

Abstract
On October 24, 2019, a marketing authorization valid through the European Union (EU) was issued for gilteritinib monotherapy for adult patients who have relapsed or refractory acute myeloid leukemia (AML) with an Fms‐like tyrosine kinase 3 (FLT3) mutation. Gilteritinib inhibits FLT3 receptor signaling and proliferation in cells exogenously expressing FLT3 including FLT3 internal tandem duplication (ITD), FLT3 D835Y, and FLT3 ITD D835Y, and it induced apoptosis in leukemic cells expressing FLT3 ITD. The recommended starting dose of gilteritinib is 120 mg (three 40 mg tablets) once daily. Gilteritinib was evaluated in one, phase III, open‐label, multicenter, randomized study of gilteritinib (n = 247, gilteritinib arm) versus salvage chemotherapy (n = 124, salvage chemotherapy arm) in patients with relapsed or refractory AML with FLT3 mutation. Overall survival (OS) was statistically significantly different between the two groups with a median OS of 9.3 months in the gilteritinib arm compared with 5.6 months for salvage chemotherapy (hazard ratio, 0.637; 95% confidence interval, 0.490–0.830; p = .0004 one‐sided log‐rank test). The most common adverse reactions with gilteritinib treatment were blood creatine phosphokinase increase, alanine aminotransferase increase, aspartate aminotransferase increase, blood alkaline phosphatase increase, diarrhea, fatigue, nausea, constipation, cough, peripheral edema, dyspnea, dizziness, hypotension, pain in extremity, asthenia, arthralgia, and myalgia. The objective of this article is to summarize the scientific review of the application leading to regulatory approval in the EU.
Implications for Practice
Xospata was approved in the European Union as monotherapy for the treatment of adult patients with relapsed or refractory acute myeloid leukemia (AML) with an Fms‐like tyrosine kinase 3 (FLT3) mutation. Gilteritinib resulted in a clinically meaningful and statistically significant improvement of overall survival compared with salvage chemotherapy. At the time of the marketing authorization of gilteritinib, there were no approved standard therapies specifically for adult patients diagnosed with relapsed or refractory AML with FLT3 mutation. In terms of safety, the overall accepted safety profile was considered manageable.
Keywords: Gilteritinib (Xospata), Acute myeloid leukemia, Fms‐like tyrosine kinase 3, European Medicines Agency
Short abstract
This article addresses development and authorization of gilteritinib monotherapy for the treatment of acute myeloid leukemia with an FLT3 mutation, summarizing the scientific review of the application leading to regulatory approval in the European Union.
Background
Acute myeloid leukemia (AML) is generally characterized by aberrant differentiation and proliferation of malignantly transformed myeloid progenitor cells but can be considered a heterogeneous disease state with various molecular and genetic abnormalities that result in variable clinical outcomes. AML accounts for approximately 80% of acute leukemia diagnosed in adults, with a median age at diagnosis of 67 years 1, 2, 3. The 5‐year overall AML survival rate is 15%, with an age‐related decrease in survival rates from 67% to 5% in patients who are 0 to 14 years of age and greater than 65 years of age, respectively 4. Certain genetic factors appear to predispose patients to poorer outcomes. Mutational status of Fms‐like tyrosine kinase 3 (FLT3), a member of the class III receptor tyrosine kinase, is now well recognized as delineating a subtype of leukemia with poor prognosis, with a higher relapse rate and a shorter duration of remission from initial therapy (6 months vs. 11.5 months for those without FLT3 internal tandem duplication [ITD] mutations), as well as reduced disease‐free survival (16% to 27% vs. 41% at 5 years) and overall survival (OS) (15% to 31% vs. 42% at 5 years) 5, 6, 7, 8, 9.
At the time of the marketing authorization of gilteritinib there were no approved standard therapies specifically for adult patients diagnosed with relapsed or refractory AML with FLT3 mutation.
