

Abstract
Introduction
Advanced gynecologic cancers have a poor prognosis and constitute a major challenge for adequate treatment strategies. By analyzing and targeting molecular alterations, molecular guided treatments may be a viable option for the treatment of advanced gynecologic cancers.
Patients and Methods
In this single‐center, real‐world retrospective analysis of our platform for precision cancer medicine (PCM), we describe the molecular profiling of 72 patients diagnosed with different types of advanced gynecologic malignancies. Tumor samples of the patients were examined by next‐generation sequencing panel and immunohistochemistry (IHC).
Results
In total, we identified 209 genetic aberrations in 72 patients. The ten most frequent alterations were TP53 (n = 42, 20%), KRAS (n = 14, 6.6%), PIK3CA (n = 11, 5.2%), PIK3R1 (n = 9, 4.3%), ATR (n = 8, 3.8%), PTEN (n = 8, 3.8%), BRCA1 (n = 6, 2.8%), NF1 (n = 4, 1.9%), NOTCH1 (n = 4, 1.9%), and POLE (n = 4, 1.9%), which account for more than half of all molecular alterations (52.6%). In 21 (29.1%) patients only one mutation could be detected, and 44 (61.1%) patients had more than one mutation. No molecular alterations were detected in seven (9.7%) patients. IHC detected expression of phosphorylated mammalian target of rapamycin and epidermal growth factor receptor in 58 (80.6%) and 53 (73.6%) patients, respectively. In over two thirds (n = 49, 68.1%), a targeted therapy was suggested, based on the identified genetic aberrations. The most frequently recommended specific treatment was the combination of everolimus with exemestane (n = 18, 25 %).
Conclusion
Based on our observations, it seems that PCM might be a feasible approach for advanced gynecologic cancers with limited treatment options.
Implications for Practice
Nowadays molecular profiling of advanced gynecologic malignancies is feasible in the clinical routine. A molecular portrait should be done for every patient with an advanced therapy‐refractory gynecologic malignancy to offer molecular‐based treatment concepts.
Keywords: Precision medicine, Gynecologic oncology, Molecular aberrations, Molecular profiling, Immunohistochemistry, Targeted agents
Short abstract
To determine the feasibility of precision cancer medicine (PCM) in gynecologic cancers, this retrospective study analyzed patients with advanced gynecologic cancers enrolled and profiled in a PCM platform, MONDTI, focusing on the technical feasibility to map the molecular profiles of advanced, pretreated, and mainly relapsed gynecologic cancers and to subsequently target the detected molecular alterations
Introduction
Despite intense research efforts and therapeutic advances, gynecologic cancers are a leading cause of cancer deaths. According to GLOBOCAN 2018, cervical cancer was the fourth most frequent cancer type in women (after breast cancer, colorectal cancer, and lung cancer) of all cancer cases diagnosed in 2018. Malignancies of the cervix uteri, corpus uteri, ovary, vulva, and vagina together contribute to 15% of all newly diagnosed cancer cases in 2018, equating to over 1.3 million cases. These cancer types accounted for over 600,000 cancer deaths, equivalent to 14.6% of all cancer deaths in 2018 [1]. Advanced gynecologic malignancies bear a dismal prognosis and constitute a major challenge for adequate treatment strategies. For instance, the 5‐year relative survival rates were calculated at 17% for metastatic cervical cancer and 30% for metastatic epithelial ovarian cancer in the U.S., based on the SEER (Surveillance, Epidemiology, and End Results) database review 1975–2015 [2].
In recent years, there has been an effort to individualize and personalize therapy concepts in many cancer entities. This approach is known as precision cancer medicine (PCM) [3]. In this context, targeted agents play a pivotal role. In contrast to conventional systemic cytotoxic chemotherapy that inhibits DNA synthesis and mitosis and causes a broad range of significant treatment‐related adverse events, targeted antitumoral agents—consisting mainly of antibodies and small molecular agents—interfere with and alter the signaling pathways of malignant cells to induce damage to the cancer cells [4]. The main rationale of PCM is to match a therapeutic agent to its corresponding molecular target, allowing a precise treatment tailored to a specific patient. It aims to achieve a better and more sustained response than more generic treatments, reducing the side effects on healthy cells and tissues [5].
Precision cancer medicine in gynecologic oncology is an evolving field. In several gynecologic malignancies, targeted therapies find its way into standard therapy regimen. For instance, inhibitors of poly ADP ribose polymerase (PARP inhibitors) like olaparib are small molecular agents that are indicated for the maintenance treatment of patients with BRCA‐mutated epithelial ovarian cancer (EOC) who showed a response to platinum‐based chemotherapy either in first line or in recurrence [6, 7, 8]. Similarly, the recently published PAOLA‐1 phase III trial demonstrated that the combination of olaparib with bevacizumab in patients with International Federation of Gynecology and Obstetrics grade III–IV EOC significantly increased progression‐free survival (PFS) in all patients in general, but particularly in patients with BRCA mutation and in patients with a homologous recombination deficiency (HRD) [9].
Likewise, the important PRIMA phase III trial tested the efficacy of niraparib in a high‐risk population with newly diagnosed advanced EOC. Again, the PARP inhibitor niraparib was effective in patients with advanced EOC in general, but particularly in patients with BRCA mutation and in patients with HRD [10].
The U.S. Food and Drug Administration (FDA) approved the monoclonal antibody pembrolizumab for patients with metastatic programmed death‐ligand 1 (PD‐L1)–positive cervical cancer refractory to chemotherapy in June 2018 [11]. And in endometrial cancer, assessment of molecular alterations to triage patients into different adjuvant treatment arms is currently investigated in the PORTEC‐4a trial (ClinicalTrials.gov identifier: NCT03469674).
To determine the feasibility of PCM in gynecologic cancers, we conducted a retrospective subgroup analysis of all patients with advanced gynecologic cancers who had been enrolled and profiled in our PCM platform MONDTI (a platform for molecular characterization of metastatic solid tumors to identify actionable genomic alterations) of the Comprehensive Cancer Centre, Medical University of Vienna. We sought to analyze specifically the technical feasibility to map the molecular profiles of advanced, pretreated, and mainly relapsed gynecologic cancers and to subsequently target the detected molecular alterations.
Materials and Methods
Patients and Design of the Precision Medicine Platform
Patients with pretreated, advanced gynecologic malignancies, who were refractory to all standard treatment options, were eligible for inclusion in our PCM platform—provided archival tissue samples were available. Patients had to have an Eastern Cooperative Oncology Group performance status of 0 or 1. Our PCM platform is not a clinical trial but intends to provide the possibility of a targeted therapy to patients for whom no standard antitumoral treatment is available. Informed consent was obtained from all patients before inclusion in our platform. Furthermore, the Institutional Ethics Committee of the Medical University of Vienna has also approved this analysis (1039/2017).
