

Abstract
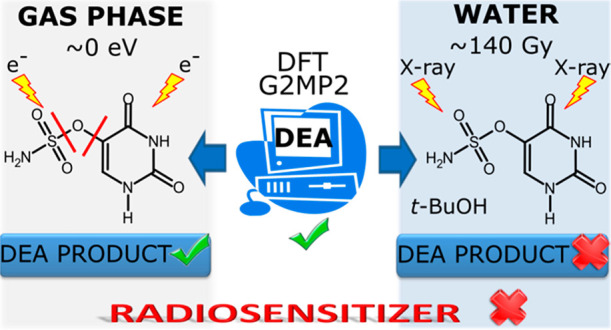
Efficient radiotherapy requires the concomitant use of ionizing radiation (IR) and a radiosensitizer. In the present work uracil-5-yl O-sulfamate (SU) is tested against its radiosensitizing potential. The compound possesses appropriate dissociative electron attachment (DEA) characteristics calculated at the M06-2X/6-31++G(d,p) level. Crossed electron–molecular beam experiments in the gas phase demonstrate that SU undergoes efficient DEA processes, and the single C–O or S–O bond dissociations account for the majority of fragments induced by electron attachment. Most DEAs proceed already for electrons with kinetic energies of ∼0 eV, which is supported by the exothermic thresholds calculated at the M06-2X/aug-cc-pVTZ level. However, in water solution under reductive conditions and physiological pH, SU does not undergo radiolysis, which demonstrates the crucial influence of aqueous environment on the radiosensitizing properties of modified nucleosides.
1. Introduction
Radiotherapy is one of the most common modalities in anticancer treatment. Indeed, around 80% of cancerous patients are exposed to ionizing radiation (IR) at certain stages of their therapy.1 This modality usually employs sparsely ionizing radiation, i.e., X-ray photons or high energy electrons delivered by linear accelerators (linacs).2 Still much less common, although already available clinically, are heavy-particle or proton beam therapies. Although such modalities seem to be less affected by tumor hypoxia, large cost related to the construction and usage of heavy particle facilities makes a serious limitation to their widespread usage.2
X-rays and beams of electrons produced by linacs generate hydroxyl radicals and secondary electrons (water radiolysis) when passing through water, which constitutes ca. 70% of the human body.2 As indicated by recent studies carried out by the Mostafavi group, ultra-short-lived prehydrated3 or conduction band4 electrons may partially account for DNA damage induced by secondary electrons in cellular environments.
However, an efficient radiotherapy should be associated with the concomitant use of IR and a radiosensitizing agent. Several classes of small-molecule radiosensitizers have been proposed and tested in the clinic so far.5 Nevertheless, as suggested by ongoing clinical trials, the number of tested radiosensitizers is relatively low.6 As far as the clinically allowed radiosensitizers are concerned, the situation is even worse. For instance, no chemicals working as radiosensitizers are used in clinical practice against gastrointestinal cancers7 and nimorazole—4-[2-(5-nitroimidazol-1-yl)ethyl]morpholine—is a rare example of an approved radiosensitizing molecule for the treatment of head and neck cancers in Denmark.8
Uridine analogues which incorporate to DNA belong to a class of radiosensitizers comprising the most thoroughly studied 5-bromo- and 5-iodo-2′-deoxyuyridines.9 It is believed that radiosensitizing properties of these molecules, while constituting a part of DNA, are related to the electron-attachment-induced dissociation of the C5–X bond that releases a halide anion leaves behind a reactive uracil-5-yl radical in the biopolymer molecule.10 Secondary reactions beginning with hydrogen atom transfer between the uracil-5-yl radical and an adjacent sugar molecule lead ultimately to a single strand break which may result in cell death if not repaired.9
It is worth emphasizing that in this mode of action the radiosensitizing uridines utilize solvated electrons, which are one of the most abundant products of water radiolysis.11 It has long been demonstrated that solvated electrons bind to nucleobases, nucleotides, and DNA almost at a diffusion controlled rate.12 However, no strand breaks are produced as a result of electron attachment to the native DNA, which was proved experimentally13 and justified theoretically.14 Only specific chemical modifications to the DNA monomeric units make them prone to dissociative electron attachment (DEA).15 The mechanism that utilizes electrons unreactive toward native DNA and is operative under hypoxia when the damaging properties of hydroxyl radical are significantly impaired prompted us to propose several new uridine radiosensitizers. To this end, 5-selenocyanato-2′-deoxyuridine (SeCNdU),16 5-trifluoromethanesulfonyl-2′-deoxyuridine (OTfdU),16 5-iodo-4-thio-2′-deoxyuridine (ISdU),17 or 5-thiocyanato-2′-deoxyuridine (SCNdU)18 can be mentioned as representative examples. In the heart of our approach lies the quantum chemically calculated DEA profile obtained for a verified nucleoside/nucleobase. Consequently, we seek for derivatives for which releasing of an anion (a leaving group) triggered by electron attachment is associated with a sufficiently large thermodynamic stimulus that makes the whole damage process spontaneous and with a tiny activation barrier preventing the protonation of the formed anion in an aqueous solution. The latter process, fast enough to be competitive with DEA, is probably responsible for the lack of DNA strand breaks in the IR irradiated aqueous solution, as opposed to a plasmid DNA bombarded with low energy electrons (LEEs) under ultrahigh vacuum,19 where strand breaks are induced by electrons with energies well below the ionization threshold of the biomolecule. The body of data concerning the radiosensitizing nucleobases/nucleosides suggests that an effective radiosensitizer has to be decomposed efficiently due to electron attachment.15,20−24 The dissociation channels that can be opened by the attachment of LEEs to a molecule in the gas phase account for the damaging potential that inhabits electron–molecule interactions. At least some of the channels, observed in the gas phase, are expected to be also operative in an aqueous solution. Hence, the analysis of ion yields induced by electron attachment in the gas phase should help one to interpret and comprehend the reactivity triggered by hydrated electrons in solution. The decomposition of a potential radiosensitizer by LEEs can be studied in the gas phase using a crossed electron–molecular beam technique, which utilizes mass spectrometry for the analysis of fragment anions formed as a result of DEA.25,26
In the current work, we report for the first time on the physicochemical characteristics of uracil-5-yl O-sulfamate (SU, NH2SO3C4H4N2O2; molecular mass 207 g·mol–1), a potential radiosensitizer. Since favorable DEA characteristics have been calculated for this compound at the DFT level, it has been chemically synthesized and its molecular structure has been confirmed crystallographically. Moreover, its propensity to electron-induced decomposition has been determined in both the gas phase and water solution. The measured energy onsets of LEE triggered dissociation in the gas phase have been supported by the quantum chemical calculations of the thermodynamic thresholds for the occurrence of the observed anions. On the other hand, the outcome of radiolysis of water solutions containing the studied compound has been interpreted in terms of measured pKa and the DEA profile calculated at the G2MP2 level.
2. Experimental and Computational Methods
2.1. Synthesis
To a stirred solution of chlorosulfonyl isocyanate (3.05 mL, 7.02 mmol) in dry dichloromethane (DCM; 12 mL) at 40 °C, a mixture of formic acid (1.36 mL, 7.21 mmol) and N,N-dimethylacetamide (DMA; 0.029 mL, 0.075 mmol) was added. After ca. 3 h of vigorous stirring, a suspension of 5-hydroxyuracil (100 mg, 0.78 mmol) in DMA (5 mL) was added. The mixture was stirred at ambient temperature for 24 h and then poured into water (20 mL). After 3 h, the formed precipitate was filtered off. Uracil-5-yl O-sulfamate was obtained as a white solid (54 mg) in a 33.4% yield. 5-Hydroxyuracil, chlorosulfonyl isocyanate, DMA, formic acid, and anhydrous DCM were commercially available from Sigma-Aldrich.
1H NMR (Figure S1 in the Supporting Information), δ: 11.45 (s, 1H), 10.95−11.05 (m, 1H), 7.93 (s, 2H), 7.52 (d, 1H). HRMS (Figure S2), m/z: [M – H]− calcd for C4H5N3O5S 207.1646; found 206.0003. UV spectrum (water; Figure S3), λmax: 268 nm.
The 1H NMR spectrum was recorded on a Bruker AVANCE III, 500 MHz spectrometer. Chemical shifts are reported in parts per million relative to the residual signal of DMSO-d6 (2.49 ppm). The MS measurements were done with use of a TripleTOF 5600+ (SCIEX, Germany), and the UV spectrum was recorded on a Dionex UltiMate 3000 System with a diode array detector.