In February 2019, Astellas Pharma Europe B.V. applied for a marketing authorization via the European Medicines Agency (EMA) centralized procedure for gilteritinib with the invented name Xospata. Gilteritinib is designated as an orphan medicinal product in the European Union (EU). The review has been conducted by the Committee for Medicinal Products for Human Use (CHMP), the scientific committee of the EMA responsible for providing a scientific opinion on the granting of a marketing authorization. The review was started on February 27, 2019, and a positive opinion was issued on September 19, 2019. The CHMP conducted accelerated assessment, which is rapid assessment of medicines in the centralized procedure that are of major interest for public health, especially ones that are therapeutic innovations. Accelerated assessment usually takes 150 evaluation days, rather than 210. Table 1 presents a summary of key regulatory steps and procedures for Xospata. The approved indication in the EU is as follows: “Xospata is indicated as monotherapy for the treatment of adult patients who have relapsed or refractory acute myeloid leukemia (AML) with a FLT3 mutation.”
Table 1.
Steps in the evaluation of the marketing authorization for Xospata
| Step/procedure | Date | Active review time, days |
|---|---|---|
| Initial marketing authorization application received | February 7, 2019 | 0 |
| Adoption of the consolidated list of questions by the CHMP | May 27, 2019 | 90 |
| Submission of responses by the applicant | June 20, 2019 | |
| The applicant addressed outstanding issues during an oral explanation to the CHMP | September 19, 2019 | 150 |
| The CHMP adopted a positive opinion for granting a marketing authorization to Xospata | September 19, 2019 | 150 |
| The European Commission granted a marketing authorization valid across the EU | October 24, 2019 |
Abbreviations: CHMP, Committee for Medicinal Products for Human Use; EU, European Union.
Nonclinical Aspects and Clinical Pharmacology
Gilteritinib fumarate is an FLT3 and AXL inhibitor (Fig. 1). Gilteritinib inhibits FLT3 receptor signaling and proliferation in cells exogenously expressing FLT3 including FLT3 ITD, FLT3 D835Y, and FLT3 ITD‐ D835Y, and it induced apoptosis in leukemic cells expressing FLT3 ITD.
Figure 1.
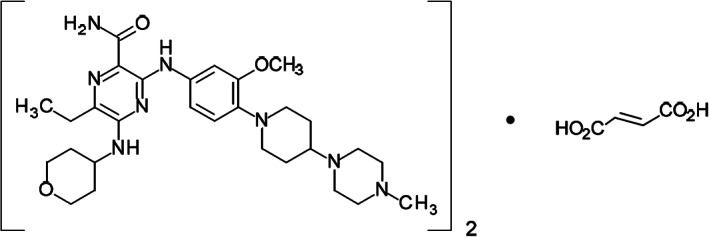
Molecular structure of gilteritinib. The molecular formula is (C29H44N8O3)2 • C4H4O4. The relative molecular mass is 1,221.50 g/mol. The chemical name of gilteritinib is (E)‐but‐2‐enedioic acid;6‐ethyl‐3‐[3‐methoxy‐4‐[4‐(4‐methylpiperazin‐1‐yl)piperidin‐1‐yl]anilino]‐5‐(oxan‐4‐ylamino)pyrazine‐2‐carboxamide.
In rats and dogs, gilteritinib‐induced adverse findings observed in the lungs, immune system, bone marrow, hematopoietic system, epithelial tissue, liver, kidney/urinary bladder, and gastrointestinal tract were considered to be pharmacologically related. Gilteritinib has the potential to be genotoxic, based on the positive in vivo micronucleus results. Embryo‐fetal toxicity was observed in rats at low exposures compared with clinical exposure and is considered clinically relevant. Based on these nonclinical findings, it is recommended that women of childbearing potential and men of reproductive potential should use effective contraception during and up to 6 months (women) or 4 months (men) after the last dose administered.