Tissue Samples
Formalin‐fixed, paraffin‐embedded tissue samples from patients with advanced gynecologic malignancies who had progressed to all standard therapy regimens were obtained from the archive of the Department of Pathology, Medical University Vienna, Vienna, Austria.
Cancer Gene Panel Sequencing
DNA was extracted from paraffin‐embedded tissue blocks with a QIAamp Tissue KitTM (Qiagen, Hilden, Germany). Ten nanograms of DNA per tissue sample was provided for sequencing. The DNA library was created by multiplex polymerase chain reaction with the Ion AmpliSeq Cancer Hotspot Panel version 2 (Thermo Fisher Scientific, Waltham, MA), which covers mutation hotspots of 50 genes. The panel includes driver mutations, oncogenes, and tumor suppressor genes. By mid‐2018, the gene panel was expanded using the 161‐gene next‐generation sequencing panel of Oncomine Comprehensive Assay version 3 (Thermo Fisher Scientific), which covers genetic alterations and gene fusions. The complete list of the gene panel is provided in the supplemental online Appendix. The Ampliseq cancer hotspot panel was sequenced with an Ion PGM (Thermo Fisher Scientific) and the Oncomine Comprehensive Assay version 3 on an Ion S5 sequencer (Thermo Fisher Scientific).
The identified genetic variants were classified according to a five‐tier system comprising the modifiers pathogenic, likely pathogenic, uncertain significance, likely benign, and benign [12]. The variants pathogenic and likely pathogenic were taken into consideration for the recommendation of targeted therapy.
Microsatellite Instability Analysis
The status of microsatellite instability (MSI) was analyzed by the MSI Analysis System, version 1.1 (Promega Corporation, Madison, WI).
Immunohistochemistry
Immunohistochemistry (IHC) was performed using 2‐μm‐thin tissue sections read by a Ventana Benchmark Ultra stainer (Ventana, Tucson, Arizona, USA). The following antibodies were applied: anaplastic lymphoma kinase (ALK; clone 1A4; Zytomed, Berlin, Germany), CD20 (clone L26; Dako), CD30 (clone BerH2; Agilent Technologies, Vienna, Austria), epidermal growth factor receptor (EGFR; clone 3C6; Ventana), estrogen receptor (clone SP1; Ventana), human epidermal growth factor receptor 2 (HER2; clone 4B5; Ventana), HER3 (clone SP71; Abcam, Cambridge, UK), C‐kit receptor (KIT; clone 9.7; Ventana), MET (clone SP44; Ventana), TRKA/B/C (clone EPR17341, Abcam), phosphorylated mammalian target of rapamycin (mTOR; clone 49F9; Cell Signaling Technology, Danvers, MA), platelet‐derived growth factor receptor alpha (PDGFRA; rabbit polyclonal; Thermo Fisher Scientific), PDGFRB (clone 28E1, Cell Signaling Technology), PD‐L1 (clone E1L3N; Cell Signaling Technology), progesterone receptor (clone 1E2; Ventana), phosphatase and tensin homolog (PTEN; clone Y184; Abcam), and ROS1 (clone D4D6; Cell Signaling Technology).
To assess the immunostaining intensity for the antigens EGFR, phosphorylated mTOR, PDGFRA, PDGFRB, and PTEN, a combinative semiquantitative score for immunohistochemistry was used. The immunostaining intensity was graded from 0 to 3 (0 = negative, 1 = weak, 2 = moderate, 3 = strong). To calculate the score, the intensity grade was multiplied by the percentage of corresponding positive cells: (maximum 300) = (% negative × 0) + (% weak × 1) + (% moderate × 2) + (% strong × 3). An IHC score of ≥100 was considered positive for these markers.
The immunohistochemical staining intensity for HER2 was scored from 0 to 3+ (0 = negative, 1+ = negative, 2+ = positive, 3+ = positive) pursuant to the scoring guidelines of the Dako HercepTestR from the company Agilent Technologies (Agilent Technologies, Vienna, Austria). In case of HER2 score 2+, a further test with HER2 in situ hybridization was performed to verify the HER2 gene amplification. For HER3 staining, 2+ and 3+ were defined as positive.
Estrogen receptor and progesterone receptor stainings were graded according to the Allred scoring system from 0 to 8. A cutoff of ≥10% positive tumor cells of any staining intensity was chosen for these two markers.
MET staining was scored from 0 to 3 (0 = negative, 1 = weak, 2 = moderate, 3 = strong) based on a paper by Koeppen et al. [13]. MET 2+ and MET 3+ were considered positive.
For PD‐L1, the tumor proportion score was calculated, which is the percentage of viable malignant cells showing membrane staining. The specimen was considered to have PD‐L1 expression if the tumor proportion score was ≥1%.
ALK, CD30, CD20, and ROS1 stainings were classified positive or negative based on the percentage of reactive tumor cells, however, without graduation of the staining intensity.
All antibodies used in this study, were validated and approved at the clinical institute of pathology of the Medical University of Vienna and are used in routine IHC staining for clinical purposes. ALK, CD30, CD20, and ROS1 staining in more than 20% of the malignant cells were considered positive.
Pan‐Trk immunohistochemical staining for TrkA/B/C was considered positive if ≥1% of tumor cells exhibited positivity at any intensity above background.
Fluorescence In Situ Hybridization
Fluorescence in situ hybridization (FISH) was applied only in selected cases to verify PTEN loss.
FISH was performed with 4‐μm‐thick formalin‐fixed, paraffin‐embedded tissue sections. The following FISH probe was utilized: PTEN (10q23.31)/Centromere 10 (ZytoVision, Bremerhaven, Germany). Two hundred cell nuclei per tumor were evaluated. The PTEN FISH was considered positive for PTEN gene loss with ≥30% of cells with only one or no PTEN signals. A chromosome 10 centromere FISH probe served as a control for ploidy of chromosome 10.
Multidisciplinary Boards (Molecular Tumor Boards for PCM)
After thorough examination of the molecular profile of each tumor sample by a qualified and competent molecular pathologist, the results and findings were reviewed in a multidisciplinary tumor board (MTB) that was held every other week.
Members of the board included molecular pathologists, radiologists, clinical oncologists, biostatisticians, basic scientists, and, for gynecooncologic patients, the respective gynecologist. The MTB recommended the targeted therapy based on the specific molecular profile of each patient. The targeted therapies included tyrosine kinase inhibitors, checkpoint inhibitors (e.g., anti–PD‐L1 monoclonal antibodies), and growth factor receptor antibodies with or without endocrine therapy. The treatment recommendations by the MTB were prioritized dependent on the level of evidence from high to low according to phase III to phase I trials.