2.2. XRD Measurements and Refinements
A good-quality single crystal of uracil-5-yl O-sulfamate has been selected for the X-ray diffraction experiments at T = 295(2) K (Table 1). It was mounted with epoxy glue at the tip of a glass capillary. The diffraction data were collected on an Oxford Diffraction Gemini R ULTRA Ruby CCD diffractometer with Mo Kα (λ = 0.710 73 Å) radiation. The lattice parameters were obtained by least-squares fit to the optimized setting angles of the reflections collected by means of CrysAlis CCD.27 The data were reduced using CrysAlis RED software27 and applying multiscan absorption corrections (empirical absorption correction using spherical harmonics, implemented in the SCALE3 ABSPACK scaling algorithm). The structural resolution procedure was carried out using the SHELX package.28 The structure was solved with direct methods that carried out refinements by full-matrix least squares on F2 using the SHELXL-2017/1 program.28 A hydrogen atom bound to the aromatic carbon atom was placed geometrically and refined using a riding model with C–H = 0.93 Å and Uiso(H) = 1.2Ueq(C). All hydrogen atoms bound to nitrogen atoms were placed geometrically and refined freely with Uiso(H) = 1.2Ueq(N). All interactions were calculated using the PLATON program.29 The ORTEPII,30 PLUTO-78,31 and Mercury32 programs were used to prepare the molecular graphics.
Table 1. Summary of the Resonance Positions, Experimental Thresholds, and Calculated Thermodynamic Thresholds for the Fragment Anions Formed upon Electron Attachment to Uracil-5-yl O-Sulfamate.
| threshold (eV) at 1.58 × 10–11 atm |
||||||||
|---|---|---|---|---|---|---|---|---|
| maxima
of peak positions (eV) |
calcd |
|||||||
| mass m/z | anion | 1 | 2 | 3 | 4 | expt 430.15 K | 298.15 K | 430.15 K |
| 127 | C4H3N2O3 | ∼0 | 0.2 | 1.5 | – | ∼0 | –0.36 | –0.90 |
| 126 | C4H2N2O3 | ∼0 | 0.1 | 0.2 | 1.2 | ∼0 | –0.25 | –0.78 |
| 99 | C3H3N2O2 | 0.1 | 0.8 | – | – | ∼0 | –0.66 | –1.70 |
| 96 | NH2SO3 | ∼0 | 0.3 | 1.3 | – | ∼0 | –0.12 | –0.65 |
| 95 | NHSO3 | ∼0 | 0.2 | – | – | ∼0 | –0.89 | –1.41 |
| 86 | C2H2N2O2 | 0.1 | 0.3 | 0.9 | – | ∼0 | – | – |
| 80 | NH2SO2 | ∼0 | – | – | – | ∼0 | –0.79 | –1.33 |
| SO3 | ∼0 | –1.76 | –2.29 | |||||
| 64 | SO2 | 1.1 | 5.8 | – | – | ∼0 | 0.09 | –0.45 |
| 62 | NSO | 0.6 | 0.8 | – | – | ∼0.3 | –0.98 | –2.01 |
| 48 | NH2O2 | 5.0 | 5.4 | – | – | ∼4.5 | 3.84 | 3.31 |
| 42 | OCN | ∼0 | 0.4 | 1.3 | – | ∼0 | – | – |
| 16 | NH2 | 4.9 | 8.3 | 11.6 | – | ∼4 | 2.63 | 2.11 |
| O | ∼4 | 2.86 | 2.40 | |||||
Full crystallographic details of the title compound have been deposited in the Cambridge Crystallographic Data Center (Deposition No. CCDC 1997918), and they may be obtained from http://www.ccdc.cam.ac.uk (email deposit@ccdc.cam.ac.uk or The Director, CCDC, 12 Union Road, Cambridge, CB2 1EZ, U.K.).
2.3. Crossed Electron–Molecular Beam Setup
The anion efficiency curves for mass-selected fragment anions were obtained with a high-resolution crossed electron–molecular beam apparatus, which consists of a hemispherical electron monochromator (HEM) combined with a quadrupole mass analyzer. The experiment is described in detail in ref (33). The sample molecules were placed in a copper oven installed in a vacuum chamber. The oven was resistively heated up to 428 K to achieve sufficient sublimation of the sample. The formed neutral effusive beam was introduced into the interaction region of the monochromator via a capillary (ϕ = 1 mm) attached to the oven. In the interaction region, the molecular beam perpendicularly crossed a well-defined electron beam. The electron beam was formed in the HEM, which was operating at an electron energy resolution of 100 meV and electron currents of 5–30 nA ensuring a reasonable balance between electron energy resolution and ion intensity. The chamber pressure was about 1.6 × 10–11 atm ensuring single-collision conditions. The anions formed in the interaction region were extracted by a weak electrostatic field into the entrance of a quadrupole mass analyzer and detected using a channel electron multiplier via a single-pulse counting mode. The electron energy resolution was determined using the well-known s-wave electron attachment to CCl4, which leads to the formation of Cl– at 0 eV. This reaction was also used to calibrate the energy scale.
2.4. Radiolysis
A water solution of SU at a concentration of 0.1 mM, in the presence of 30 mM t-BuOH, a scavenger of hydroxyl radicals (•OH), and 10 mM phosphate buffer (pH 7.0) was prepared. The mixture was saturated with argon for ca. 3 min. After that all samples were irradiated with a dose of 140 Gy. The samples containing SU and BrU (both at the concentration of 0.1 mM) were prepared using the same procedure. All radiolysis experiments were performed in a Cellrad X-ray cabinet (Faxitron X-ray Corp.). All samples were prepared at least in triplicate.
After X-ray irradiation, samples were analyzed with the RP-HPLC method. A C18 column (Wakopak Handy ODS, 4.6 × 150 mm, 5 μm in particle size and 100 Å in pore size), isocratic elution with 0.1% HCOOH, and flow rate 1 mL/min for the separation of analytes were used. The HPLC analyses were carried out using a Dionex UltiMate 3000 System with a diode array detector, which was set at 260 nm. All samples were analyzed at least in triplicate.
2.5. Potentiometric Titrations
Potentiometric titrations were performed at 298.15 K, using a Cerko Lab System microtitration unit fitted with a 5 mL Hamilton’s syringe, a pH combined electrode (Hydromet ERH-13-6) calibrated according to IUPAC recommendations,34 and a self-made measuring cell (30 mL) equipped with a magnetic stirrer. The temperature was controlled using a Lauda E100 circulation thermostat. The composition of the titrand solution was as follows: 1 mM uracil-5-yl O-sulfamate and 2.55 mM HCl. The solutions were potentiometrically titrated with a standardized 24 mM NaOH solution in the pH range from 2.5 to 11.5. The experiment consisted of injecting 0.02 mL of the titrant at 2 min intervals into the reaction cell, which initially contained 5.0 mL of the titrand solution. The dissociation constants were refined by least-squares calculations using the Hyperquad2008 (ver. 5.2.19) computer program.35
2.6. Quantum Chemical Calculations
The thermodynamic thresholds for various dissociation pathways of SU in the gas phase were calculated for its most stable conformer. These thresholds were obtained as the difference between the Gibbs free energies, ΔG, of the products and substrate in their ground states, as it was performed before,36 with eq 1a:
| 1a |
The optimized reactants were subjected to the frequency calculations with respect to both standard state (298.15 K, 1 atm) and the experimental conditions (430.15 K, 1.6 × 10–11 atm). The pressure correction to the G value for the experimental pressure was calculated with eq 1b:37
| 1b |
where Gp,T and Strans;p,T are the free energy and the translational entropy, both at pressure p and temperature T.
In these calculations, the M06-2X38 functional combined with the aug-cc-pVTZ39,40 basis set has been used. This methodology was shown to be successful for this kind of calculation,25 giving results comparable to those obtained with the G4 scheme.41
The mechanisms of the electron-attachment-induced degradation of SU were analyzed computationally, primarily at the density functional theory (DFT) level, with the use of the M06-2X38 and B3LYP42 hybrid functionals and the 6-31++G(d,p) basis set.43,44 The polarization continuum model (PCM)45 was used to mimic aqueous reaction environment. Similar methodology was successfully employed in our previous reports regarding the electron-attachment-induced degradation of uracil derivatives.16,46,47 In order to verify the DFT estimates, we also employed the composite G2MP248 method of chemical accuracy. Gas phase G2MP2 calculations were then PCM corrected to mimic aqueous solution. Discussed in the current report, Gibbs free energies (G) in water were calculated as in eq 2:
| 2 |
where Ggas stands for the G2MP2 gas phase Gibbs free energy, while GMP2,water and GMP2,gas are Gibbs free energies obtained in water solution (PCM) and gas phase, respectively, at the all-electron MP2/6-31G(d)49 level.
All calculations were carried out with the Gaussian 1650 or Gaussian 0951 suite of programs.