The effect of mild or moderate renal impairment was evaluated using a population pharmacokinetic model. Serum creatinine, a marker of renal function, was identified as a statistically significant covariate. The risk of increased exposure because of renal impairment could not be completely excluded based on the presented data. Renal excretion is a minor route of elimination, although the relative contribution compared with the hepatic route is not known. However, nonrenal drug clearance might be affected in chronic renal disease because of inhibition and/or suppression of enzymes and transporters by circulating uremic toxins. The CHMP concluded that based on the available data, impaired renal function is not expected to significantly affect gilteritinib exposure, indicating that dose adjustment is not warranted in patients with mild or moderate renal impairment. The CHMP recommended the applicant to conduct a postapproval phase I study to investigate the effect of renal impairment on the pharmacokinetics, safety, and tolerability of gilteritinib compared with patients with normal renal function.
Clinical Efficacy
The pivotal efficacy study was the ADMIRAL study (2215‐CL‐0301), a phase III open‐label, multicenter, randomized study of gilteritinib versus salvage chemotherapy in patients with relapsed or refractory (R/R) AML with FLT3 mutation 10.
Patients were required to have primary AML or AML secondary to myelodysplastic syndrome (MDS) according to World Health Organization classification 11. Patients were refractory (to at least one cycle of induction chemotherapy) or relapsed (achieved a complete remission [CR]/complete remission with incomplete hematologic recovery [CRi]/complete remission with incomplete platelet recovery [CRp] as defined by Cheson et al. 12) after first‐line AML therapy (with or without hematopoietic stem cell transplantation [HSCT]). Patients could have been enrolled if they had Eastern Cooperative Oncology Group performance status of ≤2 and FLT3‐ITD, FLT3‐TKD/D835, or FLT3‐TKD/I836 mutation. Patients were excluded from the study if they were diagnosed with acute promyelocytic leukemia, BCR‐ABL–positive leukemia, or AML secondary to prior chemotherapy for other neoplasms (except for MDS).
Patients were randomized in a 2:1 ratio to receive gilteritinib (120 mg administered orally once daily) or salvage chemotherapy including one of the following regimens.
Low‐dose cytarabine: 20 mg cytarabine was administered twice daily by s.c. or i.v. injection for 10 days.
Azacitidine: 75 mg/m2 azacitidine was administered daily by s.c. or i.v. injection for 7 days.
Mitoxantrone, etoposide, and intermediate‐dose cytarabine: mitoxantrone 8 mg/m2 per day was administered by i.v. injection for 5 days (days 1 through 5); etoposide 100 mg/m2 per day was administered by i.v. injection for 5 days (days 1 through 5); cytarabine 1,000 mg/m2 per day was administered by i.v. injection for 5 days (days 1 through 5).
Fludarabine, cytarabine, and granulocyte colony‐stimulating factor (G‐CSF) with idarubicin: G‐CSF 300 μg/m2 per day was administered by s.c. or i.v. injection for 5 days (days 1 through 5). Additional G‐CSF by s.c. or i.v. injection was recommended 7 days after completing chemotherapy until absolute neutrophil count >0.5 × 109/L; fludarabine 30 mg/m2 per day was administered by i.v. injection for 5 days (days 2 through 6); cytarabine 2,000 mg/m2 per day was administered by i.v. injection for 5 days (days 2 through 6); idarubicin 10 mg/m2 per day was administered by i.v. injection for 3 days (days 2 through 4).
The investigator preselected the specific salvage chemotherapy regimen (i.e., low or high intensity) before randomization of each patient.
Patients in the high‐intensity chemotherapy group (n = 75) were treated for one or two cycles, whereas patients in the gilteritinib arm (n = 247) and low‐intensity chemotherapy (n = 49) arm should have continued until a treatment discontinuation criterion was met. The gilteritinib dose may have been initially reduced to 80 mg per day. The gilteritinib dose could have been further reduced to 40 mg per day if the patient had already experienced clinical benefit. Dose reductions should have occurred in a stepwise manner. If the gilteritinib dose was reduced, it was not re‐escalated. Patients on a dose of 120 mg per day who did not achieve a composite complete remission (CR, CRp, or CRi) after cycle 1 may have dose escalated to 200 mg per day. No further dose escalation was allowed.