If more than one druggable molecular aberration was identified, the MTB recommended a therapy regimen to target as many molecular aberrations as possible, with special consideration to the toxicity profile of each antitumoral agent and their potential interactions. Because the majority of patients had already received the available standard treatment options for their cancer disease prior to their inclusion in our PCM platform, nearly all targeted agents were suggested as off‐label use. If the tumor profile and the clinical characteristics of a patient met the requirements of a clinical trial for targeted therapies that was conducted in our cancer center, patients were preferentially asked if they wanted to participate in this trial.
Results
From June 2013 to July 2019, 72 patients diagnosed with gynecologic cancer were included in this subgroup analysis from the cohort of the PCM project MONDTI that has so far profiled 570 patients with various highly advanced cancer types. The cohort of gynecologic malignancies comprised 44 patients with ovarian cancer, 17 patients with endometrial cancer, 7 patients with malignant mixed Müllerian tumor, 2 patients with cervical cancer, and 2 patients with vaginal cancer. All patients were of white race. The median age at first diagnosis was 52 years, ranging from 28 to 75 years, and the median age at the time when the molecular profiling was performed was 57 years, ranging from 31 to 77 years (Table 1). The tumor tissue was obtained from biopsy or during surgical intervention.
Table 1.
Patient characteristics (n = 72)
| Patient characteristics | Number |
|---|---|
| Median (range) age at fist diagnosis | 52 (28–75) |
| Median (range) age at molecular profiling | 57 (31–77) |
| White race | 72 |
| Ovarian cancer | 44 |
| Endometrial cancer | 17 |
| Malignant mixed Müllerian tumor | 7 |
| Cervical cancer | 2 |
| Vaginal cancer | 2 |
| Relapsed disease | 54 |
| Systemic chemotherapy | 72 |
| Prior chemotherapy regimens | 1–6 |
At the time of molecular profiling, all patients had an advanced and therapy‐refractory gynecological cancer in stage IV with distant metastases, mainly in the lungs, liver, and peritoneum. Sixty‐three patients had undergone a surgical intervention. The patients received a median of three lines of prior systemic chemotherapy.
In total, we identified 209 molecular aberrations in 72 patients. The predominant mutations were TP53 (20.1%), KRAS (6.7%), and PIK3CA (5.2%). Twenty‐one patients were found to have only one mutation, and 44 patients had more than one mutation. No aberrations were detected in seven patients (Table 2).
Table 2.
Molecular aberrations in advanced gynecologic malignancies
| Aberrations | n (%) |
|---|---|
| TP53 | 42 (20.10) |
| KRAS | 14 (6.70) |
| PIK3CA | 11 (5.26) |
| PIK3R1 | 9 (4.31) |
| ATR | 8 (3.83) |
| PTEN | 8 (3.83) |
| BRCA1 | 6 (2.87) |
| NF1 | 4 (1.91) |
| NOTCH1 | 4 (1.91) |
| POLE | 4 (1.91) |
| APC | 3 (1.44) |
| ARID1A | 3 (1.44) |
| ATM | 3 (1.44) |
| CDH1 | 3 (1.44) |
| FBXW7 | 3 (1.44) |
| MAP2K1 | 3 (1.44) |
| PTCH1 | 3 (1.44) |
| RB1 | 3 (1.44) |
| TSC1 | 3 (1.44) |
| TSC2 | 3 (1.44) |
| BRAF | 2 (0.96) |
| CDK12 | 2 (0.96) |
| ERBB3 | 2 (0.96) |
| FANCA | 2 (0.96) |
| FANCI | 2 (0.96) |
| FGFR1 | 2 (0.96) |
| KIT | 2 (0.96) |
| MET | 2 (0.96) |
| MLH1 | 2 (0.96) |
| MSH2 | 2 (0.96) |
| MYC | 2 (0.96) |
| NBN | 2 (0.96) |
| NOTCH2 | 2 (0.96) |
| NOTCH3 | 2 (0.96) |
| NTRK2 | 2 (0.96) |
| PMS2 | 2 (0.96) |
| RAD50 | 2 (0.96) |
| RAD51C | 2 (0.96) |
| SETD2 | 2 (0.96) |
| AKT1 | 1 (0.48) |
| ATRX | 1 (0.48) |
| BRCA2 | 1 (0.48) |
| CCND1 | 1 (0.48) |
| CCND2 | 1 (0.48) |
| CCND3 | 1 (0.48) |
| CDKN2A | 1 (0.48) |
| CHEK1 | 1 (0.48) |
| CHEK2 | 1 (0.48) |
| DDR2 | 1 (0.48) |
| FGFR2 | 1 (0.48) |
| FGFR4 | 1 (0.48) |
| IDH1 | 1 (0.48) |
| MAPK1 | 1 (0.48) |
| MED12 | 1 (0.48) |
| MSH6 | 1 (0.48) |
| MTOR | 1 (0.48) |
| NF2 | 1 (0.48) |
| NTRK1 | 1 (0.48) |
| PALB2 | 1 (0.48) |
| PDGFRA | 1 (0.48) |
| PPP2R1A | 1 (0.48) |
| PTPN11 | 1 (0.48) |
| RET | 1 (0.48) |
| RNF43 | 1 (0.48) |
| SLX4 | 1 (0.48) |
| SMAD4 | 1 (0.48) |
| SMARCA4 | 1 (0.48) |
| SMARCB1 | 1 (0.48) |
| STK11 | 1 (0.48) |
| TERT | 1 (0.48) |
| Total | 209 (100) |
IHC detected phosphorylated mTOR expression in 58 patients, with a median score of 120. Fourteen patients had a phosphorylated mTOR score between 200 and 300. The expression of EGFR was detected in 53 patients and was slightly higher than mTOR expression, with a median score of 130. PTEN expression was revealed in 43 patients. Loss of PTEN was confirmed by FISH in 18 patients. The progesterone receptor was expressed in 40 patients, and expression of the estrogen receptor was found in 31 patients, respectively. Less common and of lower levels were expression of PDGFRA and PDGFRB, which were found in eight and five patients, respectively. Tumor expression of PD‐L1 was seen in 21 tumor samples, including three cases of malignant mixed Müllerian tumor.
In two patients with ovarian cancer a gene fusion was detected: EIF3E (exon 1) ‐ RSPO2 (exon 2) and TBL1XR1 (exon 1) ‐ PIK3CA (exon 2).
MSI status was evaluated in 61 patients; however, none of the tested patients was MSI high.