3. Results and Discussion
3.1. Uracil-5-yl O-Sulfamate: A Potential Radiosensitizer
One of our previous works concerned the combination of computational and radiolytic studies on 5-trifluoromethanesulfonyl-2′-deoxyuridine (OTfdU).16 A favorable DEA profile calculated for OTfdU was confirmed by the extent of its radiolytic decomposition in water solution, which was similar to that measured for BrdU under the same experimental conditions. Considering that, similarly to aryl triflates, aryl O-sulfamates (SU) are perceived as good electrophiles,52 we decided to check the radiosensitizing properties of uracil-5-yl O-sulfamate. It is known that C–O bonds in aryl O-sulfamates are less reactive than such bonds in aryl triflates (cf. reactivity of O-sulfamates versus that of O-triflates in Suzuki53 or Kumada54 reactions), and thereby, we expected the electron-induced O–S bond cleavage rather than the C–O one. This conclusion was actually confirmed by the DEA profile of SU calculated in aqueous solution, which shows that the O–S bond breaks almost barrierlessly (see Figure 1).
Figure 1.

DEA profile calculated for uracil-5-yl O-sulfamate at the M06-2X/6-31++G(d,p) level in an aqueous solution.
Encouraged by the favorable DFT characteristics, we synthesized uracil-5-yl O-sulfamate. The compound was obtained via a reaction of 5-hydroxyuracil with sulfamoyl chloride (H2NSO2Cl), obtained in situ from chlorosulfonyl isocyanate and formic acid in the presence of a catalytic amount of DMA (Figure 2).55
Figure 2.

Synthesis of uracil-5-yl O-sulfamate.
Besides NMR and HRMS characteristics (see Methods), the X-ray analysis confirmed the expected structure of the synthesized derivative. Single-crystal X-ray diffraction measurements show that uracil-5-yl O-sulfamate crystallized in the monoclinic P21/n space group with one molecule in the asymmetric unit (Figure 3 and Table S1).
Figure 3.
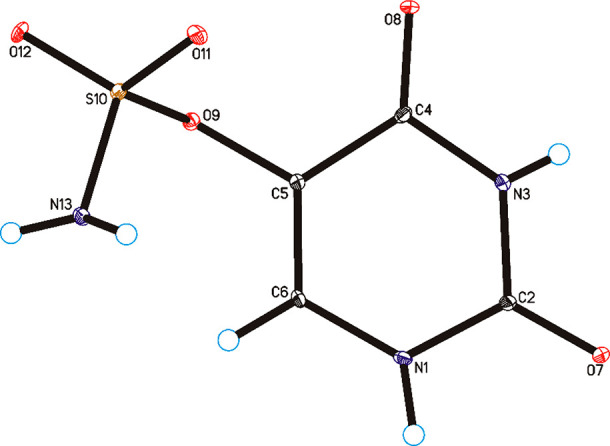
Molecular structure of uracil-5-yl O-sulfamate, showing the atom-labeling scheme. Displacement ellipsoids are drawn at the 25% probability level, and H atoms are shown as small spheres of arbitrary radius.
In the crystal of the title compound the molecules are linked via N1–H1···O7 and N3–H3···O8 hydrogen bonds to form sheets of asymmetric ribbons along the [101] direction (Table S2, Figure 4), similar to those observed in the crystal of form II of 5-fluorouracil.56 The neighboring, antiparallel ribbons are connected through N13–H13A···O8, N13–H13B···O12, and C6–H6···O12 hydrogen bonds to form a 3D framework.
Figure 4.

Crystal packing of uracil-5-yl O-sulfamate viewed along the a-axis. Hydrogen bonds are represented by dashed lines.
3.2. Dissociative Electron Attachment to Uracil-5-yl O-Sulfamate in the Gas Phase
Using the experimental setup described above, we have studied the formation of anionic fragments upon electron interaction with the synthesized potential radiosensitizer. In DEA, the resonant capture of an electron (e–) by a neutral molecule (MX) results in the formation of a transient negative ion (TNI), which subsequently relaxes by spontaneous emission of the excess electron or by dissociation, which leads to release of a fragment anion and neutral fragment(s) as shown in the following reaction:
| 3 |
where (MX)*– is TNI and M and X– represent the neutral fragment and fragment anion, respectively.
DEA to molecules may result in simple bond cleavage, such as that indicated in reaction 3, or in multiple bond cleavage including complex rearrangement involving the entire molecule. In the present study, we found 12 fragment anions upon DEA to SU in the gas phase that are discussed in sections 3.2.1–3.2.4. The results for the observed anions, summarized in Table 1, comprise the maxima of peak positions, experimentally obtained thresholds, and calculated thermodynamic thresholds. For the sake of clarity, we divide the registered DEA reactions into four main pathways leading to (a) fragment anions from the C–O bond cleavage, (b) fragment anions from the S–O bond cleavage, (c) fragment anions from the uracil-5-yl side group, and (d) fragment anions from the sulfamate side group. For all measured anion efficiency curves shown in Figures 5–10, we show both the drawn data (black lines) and cumulative multiple Gaussian fits (red lines). We just note that the overall intensity of the anion yields was about 3 orders of magnitude lower than that reported for OTfU,25 which can be explained by the substantially lower vapor pressure of uracil-5-yl O-sulfamate.
Figure 5.
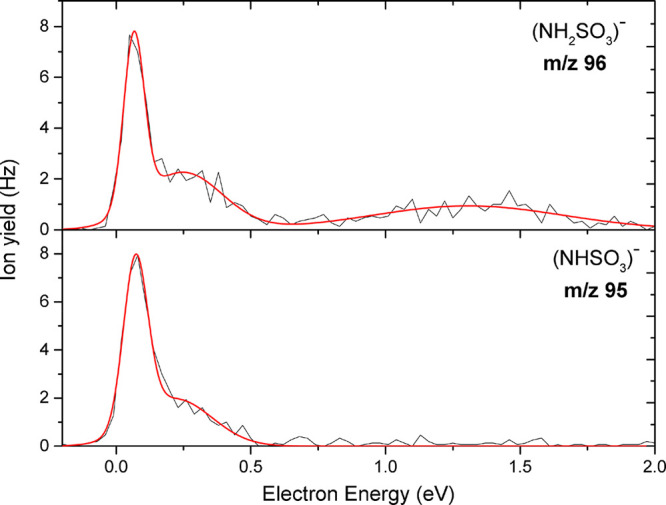
Anion efficiency curve as a function of electron energy for the fragment anions NH2SO3– at m/z 96 and NHSO3– at m/z 95 upon electron attachment to uracil-5-yl O-sulfamate.
Figure 10.
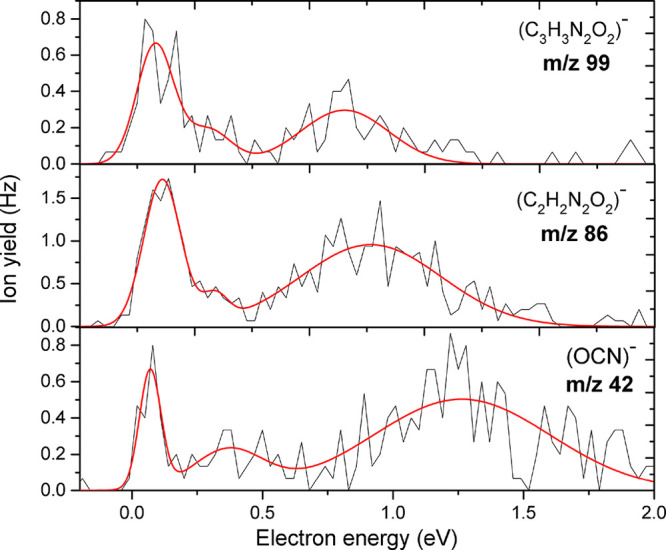
Anion efficiency curves as a function of electron energies for anions at m/z 99, m/z 86, and m/z 42 upon electron attachment to uracil-5-yl O-sulfamate.
3.2.1. Anions from Cleavage of the C5–O Bond
The C–O bond cleavage in SU upon DEA proceeds via reactions 4 and 5:
| 4 |
| 5 |
This single bond cleavage splits the molecule into two parts: the sulfamate and the uracil-5-yl moiety. We note that in the DEA experiment only the negatively charged reaction product is detected; i.e., we assigned the anion yield at m/z 96 to (NH2SO3)−. The detection of the latter anion implies a rise of the corresponding neutral radical (U-yl)•. Similarly to our previous study on OTfU,25 the reaction pathway for the formation of the (U-yl)− anion is not observed here. We, therefore, can infer that the uracilyl anion is unstable and undergoes subsequent dissociation into other anions with smaller mass, which we will discuss later. Figure 5 presents the anion efficiency curve of the anion at m/z 96, which shows a sharp resonance at 0 eV, a lower resonance at 0.25 eV, and a broad resonance at about 1.31 eV. We also observed anion yield at m/z 95 and assigned it to (NHSO3)− formed by the additional loss of a single hydrogen atom in the sulfamate anion, which occurs via exothermic reaction 5 with a predicted theoretical thermodynamic threshold of −1.41 eV. The ion yields for (NH2SO3)− and (NHSO3)− are similar except that the former anion shows an additional peak close 1 eV (see Figure 5).