The selection of the gilteritinib 120 mg once daily dosing regimen in the ADMIRAL study was based on studies 2215‐CL‐0101 and 2215‐CL‐0102 13, 14.
The primary efficacy endpoint for the second interim analysis and the final analysis was OS. OS was defined as the time from the date of randomization until the date of death from any cause. For a patient who was not known to have died by the end of study follow‐up, OS was censored at the date of last contact. OS was statistically significantly different between the two groups, and the median OS was 9.3 months in the gilteritinib arm compared with 5.6 months for the salvage chemotherapy (hazard ratio [HR], 0.637; 95% confidence interval [CI], 0.490–0.830; p = .0004 one‐sided log‐rank test; Fig. 2).
Figure 2.

Kaplan‐Meier plot of overall survival by treatment arm (study 2215‐CL‐0301). Includes all patients who were randomized (intention to treatment set). One‐sided p value is from stratified log‐rank test.Abbreviations: ASP2215, gilteritinib; Chemo, chemotherapy; CI, confidence interval; HR, hazard ratio.
In addition, for the first interim analysis, CR/complete remission with partial hematologic recovery (CRh) rate was a coprimary endpoint. CR/CRh was defined as the number of patients who achieved either CR or CRh at any of the postbaseline visits divided by the number of patients in the analysis population.
The CR/CRh rate was 34.0% (95% CI, 28.1–40.3) in gilteritinib arm versus 15.3% (95% CI, 9.5–22.9) in the salvage chemotherapy arm. The CR rate was 21.1% (95% CI, 16.1–26.7) in the gilteritinib arm versus 10.5% (95% CI, 5.7–17.3) in the salvage chemotherapy arm, with a treatment difference of 10.6% (95% CI, 2.7–18; data not shown).
There was a numerical increase in event‐free survival (EFS) in the gilteritinib arm, compared with the salvage chemotherapy arm (median EFS 2.8 months vs. 0.7 months, respectively), but the EFS endpoint did not meet the prespecified criteria for statistical significance (HR, 0.793; 95% CI, 0.577–1.089; p = .0415 one‐sided log‐rank test).
Transplantation rate was higher in the gilteritinib arm compared with the salvage chemotherapy arm (25.5% vs. 15.3%, unstratified p = .033). Posttransplantation outcomes were generally comparable between treatment arms, with the majority of patients being in remission (65% in the gilteritinib arm vs. 68% in the chemotherapy arm).
A summary of the key favorable effects observed in the ADMIRAL study is displayed in Table 2.
Table 2.
Key favorable and unfavorable effects for gilteritinib monotherapy for adult patients with relapsed or refractory acute myeloid leukemia with FLT3 mutation (study 2215‐CL‐0301, cutoff date: September 17, 2018)
| Effect | Treatment: gilteritinib (n = 247) | Control: chemotherapy (n = 124) | Uncertainties, strength of evidence |
|---|---|---|---|
| Favorable effects | |||
| OS (median time from randomization until death by any cause), months |
9.3 (7.7–10.7) |
5.6 (4.7–7.3) |
HR, 0.637 95% CI, 0.490–0.830 One‐sided p = .0004 |
| Unfavorable effects,a % | |||
| TEAE | |||
| All grades | 83.1 | 65.1 | N/A |
| Grade ≥3 | 60.2 | 52.3 | |
| ALT increased | |||
| All grades | 82.1 | 47.7 | |
| Grade ≥3 | 12.9 | 2.8 | |
| Diarrhea | |||
| All grades | 35.1 | 29.4 | |
| Grade ≥3 | 4.1 | 2.8 | |
| Nausea | |||
| All grades | 29.8 | 33 | |
| Grade ≥3 | 1.9 | 0 | |
| Fatigue | |||
| All grades | 30.4 | 12.8 | |
| Grade ≥3 | 3.1 | 0 | |
| QT prolongation | |||
| All grades | 8.8 | 0 | |
| Grade ≥3 | 2.5 | 0 | |
| Myalgia | |||
| All grades | 12.5 | 0 | |
| Grade ≥3 | 0.3 | 0 |
Integrated 120 mg gilteritinib population, n = 319; and chemotherapy, n = 109.