In over two thirds (n = 49, 68.1%) of the 72 patients, a targeted therapy was suggested based on the identified genetic aberrations. The most frequently recommended specific treatment was the combination of the mTOR inhibitor everolimus with exemestane (n = 18). Further common recommendations were checkpoint inhibitors (n = 12), everolimus alone (n = 7), and aromatase inhibitors (n = 4), which were suggested in combination with imatinib in one case and with sunitinib in another. Olaparib and the combination of trastuzumab and pertuzumab were considered as targeted therapy in two patients each. Nintedanib, tamoxifen, trametinib, and vemurafenib were each proposed in one case. In five tumor specimens, IHC failed because of insufficient tumor material, and in two cases, gene sequencing was not possible because of technical issues and was not repeated because of the poor quality of the tumor sample remnant left. Thus, the analysis rate for the genomic sequencing was at 97%, and the analysis rate for IHC was at 93%.
Sixty‐three patients had undergone a surgical intervention. In 50 patients, the tumor sample obtained during surgical resection was used for the molecular profiling. The other 13 patients were rebiopsied for the purpose of molecular analysis. Nine patients were diagnosed with an unresectable gynecologic malignancy and underwent biopsy for diagnostic confirmation of their cancer disease. The median turnaround time between surgical resection of the tumor and molecular analysis was 24.4 months. The median turnaround time between initiation of molecular profiling and discussion in MTB and therapy initiation for all 72 patients was 34 and 43 days, respectively.
For those 13 patients who underwent rebiopsy the median turnaround time from biopsy to discussion in MTB was 20 days. The median turnaround time from biopsy to therapy initiation was 26 days.
One patient died after inclusion in our PCM and after performance of molecular analysis but before her case could be discussed in our MTB. Four patients died before a targeted therapy could be initiated. Eventually, 17 patients received the recommended targeted therapy. The applied targeted therapies consisted of exemestane plus everolimus, pembrolizumab, everolimus monotherapy, olaparib, imatinib, and sunitinib (Tables 3 and 4). However, 8 of the 17 patients died before restaging. Of the remaining nine patients, seven patients achieved a clinical benefit: two had a partial response, and five patients had a stable disease. Two patients experienced progressive disease. Figure 1 depicts patient flow.
Table 3.
Rationale for targeted therapy recommendations
| Therapeutic agent (trading name) | Targets | Overview of current FDA approval in different entities | Overview of current EMA approval in different entities |
|---|---|---|---|
|
Exemestane (Aromasin) |
Aromatase |
Estrogen receptor breast cancer |
Estrogen receptor breast cancer |
| Bevacizumab (Avastin) | VEGF‐A | Metastatic CRC, metastatic RCC, NSCLC; glioblastoma, cervical cancer | Metastatic CRC, metastatic breast cancer, NSCLC, metastatic RCC, epithelial ovarian, fallopian tube and primary peritoneal cancer, cervical cancer |
|
Tamoxifen (Nolvadex) |
Estrogen receptor | Breast cancer | Breast cancer |
| Pembrolizumab (Keytruda) |
PD‐1, hypermutability |
Melanoma, NSCLC, HNSCC, HL, urothelial carcinoma, microsatellite instability‐high cancer, gastric cancer, cervical cancer | Melanoma, NSCLC, HNSCC, HL, urothelial carcinoma |
| Everolimus (Afinitor) | mTOR expression | Breast cancer, PNET, RCC, renal angiomyolipoma, SEGAs with TSC | Breast cancer, RCC, neuroendocrine tumors of pancreatic, gastrointestinal or lung origin |
|
Imatinib (Gleevec) (n = 1) |
PDGFR, KIT | Ph+ CML, KIT+ GIST, MDS/MPD associated with PDGFR, Ph+ ALL | Ph+ CML, KIT+ GIST, MDS/MPD associated with PDGFR, Ph+ ALL |
|
Sunitinib (Sutent) |
PDGFR, KIT, VEGFR, RET, FLT3 | RCC, PDAC, GIST | RCC, PDAC, GIST |
| Olaparib (Lynparza) | BRCA1/2, ATM, CHEK2, PALB2 | Ovarian cancer, breast cancer | Ovarian, fallopian tube, or primary peritoneal cancer |
| Nintedanib (Vargatef, Ofev) | FLT3, FGFR, PDGFR, VEGFR | Idiopathic pulmonary fibrosis | NSCLC |
| Vemurafenib | BRAF V600E | Melanoma with BRAF V600E mutation | Melanoma with BRAF V600E mutation |
| Dabrafenib/ Trametinib (Tafinlar/ Mekinist) | BRAF V600E | BRAF V600E melanoma or NSCLC | BRAF V600E melanoma or NSCLC |
| Trastuzumab (Herceptin) | HER2 | HER2+ breast cancer and gastric cancer | HER2+ breast cancer and gastric cancer |
|
Pertuzumab (Perjeta) |
HER2 | HER2+ breast cancer | HER2+ breast cancer |
Abbreviations: ALL, acute lymphatic leukemia; CML, chronic myeloid leukemia; CRC, colorectal cancer; EMA, European Medicines Agency; FDA, U.S. Food and Drug Administration; FLT3, fms like tyrosine kinase 3; GIST, gastrointestinal stromal tumor; HER2, human epidermal growth factor receptor 2; HL, Hodgkin lymphoma; HNSCC, head and neck squamous cell carcinoma; KIT, C‐kit receptor; MDS/MPD, myelodysplastic syndromes/ myeloproliferative disorder; mTOR, mammalian target of rapamycin; NSCLC, non‐small cell lung carcinoma; PD‐1, programmed cell death protein 1; PDAC, pancreatic ductal adenocarcinoma; PDGFR, platelet‐derived growth factor receptor; Ph+: Philadelphia chromosome positive; PNET, primitive neuroectodermal tumor; RCC, renal cell carcinoma; RET, rearranged during transfection; SEGA, subependymal giant cell astrocytoma; TSC, tuberous sclerosis complex; VEGFR, vascular endothelial growth factor.
Table 4.
Overview of targeted therapy recommendations
| Therapy | Number of targeted therapy recommendations | Number of patients who received the targeted therapy n (%) | Disease entities suggested for | Response after application of the recommended targeted therapy |
|---|---|---|---|---|
| Exemestane + everolimus | 18 | 7 (389%) | Ovarian cancer, endometrial cancer | 1 PR, 2 SD, 1 PD, three patients died before restaging |
| Pembrolizumab | 12 | 6 (153%) | Ovarian cancer, endometrial cancer, vaginal cancer |
SD (n = 3), PD (n = 2), One patient died before restaging |
| Everolimus monotherapy | 7 | 1 (58%) | Ovarian cancer | Patient died before restaging |
| Aromatase inhibitors | 2 | 0 | Ovarian cancer | — |
| Olaparib | 2 | 1 (58%) | Ovarian cancer | PR |
| Trastuzumab + pertuzumab | 2 | 0 | Endometrial cancer | — |
| Vemurafenib | 2 | 0 | Malignant mixed Müllerian tumor, Endometrial cancer | — |
| Imatinib + aromatase inhibitor | 1 | 1 (58%) | Malignant mixed Müllerian tumor | Patient died before restaging |
| Sunitinib + aromatase inhibitor | 1 | 1 (58%) | Ovarian cancer | Patient died before restaging |
| Nintedanib | 1 | 0 | Malignant mixed Müllerian tumor | — |
| Tamoxifen | 1 | 0 | Endometrial cancer | — |
| Total number | 49 | 17 (100%) | All entities |
Abbreviations: PD, progressive disease; PR, partial response; SD, stable disease.