3.2.2. Anions from S–O Bond Cleavage and Loss of a Hydrogen Atom
The single bond cleavage of the S–O bond results in the formation of two complementary fragment anions as shown in Figure 6. The anion formation proceeds via reactions 6–8:
| 6 |
| 7a |
| 7b |
| 8 |
Figure 6.
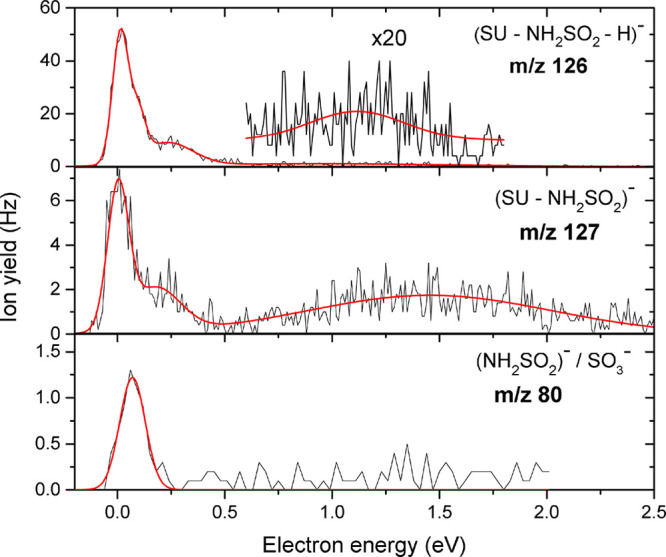
Anion efficiency curves as functions of electron energies for the fragment anions observed at m/z 126, 127, and 80 formed from the single S–O bond cleavage upon electron attachment to uracil-5-yl O-sulfamate.
We observed a fragment anion at m/z 127, which we assign to (UO)−/(SU–NH2SO2)−. The corresponding neutral radical is NH2SO2• with mass of 80 u. The anion efficiency curves for the formation of the fragment anions via reactions 6, 7a, and 7b are shown in Figure 6. The anion efficiency curve for the (UO)− anion at m/z 127 shows an intense resonance close to 0 eV. In addition to the peak at 0 eV, we registered two resonances between 0.3 and 3 eV with a broad resonance peaking around 1.5 eV. The experimental threshold observed at 0 eV is in agreement with the calculated one of −0.90 eV (Table 1). On the other hand, we observed another fragment anion at m/z 80, which we assign to either of the two isobaric species NH2SO2– or SO3– formed via the exothermic reaction 7a or 7b, respectively. The formation of each anion begins at the experimental threshold of 0 eV, which is in line with the theoretically determined thermodynamic thresholds of −1.33 eV for NH2SO2– and −2.29 eV for SO3– (Table 1).
Furthermore, we observed as the most abundant reaction channel the fragment anion at m/z 126, which we assigned to (UO–H)−/(SU–NH2SO2–H)− formed due to the loss of hydrogen atom in the (UO)− anion. The predicted thermodynamic threshold of −0.78 eV (Table 1) at the most favorable site, N3–H,25 is exothermic, which remains in accordance with the experimentally determined value of 0 eV. We observed four resonance positions for this anion, dominated by a sharp peak at 0 eV, followed by two weak resonances at 0.1 and 0.2 eV and a broad resonance at 1.1 eV.
3.2.3. Fragment Anions from Single and Multiple Bond Cleavage in the Sulfamate Group
In addition to the aforementioned fragment anions, we have observed anions formed due to single and multiple bond cleavages in the sulfamate side group. Five different fragment anions were recorded, which proceeded via the following reaction pathways:
| 9 |
| 10 |
| 11 |
| 12a |
| 12b |
Figures 7 and 8 show the anion yield curves for the anions at m/z 64 and 48, and at m/z 62, respectively. The anion at m/z 64 is assigned to SO2–, which arises from multiple bond cleavages and rearrangement in the sulfamate side group of the parent molecule via reaction 9 with UONH2 as the neutral fragment. We observed for this anion two major resonances peaks close to 1.1 eV and a broad resonance at higher energy around 5.8 eV. The calculated thermodynamic threshold of −0.45 eV matches the experimental threshold of 0 eV. On the other hand, we also observed the anion at m/z 48 which we assign to NH2O2–. Unlike most of the other anions, no peak was observed at low electron energies. The anion is only formed above a threshold of about 4.5 eV. Our calculations predict an endothermic reaction with a threshold of 3.31 eV (Table 1), which agrees with the absence of a peak at low energies. The dissociation of the sulfamate group through multiple-bond cleavage and rearrangement showed by reaction 10 leads to the NSO– anion at m/z 62. This reaction is accompanied by the release of H2O and UO• as the counterpart radical. The anion was detected as the second most abundant fragment anion. The calculated exothermic thermochemical threshold was found to be −2.01 eV (Table 1) in agreement with the experimental threshold of about 0 eV. As shown in Figure 8, two narrow closely spaced resonances with relatively high intensity were found with maxima at 0.6 and 0.8 eV.
Figure 7.

Anion efficiency curves as functions of electron energies for the fragment anions formed at m/z 64 and 48 upon electron attachment to uracil-5-yl O-sulfamate.
Figure 8.

Anion efficiency curve of the fragment anion observed at m/z 62 as a function of electron energy upon electron attachment to uracil-5-yl O-sulfamate.
Figure 9 shows the yield curve of the anion at m/z 16. We note that two isobaric fragment anions—O– formed via reaction 12a and NH2– formed via reaction 12b—have the same nominal m/z. It was not possible to separate these isobaric anions with the quadrupole mass spectrometer. Our M06-2X/aug-cc-pVTZ calculations indicate that both anions are possible to form with thermodynamic threshold values of 2.11 eV for NH2– (S–NH2 bond cleavage) and 2.40 eV for O– (S–O bond cleavage), respectively (Table 1). Therefore, the threshold values cannot be used to assign the peaks shown in Figure 9 to the exact sites of bond cleavage. However, a comparison with results from previous DEA studies for compounds containing oxygen atoms and/or amino groups may allow a tentative assignment.
Figure 9.

Anion efficiency curve as a function of electron energy for the fragment anion observed at m/z 16 upon electron attachment to uracil-5-yl sulfamate.
In a previous study Denifl et al.57 reported a resonance at 4.5 eV in the anion efficiency curve of O– upon electron attachment to uracil in the gas phase. The peak showed a similar onset of about 4 eV as obtained here. Besides, the results from the DEA study by Alizadeh et al.58 on alanine anhydride with a peak at 4.4 eV is not far from the aforementioned results on O–. The resonance contributions of the two isobaric anions O– and NH2– were distinguished upon electron attachment to the amino acid valine59 using a high-resolution VG-ZAB mass spectrometer. It was reported that the O– anion is formed at electron energies of 4.4 and 8.3 eV while NH2– is observed at 6 eV. Moreover, the recently published work by Ameixa et al.60 on the formation of fragment anions upon electron attachment to benzaldehyde showed the formation of O– via the cleavage of C=O bond in a characteristic resonance between 8 and 9 eV. In our present study, the second resonance position of 8.3 eV is in the same energy range. Hence, by analogy we can infer that our experimentally found peaks at 4.9 and 8.3 eV can be ascribed to oxygen anion. The presented data also indicate a third resonance around 11.6 eV at m/z 16, where we omitted an assignment to either O– or NH2–.
3.2.4. Fragment Anions from Single and Multiple Bond Cleavages in the Uracil-5-yl Moiety
Single and multiple bond cleavages in the uracil-5-yl group result in the formation of three other anionic species via the following reactions:
| 13 |
| 14 |
| 15 |
We show the anion yield curves for the anions occurring via reactions 13–15 in Figure 10. The formation of C3H3N2O2– at m/z 99 is formed above the experimental threshold of 0 eV. The suggested fragmentation reaction 13 leads to a ring opening of the uracil-5-yl moiety with the formation of NH2SO2• and C=O as radical and neutral fragments. The respective thermodynamic threshold for this channel was predicted to be exothermic (−1.70 eV). We also observed (C2H2N2O2)− at m/z 86 with an experimental threshold of 0 eV, formed by the loss of C2H in the dissociation of the uracil-5-yl radical. We report for this anion three resonances at 0.1, 0.3, and 0.9 eV. The calculated thermodynamic threshold for the formation of this anion was found to be 3.31 eV (p = 1 atm, T = 198.15 K) indicating an endothermic reaction pathway, which disagrees with the experimental thresholds obtained. Even though we investigated certain reactions computationally, we could not account for this difference regardless of the pressure and temperature. Thus, we conclude that the large disagreement indicates that other reaction pathways will probably occur.
