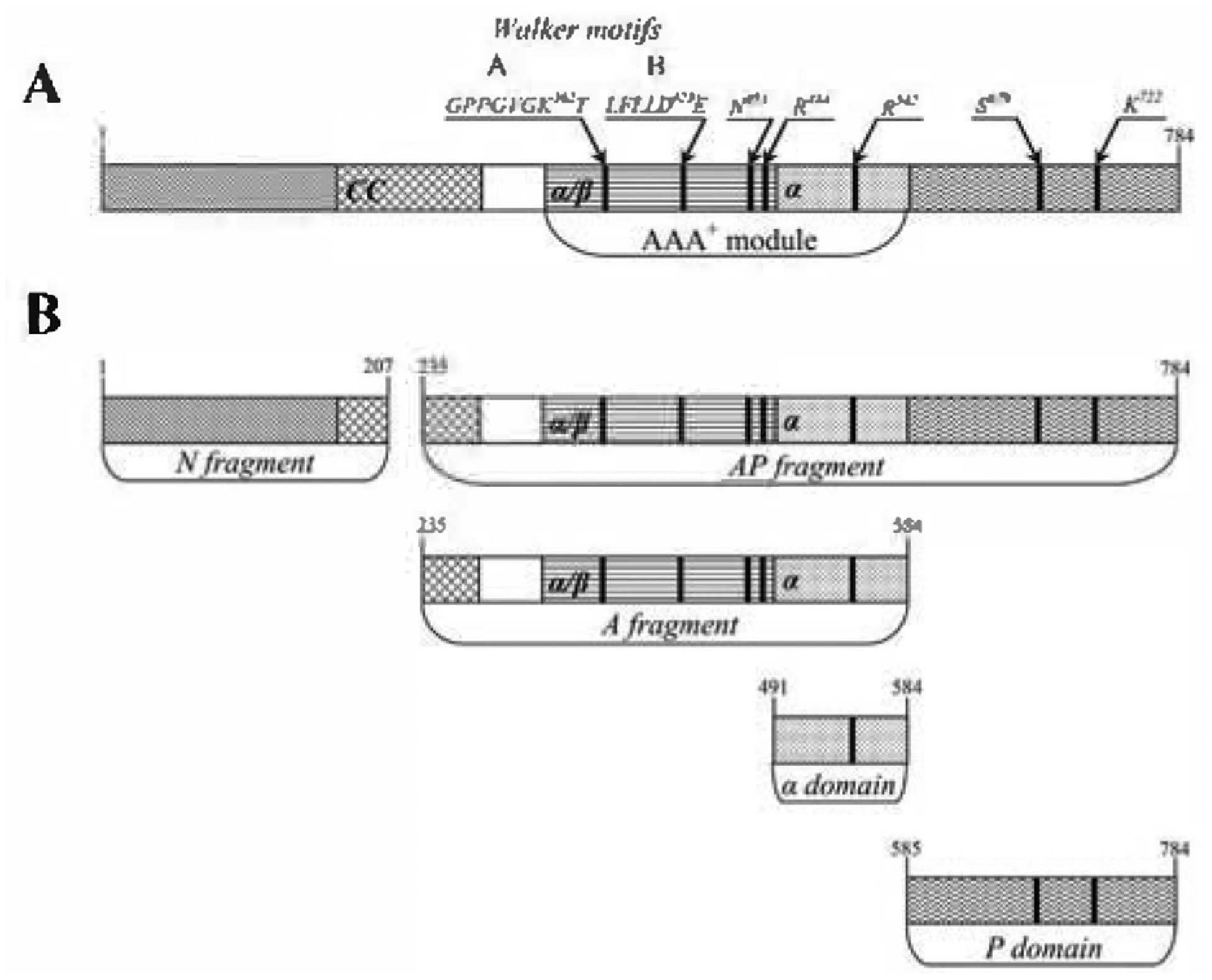
The predicted coiled-coil (CC) region, the characteristic elements of the AAA+ module (Walker motifs A and B, and residues including sensor-1 (N473), “arginine finger” (R484) of the nucleotide binding (α/β) domain, and sensor-2 (R542) of the α helical (α) domain), as well as the catalytic residues S679 and K722 of the enzyme proteolytic site, are marked.
The presence of a nucleotide substantially stabilizes the ATPase domain of the enzyme (Fig. 1A), thus making it less susceptible to cleavage by Chymotrypsin. Variations in the concentration of ADP within a wide range (50 μM to 5 mM, lanes 3–5) with a fixed concentration of Lon in the reaction mixture (about 10 μM) did not lead to any differences in the yield of the A fragment. This observation indicates that the binding constant of ADP to the ATPase active site does not exceed 50 μM. Besides the A fragment, chymotryptic cleavage of Lon in the presence of nucleotides yields the N fragment and the P domain as well (Fig. 1A, lanes 3–5).
Digestion of Lon at a low ADP concentration ≤ 1 μM (Fig. 1A, lane 2) or in the absence of any effector (not shown) also results in formation of the N and P fragments. At the same time, the α/β domain of the AAA+ module undergoes complete degradation, leading to formation of low-molecular-mass peptides, whereas the α domain remains as the sole high-molecular-mass fragment of the ATPase domain (Fig. 2B). The same set of fragments (N, P, α) is formed in the presence of magnesium ions and without nucleotides (Fig. 1B, lanes 1–3). This suggests that magnesium ions are not stand-alone effectors which could cause considerable structural alterations by binding to the enzyme, as it should be detected by limited proteolysis.
The simultaneous presence of magnesium ions and ADP changes the pattern of digestion of Lon and leads to stabilization of the resulting AP fragment. As shown in Fig. 1B (lanes 4–6), the maximal yield of the AP fragment is observed at a considerable excess of magnesium ions (keeping the concentration of ADP fixed at 1 mM). Under these conditions, most of the ADP must be in the form of a nucleotide-magnesium complex, since the dissociation constant for the complex between ADP and Mg2+ is about 0.5 mM (O’Sullivan & Smither, 1979). Thus, the stabilization of the AP fragment is determined primarily by binding of the nucleotide-magnesium complex. We may conclude that binding of either nucleotides or nucleotide-magnesium complexes leads to different conformational states of the interface between the α-domain of the AAA+ module and the proteolytic domain of Lon (Fig. 2A).
Proteolytic cleavage of Lon complexed with various nucleotides or analogs (ADP, ATP, or AMPPNP) results in similar digestion patterns (Fig. 1C, lanes 4–9). Whereas the A fragment is present in all digests, the AP fragment is only found in the presence of magnesium ions. Since sequencing of all the fragments that contain the A domain (Fig. 1C) identified Lys235 at the N-terminus, we may conclude that binding of different nucleotides does not lead to essential structural changes of the region located between the N and A domains of Lon regardless of the presence of magnesium ions. In experiments performed under similar conditions, identical fragments were obtained from both the active Lon and its proteolytically inactive S679A mutant.
Application of the technique of limited proteolysis allows differentiation between the forms of Lon that do or do not contain a bound nucleotide at different purification steps of the enzyme (Fig. 3); it is significant for the understanding of some properties of Lon, in particular, its non-processive proteolytic activity (see Activity assays section). The presence or absence of the A and/or the AP fragments among the products of chymotryptic digestion of Lon indicates that:
Figure 3. Chymotryptic digestion of differently treated samples of EcLon performed in the absence of nucleotides.

Lanes: 1 - intact Lon; 2 - cleavage of Lon eluted from Heparin-Sepharose; 3–6 - cleavage of Lon after chromatography on Sephacryl S-300 in 50 mM Tris/HCl, pH 7.5, 0.2 M NaCl. Concentrating of Lon samples before size-exclusion chromatography was performed in the absence of effectors (3), in the presence of 20 mM MgCl2 (4, 6), or in the presence of 1 mM ADP (5, 6). Conditions of the reactions of limited proteolysis described in the legend to Fig. 1.