Figure 1.
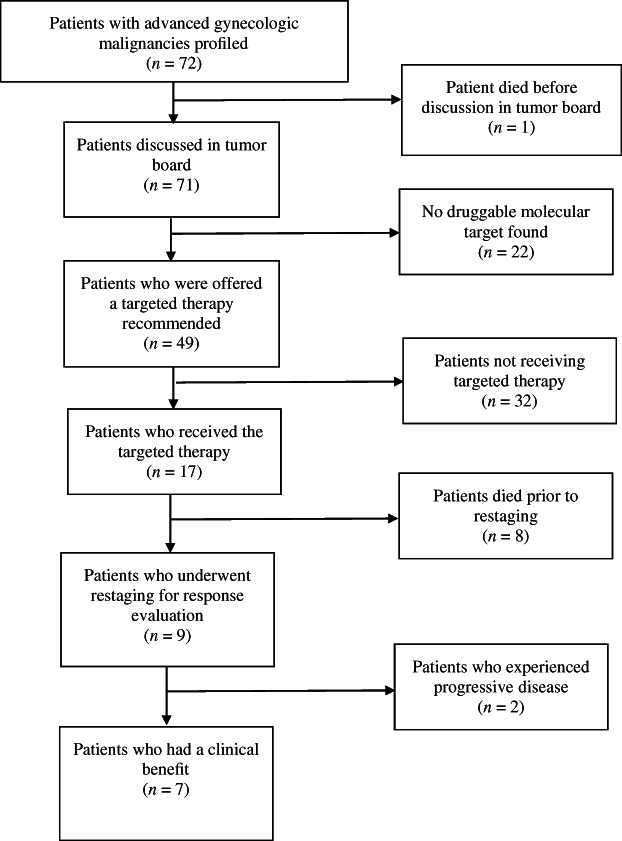
Flow chart of the 72 patients with cancer.
It is worth mentioning that there was an over sevenfold increase regarding the number of targeted therapy recommendations from July 2013 to 2018, from only two treatment suggestions in 2013 to 15 suggestions in 2018 (Table 5).
Table 5.
Increase in number of molecular guided targeted therapy recommendations per year
| July 2013 to December 2013 | 2014 | 2015 | 2016 | 2017 | 2018 | January 2019 to July 2019 | |
|---|---|---|---|---|---|---|---|
| Number recommended | 2 | 2 | 2 | 7 | 15 | 15 | 6 |
Discussion
In this retrospective single‐center real‐life analysis, we present the molecular profiling of all patients with gynecologic malignancies discussed in our MTB. Their disease was therapy‐refractory and advanced. A treatment recommendation was derived for 49 (68%) patients from the MTB.
Only a few studies have investigated the potential of precision medicine in the field of gynecologic oncology. Rodriguez‐Rodriguez et al. applied genomic profiling in 69 patients with either rare or therapy‐refractory gynecologic malignancies and reported that a treatment suggestion was made in 64 patients (92.7%) based on either clinical or genomic data. This recommendation rate might be impressive; however, there are three important issues: The therapy suggestion was not exclusively genomic driven, because therapy recommendations in the tumor board were also made in cases (based on clinical data) in which no actionable genomic mutation was found. Secondly, only 13 patients had an advanced disease and were classified as stage IV. Fourteen patients were diagnosed with stage I or II disease. Furthermore, the recommended antitumoral agents were not mentioned [14].
Similar to the previous study, Gunderson et al. published a pilot study about the use of a precision medicine program in gynecologic oncology. They enrolled 62 patients with metastatic or recurrent gynecologic malignancies and generated the genomic profile of these patients. However, in only four patients (6%) a genomic‐based targeted therapy was recommended [15]. Another study by Soumerai et al. from the Memorial Sloan Kettering Cancer Center explored the molecular characterization of 189 patients with advanced endometrial cancer. According to this study, 68% of these patients (n = 127) harbored potentially actionable mutations. This rate is in line with our recommendation rate in this study. More than one quarter (27%, n = 34) of these patients were enrolled to matched clinical trials. Nearly half of the enrolled patients (47%, n = 16) experienced a clinical benefit. In total, 8.5% (16/189) of patients achieved clinical benefit to matched targeted therapy when accounting for all patients sequenced. This rate of clinical benefit is also comparable to the rate described in this study (9.7%) [16].
The unique feature of the MONDTI platform and thus the added value of this analysis is that apart from the genomic sequencing, RNA sequencing, IHC, and cytogenetics with FISH were also performed to achieve a comprehensive molecular profile. Together these combined techniques formed a solid base for the recommendation of molecular guided targeted agents for the patients. The analysis rate was 97% for genomic sequencing and 93% for IHC. Out of the 49 therapy recommendations, 39 (80%) were derived from the information provided by IHC. Only in 10 cases (20%) the therapy suggestion was primarily based on the genomic mutations delivered by next‐generation sequencing. Thus, IHC plays a decisive role in molecular‐driven targeted therapy approaches.
Another important characteristic of the MONDTI platform is that it is an open platform that enrolls all patients with solid tumors with no further standard treatment options. Thus, unlike a clinical trial, MONDTI provides real‐life data that are relatively unbiased.
Interestingly, there was an over sevenfold rise in numbers of targeted therapy recommendations from July 2013 to 2018. This trend indicates the increasing role, relevance, and potential of PCM in the clinical routine.
Although this analysis shows that PCM is implementable in daily clinical routine, only seven patients had a clinical benefit from this therapy approach. Of note, 8 of the 17 patients died before restaging. One reason may be the long median turnaround time of 43 days for all patients from molecular analysis to therapy initiation. A shorter turnaround time may help to start earlier with the targeted therapy and to control the cancer disease. Liquid biopsy may be a viable alternative to shorten the turnaround time, to monitor the disease, and to evaluate the therapy response. Another reason may be the complexity of gynecologic malignancies. Our analysis shows that these malignancies exhibit a remarkable genetic heterogeneity [17, 18].