In the case of m/z 42 represented by reaction 15, the formation of OCN– may proceed via different reaction channels depending on the site of bond cleavage (Figure S4). Due to the variety of possible reaction channels, we omitted further calculation on the threshold of OCN–. We observed that the experimental findings are similar to the results obtained for the formation of NCO– from DEA to the potential radiosensitizer hydroxyurea (which has the structural formula CH4N2O2 or OH–NH–CO–NH2)61 using the same experimental setup. It is worth noting that the involved carbon atom has a bonding environment (HN–CO–NH) similar to that in the current molecule. Three peaks were found at positions close to 0, 0.4, and 1.2 eV, respectively. The resonance positions for NCO– are similar for SU and hydroxyurea, except for an additional peak at 0.1 eV that was reported for the latter compound. We further note that (C2H2N2O2)− could be an intermediate reaction product in the dissociation pathway leading to OCN– by the loss of two hydrogen atoms from (C2H2N2O2)− followed by subsequent C–N bond cleavage. OCN– was the second most abundant fragment anion after the dehydrogenated parent anion in DEA to the nucleobases uracil and thymine.62 It was shown for these compounds that OCN– forms in a sequential dissociation process with initial H-loss from one of the nitrogen sites of the nucleobase anion.62,63 Since we do not observe the dehydrogenated parent anion of uracil-5-yl O-sulfamate within the detection limit of the apparatus, we may rule out this reaction pathway found for nucleobases.
3.3. Radiolysis of SU under Reductive Conditions
The results described in section 3.2 demonstrate that several dissociative channels are triggered by LEE attachment to SU. To define the potential of the studied compound as a radiosensitizer, steady state radiolysis was also performed. Water solutions containing SU, free hydroxyl radical scavenger (t-BuOH), and phosphate buffer were irradiated with a dose of 140 Gy. To avoid scavenging of electrons by oxygen, all samples were deoxygenated before exposure to IR. Irradiated and nonirradiated samples were analyzed using the HPLC methods (Figure 11). To our surprise, no product of electron-induced degradation of SU was observed. It is worth noting that the change in solution pH (5.6, 8.0–10 mM phosphate buffer and 4.0, 4.9–10 mM formate buffer) and the use of higher radiation dose (280 Gy) did not affect the radiolysis process. None of the examined radiolysis conditions led to the appearance of any radioproducts from SU.
Figure 11.

HPLC traces for a solution of uracil-5-yl O-sulfamate before (black) and after irradiation (orange) with a dose of 140 Gy.
3.4. Rationalization of the Results of Radiolysis Observed for SU Solutions
Striking differences between the complex DEA picture and the lack of SU reactivity in the radiolyzed samples may result from a number of reasons. To continue our quest for searching of efficient radiosensitizing nucleosides, we have to understand why the so far successful DFT model turned out to be abortive in the case of SU which, in spite of favorable computational characteristics, turned out to have no radiosensitizing properties.
In order to eliminate possible experimental errors, we carried out X-ray irradiations of solutions containing both SU and BrU (Figure 12). In this way the radiolysis of both substances was carried out under identical conditions. As indicated by Figure 12 for a dose of 140 Gy, BrU decomposes with a yield equal to 15.83 ± 0.56%; a similar decay (15.78 ± 0.93%) was observed in another independent experiment, while practically no decomposition of SU is observed.
Figure 12.

HPLC traces for a solution containing uracil-5-yl O-sulfamate and 5-bromouracil before (black chromatogram) and after irradiation with a dose of 140 Gy (orange chromatogram).
The results depicted in Figure 12 suggest that SU is not only unreactive to solvated electrons but also does not bind them. Indeed, the presence of equimolar amounts of SU and BrU in the solution does not affect the decomposition yield of BrU (see above).
In our first attempt to explain this observation, we assumed that in water yet before irradiation SU primarily exists as an anion originating from the deprotonation of the NH2 group. In fact, the formation of such anion could prevent attachment of an electron due to repulsion between the negative charge of electron and the molecular anion which, in turn, would explain the results of radiolysis depicted in Figures 11 and 12. The DEA process to the above-described anion is depicted in Scheme S1 and discussed in the Supporting Information. In order to verify the above-mentioned assumption, the acidic dissociation constants of SU were obtained by taking into account two equilibria depicted in Figure 13. This model has provided very good fitting of the calculated data to the experimental ones (Figure S5). The potentiometric titration reveals that SU can be considered as a weak acid which dissociates in two steps shown in Figure 13. The calculated dissociation constants are equal to pKa1 = 7.52 (±0.06) and pKa2 = 9.64 (±0.04), while the relative concentrations of the species existing in SU solution as a function of pH, obtained employing these pKa values and the HySS program,64 are depicted in Figure 14.
Figure 13.

Proton dissociation scheme for uracil-5-yl O-sulfamate.
Figure 14.

Concentration distribution of species as a function of pH in uracil-5-yl O-sulfamate solution.
Hence, it is clear that our tentative hypothesis about existence of anionic forms of SU before irradiation does not hold. Indeed, at pH 7 most of SU (around 80%, see Figure 13) is in the neutral, H2L form. Nevertheless, we carried out additional radiolytic experiments in solutions of various pHs and determined the yield of SU decomposition. Regardless of pH (4 < pH < 8) the results of irradiation were always the same: SU did not decompose, even at larger doses of X-rays (up to 280 Gy). On the other hand, it is well-known that nucleobases bind solvated electrons with an almost diffusion controlled rate.12 Thus, the same yield of BrU decomposition observed for a solution containing exclusively BrU and equimolecular amounts of BrU and SU only seemingly suggests that solvated electrons do not attach to SU. Indeed, the adiabatic electron affinities calculated for both molecular systems are pretty similar (2.31 and 2.43 eV, for BrU and SU, respectively, G2MP2 free energy level). Hence, the yield of BrU decomposition in solutions containing both BrU and SU indicates that some electrons are temporarily captured by SU and then transferred to BrU, where they are ultimately consumed in the DEA reaction. In consequence, these results suggest that SU does not undergo electron-induced degradation in water.
All the above-mentioned facts imply that the discrepancy between the experimental picture and theoretical predictions may lie in our computational model. In order to check this hypothesis, we calculated the DEA profiles for SU using also the G2MP2 method having chemical accuracy (errors below 1 kcal/mol). To make sure that a possible discrepancy between the M06-2X level and a more accurate approach originates from the DFT inaccuracies for the SU DEA profile, the G2MP2 calculations were repeated for two other uracil derivatives, which represent a close correspondence between the theoretical and actual radiolytic behaviors (Figure 15, SCNU and BrU).
Figure 15.

Structures of uracil derivatives along with their abbreviated names: uracil-5-yl O-sulfamate (SU), 5-thiocyanatouracil (SCNU), and 5-bromouracil (BrU). Arrows indicate bonds possible to break during DEA process. Two DEA possible paths are marked: path A (the bond cleavage between uracil and its substituent) and path B (the bond cleavage within the substituent).
Table 2 summarizes computational characteristics obtained at the DFT and G2MP2 levels. G2MP2, as opposed to the M06-2X method, seems to support the results of radiolytic experiments where no electron-induced degradation of SU was found. Namely, the crucial activation barrier, i.e., breaking of the O–S bond (path B; Figure 15), is significantly underestimated at the M06-2X level, as it has risen from 2.3 to 7.5 kcal/mol (G2MP2). Thus, the activation barrier calculated with the high accuracy method explains the experimental observations. Indeed, as indicated in our recent studies on radiosensitizing properties of 5-iodo-4-thio-2′-deoxyuridine17 and 5-bromo-4-thio-2′-deoxyuridine,18 the activation energy of ca. 7 kcal/mol is sufficient to completely quench the electron-attachment-induced release of the halide anion from the above-mentioned thiouridines. Hence, the activation barriers of 11.1 and 7.5 kcal/mol for the dissociation of the C–O and O–S bonds, respectively, obtained at the G2MP2 level, justify the observed stability of the SU anion in the radiolytic experiments. These dissociation paths are opened in the gas phase even for 0 eV electrons (see sections 3.2.1 and 3.2.2) since electron attachment (adiabatic electron affinity (AEA) = 56.0 kcal/mol at the G2MP2 free energy level, in solution) delivers the amount of energy that several times overcomes the calculated kinetic barriers. The discussed dissociations are exothermic, in both the gas and aqueous phases, but in the latter environment the energy released due to electron attachment is, unlike in the gas phase, swiftly dissipated to the solvent. Therefore, in a liquid phase, the formed SU anions are kinetically stable, while the medium barriers do not constitute any obstacle for the respective bond cleavage in the gas phase at low pressure.
Table 2. Thermodynamic (ΔG) and Kinetic (ΔG*) Barriers Calculated for DEA Degradation Reactions of Anion Radical Uracil Derivativesa.
| thermodynamics, ΔG |
activation barriers, ΔG* |
||||||
|---|---|---|---|---|---|---|---|
| substance | degradation path | G2MP2 | M06-2X | B3LYP | G2MP2 | M06-2X | B3LYP |
| SU | C–O (path A) | –12.1 | –7.7b | –12.4 | 11.1 | 10.4b | 11.7 |
| O–S (path B) | –17.4 | –39.4b | –42.6 | 7.5 | 2.3b | 0.96 | |
| SCNU | C–S (path A) | 3.6 | –1.6c | –3.7d | 7.9 | 8.7c | 3.4d |
| S–C (path B) | –12.1 | –16.2c | – | 1.6 | 4.1c | – | |
| BrU | C–Br (path A) | –7.5 | – | –8.0e | 1.8 | – | 2.5e |
All values given in kcal/mol. All calculations conducted with use of the PCM solvation model; for DFT methods 6-31++G(d,p) basis set was used.