(i) Lon protease must retain bound ADP even after the removal of free nucleotide by gel filtration when it is saturated with ADP just before that purification step (Fig. 3, compare lanes 3 and 5);
(ii) magnesium ions display low affinity to the preformed enzyme-nucleotide complex (at least in the case of Lon-ADP complex) and exchange rapidly with the free ions in the solution, since the AP fragment is not detected after digestion of the enzyme that was concentrated in the presence of ADP and Mg2+ ions and subjected to gel filtration in their absence (compare Fig. 3, lane 6 and Fig. 1C, lane 8);
(iii) limited proteolysis of Lon after the chromatography on Heparin-Sepharose indicates a “nucleotide free” state of the enzyme based on the complete hydrolysis of the A domain (Fig. 3, lane 2). Neither the gel filtration process itself nor the preliminary saturation of the enzyme with Mg2+ ions causes structural alterations in Lon, since the digestion products presented in lanes 3 and 4 of Fig. 3 are identical.
Comparison of the results of limited proteolysis of EcLon by chymotrypsin and by other proteases
Table 1 summarizes the results of limited proteolysis of EcLon by chymotrypsin, glutamyl endopeptidase, or trypsin. These data are derived either from the experiments described here or from previous publications (Vasilyeva et al., 2002; Patterson et al., 2004).
Table 1.
Fragmentation of EcLon by proteolytic enzymes of different specificity
| Domain or fragment | Chymotrypsin (present study) | Trypsina | Glutamyl endopeptidaseb | ||||
|---|---|---|---|---|---|---|---|
| No. Nuc (+/− Mg2+) | ATP/ADP | ATP/ADP (+ Mg2+) | No. Nuc (+ Mg2+) | ATP/ADP (+ Mg2+) | No. Nucd (+/− Mg2+) | ATP/ADP (+/− Mg2+) | |
| N | 1–207 | 1–207 | 1–207 | 1/6–235/239 | 1/6–235/239 | 5–240 | 5–240 |
| A | 235–584 | 235–584 | |||||
| AP | 235–784 | 236/240–784C | 236/240–784 | 241–784 | |||
| α | 491–584 | ||||||
| αP | 490–784 | 487–784 | |||||
| P | 585–784 | 585–784 | 585–784 | ||||
Nue - nucleotides.
According to literature data (Patterson et al., 2004; Vasilyeva et al., 2002). Some incorrect numbers of amino-acid residues flanking the corresponding tryptic fragments were changed according to the sequencing data presented in the same paper (Patterson et al., 2004).
Unstable intermediate fragment.
identical results were obtained in the presence of non-hydrolyzable ATP analogs or non-adenosine nucleotides.
It is seen that application of different enzymes leads to non-identical sets of fragments. The most highly invariant Lon fragment that is formed under all conditions is the N-terminal part, with the maximum length of about 240 amino acids. Limited proteolysis with chymotrypsin yields a slightly shorter form (207 amino acids), due to the additional cleavage at the Leu207-Gln208 site, as mentioned above. Thus, we can conclude that the end of the N-terminal domain is located somewhere between residues 207 and 240.
In the absence of a bound nucleotide, the α domain of the AAA+ module (amino acids 491–584) and the proteolytic P domain (585–784) were only detected after digestion with chymotrypsin. The corresponding domains could not be seen as stable fragments after trypsin treatment, and they were also not formed during treatment of Lon with glutamyl endopeptidase (V8 protease). A stable αP fragment (487–784) was observed after V8 digestion, but it was not seen after digestion with chymotrypsin; the tryptic αP fragment (490–784) did not appear to be very stable and was further degraded by trypsin. It should be noted that, in the absence of nucleotides, none of the proteases utilized in these experiments (Table 1) yield either the α/β domain of the AAA+ module or its stable combination with other neighboring fragments (N and/or α domains). Removal of the α-helical domain and/or the N-terminal part of EcLon seems to decrease the structural stability of the α/β domain, followed by its rapid proteolytic cleavage. On the other hand, effective degradation of the α/β domain as part of the AAA+ module could be caused by its own conformational lability in the absence of a bound nucleotide.
Limited cleavage of Lon by any proteolytic enzyme in the presence of ATP/ADP-Mg results in preferential removal of the N-terminal part only, yielding an AP fragment (235/236/240/241–784) as a combination of the ATPase and proteolytic domains (Table 1). The highest stability of the AP fragment during tryptic hydrolysis in the presence of magnesium ions was shown solely for adenosine nucleotides (Patterson et al., 2004). A study of the proteolytic products that resulted from digestion of EcLon by glutamyl endopeptidase showed that only ATP and ADP are crucial for stabilization of an AP fragment, irrespective of the presence of magnesium ions. Limited proteolysis in the presence of other nucleotides, including a number of non-hydrolyzable analogs of ATP, leads to complete digestion of the ATPase fragment (Vasilyeva et al., 2002). It is noteworthy that, under chymotryptic digestion, the presence of the magnesium ions substantially enhances the stability of the AP fragment as the binding of the nucleotide alone results in stabilization of the ATPase domain of the enzyme (Table 1).
To summarize, our analysis illustrates common sensitivity of the cleavage site located between the α and P domains to the state of the ATPase active site, such as the presence of nucleotides, their nature, as well as the presence or absence of magnesium ions. In addition, the choice of the enzyme selected to perform limited proteolysis will influence the nature of the resulting fragments and the outcome in a significant way. At the same time, despite the major differences in the specificity of the proteases used for the reaction, the principal cleavage sites of Lon are concentrated in four areas of the enzyme’s peptide chain (Boxes I-IV in Fig. 4).
Figure 4. Secondary structure of EcLon subunit and the sites of its proteolytic cleavage.

Arrows indicate PI residues of peptide bonds cleaved by chymotrypsin (blue), trypsin (red) (Patterson et al., 2004) and glutamyl endopeptidase (green) (Vasilyeva et al., 2002). The site of the autocatalytic cleavage of full-length Lon is marked by thin black arrow. A bold black arrow corresponds to the site of auto-cleavage of the AP fragment. Boxes I-IV represent regions sensitive to proteolytic digestion leading to the formation of high-molecular-mass intermediates of Lon degradation. Secondary structure elements shown are based on crystal data (underlined) (Botos et al., 2004a; 2004b; Li et al., 2005) and predictions (McGuffin et al., 2000) (http://bioinf.cs.ucl.ac.uk/psipred) - α-helices (red), 310-helix (pink), β-strands (blue), and coils (black). The predicted coiled-coil region of the enzyme (Lupas et al., 1991) (http://www.ch.embnet.org) is highlighted in yellow.
A comparison of the locations of the cleavage sites with the structural data as well as with secondary structure predictions indicates that the cleavage sites which separate the N-terminal fragment from the rest of the molecule are located in highly helical areas (Boxes I, II). By contrast, the cleavage sites flanking the α domain (Boxes III and IV) are found in less structured regions. It is important to note that Box III (Fig. 4) contains not only the common cleavage sites for all proteases discussed here, but also the preferred autolysis site of the full-length Lon.
The A fragment which is formed during limited proteolysis contains about 350 amino-acid residues, flanked by sites located within Boxes II and III. It is about 100 residues longer than a typical AAA+ module, whose average size is between 220 and 250 amino acids (Neuwald et al., 1999). This N-terminal extension of the AAA+ module may correspond to the extra domain found in the majority of AAA+ proteins (Iyer et al., 2004). The structural and functional importance of that extension, as well as of the N-terminal part of the full-length Lon (first about 240 residues), are not clear at this time. Based on secondary structure prediction, residues extending from about 170/190 to about 280/300 may form a coiled-coil (CC) domain (Fig. 4, highlighted in yellow). Thus the A and AP fragments of Lon formed during limited proteolysis contain approximately half of the CC domain of the intact enzyme (Fig. 2B).