Abbreviations: ALT, alanine aminotransferase; CI, confidence interval; HR, hazard ratio; N/A, not applicable; OS, overall survival; TEAE, treatment‐emergent adverse event.
Supportive study 2215‐CL‐0101 was a phase I/II open‐label, dose escalation study investigating the safety, tolerability, pharmacokinetics, and pharmacodynamics of gilteritinib in patients with relapsed or refractory AML. In the gilteritinib 120 mg treatment arm of the study (n = 56), patients were treated with either one (n = 14) or more than one (n = 42) prior lines of treatment. In this patient population, median OS in patients with one prior line of therapy was 10.3 months (95% CI, 3.1–17.5) versus 7.2 (95% CI, 4.3–9.4) months in patients with more than one prior line of therapy. The CR/CRh rate in patients with one prior line of therapy was 28.6% versus 21.4% in patients with more than one prior line of therapy.
Clinical Safety
The safety database (integrated R/R AML safety population) included a total of 522 patients who received at least one dose of gilteritinib, composed of 252 patients from study 2215‐CL‐0101, 24 patients from study 2215‐CL‐0102, and 246 patients from study 2215‐CL‐0301. Of these 522 patients, 319 received a starting dose of gilteritinib 120 mg (including all 246 patients from study 2215‐CL‐0301).
In the integrated gilteritinib 120 mg group, the median duration of exposure was 111 days (ranging from 4 to 1,320 days). The median relative dose intensity was 100%. Dose modifications of gilteritinib were frequent; in the integrated gilteritinib 120 mg group, dose increases to 200 mg were experienced by 35.4% (113/319) of patients because of lack of efficacy. Dose decreases were experienced by 25.7% (82/319) of patients. Overall, 47.3% (151/319) of patients experienced at least 1 day of gilteritinib dose interruption. Treatment‐emergent adverse events leading to discontinuation of gilteritinib were experienced by 21.9% (70/319) of patients; such events were considered drug‐related in 10.0% (32/319) of patients.
Treatment‐emergent adverse event (TEAE) was defined as an adverse event observed after starting administration of the study drug (gilteritinib or salvage chemotherapy). In the integrated R/R AML safety population, 99.4% of patients experienced at least one TEAE in the integrated gilteritinib group and 98.2 % in the chemotherapy group, of which 83.1% and 65.1%, respectively, were drug related.
The most frequent TEAEs with gilteritinib were blood creatine phosphokinase increased (53.9%), alanine aminotransferase (ALT) increased (82.1%), aspartate aminotransferase (AST) increased (80.6%), blood alkaline phosphatase increased (68.7%), diarrhea (35.1%), fatigue (30.4%), nausea (29.8%), constipation (28.2%), cough (28.2%), peripheral edema (24.1%), dyspnea (24.1%), dizziness (20.4%), hypotension (17.2%), pain in extremity (14.7%), asthenia (13.8%), arthralgia (12.5%), and myalgia (12.5%).
The incidence of grade 3 or higher drug‐related TEAEs was 60.2% in the integrated gilteritinib group and 52.3% in the chemotherapy group. The most frequently reported grade 3 or higher TEAEs for gilteritinib group included anemia (16.9%), febrile neutropenia (12.2%), thrombocytopenia (11.6%), and platelet count decreased (11.3%). In the integrated gilteritinib 120 mg group, 80.9% (258/319) of patients experienced at least one serious adverse event (SAE), which was drug‐related in 33.9% (108/319) of patients. The most frequently reported SAEs were febrile neutropenia (29.8% [95/319]), AML (13.5% [43/319]), pyrexia (13.2% [42/319]), and pneumonia (12.2% [39/319]). The most frequently reported drug‐related SAEs were febrile neutropenia (7.5% [24/319]), ALT increased (3.4% [11/319]), and AST increased (3.1% [10/319]).