Calculated for 1-methyl-5-sulfamateuracil (MeOSOU).
Calculated for 5-thiocyanato-2′-deoxyuridine (SCNdU).46
Calculated for 1-methyl-5-thiocyanatouracil (MetSCNU).
Calculated for 5-bromo-1-methyluracil (MetBrU).
It is worth noticing that the discrepancy between the DFT and G2MP2 models does not exist for the other two derivatives: 5-thiocyanatouracil and 5-bromouracil (see Figure 15). For both systems the DFT data (M06-2X for SCNU and B3LYP for BrU) are in good agreement with the radiolytic results. The G2MP2 estimates reveal that the crucial activation barriers change only slightly for the degradation of BrU (from 2.5 kcal/mol at the B3LYP level to 1.8 kcal/mol at G2MP2) and SCNU (from 8.7 kcal/mol at the M06-2X level to 7.9 kcal/mol at G2MP2 for path A, and from 4.1 kcal/mol at M06-2X to 1.6 kcal/mol at G2MP2 for path B; see Table 2). A similar conclusion can be drawn for the thermodynamic data. The free energies of dissociation for all bonds except S–O differ no more than by several kilocalories per mole (see Table 2). Only the thermodynamic stimulus associated with the S–O bond cleavage in the SU anion is overestimated at the M06-2X and B3LYP levels by as much as 22 and 25.2 kcal/mol, respectively. The inaccurate estimation of activation barriers and thermodynamics, calculated for the S–O bond scission, seem to be reminiscent of the semiempirical nature of DFT methodology.65 Probably, the M06-2X characteristics calculated for the dissociation of S–O bonds in radical anions can be considered as artifacts of this DFT approach.
4. Conclusions
The number of radiosensitizers approved for clinical use is still very low although hypoxia present in all solid tumors makes cancer cells resistant to IR exposure, which significantly lowers the efficacy of the commonly used radiotherapy. Therefore, efforts aiming at working out and introducing hypoxic radiosensitizers into clinical practice are well justified.
In the quest for such chemicals, we proposed a uracil derivative—uracil-5-yl O-sulfamate—with promising DEA characteristics calculated in an aqueous solution at the M06-2X/6-31++G(d,p) level. This compound turned out to be prone to DEA processes in the gas phase, and products of simple dissociation of the C–O and S–O bonds induced already by 0 eV electrons prevail among the recorded anionic fragments. The most abundant DEA products originate from highly exothermic reactions; e.g., the release of the NSO– anion is related to the second most exothermic process (Table 1). Similarly, the most efficient fragmentation, i.e., the formation of anion with m/z equal to 126, is related to reaction 8, whose exothermicity amounts to −0.75 eV at the M06-2X/aug-cc-pVTZ level. We have observed also a number of complex fragmentations associated with the simultaneous cleavage of several bonds in the sulfamate or uracil-5-yl group. These processes are, however, significantly less pronounced as indicated by the experimental anion yields.
It might seem that promising results of DEA experiments in the gas phase, where one observes several dissociative channels induced by LEE, should be mirrored in steady state radiolysis performed in water. However, the results of the two type of experiments carried out for O-sulfamate do not correlate with each other. Although the scission of the S–O or C5–O bonds in the SU anion is highly exothermic, no reactivity under reductive conditions is observed in radiolytic experiments. Neither variation of pH nor increase of the dose of incident radiation changes this experimental picture. We traced back the observed lack of reactivity of SU in the IR irradiated water solutions to the inaccuracies of the adopted DFT model. In particular, the comparison of energetic characteristics obtained at the M06-2X and G2MP2 levels allows the activation barrier and thermodynamic stimulus for the dissociation of the S–O bond calculated at the M06-2X level to be regarded as an artifact of DFT methodology. In solution, the medium activation barriers prevent electron-induced decomposition of SU, which simultaneously leads to questions of its radiosensitizing potential. At the same time, these barriers do not prevent the nucleobase fragmentation in the gas phase due to the large electron affinity of SU that allows the barriers to be easily surmounted under such conditions.
The current work enables certain drawbacks of our theoretical model for potential radiosensitizers to be understood and overcome. These findings cannot be overestimated from the practical reasons since, when properly implemented in the computational tool, they will prevent time- and cost-consuming synthesis and physicochemical experiments on nonradiosensitizing systems. Moreover, our results emphasize the crucial influence of a water environment on the electron-induced degradation processes and prove that efficient DEA in the gas phase does not guarantee adequate degradation in water.
Acknowledgments
This work was supported by the Polish National Science Center (NCN) under Grant No. UMO-2014/14/A/ST4/00405 (J.R.) and by the FWF, Vienna (P30332) (S.D.). Calculations have been carried out in the Wroclaw Center for Networking and Supercomputing (wcss.wroc.pl), Grant No. 209, and at a local cluster.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpcb.0c03844.
Crystal data and structure refinement parameters for uracil-5-yl O-sulfamate, hydrogen bonding interactions in the crystal structure of SU, 1H NMR spectrum, high resolution mass spectrum, and UV spectrum of uracil-5-yl O-sulfamate, possible channels leading to the formation of the OCN– anion, representative titration curve, dissociative electron attachment process calculated for the deprotonated anionic form of uracil-5-yl O-sulfamate at the M06-2X/6-31++G(d,p) level, PCM water solution, and complete refs (50) and (51) (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Gaba N.Radioprotectors and Radiosensitizers. Chemical Modifiers; Lap Lambert Academic Publishing GmbH & Co. KG: Saarbrücken, Germany, 2011. [Google Scholar]
- Ogawa Y. Paradigm Shift in Radiation Biology/Radiation Oncology – Exploitation of the “H2O2 Effect” for Radiotherapy Using Low-LET (Linear Energy Transfer) Radiation such as X-rays and High-Energy Electrons. Cancers 2016, 8, 28. 10.3390/cancers8030028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.; Wang F.; Denisov S. A.; Adhikary A.; Mostafavi M. Reactivity of Prehydrated Electrons Toward Nucleobases and Nucleotides in Aqueous Solution. Sci. Adv. 2017, 3, e1701669. 10.1126/sciadv.1701669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J.; Kumar A.; Muroya Y.; Yamashita S.; Sakurai T.; Denisov S. A.; Sevilla M. D.; Adhikary A.; Seki S.; Mostafavi M. Observation of Dissociative Quasi-Free Electron Attachment to Nucleoside via Excited Anion Radical in Solution. Nat. Commun. 2019, 10, 102. 10.1038/s41467-018-08005-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams G. E. Chemical Radiosensitization of Hypoxic Cells. Br. Med. Bull. 1973, 29, 48–53. 10.1093/oxfordjournals.bmb.a070956. [DOI] [PubMed] [Google Scholar]
- Wang H.; Mu X.; He H.; Zhang X.-D. Cancer Radiosensitizers. Trends Pharmacol. Sci. 2018, 39, 24–48. 10.1016/j.tips.2017.11.003. [DOI] [PubMed] [Google Scholar]