The cleavage within the N-terminal extension of the AAA+ module was not detected upon treatment with the proteases discussed here (Table 1), but occurs during the early stages of the unusual autolysis of the AP fragment which takes place upon ATP hydrolysis (see the Enzymatic activity of fragments of EcLon section). The site of the autocatalytic cleavage of AP (Ala286-Glu287), located in the methionine-rich area of the protein sequence, corresponds approximately to the C-terminal boundary of the predicted CC domain (Fig. 4) and possibly defines the N-terminal boundary of the ATPase domain of Lon.
Oligomeric state of EcLon and its fragments
The oligomeric state of active LonA proteases is still unclear. Sedimentation studies of Lon protease from Mycobacterium smegmatis (Rudyak et al., 2001) and size-exclusion chromatography data for the Brevibacillus thermoruber enzyme (Lee et al., 2004b) indicated the presence of hexameric assemblies. By contrast, based both on sedimentation analysis and electron microscopy, mitochondrial Lon protease from Saccharomyces cerevisiae was assumed to form heptamers (Stahlberg et al., 1999). Earlier data indicated that EcLon exists in solution as a tetramer or an octamer (Chung & Goldberg, 1981; Rivett, 1989; Goldberg et al., 1994); the tetrameric state of EcLon was confirmed in 2004 (Vasilyeva et al., 2004). However, recent electron microscopy data suggest that EcLon is a hexamer, stabilized solely by magnesium ions but not by nucleotides (Park et al., 2005).
Our data on the oligomeric state of EcLon are not completely conclusive since they seem to be dependent on a number of factors, such as the method of enzyme purification, temperature, the presence of ligands, protein concentration, and solution viscosity. In particular, size-exclusion chromatography on Sephacryl S-300 performed on a preparative scale suggests that the oligomeric state of full-length enzyme (both wild type Lon and the inactive mutant Lon-S679A) could be a dodecamer or possibly an oblong, non-globular octamer with increased mobility. At the same time, the accuracy of interpretation of the size-exclusion chromatography data (Table 2) is not high for the area of EcLon elution, since the value of the availability constant (Kav) is less than 0.1.
Table 2.
Oligomeric state of full-length Lon and its chymotryptic fragments
| Protein (aa) | molecular mass, kDa (monomer) | Size-exlusion chromatography dataa; molecular mass, kDa (n) | Velocity sedimentation datae | Crystallo-graphic dataf (n) | |||
|---|---|---|---|---|---|---|---|
| Superdex 75 16/60 | Sephacryl S-300 26/60 | S20, w′s | molecular mass, kDa (n) | RQ% | |||
| N (1–207) | 23.2 | 27.5 (1) | ND | 1.93 | 23.6 (1) | 100 | - |
| Ab(235–584) | 39.4 | 42.3 (1) | 45 (1) | ND | - | - | - |
| AP-w.t. (235–784) | 60.8 | ND | 220 (4)C 125 (2)d |
3.75 8.80 21.50 |
64.0 (1) 230.0 (4) 878.4 (ass) |
50 20 30 |
- |
| AP-S679A (235–784) | 60.8 | ND | 220 (4)C 125 (2)d |
ND | - | - | - |
| α (491–584) | 10.8 | 10.0 (1) | ND | ND | - | - | (1) |
| P-w.t. or 5679A (585–784) | 21.4 | 23.8 (1) | ND | 1.75 | 20.4 (1) | 100 | (6) |
| Lon-w.t. or 5679A (1–784) | 87.4 | ND | ~ 1000 (?) | ~ 50–100 | ND | ||
(n) - Number of subunits; RQ - relative quantity; ND - not determined; ass - associates.
Chromatography was performed at 4°C in 50 mM Tris/HCl buffer, pH 7.5, containing 0.2 M NaCl. Protein concentration in all samples was similar and about 15 mg/ml. Molecular mass (Mr) was derived from the equation logXav= a - b logMr, were Kav is availability constant determined as (Ve-Vo)/(Vt-Vo); Ve - protein elution volume, Vo and Vt - column void and total volume, respectively; a and b are experimental constants estimated from the corresponding calibration plot.
identical results were observed in the presence of 0.5 mM ADP in the column buffer, irrespective saturation of protein sample with nucleotide before size-exclusion chromatography.
Protein was saturated with 1 mM ADP or AMPPNP on the concentrating step before chromatography performed in the absence of nucleotides.
Protein was concentrated without nucleotides before chromatography.
Data obtained at 25°C without additives of nucleotides.
Published in (Botos et al., 2004a; 2004b).
Size-exclusion chromatography experiments suggest that the high-molecular-mass oligomer of Lon appears to be stable and does not tend to dissociate when enzyme concentration is considerably decreased in recurring experiments. Velocity sedimentation studies of EcLon performed after size-exclusion chromatography reveal a complex picture of the state of the protein in solution at room temperature. Different forms of high-molecular-mass assemblies with sedimentation coefficients as high as 50–100S were observed to coexist with entities as small as dimers or monomers (7.6 and 4.7S, respectively). The addition of magnesium ions, nucleotides, or nucleotide-magnesium complexes does not change the picture significantly. The ultracentrifugation data indicate the sensitivity of Lon to hydrodynamic force under the experimental conditions.
It is clear that much more extensive investigation of the oligomeric state of EcLon in solution is necessary. It is likely that the predisposition of Lon to form hetero-aggregates (50–100S), observed at decreased temperature and/or increased concentration of protein, might be the reason for the difficulties encountered in crystallization of the full-length EcLon. It is important to note that such aggregation might not be happening under conditions that are routinely utilized for in vitro investigation of the enzymatic functions of Lon (low enzyme concentrations, physiological temperature, a full set of effectors, the presence of a protein substrate, and ATP hydrolysis). However, investigation of the quaternary structure of EcLon in its functional form provides special challenges, since the physical methods used for that purpose are not fully compatible with the dynamic nature of the enzymatic processes.
Although the oligomeric state of the active, full-length EcLon is still somewhat ambiguous, a much clearer picture was obtained for the fragments generated by limited proteolysis with chymotrypsin (Table 2, Fig. 2). The data show that the AP fragment (residues 235–784) is the smallest part of Lon capable of forming oligomers. The N-terminal and the ATPase fragments, as well as the α domain of the AAA+ module and the P domain of Lon remain monomeric in solution, and none of the tested proteins changed its oligomeric state in the presence of magnesium ions. Additional experiments have shown that, during low-temperature gel filtration, the ATPase fragment retains its monomeric state also in the presence of nucleotides.
The P domain, however, appears to create hexameric structures in the crystals of not only EcLon (a member of the LonA subfamily), but also of LonB from Archaeoglobus fulgidus (Botos et al., 2005), although only dimers have been seen in the crystals of the P domain of LonB from Methanococcus jannaschii (Im et al.f 2004). These results may indicate a predisposition to oligomerization of the P domains of Lon proteases of both subfamilies at the high protein concentration needed to grow crystals, but they have to be interpreted with some caution, since the oligomeric state in the crystals is sometimes unrelated to the one seen under physiological conditions. The ATPase fragment of EcLon is monomeric by itself, while it is di- or tetrameric when linked to the P domain, forming the AP fragment (Table 2). This result indicates that the P domain could be directly involved in the development of the quaternary structure through interactions with the ATPase domain of an adjacent subunit of the oligomer.
The inability of the proteolytically obtained ATPase fragment of EcLon that contains all characteristic elements of ring-shaped AAA+ proteins to form oligomers is very surprising. In particular, this fragment includes the N-terminal extension of the AAA+ module (about 100 aa) together with the region which was postulated to be an oligomerization domain (OD) of Lon proteases (Lee et al., 2004a). Thus it is currently not possible to conclude that the OD region is predominantly responsible for the formation of the oligomeric structure of Lon.