This finding is consistent with the well‐described extreme and complex intratumoral heterogeneity in gynecologic malignancies that occurs within the same tumor tissue; vascularization, proliferation, and subclones are all known to be highly variable. The pattern of genetic and epigenetic aberrations changes both spatially and temporally. The tumor biology at metastatic sites is different from the primary site and differs again at the time point of relapse.
Sixty‐three patients had undergone a surgical intervention. In 50 patients, the tumor sample obtained during surgical resection was used for the molecular profiling.
An important limitation of this study is that the median turnaround time between surgical resection of the tumor and molecular analysis was 24.4 months. In this time interval, the molecular landscape of the cancer cells may have changed. Furthermore, a growing body of literature suggests that the antitumoral therapy itself can drive and change the molecular portrait of malignant cells by creating new driver mutations in subclones that become insensitive to drugs [19, 20, 21]. Thus, the molecular profile may differ at the time of therapy recommendation from the time of surgical resection. Liquid biopsy may help in future to detect the molecular aberrations and alterations in real time to tailor a targeted therapy that matches the current molecular landscape.
Despite the costs of extensive molecular profiling, the outcome of this study with only seven patients experiencing a clinical benefit was modest. However, personalized medicine in oncology is still in its infancy in terms of both diagnostic procedures and targeted therapy strategies and warrants further financial investment and studies and clinical trials to discover its full potential. Currently, the costs of molecular profiling are directly covered by the General Hospital of Vienna provided that the patients have no further standard treatment options.
The most frequent targeted therapy recommended by the MTB was the combination of exemestane with everolimus, which was recommended in 18 patients from 72 patients (25%) who were pretreated with nonsteroidal aromatase inhibitors. Until now, several studies have investigated the antitumoral activity of exemestane and everolimus in breast cancer. The pivotal phase III BOLERO‐2 trial investigated the combination of everolimus and exemestane versus exemestane alone in in postmenopausal women with hormone receptor–positive, HER2‐negative advanced breast cancer whose disease was refractory to nonsteroidal aromatase inhibitor. Beaver and Park showed in this trial that the combination therapy is superior to exemestane alone because the combination regimen resulted in a twofold increase in median progression‐free survival: 7.8 months versus 3.2 months. However, BOLERO‐2 did not meet its secondary endpoint, which was overall survival [22]. The follow‐up phase II BOLERO‐6 trial also underscored the efficacy of exemestane plus everolimus versus everolimus alone [23].
The phase IIIB 4EVER trial in Germany, the EVA study in Italy, and the phase IIIb BALLET trial in Spain all three evaluated the safety of exemestane and everolimus in the same patient population and confirmed the safety of this therapy regimen [24, 25, 26]. However, this combination has not yet been tested in gynecologic cancers, for example, ovarian cancer or endometrial cancer.
Everolimus alone was suggested for seven patients whose tumor tissue samples did not express HER2 or hormone receptors.
Pembrolizumab was recommended in 11 patients pretreated with platinum‐based chemotherapy. The efficacy of pembrolizumab was tested in the phase Ib KEYNOTE‐028 trial in 26 patients with heavily pretreated PD‐L1–positive advanced ovarian cancer. After a median follow‐up duration of 15.4 months, pembrolizumab achieved complete response in one, partial response in two, and stable disease in seven patients, resulting in an overall response rate (ORR) of 11.5%. Median PFS and median overall survival were 1.9 and 13.8 months, respectively [27]. Based on these promising results, pembrolizumab was examined in the phase II KEYNOTE‐100 trial in women with advanced or recurrent ovarian cancer after front‐line platinum‐based therapy. Of the 376 patients included, 8% had a response and 37.2%, experienced disease control. PFS was 2.1 months. ORR was 10.2% and 17.1% for patients with PD‐L1 combined positive score ≥ 1% and ≥ 10%. Thus, a higher PD‐L1 combined positive score was correlated with a higher response rate [28]. The phase III KEYNOTE‐775 trial is currently testing the clinical efficacy of pembrolizumab in combination with lenvatinib versus chemotherapy in second line endometrial cancer. The results are still awaited [29]. In June 2018—based on the practice‐changing data of the phase II KEYNOTE‐158 trial—pembrolizumab was granted accelerated approval by FDA for patients with recurrent or metastatic PD‐L1–positive cervical cancer [30].
Sunitinib is an oral, multitargeted receptor tyrosine kinase inhibitor. Sunitinib was applied in a phase II trial in 34 women with advanced endometrial cancer. Partial response and stable disease each were reported in six women. In total, ten patients (30%) experienced disease control for at least 6 months [31]. In another phase II trial, the potential clinical benefit of sunitinib was studied in 19 women with locally advanced or metastatic cervical carcinoma. Sixteen patients achieved stable disease; however, no objective response was reported [32].
Nintedanib was recommended for a patient with advanced malignant mixed Müllerian tumor because of FGFR1 mutation. Like sunitinib, nintedanib is also an oral, multitargeted receptor tyrosine kinase inhibitor. It was tested in a phase II trial in patients with bevacizumab‐resistant recurrent epithelial ovarian cancer. Only two patients had partial response. Stable disease was seen in 10 women. Fifteen patients experienced progressive disease. Thus, nintedanib exhibited only minimal activity in this patient population [33]. In contrast, in the large‐scale randomized phase III AGO‐OVAR trial, nintedanib showed antitumoral activity in patients with advanced ovarian cancer. Nintedanib was tested in combination with carboplatin and paclitaxel in first‐line against placebo and significantly improved the median PFS: 17.2 months versus 16.6 months in the placebo group [34].
Imatinib is another tyrosine kinase inhibitor and was recommended for a patient with advanced epithelial ovarian cancer who expressed PDGFRA. Safra et al. tested weekly paclitaxel with the intermittent application of imatinib in 14 women with epithelial ovarian cancer. Four patients achieved an objective response [35].
Based on the data of the phase III trials CLEOPATRA and PERUSE, the addition of pertuzumab to trastuzumab and a taxane backbone is currently the gold standard in the management of HER2‐positive metastatic breast cancer. This therapy regimen was offered for two patients with HER2‐positive metastatic epithelial ovarian cancer [36, 37].
Vemurafenib was offered for two patients with malignant mixed Müllerian tumor and endometrial cancer, respectively, who both harbored the BRAF V600E mutation. In a phase II basket trial, Hyman et al. investigated the potential benefit of vemurafenib in BRAF V600E mutation–positive nonmelanoma cancers. In patients with non‐small cell lung cancer, vemurafenib achieved a response rate of 42% and median PFS of 7.3 months. However, in patients with other malignancies, including colorectal or ovarian cancer, only anecdotal responses were observed. Further studies are warranted to evaluate the efficacy of vemurafenib [38].