The percentage of patients in study 2215‐CL‐0301 with a TEAE leading to death that was considered drug‐related was similar in both treatment arms (4.1% in the gilteritinib arm compared with 4.6% of patients in the salvage chemotherapy arm).
Similar to other tyrosine kinase inhibitors (TKIs), QT prolongation was observed 7. In the integrated gilteritinib 120 mg group, the majority of patients experienced an increase in QTcF value from baseline; although the mean value of QTc showed little change with use of gilteritinib, more patients had abnormally high values while taking gilteritinib than at baseline. The most frequent TEAEs by preferred term within this category were electrocardiogram QT prolonged (8.8%; drug‐related in 6.3%) and syncope (5.0%; drug‐related in 0.6%). Serious TEAEs in this category were experienced by 17 patients (5.3%) and considered drug‐related SAEs in 6 patients (1.9%). The results of in vitro and in vivo nonclinical studies did, however, not provide a clear indication of a strong potential to prolong QT in humans. Based on the literature reporting, in some cases, 10% to 20% inhibition of the human ether‐a‐go‐go‐related gene (hERG) can be related to QT prolongation in vivo 8, 9. Taken together there may be multiple factors contributing to gilteritinib‐induced QT prolongation seen in patients, such as a weak hERG current suppression and increased CaV1.2 current.
Differentiation syndrome was reported for 3% of patients. Of the 11 patients who experienced differentiation syndrome, 9 recovered after treatment or after dose interruption of gilteritinib. No SAEs were reported, but one patient experienced a drug‐related grade 3 or higher event.
In the integrated gilteritinib 120 mg group, 0.6% of patients experienced the TEAE of posterior reversible encephalopathy syndrome (PRES). These TEAEs were serious and grade ≥ 3; however, none of them resulted in death. The events of PRES resolved after drug discontinuation and routine medical management.
The risk management plan agreed upon at the time of approval includes identified risks, namely, PRES and differentiation syndrome. Potential risks included torsades de pointes (TdP), serious gastrointestinal disorders, eye disorders, pulmonary adverse events, pancreatitis, embryo‐fetal lethality, suppressed fetal growth, and teratogenicity. Important missing information includes safety in patients with renal impairment and long‐term safety. In addition, a health care professional survey study to evaluate awareness and clinical knowledge of health care professionals on differentiation syndrome, PRES, and TdP will be conducted by the applicant company.
Detailed warnings about these risks are available in the product information.
A summary of the key unfavorable effects observed the integrated R/R AML safety population is displayed in Table 2.
Benefit‐Risk Assessment
The CHMP concluded that the ADMIRAL study provided convincing evidence of clinical efficacy of gilteritinib in terms of the primary endpoint OS, compared with salvage chemotherapy in patients with R/R AML with FLT3 mutation (Table 2).
Initially, the benefit‐risk balance was uncertain in patients receiving more than one prior treatment. Although the pivotal study included patients with R/R AML previously treated with only one prior line of therapy, the therapeutic indication applied by the sponsor was broad, and it did not reflect this specific inclusion criterion. Nevertheless, the results from the supportive study 2215‐0101 indicated that responses, including CR and composite complete remission, were achieved also for patients in later treatment lines. Furthermore, the reported median OS generally exceeded that observed with chemotherapy both in the clinical pivotal study and when compared with a published historical data set. In addition, there was no clear indication from the pharmacodynamic data on potential resistance mechanisms to suggest a reduced benefit of gilteritinib versus chemotherapy in later treatment lines. From a safety perspective, when analyzing all doses combined, there were no apparent clinically meaningful differences with regard to prior lines of chemotherapy. Based on the above considerations, the CHMP agreed that the benefit‐risk ratio of gilteritinib was considered positive also in patients with more than one prior treatment.