The data presented here do not directly support the existence of a structural core of Lon formed by hexamers of AAA+ module, an arrangement typical for heterooligomeric ATP-dependent proteases and other representatives of the proteins belonging to the AAA+ superfamily. First, an isolated A fragment is a monomer under moderate salt concentration (0.2 M NaCl), and, second, it appears that an isolated AP fragment forms a tetramer or a dimer (Table 2) rather than a hexamer.
The differences in the oligomeric states of the AP fragments obtained by chymotryptic digestion of full-length Lon with either intact or mutated proteolytic center are strongly dependent on the procedure utilized for preparing protein samples for gel filtration. In particular, AP behaves as a dimer if concentrated without any effectors (it is worthy to note that the dimeric state was found also for the AP fragment that was prepared by digestion of EcLon with glutamyl endopeptidase (Vasilyeva et al., 2004)). The presence of nucleotides (ADP or AMPPNP), irrespective of addition of magnesium ions, leads to stabilization of a tetrameric form of AP. One may suggest that oligomerization of the full-length Lon might proceed through assembly of dimers.
It should be noted that AP oligomers completely dissociate and form individual subunits under the conditions of gel filtration in a high-salt buffer (1 M NaCl, not shown), confirming the main impact of ionic intersubunit interactions for stabilization of the oligomers.
Direct participation of the N-terminal part of EcLon in oligomerization is not fully proven, as none of the fragments obtained by digestion with chymotrypsin contains the intact putative CC domain of the enzyme (aa ~180 – ~280). At the same time, different oligomeric states of the N fragment (1–207), AP fragment (235–784), as well as full-length Lon suggest that the CC domain should play a major role in the formation of the stable oligomeric structure of the enzyme. On the other hand, this segment of Lon may represent a special problem in a structural study, due to its potential lability and irregularity of the coiled-coil structure. In particular, the CC domain may be involved in the formation of the irregular oligomers of the enzyme detected in vitro through sedimentation analysis and size exclusion chromatography. These higher-level assemblies are most likely composed of preformed oligomers of the enzyme. Nevertheless, it is known that crystallization of proteins which possess extensive irregularities in their structure is still possible (Takeda et al., 2003).
Based on the results presented here, it is clear that further investigation of the oligomerization of EcLon should be augmented by studies of recombinant N, AP, and NA fragments of the enzyme that have overlapping primary structure and include the complete CC region.
Enzymatic activity of fragments of EcLon
The fragments containing the catalytic sites for both enzymatic functions of Lon (ATPase and proteolytic) were purified to homogeneity and then tested for their respective activities. The A and AP fragments obtained from wild-type EcLon or its proteolytically inactive form EcLon-S679A were tested for the presence of ATPase activity, whereas the AP and P fragments of the enzyme with intact proteolytic site were tested for their ability to hydrolyze the low-molecular-mass tripeptide thioester Suc-Phe-Leu-Phe-SBzl (Melnikov et al., 2001), as well as β-casein, a model protein substrate. All protein fragments obtained by limited proteolysis, including the non-catalytic N fragment and the α domain of the AAA+ module, were also tested for the presence of possible traces of α-chymotrypsin using Suc-Ala-Ala-Phe-pNA, an efficient substrate of chymotrypsin that is not cleaved by EcLon. The results of such tests did not indicate the presence of any chymotryptic activity in the purified samples of the fragments of EcLon.
None of the fragments containing the catalytic sites exhibit the level of activity of full-length Lon (Table 3). It is important to stress that the A fragment and the P domain that have been excised out of wild type EcLon are not capable of hydrolyzing their typical substrates (ATP and casein, respectively). Nevertheless, the A fragment that is monomeric in solution under all tested conditions (see above), is still capable of binding nucleotides, as was shown by its stabilization in their presence upon limited proteolysis of Lon (Figs. 1 and 3), and was finally confirmed by the chymotryptic digestion of its purified preparation (Fig. 5).
Table 3.
Enzymatic activity of Lon fragments containing catalytic sites and of full-length enzyme
| Enzyme or fragment | ATPasea | Thioesterase | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| (-) β-casein | (+) β-casein | |||||||||
| I | II | II | (−) ATP/Mg | (+) ATP/Mg | ||||||
| kcat’ rnin−1 | km’ rnM | kcat’ mm−1 | km’mM | kcat’ mm−1 | km’ mM | kcat’ s−1 | km’ mM | kcat’ s−1 | km’ mM | |
| Lon-w.t. | 110 ± 8 | 0.60 ± 0.05 | 40 ± 5 | 0.20 ± 0.04 | 120 ± 10 | 0.20 ± 0.05 | 4.2 ± 0.4 | 3.5 ± 0.01 | 50 ± 4 | 3.0 ± 0.02 |
| Lon-S679A | 100 ± 5 | 0.50 ± 0.07 | 33 ± 3 | 0.15 ± 0.05 | 100 ± 8 | 0.20 ± 0.04 | inactive | |||
| A | inactive | |||||||||
| AP-w.t. | 33 ± 2 | 0.30 ± 0.06 | 35 ± 2 | 0.30 ± 0.04 | 30 ± 2 | 0.25 ± 0.06 | 0.7 ± 0.1 | 0.4 ± 0.07 | 13 ± 1 | 0.4 ± 0.06 |
| AP-S679A | 25 ± 3 | 0.25 ± 0.08 | 30 ± 4 | 0.20 ± 0.05 | 28 ± 2 | 0.30 ± 0.06 | inactive | |||
| P-w.t. | 0.04 + 0.01 | 0.30 + 0.08 | ||||||||
ATPase activity determined at the ATP-Mg ratio 1:1 (condition I) or in the presence of constant excess of magnesium ions (15 mM concentration, condition II). Kinetic parameters were derived from linear double-reciprocal plots 1/V-1/[S]. The values of the apparent catalytic constant kcat were calculated using concentration of protein (as monomer) estimated by the predicted molar absorbance coefficient (http://au.expasy.org/tools/protparam.html) and the absorbance at 280 nm.
Figure 5. Stabilization of the purified A fragment of EcLon with ADP upon chymotryptic digestion.

Fragment A was obtained according to the scheme described in Materials and Methods. Reaction conditions were standard (see legend to Fig. 1). Concentrations: fragment A − 1 mg/ml, chymotrypsin − 10 μg/ml, ADP −1 mM.
The proteolytically-inactive monomeric P domain displays a thioesterase activity (based on the kcat value, Table 3). Although such activity is two orders of magnitude lower than the activity of full-length Lon in the absence of effectors, it is still detectable. According to earlier data (Rasulova et al., 1998), the P domain should also be able to hydrolyze with low efficiency an oligopeptide substrate (melittin). The essential role of allosteric activation of the proteolytic domains seems to be a common feature of both the Lon A and LonB subfamilies. Crystal structures of isolated P domains of two members of the latter subfamily have shown that the active sites assume an inactive conformation in the absence of other domains and/or substrates (Im et al., 2004; Botos et al., 2005; Dauter et al., 2005). In contrast to the inactive counterparts, the AP fragment exhibits both the ATPase and proteolytic activities, albeit distinct from the activities of the full-length enzyme.
It has been shown previously that the presence of a protein substrate leads to a several-fold increase in the rate of ATP hydrolysis by both wild-type EcLon (Menon & Goldberg, 1987; Melnikov et al., 2000) and its proteolytically inactive mutant Lon-S679A (Fischer & Glockshuber, 1993). These results indicated that activation of ATP hydrolysis is not directly linked to the degradation of a protein target, but instead depends on substrate-enzyme interactions that might take place in additional binding sites of EcLon. The reduced ATPase activity of Lon in the absence of a protein substrate was shown (Melnikov et al., 2000) to be mainly caused by the influence of excess magnesium ions present in the reaction solution along with the ATP-Mg complex (Table 3). This inhibitory effect seems to be connected to the peculiarities of cooperative ATP hydrolysis and nucleotide exchange exhibited at the level of the quaternary structure of Lon. These magnesium-dependent aspects of the function of Lon have not been extensively discussed in the investigations of enzyme kinetics (Vineyard et al., 2006). However, our interpretation (Melnikov et al., 2000; 2001) of the role of magnesium ions in the activity of Lon appears to resemble the one discussed in a recent paper (Rosenfeld et al., 2005) dealing with the study of the processive mechanism of myosin V. The inhibitory properties of excess magnesium discussed here are also in good agreement with recent structural data on helicases complexed with ADP (Lee & Yang, 2006; Enemark & Joshua-Tor, 2006). A mimic of the intermediate step of ATP hydrolysis that involved ADP and Mg2+ ions occupying the position of the γ-phosphate of the nucleotide was trapped in the crystals, thus slowing down the turnover of ATP.