In our PCM platform, we also identified two patients with recurrent advanced ovarian cancer: one with BRCA1 and the other with BRCA2 mutation. For these patients, we recommended olaparib before approval. Presently, olaparib has an FDA approval for treatment of germline BRCA‐mutated advanced ovarian cancer. Olaparib is an orally active inhibitor of the enzyme PARP that is of pivotal importance in the homologous recombination repair of the DNA [6].
Conclusion
Taken together, the complex tumor biology and the long turnaround time for the mapping of the molecular profile pose major challenges in the clinical implementation of PCM for the management of gynecologic malignancies. Further research is needed to better understand the tumor biology, to reduce the turnaround time, and to develop more sophisticated molecular guided therapy regimens.
This subgroup analysis shows that profiling of gynecologic cancers is possible and feasible in the daily clinical routine of a university hospital setting. However, there is room for improvement for the development of molecular‐driven targeted agents and in terms of availability and implementation of molecular profiling in main and community hospitals. PCM has the potential to become more widely used in cancer drug development and therapy planning and strategy for gynecological tumors [39, 40].
Author Contributions
Conception/design: Hossein Taghizadeh, Robert M. Mader, Leonhard Müllauer, Stefanie Aust, Stephan Polterauer, Heinz Kölbl, Veronika Seebacher, Christoph Grimm, Alexander Reinthaller, Gerald W. Prager
Provision of study material or patients: Hossein Taghizadeh, Robert M. Mader, Leonhard Müllauer, Stefanie Aust, Stephan Polterauer, Heinz Kölbl, Veronika Seebacher, Christoph Grimm, Alexander Reinthaller, Gerald W. Prager
Collection and/or assembly of data: Hossein Taghizadeh, Robert M. Mader, Leonhard Müllauer, Stefanie Aust, Stephan Polterauer, Heinz Kölbl, Veronika Seebacher, Christoph Grimm, Alexander Reinthaller, Gerald W. Prager
Data analysis and interpretation: Hossein Taghizadeh, Robert M. Mader, Leonhard Müllauer, Stefanie Aust, Stephan Polterauer, Heinz Kölbl, Veronika Seebacher, Christoph Grimm, Alexander Reinthaller, Gerald W. Prager
Manuscript writing: Hossein Taghizadeh, Robert M. Mader, Leonhard Müllauer, Stefanie Aust, Stephan Polterauer, Heinz Kölbl, Veronika Seebacher, Christoph Grimm, Alexander Reinthaller, Gerald W. Prager
Final approval of manuscript: Hossein Taghizadeh, Robert M. Mader, Leonhard Müllauer, Stefanie Aust, Stephan Polterauer, Heinz Kölbl, Veronika Seebacher, Christoph Grimm, Alexander Reinthaller, Gerald W. Prager
Disclosures
Christoph Grimm: AstraZeneca, Celgene, Merck Sharp & Dohme, PharmaMar, Roche, GlaxoSmithKline/Tesaro, Vifor Pharma, Clovis (C/A), Amgen, AstraZeneca, Merck Sharp Dohme, PharmaMar, Roche, GlaxoSmithKline/Tesaro (ET), Meda Pharma, Roche Diagnostics (RF). The other authors indicated no financial relationships.
(C/A) Consulting/advisory relationship; (RF) Research funding; (E) Employment; (ET) Expert testimony; (H) Honoraria received; (OI) Ownership interests; (IP) Intellectual property rights/inventor/patent holder; (SAB) Scientific advisory board
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Appendix S1: Supporting infotmation
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact Commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
References
- 1. Bray F, Ferlay J, Soerjomataram I et al. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 2. SEER Cancer Statistics Review (CSR) 1975‐2016. National Cancer Institute Web site. Available from https://seer.cancer.gov/csr/1975_2016/. Accessed March 4, 2020.
- 3. Von Hoff DD, Stephenson JJ Jr, Rosen P et al. Pilot study using molecular profiling of patients’ tumors to find potential targets and select treatments for their refractory cancers. J Clin Oncol 2010;28:4877–4883. [DOI] [PubMed] [Google Scholar]
- 4. Joo WD, Visintin I, G Mor G. Targeted cancer therapy–Are the days of systemic chemotherapy numbered? Maturitas 2013;76:308–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Chen HZ, Bonneville R, Roychowdhury S. Implementing precision cancer medicine in the genomic era. Semin Cancer Biol 2019;55:16–27. [DOI] [PubMed] [Google Scholar]
- 6. Ma J, Deng H, Li J et al. Efficacy and safety of olaparib maintenance therapy in platinum‐sensitive ovarian cancer patients with BRCA mutations: A meta‐analysis on randomized controlled trials. Cancer Manag Res 2019;11:3061–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Moore K, Colombo N, Scambia G et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med 2018;379:2495–2505. [DOI] [PubMed] [Google Scholar]
- 8. Friedlander M, Gebski V, Gibbs E et al. Health‐related quality of life and patient‐centred outcomes with olaparib maintenance after chemotherapy in patients with platinum‐sensitive, relapsed ovarian cancer and a BRCA1/2 mutation (SOLO2/ENGOT Ov‐21): A placebo‐controlled, phase 3 randomised trial. Lancet Oncol 2018;19:1126–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ray‐Coquard IL, Harter P, Gonzalez Martin A et al. PAOLA‐1: An ENGOT/GCIG phase III trial of olaparib versus placebo combined with bevacizumab as maintenance treatment in patients with advanced ovarian cancer following first‐line platinum‐based chemotherapy plus bevacizumab. J Clin Oncol 2016;34(suppl 15):TPS5607a. [Google Scholar]
- 10. González‐Martin A, Pothuri B, Vergote I et al.; PRIMA/ENGOT‐OV26/GOG‐3012 Investigators. Niraparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med 2019. [DOI] [PubMed] [Google Scholar]
- 11. Liu Y, Wu L, Tong R et al. PD‐1/PD‐L1 inhibitors in cervical cancer. Front Pharmacol 2019;10:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Richards S, Aziz N, Bale S et al.; ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med 2015;17:405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Koeppen H, Yu W, Zha J et al. Biomarker analyses from a placebo‐controlled phase II study evaluating erlotinib ± onartuzumab in advanced non‐small cell lung cancer: MET expression levels are predictive of patient benefit. Clin Cancer Res 2014;20:4488–4498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rodriguez‐Rodriguez L, Hirshfield KM, Rojas V et al. Use of comprehensive genomic profiling to direct point‐of‐care management of patients with gynecologic cancers. Gynecol Oncol 2016;141:2–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Gunderson CC, Rowland MR, Wright DL et al. Initiation of a formalized precision medicine program in gynecologic oncology. Gynecol Oncol 2016;141:24–28. [DOI] [PubMed] [Google Scholar]