The transplantation rate was 25.5% (63/247) in the gilteritinib arm and 15.3% (19/124) in the salvage chemotherapy arm. Among the gilteritinib‐treated patients receiving HSCT postrandomization, the majority reinitiated gilteritinib treatment post‐HSCT (40/63, 63%). Because of the lack of rerandomization after HSCT, the additional benefit conferred by post‐HSCT gilteritinib could not be determined. However, the CHMP concluded that because continuation of treatment post‐HSCT constituted the overall treatment strategy for gilteritinib and there were no comparative data to substantiate long‐term benefit in patients not receiving posttransplant treatment, the option to reinitiate gilteritinib in patients after HSCT is included in section 4.2 of the product information.
Regarding the secondary endpoint EFS, the early steep drop in the Kaplan‐Meier plot (Fig. 3) was due to the definition of treatment failures in the analysis (failure to achieve any of the response of CR, CRp, or CRi during the treatment) with the event date assigned to randomization date. Treatment failures assigned to randomization date constituted a high proportion (38%–39%) of the total EFS events, and by month 3 there were only 4 versus 108 patients included in the number at risk for the chemotherapy and gilteritinib arms, respectively. The relapse assessments based on bone marrow evaluations were only conducted for the first 2 months (expected treatment duration) in the high‐intensity chemotherapy group, whereas in the low‐intensity chemotherapy group and the gilteritinib arm, response/relapse assessments were undertaken until disease progression. In conclusion, results from the secondary endpoint EFS was considered of limited value because of methodological flaws. Like the EFS analysis, the results of the CR were of limited value because of lack of long‐term follow‐up of response and relapse status for patients in the high‐intensity chemotherapy group (beyond 2 months) and the large proportion of patients with no evaluable postbaseline response assessments. However, despite the lack of support from the EFS and CR endpoints, the CHMP concluded that the robust and clinically relevant benefit on OS was sufficient to establish the benefit of gilteritinib.
Figure 3.

Kaplan‐Meier plot of event‐free survival by treatment arm (study 2215‐CL‐0301).Abbreviations: ASP2215, gilteritinib; Chemo, chemotherapy; CI, confidence interval; HR, hazard ratio; NE, not evaluable.
Dose escalation was part of the treatment strategy in the pivotal study, and 31% (78/247) of the patients in the gilteritinib arm were dose escalated to the 200 mg dose per day. There is an uncertainty with regard to the benefit of the increase of the gilteritinib dose in patients with lack of response after one treatment cycle, as it cannot be determined whether the observed increased response rates in the pivotal study were due to the increase in gilteritinib dose or the longer treatment duration.
Despite the higher frequencies of grade ≥ 3 TEAEs and SAEs in the dose‐escalated group compared with the non–dose‐escalated group, the frequency rates of dose interruption and treatment discontinuation were similar across the groups. Although a limited number of patients escalated to 200 mg, these results suggest that the dose is tolerable. Based on the above, the scientific review concluded that in the absence of a response after 4 weeks of treatment, the dose can be increased to 200 mg once daily, if tolerated and clinically warranted.
The safety profile of gilteritinib at the proposed therapeutic dose of 120 mg was manageable in the population of patients with R/R AML studied considering the disease, and the most commonly occurring adverse events were generally associated with the known pathophysiology of AML and known toxicity from other TKIs.
Conclusion
Based on the review of data on quality, safety, and efficacy, the EMA CHMP concluded by consensus that the risk‐benefit balance of gilteritinib monotherapy for adult patients with relapsed or refractory acute myeloid leukemia with FLT3 mutation was favorable, and hence recommended the granting of the marketing authorization.
The most current information on this medicinal product is available on the EMA Web site (https://www.ema.europa.eu/en/medicines).