- Buckley A. M.; Lynam-Lennon N.; O’Neill H.; O’Sullivan J. Targeting Hallmarks of Cancer to Enhance Radiosensitivity in Gastrointestinal Cancers. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 298–313. 10.1038/s41575-019-0247-2. [DOI] [PubMed] [Google Scholar]
- Overgaard J. Improving Radiotherapy of Squamous Cell Carcinoma of the Head and Neck (HNSCC) Through a Continuous Process of Biological Based Clinical Trials. A 40-Year Experience from the Danish Head and Neck Cancer Group-DAHANCA. Eur. J. Cancer 2017, 72, S102. 10.1016/S0959-8049(17)30416-1. [DOI] [Google Scholar]
- Rak J.; Chomicz L.; Wiczk J.; Westphal K.; Zdrowowicz M.; Wityk P.; Żyndul M.; Makurat S.; Golon Ł. Mechanisms of Damage to DNA Labeled with Electrophilic Nucleobases Induced by Ionizing or UV Radiation. J. Phys. Chem. B 2015, 119, 8227–8238. 10.1021/acs.jpcb.5b03948. [DOI] [PubMed] [Google Scholar]
- Cecchini S.; Masson C.; La Madeleine C.; Huels M. A.; Sanche L.; Wagner J. R.; Hunting D. J. Interstrand Cross-Link Induction by UV Radiation in Bromodeoxyuridine-Substituted DNA: Dependence on DNA Conformation. Biochemistry 2005, 44, 16957–16966. 10.1021/bi050799x. [DOI] [PubMed] [Google Scholar]
- von Sonntag C.Free-Radical-Induced DNA Damage and Its Repair; Springer-Verlag: Heidelberg, Germany, 2006. [Google Scholar]
- Steenken S. Purine Bases, Nucleosides, and Nucleotides: Aqueous Solution Redox Chemistry and Transformation Reactions of Their Radical Cations and e- and OH Adducts. Chem. Rev. 1989, 89, 503–520. 10.1021/cr00093a003. [DOI] [Google Scholar]
- Nabben F. J.; Karman J. P.; Loman H. Inactivation of Biologically Active DNA by Hydrated Electrons. Int. J. Radiat. Biol. Relat. Stud. Phys., Chem. Med. 1982, 42, 23–30. 10.1080/09553008214550881. [DOI] [PubMed] [Google Scholar]
- Kohanoff J.; McAllister M.; Tribello G. A.; Gu B. Interactions Between Low Energy Electrons and DNA: a Perspective from First-Principles Simulations. J. Phys.: Condens. Matter 2017, 29, 383001. 10.1088/1361-648X/aa79e3. [DOI] [PubMed] [Google Scholar]
- Westphal K.; Wiczk J.; Miloch J.; Kciuk G.; Bobrowski K.; Rak J. Irreversible Electron Attachment - a Key to DNA Damage by Solvated Electrons in Aqueous Solution. Org. Biomol. Chem. 2015, 13, 10362–10369. 10.1039/C5OB01542A. [DOI] [PubMed] [Google Scholar]
- Makurat S.; Zdrowowicz M.; Chomicz-Mańka L.; Kozak W.; Serdiuk I. E.; Wityk P.; Kawecka A.; Sosnowska M.; Rak J. 5-Selenocyanato and 5-Trifluoromethanesulfonyl Derivatives of 2′-Deoxyuridine: Synthesis, Radiation and Computational Chemistry as Well as Cytotoxicity. RSC Adv. 2018, 8, 21378–21388. 10.1039/C8RA03172J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makurat S.; Spisz P.; Kozak W.; Rak J.; Zdrowowicz M. 5-Iodo-4-thio-2′-Deoxyuridine as a Sensitizer of X-ray Induced Cancer Cell Killing. Int. J. Mol. Sci. 2019, 20, 1308. 10.3390/ijms20061308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spisz P.; Zdrowowicz M.; Makurat S.; Kozak W.; Skotnicki K.; Bobrowski K.; Rak J. Why Does the Type of Halogen Atom Matter for the Radiosensitizing Properties of 5-Halogen Substituted 4-Thio-2′-Deoxyuridines?. Molecules 2019, 24, 2819. 10.3390/molecules24152819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boudaïffa B.; Cloutier P.; Hunting D.; Huels M. A.; Sanche L. Resonant Formation of DNA Strand Breaks by Low-Energy (3 to 20 eV) Electrons. Science 2000, 287, 1658–1660. 10.1126/science.287.5458.1658. [DOI] [PubMed] [Google Scholar]
- Cecchini S.; Girouard S.; Huels M. A.; Sanche L.; Hunting D. J. Interstrand Cross-Links: A New Type of γ-Ray Damage in Bromodeoxyuridine-Substituted DNA. Biochemistry 2005, 44, 1932–1940. 10.1021/bi048105s. [DOI] [PubMed] [Google Scholar]
- Chomicz L.; Rak J.; Storoniak P. Electron-Induced Elimination of the Bromide Anion from Brominated Nucleobases. A Computational Study. J. Phys. Chem. B 2012, 116, 5612–5619. 10.1021/jp3008738. [DOI] [PubMed] [Google Scholar]
- Wieczór M.; Wityk P.; Czub J.; Chomicz L.; Rak J. A First-Principles Study of Electron Attachment to the Fully Hydrated Bromonucleobases. Chem. Phys. Lett. 2014, 595, 133–137. 10.1016/j.cplett.2014.01.052. [DOI] [Google Scholar]
- Park Y.; Polska K.; Rak J.; Wagner J. R.; Sanche L. Fundamental Mechanisms of DNA Radiosensitization: Damage Induced by Low-Energy Electrons in Brominated Oligonucleotide Trimers. J. Phys. Chem. B 2012, 116, 9676–9682. 10.1021/jp304964r. [DOI] [PubMed] [Google Scholar]
- Polska K.; Rak J.; Bass A. D.; Cloutier P.; Sanche L. Electron Stimulated Desorption of Anions from Native and Brominated Single Stranded Oligonucleotide Trimers. J. Chem. Phys. 2012, 136, 075101. 10.1063/1.3685587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ameixa J.; Arthur-Baidoo E.; Meißner R.; Makurat S.; Kozak W.; Butowska K.; Ferreira da Silva F.; Rak J.; Denifl S. Low-Energy Electron-Induced Decomposition of 5-Trifluoromethanesulfonyl-Uracil: A Potential Radiosensitizer. J. Chem. Phys. 2018, 149, 164307. 10.1063/1.5050594. [DOI] [PubMed] [Google Scholar]
- Meißner R.; Makurat S.; Kozak W.; Limão-Vieira P.; Rak J.; Denifl S. Electron-Induced Dissociation of the Potential Radiosensitizer 5-Selenocyanato-2′-deoxyuridine. J. Phys. Chem. B 2019, 123, 1274–1282. 10.1021/acs.jpcb.8b11523. [DOI] [PubMed] [Google Scholar]
- CrysAlis CCD and CrysAlis RED, ver. 1.171.36.24; Oxford Diffraction Ltd.: Yarnton, England, 2012. [Google Scholar]
- Sheldrick G. M. Crystal Structure Refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015, C71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spek A. L. Structure Validation in Chemical Crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2009, D65, 148–155. 10.1107/S090744490804362X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson C. K.ORTEP II; Report ORNL-5138; Oak Ridge National Laboratory: Oak Ridge, TN, USA, 1976. [Google Scholar]
- Motherwell S.; Clegg S.. PLUTO-78, Program for Drawing and Molecular Structure; University of Cambridge: Cambridge, U.K., 1978. [Google Scholar]
- Macrae C. F.; Bruno I. J.; Chisholm J. A.; Edgington P. R.; McCabe P.; Pidcock E.; Rodriguez-Monge L.; Taylor R.; van de Streek J.; Wood P. A. Mercury CSD 2.0–New Features for the Visualization and Investigation of Crystal Structures. J. Appl. Crystallogr. 2008, 41, 466–470. 10.1107/S0021889807067908. [DOI] [Google Scholar]
- Meißner R.; Kočišek J.; Feketeová L.; Fedor J.; Fárník M.; Limão-Vieira P.; Illenberger E.; Denifl S. Low-Energy Electrons Transform the Nimorazole Molecule into a Radiosensitiser. Nat. Commun. 2019, 10, 2388. 10.1038/s41467-019-10340-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brandariz I.; Barriada J.; Vilarino T.; Sastre de Vicente M. E. Comparison of Several Calibration Procedures for Glass Electrodes in Proton Concentration. Monatsh. Chem. 2004, 135, 1475–1488. 10.1007/s00706-004-0239-x. [DOI] [Google Scholar]
- Gans P.; Sabatini A.; Vacca A. Investigation of Equilibria in Solution. Determination of Equilibrium Constants with the HYPERQUAD Suite of Programs. Talanta 1996, 43, 1739–1753. 10.1016/0039-9140(96)01958-3. [DOI] [PubMed] [Google Scholar]
- Ribar A.; Huber S. E.; Śmiałek M. A.; Tanzer K.; Neustetter M.; Schürmann R.; Bald I.; Denifl S. Hydroperoxyl Radical and Formic Acid Formation from Common DNA Stabilizers Upon Low Energy Electron Attachment. Phys. Chem. Chem. Phys. 2018, 20, 5578–5585. 10.1039/C7CP07697E. [DOI] [PubMed] [Google Scholar]
- Ochterski J. W.Thermochemistry in Gaussian; Gaussian Inc.: 2000; pp 1–19. [Google Scholar]