- 16. Soumerai TE, Dononghue MTA, Bandlamudi C et al. Clinical utility of prospective molecular characterization in advanced endometrial cancer. Clin Cancer Res 2018;24:5939–5947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Iwahashi N, Sakai K, Noguchi T et al. Liquid biopsy‐based comprehensive gene mutation profiling for gynecological cancer using cancer personalized profiling by deep sequencing. Sci Rep 2019;9:10426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chang L, Ni J, Zhu Y et al. Liquid biopsy in ovarian cancer: Recent advances in circulating extracellular vesicle detection for early diagnosis and monitoring progression. Theranostics 2019;9:4130–4140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lambrechts S, Smeets D, Moisse M et al. Genetic heterogeneity after first‐line chemotherapy in high‐grade serous ovarian cancer. Eur J Cancer 2016;53:51–64. [DOI] [PubMed] [Google Scholar]
- 20. Bai H, Cao D, Yang J et al. Genetic and epigenetic heterogeneity of epithelial ovarian cancer and the clinical implications for molecular targeted therapy. J Cell Mol Med 2016;20:581–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang YC, Li XP. Clinical significance of intratumor heterogeneity for gynecological carcinoma. Chronic Dis Transl Med 2015;1:14–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Beaver JA, Park BH. The BOLERO‐2 trial: The addition of everolimus to exemestane in the treatment of postmenopausal hormone receptor‐positive advanced breast cancer. Future Oncol 2012;8:651–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jerusalem G, de Boer RH, Hurvitz S et al. Everolimus plus exemestane vs everolimus or capecitabine monotherapy for estrogen receptor‐positive, HER2‐negative advanced breast cancer: The BOLERO‐6 randomized clinical trial. JAMA Oncol 2018;4:1367–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tesch H, Stoetzer O, Decker T et al. Efficacy and safety of everolimus plus exemestane in postmenopausal women with hormone receptor‐positive, human epidermal growth factor receptor 2‐negative locally advanced or metastatic breast cancer: Results of the single‐arm, phase IIIB 4EVER trial. Int J Cancer 2019;144:877–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Cazzaniga ME, Airoldi M, Arcangeli V et al.; EVA Study Group. Efficacy and safety of everolimus and exemestane in hormone‐receptor positive (HR+) human‐epidermal‐growth‐factor negative (HER2‐) advanced breast cancer patients: New insights beyond clinical trials. The EVA study. Breast 2017;35:115–121. [DOI] [PubMed] [Google Scholar]
- 26. Ciruelos E, Vidal M, Martínez de Dueñas E et al. Safety of everolimus plus exemestane in patients with hormone‐receptor‐positive, HER2‐negative locally advanced or metastatic breast cancer: Results of phase IIIb BALLET trial in Spain. Clin Transl Oncol 2018;20:753–760. [DOI] [PubMed] [Google Scholar]
- 27. Frenel JS, Le Tourneau C, O'Neill B et al. Safety and efficacy of pembrolizumab in advanced, programmed death ligand 1‐positive cervical cancer: Results from the phase Ib KEYNOTE‐028 trial. J Clin Oncol 2017;35:4035–4041. [DOI] [PubMed] [Google Scholar]
- 28. Varga A, Piha‐Paul S, Ott PA et al. Pembrolizumab in patients with programmed death ligand 1‐positive advanced ovarian cancer: Analysis of KEYNOTE‐028. Gynecol Oncol 2019;152:243–250. [DOI] [PubMed] [Google Scholar]
- 29. Charo LM, Plaxe SC. Recent advances in endometrial cancer: A review of key clinical trials from 2015 to 2019. F1000Res 2019;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chung HC, Ros W, Delord JP et al. Efficacy and safety of pembrolizumab in previously treated advanced cervical cancer: Results from the phase II KEYNOTE‐158 study. J Clin Oncol 2019;37:1470–1478. [DOI] [PubMed] [Google Scholar]
- 31. Castonguay V, Lheureux S, Welch S et al. A phase II trial of sunitinib in women with metastatic or recurrent endometrial carcinoma: A study of the Princess Margaret, Chicago and California Consortia. Gynecol Oncol 2014;134:274–280. [DOI] [PubMed] [Google Scholar]
- 32. Mackay HJ, Tinker A, Winquist E et al. A phase II study of sunitinib in patients with locally advanced or metastatic cervical carcinoma: NCIC CTG trial IND184. Gynecol Oncol 2010;116:163–167. [DOI] [PubMed] [Google Scholar]
- 33. Secord AA, McCollum M, Davidson BA et al. Phase II trial of nintedanib in patients with bevacizumab‐resistant recurrent epithelial ovarian, tubal, and peritoneal cancer. Gynecol Oncol 2019;153:555–561. [DOI] [PubMed] [Google Scholar]
- 34. du Bois A, Kristensen G, Ray‐Coquard I et al.; AGO Study Group–led Gynecologic Cancer Intergroup/European Network of Gynaecologic Oncology Trials Group Intergroup Consortium. Standard first‐line chemotherapy with or without nintedanib for advanced ovarian cancer (AGO‐OVAR 12): A randomised, double‐blind, placebo‐controlled phase 3 trial. Lancet Oncol 2016;17:78–89. [DOI] [PubMed] [Google Scholar]
- 35. Safra T, Andreopoulou E, Levinson B et al. Weekly paclitaxel with intermittent imatinib mesylate (Gleevec): Tolerance and activity in recurrent epithelial ovarian cancer. Anticancer Res 2010;30:3243–3247. [PubMed] [Google Scholar]
- 36. Baselga J, Swain SM. CLEOPATRA: A phase III evaluation of pertuzumab and trastuzumab for HER2‐positive metastatic breast cancer. Clin Breast Cancer 2010;10:489–491. [DOI] [PubMed] [Google Scholar]
- 37. Bachelot T, Ciruelos E, Schneeweiss A et al.; PERUSE Investigators. Preliminary safety and efficacy of first‐line pertuzumab combined with trastuzumab and taxane therapy for HER2‐positive locally recurrent or metastatic breast cancer (PERUSE). Ann Oncol 2019;30:766–773. [DOI] [PubMed] [Google Scholar]
- 38. Hyman DM, Puzanov I, Subbiah V et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373:726–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Klughammer J, Kiesel B, Roetzer T et al. The DNA methylation landscape of glioblastoma disease progression shows extensive heterogeneity in time and space. Nat Med 2018;24:1611–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Xiu J, Piccioni D, Juarez T et al. Multi‐platform molecular profiling of a large cohort of glioblastomas reveals potential therapeutic strategies. Oncotarget 2016;7:21556–21569. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Appendix S1: Supporting infotmation