The ATPase activity of the AP fragments obtained from either the wild type or inactive S679A mutant of full-length Lon is decreased relative to that of the intact enzyme (Table 3), insensitive to the presence of free magnesium ions, and not increased in the presence of a protein substrate (an observation that possibly provides additional evidence for the presumed role of the N domain as an additional substrate-binding region of Lon (Ebel et al., 1999)). These results might suggest that the cooperative mechanism of the ATP hydrolysis by the AP fragment is altered due to the removal of the N-terminal part of Lon (residues 1–234).
Nevertheless, the AP fragment of wild type Lon is capable of hydrolyzing a low-molecular mass thioester, Suc-Phe-Leu-Phe-SBzl, in an ATP-dependent manner, not distinguishable from the full-length Lon (Table 3). Thus the catalytic constants for the thioesterase activity of AP and wild type Lon do not differ dramatically, although the apparent Km parameters that are characteristic for AP are more strongly affected (Table 3).
Testing the proteolytic activity of the AP fragment of wild type Lon revealed some unexpected properties of this truncated form of the enzyme (Fig. 6A). The initial experiments, performed under standard reaction conditions and during relatively short-term incubation of the reaction components (about 2 h), did not detect any ability of the AP fragment to hydrolyze protein substrates. However, in several special cases, it was possible to detect proteolytic activity of AP during prolonged incubation with a substrate (about 16 h). These conditions included either the absence of nucleotides, regardless of the presence of magnesium ions (Fig. 6A, lanes 2, 6), or ATP hydrolysis (Fig. 6A, lane 7), or activation of AP with a non-hydrolyzable analog of ATP (AMPPNP) in the presence of magnesium ions (Fig. 6A, lane 9).
Figure 6. Comparison of the proteolytic properties of the AP fragment (A) and of full-length Lon protease (B).

Tested proteins were not saturated with any nucleotide before final purification step on size-exclusion column (see Materials and Methods). Lane 1 - β-casein. Reactions were performed at 37°C during 16 h for AP, and 2 h for Lon. For other conditions see legend to Fig. 1. Concentrations: AP and Lon − 0.1 mg/ml, β-casein − 1 mg/ml, nucleotides −1 mM, MgCl2 − 20 mM.
As shown in Fig. 6A, the proteolytic activity of AP is not processive in all cases, as it is accompanied by the formation of high-molecular mass intermediates of the protein substrate degradation. Along with that, the autocatalytic degradation of the AP fragment also persists, with the exception of the reaction performed in the presence of AMPPNP-Mg. The presence of ATP or its analog in the reaction media in the absence of magnesium ions (Fig. 6A, lanes 3, 5), or the presence of ADP regardless of the presence of magnesium ions (Fig. 6A, lanes 4, 8), not only suppress the proteolytic activity but also prevent autolysis of the AP fragment. The protective effect of some nucleotides against self-degradation of AP is similar to that described for the yeast Lon protease (Stahlberg et al., 1999).
The non-processive degradation of protein substrates is not a unique feature of the AP fragment, but also of the full-length enzyme (Fig. 6B). However, its in vitro manifestation strictly depends on the procedures used for isolation and purification of the intact Lon. The necessary prerequisite for the manifestation of this property is the removal of the intracellular nucleotides from the enzyme samples, and most of all ADP, which is simultaneously a Lon-specific ligand, a product of ATP hydrolysis, and an allosteric inhibitor of the peptidase activity of Lon.
The presence of variable amounts of ADP in the enzyme preparations obtained by different procedures is the most likely reason for the variability of the ATP-independent peptide hydrolase activity. In particular, data presented in Table 3 characterize the thioesterase activity displayed by wild type Lon that was saturated with ADP before the final step of purification by size-exclusion chromatography. Omitting the stage of saturation with the nucleotide resulted in an increase of kcat up to about 35–40 s−1 in the absence of ATP-Mg. This nucleotide-free form of the enzyme is characterized by very high level of non-processive proteolytic activity (Fig. 6B, lanes 2, 6); however, the enzyme retains its main function as a processive protease upon ATP hydrolysis (Fig. 6B, lane 7).
The non-processive degradation of protein substrates by Lon also takes place under conditions of static activation of the enzyme when the activator does not undergo any transformation (e.g. in the presence of AMPPNP and magnesium ions). As reported earlier (Edmunds & Goldberg, 1986) but not discussed in detail, such non-processive proteolytic activity of Lon, revealed in the presence of non-hydrolyzable ATP analogs, is unique to this enzyme.
The non-processive proteolytic activity of Lon, observed at low concentration of the enzyme, in the absence of nucleotides, or at an elevated temperature may indicate that the quaternary structure of Lon is not very stable. In the low-molecular-mass oligomers of Lon the proteolytic sites are sterically open, and the enzyme is capable of hydrolyzing protein substrates in a non-processive manner, acting as a common protease.
It is necessary to emphasize that the AP fragment is actually a proteolytically active enzyme capable of hydrolyzing a protein substrate in a non-processive manner (Fig. 6A, lane 9). At the same time, a proteolytic activity of the autolysis products of AP cannot be completely ruled out (Fig. 6A, lanes 2, 6, 7). Stabilization of AP under conditions of static activation (by AMPPNP-Mg), and self-degradation of AP under conditions of dynamic activation (upon ATP hydrolysis), which is not typical for a full-length Lon, allows one to differentiate between the conformational or oligomeric states of AP in the different activation systems (AMPPNP-Mg or ATP-Mg).
The initial stage of AP self-degradation in the absence of a protein substrate depends on the presence of the ATP-Mg complex (Fig. 7). Interestingly, the apparent rate of AP autolysis under conditions of ATP hydrolysis is not altered in the presence of a protein substrate (not illustrated), which may indicate the intramolecular nature of the process taking place in this case. However, the final conclusion about the nature of this process should be based on the investigation of the kinetic order of the autolytic reaction.
Figure 7. Autolysis of the AP fragment in the absence of nucleotides and upon ATP hydrolysis.