- Zhao Y.; Truhlar D. G. The M06 Suite of Density Functionals for Main Group Thermochemistry, Thermochemical Kinetics, Noncovalent Interactions, Excited States, and Transition Elements: Two New Functionals and Systematic Testing of Four M06-Class Functionals and 12 Other Functionals. Theor. Chem. Acc. 2008, 120, 215–241. 10.1007/s00214-007-0310-x. [DOI] [Google Scholar]
- Kendall R. A.; Dunning T. H.; Harrison R. J. Electron Affinities of the First-Row Atoms Revisited. Systematic Basis Sets and Wave Functions. J. Chem. Phys. 1992, 96, 6796–6806. 10.1063/1.462569. [DOI] [Google Scholar]
- Woon D. E.; Dunning T. H. Gaussian Basis Sets for Use in Correlated Molecular Calculations. III. J. Chem. Phys. 1993, 98, 1358–1371. 10.1063/1.464303. [DOI] [Google Scholar]
- Curtiss L. A.; Redfern P. C.; Raghavachari K. Gaussian-4 Theory. J. Chem. Phys. 2007, 126, 084108. 10.1063/1.2436888. [DOI] [PubMed] [Google Scholar]
- Becke A. D. Density-Functional Exchange-Energy Approximation with Correct Asymptotic Behavior. Phys. Rev. A: At., Mol., Opt. Phys. 1988, 38, 3098–3100. 10.1103/PhysRevA.38.3098. [DOI] [PubMed] [Google Scholar]; Becke A. D. Density-Functional Thermochemistry. III. The Role of Exact Exchange. J. Chem. Phys. 1993, 98, 5648–5652. 10.1063/1.464913. [DOI] [Google Scholar]; Lee C.; Yang W.; Parr R. G. Development of the Colle-Salvetti Correlation-Energy Formula into a Functional of the Electron Density. Phys. Rev. B: Condens. Matter Mater. Phys. 1988, 37, 785–789. 10.1103/PhysRevB.37.785. [DOI] [PubMed] [Google Scholar]
- Ditchfield R.; Hehre W. J.; Pople J. A. Self-Consistent Molecular Orbital Methods. IX. Extended Gaussian-Type Basis for Molecular-Orbital Studies of Organic Molecules. J. Chem. Phys. 1971, 54, 724–728. 10.1063/1.1674902. [DOI] [Google Scholar]
- Hehre W. J.; Ditchfield R.; Pople J. A. Self-Consistent Molecular Orbital Methods. XII. Further Extensions of Gaussian-Type Basis Sets for Use in Molecular Orbital Studies of Organic Molecules. J. Chem. Phys. 1972, 56, 2257–2261. 10.1063/1.1677527. [DOI] [Google Scholar]
- Tomasi J.; Mennucci B.; Cammi R. Quantum Mechanical Continuum Solvation Models. Chem. Rev. 2005, 105, 2999–3093. 10.1021/cr9904009. [DOI] [PubMed] [Google Scholar]
- Zdrowowicz M.; Chomicz L.; Żyndul M.; Wityk P.; Rak J.; Wiegand T. J.; Hanson C. G.; Adhikary A.; Sevilla M. D. 5-Thiocyanato-2′-Deoxyuridine as a Possible Radiosensitizer: Electron- Induced Formation of Uracil-C5-thiyl Radical and its Dimerization. Phys. Chem. Chem. Phys. 2015, 17, 16907–16916. 10.1039/C5CP02081F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makurat S.; Chomicz-Mańka L.; Rak J. Electrophilic 5-Substituted Uracils as Potential Radiosensitizers: A Density Functional Theory Study. ChemPhysChem 2016, 17, 2572–2578. 10.1002/cphc.201600240. [DOI] [PubMed] [Google Scholar]
- Curtiss L. A.; Redfern P. C.; Raghavachari K.; Rassolov V.; Pople J. A. Gaussian-3 Theory Using Reduced Møller-Plesset Order. J. Chem. Phys. 1999, 110, 4703–4709. 10.1063/1.478385. [DOI] [Google Scholar]
- Frisch M. J.; Head-Gordon M.; Pople J. A. Direct MP2 Gradient Method. Chem. Phys. Lett. 1990, 166, 275–280. 10.1016/0009-2614(90)80029-D. [DOI] [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Petersson G. A.; Nakatsuji H.; et al. Gaussian 16, rev. C.01; Gaussian, Inc.: Wallingford, CT, 2019. [Google Scholar]
- Frisch M. J.; Trucks G. W.; Schlegel H. B.; Scuseria G. E.; Robb M. A.; Cheeseman J. R.; Scalmani G.; Barone V.; Mennucci B.; Petersson G. A.; et al. Gaussian 09, rev. D.01; Gaussian, Inc.: Wallingford, CT, 2013. [Google Scholar]
- Rosen B. M.; Quasdorf K. W.; Wilson D. A.; Zhang N.; Resmerita A.-M.; Garg N. K.; Percec V. Nickel-Catalyzed Cross-Couplings Involving Carbon–Oxygen Bonds. Chem. Rev. 2011, 111, 1346–1416. 10.1021/cr100259t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quasdorf K. W.; Antoft-Finch A.; Liu P.; Silberstein A. L.; Komaromi A.; Blackburn T.; Ramgren S. D.; Houk K. N.; Snieckus V.; Garg N. K. Suzuki–Miyaura Cross-Coupling of Aryl Carbamates and Sulfamates: Experimental and Computational Studies. J. Am. Chem. Soc. 2011, 133, 6352–6363. 10.1021/ja200398c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macklin T. K.; Snieckus V. Directed Ortho Metalation Methodology. The N,N-Dialkyl Aryl O-Sulfamate as a New Directed Metalation Group and Cross-Coupling Partner for Grignard Reagents. Org. Lett. 2005, 7, 2519–2522. 10.1021/ol050393c. [DOI] [PubMed] [Google Scholar]
- Geisler J.; Schneider F.; Lovis K.; Lopez Holguin F.. US 20030171346, 2003.
- Barnett S. A.; Hulme A. T.; Issa N.; Lewis T. C.; Price L. S.; Tocher D. A.; Price S. L. The Observed and Energetically Feasible Crystal Structures of 5-Substituted uracils. New J. Chem. 2008, 32, 1761–1775. 10.1039/b806763e. [DOI] [Google Scholar]
- Denifl S.; Ptasińska S.; Hanel G.; Gstir B.; Probst M.; Scheier P.; Märk T. D. Electron Attachment to Gas-Phase Uracil. J. Chem. Phys. 2004, 120, 6557–6565. 10.1063/1.1649724. [DOI] [PubMed] [Google Scholar]
- Alizadeh E.; Gschliesser D.; Bartl P.; Hager M.; Edtbauer A.; Vizcaino V.; Mauracher A.; Probst M.; Märk T. D.; Ptasińska S.; et al. Bond Dissociation of the Dipeptide Dialanine and Its Derivative Alanine Anhydride Induced by Low Energy Electrons. J. Chem. Phys. 2011, 134, 054305. 10.1063/1.3544217. [DOI] [PubMed] [Google Scholar]
- Denifl S.; Flosadóttir H. D.; Edtbauer A.; Ingólfsson O.; Märk T. D.; Scheier P. A. Detailed Study on the Decomposition Pathways of the Amino Acid Valine upon Dissociative Electron Attachment. Eur. Phys. J. D 2010, 60, 37–44. 10.1140/epjd/e2010-00060-5. [DOI] [Google Scholar]
- Ameixa J.; Arthur-Baidoo A.; Pereira-da-Silva J.; Ryszka M.; Carmichael I.; Cornetta L. M.; do N. Varella M. T.; Ferreira da Silva F.; Ptasińska S.; Denifl S. Formation of Resonances and Anionic Fragments upon Electron Attachment to Benzaldehyde. Phys. Chem. Chem. Phys. 2020, 22, 8171–8181. 10.1039/D0CP00029A. [DOI] [PubMed] [Google Scholar]
- Huber S. E.; Śmiałek M. A.; Tanzer K.; Denifl S. Dissociative Electron Attachment to the Radiosensitizing Chemotherapeutic Agent Hydroxyurea. J. Chem. Phys. 2016, 144, 224309. 10.1063/1.4953579. [DOI] [PubMed] [Google Scholar]
- Da Silva F. F.; Matias C.; Almeida D.; García G.; Ingólfsson O.; Flosadóttir H. D.; Ómarsson B.; Ptasińska S.; Puschnigg B.; Scheier P.; et al. NCO–, a Key Fragment upon Dissociative Electron Attachment and Electron Transfer to Pyrimidine Bases: Site Selectivity for a Slow Decay Process. J. Am. Soc. Mass Spectrom. 2013, 24, 1787–1797. 10.1007/s13361-013-0715-9. [DOI] [PubMed] [Google Scholar]
- Denifl S.; Zappa F.; Mauracher A.; Ferreira Da Silva F.; Bacher A.; Echt O.; Märk T. D.; Bohme D. K.; Scheier P. Dissociative Electron Attachment to DNA Bases near Absolute Zero Temperature: Freezing Dissociation Intermediates. ChemPhysChem 2008, 9, 1387–1389. 10.1002/cphc.200800245. [DOI] [PubMed] [Google Scholar]
- Alderighi L.; Gans P.; Ienco A.; Peters D.; Sabatini A.; Vacca A. Hyperquad Simulation and Speciation (HySS): a Utility Program for the Investigation of Equilibria Involving Soluble and Partially Soluble Species. Coord. Chem. Rev. 1999, 184, 311–318. 10.1016/S0010-8545(98)00260-4. [DOI] [Google Scholar]
- Jensen F.Introduction to Computational Chemistry; John Wiley & Sons Ltd.: Chichester, U.K., 2007. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.