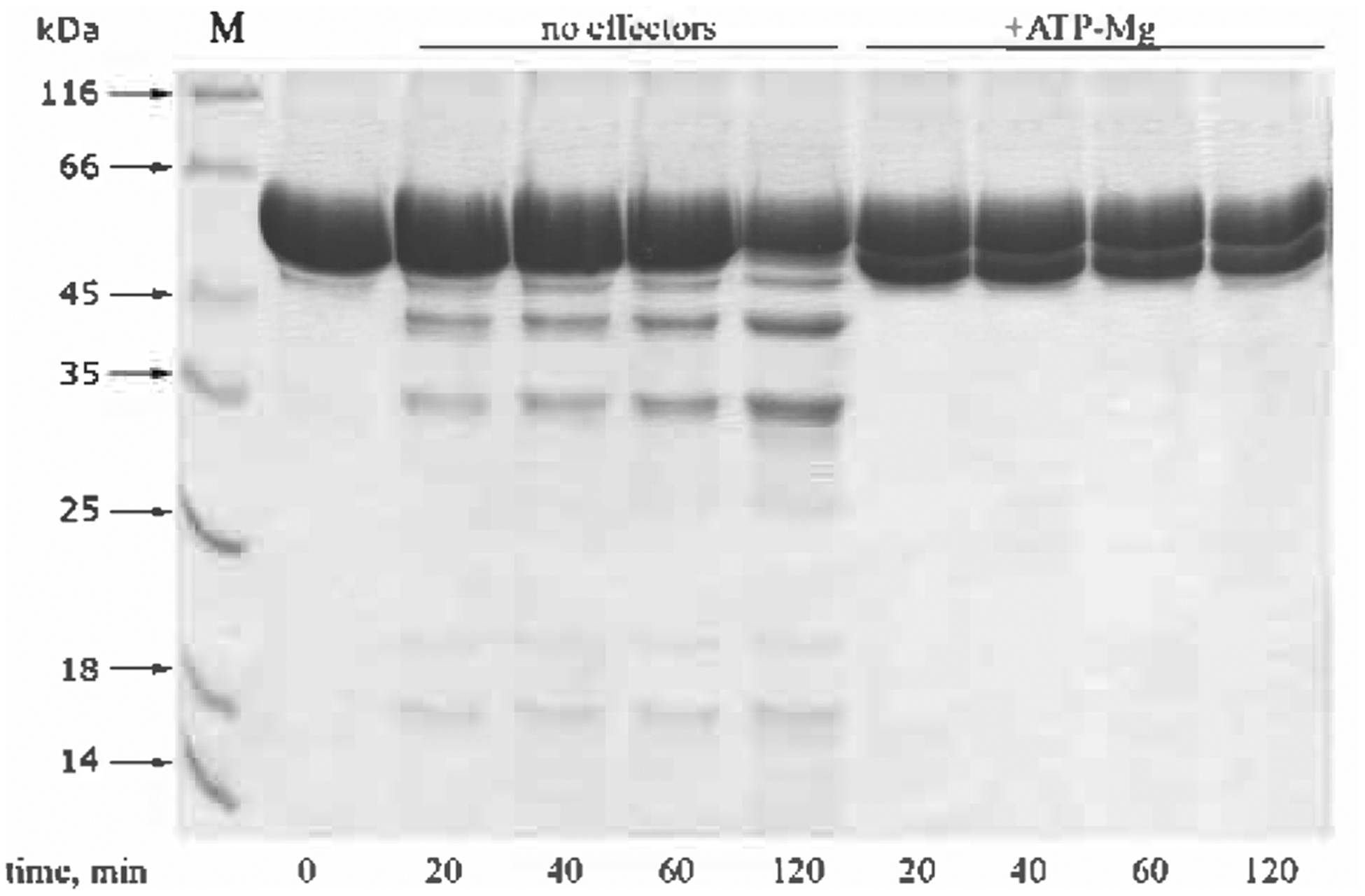
Reactions were performed at 37°C, other conditions were standard (see legend to Fig. 1). Concentrations: AP − 1 mg/ml, ATP − 1 mM, MgCl2 − 20 mM.
The products of autocatalytic cleavage of AP have not been studied in much detail. However, N-terminal analysis of the major product of such self-degradation obtained under the conditions of ATP hydrolysis (Fig. 7) indicated cleavage of the bond between Ala286 and Glu287, near the C-terminal end of the predicted CC domain of the enzyme (Figs. 2 and 4). No cleavage close to this site was observed during digestion of Lon by proteases of different specificity (Fig. 4), and it might be assumed that this part of the structure is buried within the Lon oligomer and involved in direct interactions with the proteolytic domain of the enzyme.
A comparison of the enzymatic properties of the full-length Lon and its truncated AP form indicates that the removal of the N-terminal segment (amino acids 1–234) decreases the degree of oligomerization, thus creating an enzymatic form with an altered pattern of cooperative ATP hydrolysis, incapable of processive degradation of protein substrates, and susceptible to the autocatalytic cleavage.
CONCLUSIONS
A study of the oligomeric state and properties of the EcLon fragments that were obtained by limited proteolysis revealed the contribution of individual domains to the quaternary structure of the full-length enzyme and clarified the relationship between the quaternary structure and the two different enzymatic activities of this enzyme. Four regions in Lon structure that are sensitive to proteolytic digestion have been located and the boundaries of the α-helical and proteolytic domains were confirmed. True boundaries of the N-terminal and ATPase domains need to be defined more precisely. The conformational state of the interface between the α and the proteolytic domain of Lon complexed with nucleotides has been shown to be influenced by the presence of magnesium ions. However, no essential structural changes were revealed in the region located between the N and A domains of Lon regardless of the presence of effectors.
All chymotryptic fragments and domains of Lon, with the exception of AP, have been shown to be monomeric in solution. Unlike the ATPase subunits of the heterooligomeric AAA+ proteases, the isolated ATPase fragment of Lon exhibits no ATPase activity and does not change its monomeric state in solution, although it can still bind nucleotides. The individual P domain appears to have no proteolytic activity at all.
In contrast to the monomeric and inactive A fragment and P domain, the AP fragment exists as an oligomer and exhibits both the ATPase and proteolytic activities. This means that both counterparts of the AP fragment play a significant role in the formation of the quaternary structure of the enzyme. The AP fragment is a non-processive protease and undergoes unusual self-degradation upon ATP hydrolysis. The self-cleavage site is located at the C-terminal end of the predicted coiled-coil region of the enzyme. We postulate that this part of the structure is buried within the Lon oligomer and is involved in direct interactions with the proteolytic domain of the enzyme.
Taken together, these results reveal the crucial role played by the non-catalytic N-terminal part of EcLon in enabling enzymatic functionality. The low-molecular-mass forms of EcLon appear to possess the ability for non-processive degradation of protein substrates. The data presented here could also reflect the important impact of the coiled-coil region of Lon on the formation of the quaternary structure of the enzyme. It will be necessary to continue studies of various recombinant fragments whose primary structures overlap and contain the full coiled-coil area of the enzyme.
Acknowledgements
This work was supported in part by grant 05-04-48383 from the Russian Foundation for Basic Research, by the US Civilian Research and Development Foundation grant RB1-2505-M0-03, and by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Abbreviations:
- A
ATPase fragment of Lon (aa 235–584)
- aa
amino acid
- AMPPNP
adenyl-5’-yl-imidodiphosphate
- α
α-helical domain of AAA+ module of Lon (aa 491–584)
- AP
truncated Lon (aa 235–784) lacking N-terminal part
- CC
coiled-coils
- N
N-terminal fragment of Lon (aa 1–207)
- P
proteolytic domain of Lon (aa 585–784)
- PMSF
phenylmethylsulfonyl fluoride
REFERENCES
- Amerik A Yu, Antonov VK, Ostr oumova NI, Rotanova TV, Chistiakova LG (1990) Cloning, structure and expression of the full-size lon gene in Escherichia coli coding for ATP-dependent La-protease. Bioorg Khim 16: 869–880. [PubMed] [Google Scholar]
- Amerik AYu, Antonov VK, Gorbalenya AE, Kotova SA, Rotanova TV, Shimbarevich EV (1991) Site-directed mutagenesis of La protease. A catalytically active serine residue. FEBS Lett 287: 211–214. [DOI] [PubMed] [Google Scholar]
- Bencini DA, Wild JR, O’Donovan GA (1983) Linear one-step assay for the determination of orthophosphate. Anal Biochem 132: 254–258. [DOI] [PubMed] [Google Scholar]
- Botos I, Melnikov EE, Cherry S, Khalatova AG, Rasulova FS, Tropea JE, Maurizi MR, Rotanova TV, Gust china A, Wlodawer A (2004a) Crystal structure of the AAA+ α domain of E. coli Lon protease at 1.9Å resolution. J Struct Biol 146: 113–122. [DOI] [PubMed] [Google Scholar]
- Botos I, Melnikov EE, Cherry S, Tropea JE, Khalatova AG, Rasulova F, Dauter Z, Maurizi MR, Rotanova TV, Wlodawer A, Gustchina A (2004b) The catalytic domain of Escherichia coli Lon protease has a unique fold and a Ser-Lys dyad in the active site. J Biol Chem 279: 8140–8148. [DOI] [PubMed] [Google Scholar]
- Botos I, Melnikov EE, Cherry S, Kozlov S, Makhovskaya OV, Tropea JE, Gustchina A, Rotanova TV, Wlodawer A (2005) Atomic-resolution crystal structure of the proteolytic domain of Archaeoglobus fulgidus Lon reveals the conformational variability in the active sites of Lon proteases. J Mol Biol 351: 144–157. [DOI] [PubMed] [Google Scholar]
- Burton RE, Baker TA, Sauer RT (2005) Nucleotide-depend-ent substrate recognition by the AAA+ HslUV protease. Nat Struct Mol Biol 12: 245–251. [DOI] [PubMed] [Google Scholar]
- Charette MF, Henderson GW, Markovitz A (1981) ATP hy-drolysis-dependent protease activity of the lon (capR) protein of Escherichia coli K-12. Proc Natl Acad Sci USA 78: 4728–4732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung CH, Goldberg AL (1981) The product of the lon (capR) gene in Escherichia coli is the ATP-dependent protease, protease La. Proc Natl Acad Sci USA 78: 4931–4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dauter Z, Botos I, LaRonde-LeBlanc N, Wlodawer A (2005) Pathological crystallography: case studies of several unusual macromolecular crystals. Acta Crystallogr D Biol Crystallogr 61: 967–975. [DOI] [PubMed] [Google Scholar]
- Ebel W, Skinner MM, Dierksen KP, Scott JM, Trempy JE (1999) A conserved domain in Escherichia coli Lon protease is involved in substrate discriminator activity. J Bacteriol 181: 2236–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edmunds T, Goldberg AL (1986) Role of ATP hydrolysis in the degradation of proteins by protease La from Escherichia coli. J Cell Biochem 32: 187–191. [DOI] [PubMed] [Google Scholar]
- Enemark EJ, Joshua-Tor L (2006) Mechanism of DNA translocation in a replicative hexameric helicase. Nature 442: 270–275. [DOI] [PubMed] [Google Scholar]
- Erlanger BF, Kokowsky N, Cohen W (1961) The preparation and properties of two new chromogenic substrates of trypsin. Arch Biochem Biophys 95: 271–278. [DOI] [PubMed] [Google Scholar]
- Fischer H, Glockshuber R (1993) ATP hydrolysis is not stoichiometrically linked with proteolysis in the ATP-dependent protease La from Escherichia coli. J Biol Chem 268: 22502–22507. [PubMed] [Google Scholar]
- Frickey T, Lupas AN (2004) Phylogenetic analysis of AAA proteins. J Struct Biol 146: 2–10. [DOI] [PubMed] [Google Scholar]
- Goldberg AL (1992) The mechanism and functions of ATP-dependent proteases in bacterial and animal cells. Eur J Biochem 203: 9–23. [DOI] [PubMed] [Google Scholar]
- Goldberg AL, Moerschell RP, Chung CH, Maurizi MR (1994) ATP-dependent protease La (lon) from Escherichia coli. Methods Enzymol 244: 350–375. [DOI] [PubMed] [Google Scholar]
- Gottesman S (2003) Proteolysis in bacterial regulatory circuits. Annu Rev Cell Dev Biol 19: 565–587. [DOI] [PubMed] [Google Scholar]
- Gottesman S, Wickner S, Jubete Y, Singh SK, Kessel M, Maurizi M (1995) Selective, energy-dependent proteolysis in Escherichia coli. Cold Spring Harb Symp Quant Biol 60: 533–548. [DOI] [PubMed] [Google Scholar]
- Halsall HB (1967) Atassi-Gandhi sedimentation coefficient and molecular weight relationships. Nature 215: 880–881. [Google Scholar]
- Im YJ, Na Y, Kang GB, Rho SH, Kim MK, Lee JH, Chung CH, Eom SH (2004) The active site of a Lon protease from Methanococcus jannaschii distinctly differs from the canonical catalytic dyad of Lon proteases. J Biol Chem 279: 53451–53457. [DOI] [PubMed] [Google Scholar]
- Iyer LM, Leipe DD, Koonin EV, Aravind L (2004) Evolutionary history and higher order classification of AAA+ ATPases. J Struct Biol 146: 11–31. [DOI] [PubMed] [Google Scholar]
- Joshi SA, Hersch GL, Baker TA, Sauer RT (2004) Communication between ClpX and ClpP during substrate processing and degradation. Nat Struct Mol Biol 11: 404–411. [DOI] [PubMed] [Google Scholar]
- Kenniston JA, Baker TA, Sauer RT (2005) Partitioning between unfolding and release of native domains during ClpXP degradation determines substrate selectivity and partial processing. Proc Natl Acad Sci USA 102: 1390–1395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JY, Yang W (2006) UvrD helicase unwinds DNA one base pair at a time by a two-part power stroke. Cell 127: 1349–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AY, Hsu CH, Wu SH (2004a) Functional domains of Brevibacillus thermoruber Lon protease for oligomerization and DNA binding: role of N-terminal and sensor and substrate discrimination domains. J Biol Chem 279: 34903–34912. [DOI] [PubMed] [Google Scholar]
- Lee AY, Tsay SS, Chen MY, Wu SH (2004b) Identification of a gene encoding Lon protease from Brevibacillus thermoruber WR-249 and biochemical characterization of its thermostable recombinant enzyme. Eur J Biochem 271: 834–844. [DOI] [PubMed] [Google Scholar]
- Li M, Rasulova F, Melnikov EE, Rotanova TV, Gustchina A, Maurizi MR, Wlodawer A (2005) Crystal structure of the N-terminal domain of E. coli Lon protease. Protein Sci 14: 2895–2900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupas AN, Martin J (2002) AAA proteins. Curr Opin Struct Biol 12: 746–753. [DOI] [PubMed] [Google Scholar]
- Lupas A, Van Dyke M, Stock J (1991) Predicting coiled coils from protein sequences. Science 252: 1162–1164. [DOI] [PubMed] [Google Scholar]
- Martin A, Baker TA, Sauer RT (2005) Rebuilt AAA+ motors reveal operating principles for ATP-fuelled machines. Nature 437: 1115–1120. [DOI] [PubMed] [Google Scholar]
- Maupin-Furlow JA, Gil MA, Humbard MA, Kirkland PA, Li W, Reuter CJ, Wright AJ (2005) Archaeal proteas-omes and other regulatory proteases. Curr Opin Microbiol 8: 720–728. [DOI] [PubMed] [Google Scholar]
- Maurizi MR, Li CC (2001) AAA proteins: in search of a common molecular basis. International Meeting on Cellular Functions of AAA Proteins. EMBO Rep 2: 980–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffin LJ, Bryson K, Jones DT (2000) The PSIPRED protein structure prediction server. Bioinformatics 16: 404–405. [DOI] [PubMed] [Google Scholar]
- Melnikov EE, Tsirulnikov KB, Rotanova TV (2000) Coupling of proteolysis with ATP hydrolysis by Escherichia coli Lon protease. I. Kinetic aspects of ATP hydrolysis. Bioorg Khim 26: 530–538. [PubMed] [Google Scholar]
- Melnikov EE, Tsirulnikov KB, Rotanova TV (2001) Coupling of proteolysis and hydrolysis of ATP upon functioning of Lon protease of Escherichia coli. II. Hydrolysis of ATP and activity of peptide hydrolase sites of the enzyme. Bioorg Khim 27: 120–129. [DOI] [PubMed] [Google Scholar]
- Menon AS, Goldberg AL (1987) Protein substrates activate the ATP-dependent protease La by promoting nucleotide binding and release of bound ADP. J Biol Chem 262: 14929–14934. [PubMed] [Google Scholar]
- Neuwald AF, Aravind L, Spouge JL, Koonin EV (1999) AAA+: A class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexes. Genome Res 9: 27–43. [PubMed] [Google Scholar]
- Ogura T, Whiteheart SW, Wilkinson AJ (2004) Conserved arginine residues implicated in ATP hydrolysis, nucle-otide-sensing, and inter-subunit interactions in AAA and AAA+ ATPases. J Struct Biol 146: 106–112. [DOI] [PubMed] [Google Scholar]
- Ogura T, Wilkinson AJ (2001) AAA+ superfamily ATPases: common structure — diverse function. Genes Cells 6: 575–597. [DOI] [PubMed] [Google Scholar]
- O’Sullivan WJ, Smither GW (1979) Stability constants for biologically important metal-ligand complexes. Methods Enzymol 63: 295–325. [DOI] [PubMed] [Google Scholar]
- Park SC, Jia B, Yang JK, Van D, Shao YG, Han SW, Jeon YJ, Chang CH, Cheong GW (2005) Oligomeric structure of the ATP-dependent protease La (Lon) of Escherichia coli. Mol Cells 21: 129–134. [PubMed] [Google Scholar]
- Patterson J, Vineyard D, Thomas-Wohlever J, Behshad R, Burke M, Lee I (2004) Correlation of an adenine-spe-cific conformational change with the ATP-dependent peptidase activity of Escherichia coli Lon. Biochemistry 43: 7432–7442. [DOI] [PubMed] [Google Scholar]
- Ramachandran R, Hartmann C, Song HK, Huber R, Bo-chtler M (2002) Functional interactions of HslV (ClpQ) with the ATPase HslU (ClpY). Proc Natl Acad Sci USA 99: 7396–7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasulova FS, Dergousova NI, Starkova NN, Melnikov EE, Rumsh LD, Ginodman LM, Rotanova TV (1998) The isolated proteolytic domain of Escherichia coli ATP-de-pendent protease Lon exhibits the peptidase activity. FEBS Lett 432: 179–181. [DOI] [PubMed] [Google Scholar]
- Reid BG, Fenton WA, Horwich AL, Weber-Ban EU (2001) ClpA mediates directional translocation of substrate proteins into the ClpP protease. Proc Natl Acad Sci USA 98: 3768–3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivett AJ (1989) High molecular mass intracellular proteases. Biochem J 263: 625–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenfeld SS, Houdusse A, Sweeney HL (2005) Magnesium regulates ADP dissociation from myosin V. J Biol Chem 280: 6072–6079. [DOI] [PubMed] [Google Scholar]
- Rotanova TV (1999) Structural and functional characteristics of ATP-dependent Lon-proteinase from Escherichia coli. Bioorg Khim 25: 883–891. [PubMed] [Google Scholar]
- Rotanova TV (2002) Peptide hydrolases with catalytic dyad Ser-Lys. Similarity and distinctions of the active centers of ATP-dependent Lon proteases, LexA repressors, signal peptidases and C-terminal processing proteases. Vopr Med Khim 48: 541–552. [PubMed] [Google Scholar]
- Rotanova TV, Melnikov EE, Tsirulnikov KB (2003) Catalytic dyad Ser-Lys at the active site of Escherichia coli ATP-dependent Lon-proteinase. Bioorg Khim 29: 97–99. [DOI] [PubMed] [Google Scholar]
- Rotanova TV, Melnikov EE, Khalatova AG, Makhovskaya OV, Botos I, Wlodawer A, Gustchina A (2004) Classification of ATP-dependent proteases Lon and comparison of the active sites of their proteolytic domains. Eur J Biochem 271: 4865–4871. [DOI] [PubMed] [Google Scholar]
- Rouiller I, DeLaBarre B, May AP, Weis WI, Brunger AT, Milligan RA, Wilson-Kubalek EM (2002) Conformational changes of the multifunction p97 AAA ATPase during its ATPase cycle. Nat Struct Biol 9: 950–957. [DOI] [PubMed] [Google Scholar]
- Rudyak SG, Brenowitz M, Shrader TE (2001) Mg2+-linked oligomerization modulates the catalytic activity of the Lon (La) protease from Mycobacterium smegmatis. Biochemistry 40: 9317–9323. [DOI] [PubMed] [Google Scholar]
- Sauer RT, Bolon DN, Burton BM, Burton RE, Flynn JM, Grant RA, Hersch GL, Joshi SA, Kenniston JA, Levchenko I, Neher SB, Oakes ES, Siddiqui SM, Wah DA, Baker TA (2004) Sculpting the proteome with AAA+ proteases and disassembly machines. Cell 119: 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schechter I, Berger A (1967) On the size of the active site in proteases. I. Papain. Biochem Biophys Res Commun 27: 157–162. [DOI] [PubMed] [Google Scholar]
- Schmidt M, Lupas AN, Finley D (1999) Structure and mechanism of ATP-dependent proteases. Curr Opin Chem Biol 3: 584–591. [DOI] [PubMed] [Google Scholar]
- Stahlberg H, Kutejova E, Suda K, Wolpensinger B, Lustig A, Schatz G, Engel A, Suzuki CK (1999) Mitochondrial Lon of Saccharomyces cerevisiae is a ring-shaped protease with seven flexible subunits. Proc Natl Acad Sci USA 96: 6787–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swamy KH, Goldberg AL (1981) E. coli contains eight soluble proteolytic activities, one being ATP dependent. Nature 292: 652–654. [DOI] [PubMed] [Google Scholar]
- Takeda S, Yamashita A, Maeda K, Maeda Y (2003) Structure of the core domain of human cardiac troponin in the Ca2+-saturated form. Nature 424: 35–41. [DOI] [PubMed] [Google Scholar]
- Tsilibaris V, Maenhaut-Michel G, Van Melderen L (2006) Res Microbiol 157: 701–713. [DOI] [PubMed] [Google Scholar]
- Vale RD (2000) AAA proteins. Lords of the ring. J Cell Biol 150: 13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Holde KE, Weischet WO (1978) Boundary analysis of sedimentation-velocity experiments with monodisperse and paucidisperse solutes. Biopolymers 17: 1387–1403. [Google Scholar]
- Vasilyeva OV, Kolygo KB, Leonova YF, Potapenko NA, Ovchinnikova TV (2002) Domain structure and ATP-induced conformational changes in Escherichia coli protease Lon revealed by limited proteolysis and autolysis. FEBS Lett 526: 66–70. [DOI] [PubMed] [Google Scholar]
- Vasilyeva OV, Martynova NIu, Potapenko NA, Ovchinnikova TV (2004) Isolation and characterization of fragments of ATP-dependent protease Lon from Escherichia coli obtained by limited proteolysis. Bioorg Khim 30: 341–349. [PubMed] [Google Scholar]
- Vineyard D, Zhang X, Lee I (2006) Transient kinetic experiments demonstrate the existence of a unique catalytic enzyme form in the peptide-stimulated ATPase mechanism of Escherichia coli Lon protease. Biochemistry 45: 11432–11443. [DOI] [PubMed] [Google Scholar]
- Weber-Ban EU, Reid BG, Miranker AD, Horwich AL (1999) Global unfolding of a substrate protein by the HsplOO chaperone ClpA. Nature 401: 90–93. [DOI] [PubMed] [Google Scholar]
- Wickner S, Maurizi MR, Gottesman S (1999) Posttransla-tional quality control: folding, refolding, and degrading proteins. Science 286: 1888–1893. [DOI] [PubMed] [Google Scholar]
- Zehnbauer BA, Foley EC, Henderson GW, Markovitz A (1981) Identification and purification of the Lon+ (capR+) gene product, a DNA-binding protein. Proc Natl Acad Sci USA 78: 2043–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]