Author Contributions
Data analysis and interpretation: Hilde Røshol, Helga Haugom Olsen, Ida B. Aas, Marianne Løiten Dalhus, Gro Dahlseng Håkonsen, Laila Sortvik Nilssen, Vibeke Lindberg, Mats Økvist, Bjørg Bolstad, Irēna Rogovska, Natalja Karpova
Manuscript writing: Kyriaki Tzogani, Hilde Røshol, Helga Haugom Olsen, Ida B. Aas, Marianne Løiten Dalhus, Gro Dahlseng Håkonsen, Laila Sortvik Nilssen, Vibeke Lindberg, Mats Økvist, Bjørg Bolstad, Irēna Rogovska, Natalja Karpova, Francesco Pignatti
Final approval of manuscript: Kyriaki Tzogani, Hilde Røshol, Helga Haugom Olsen, Ida B. Aas, Marianne Løiten Dalhus, Gro Dahlseng Håkonsen, Laila Sortvik Nilssen, Vibeke Lindberg, Mats Økvist, Bjørg Bolstad, Irēna Rogovska, Natalja Karpova, Harald Enzmann, Christian Gisselbrecht, Francesco Pignatti
Disclosures
The authors indicated no financial relationships.
Acknowledgments
The scientific assessment summarized in this report is based on important contributions from the rapporteur and co‐rapporteur assessment teams, CHMP members, and additional experts after the application for a marketing authorization from the company. This publication is a summary of the European Public Assessment Report, the summary of product characteristics, and other product information as published on the EMA Web site (https://www.ema.europa.eu/en/medicines). For the most current information on this marketing authorization, please refer to the EMA Web site. The authors of this article remain solely responsible for the opinions expressed in this publication.
Disclosures of potential conflicts of interest may be found at the end of this article.
No part of this article may be reproduced, stored, or transmitted in any form or for any means without the prior permission in writing from the copyright holder. For information on purchasing reprints contact Commercialreprints@wiley.com. For permission information contact permissions@wiley.com.
References
- 1. Maksimovic N, Zaric M, Gazibara T et al. Incidence and mortality patterns of acute myeloid leukemia in Belgrade, Serbia (1999‐2013). Medicina (Kaunas) 2018;54:E5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yamamoto JF, Goodman MT. Patterns of leukemia incidence in the United States by subtype and demographic characteristics, 1997–2002. Cancer Causes Control 2008;19:379–390. [DOI] [PubMed] [Google Scholar]
- 3. Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin 2017;67:7–30. [DOI] [PubMed] [Google Scholar]
- 4. Visser O, Trama A, Maynadié M et al.; RARECARE Working Group. Incidence, survival and prevalence of myeloid malignancies in Europe. Eur J Cancer 2012;48:3257–3266. [DOI] [PubMed] [Google Scholar]
- 5. Gale RE, Green C, Allen C et al.; Medical Research Council Adult Leukaemia Working Party. The impact of FLT3 internal tandem duplication mutant level, number, size, and interaction with NPM1 mutations in a large cohort of young adult patients with acute myeloid leukemia. Blood 2008;111:2776–2784. [DOI] [PubMed] [Google Scholar]
- 6. Yanada M, Matsuo K, Suzuki T et al. Prognostic significance of FLT3 internal tandem duplication and tyrosine kinase domain mutations for acute myeloid leukemia: A meta‐analysis. Leukemia 2005;19:1345–1349. [DOI] [PubMed] [Google Scholar]
- 7. Tiesmeier J, Müller‐Tidow C, Westermann A et al. Evolution of FLT3‐ITD and D835 activating point mutations in relapsing acute myeloid leukemia and response to salvage therapy. Leuk Res 2004;28:1069–1074. [DOI] [PubMed] [Google Scholar]
- 8. Patel JP, Gönen M, Figueroa ME et al. Prognostic Relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med 2012;366:1079–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Moreno I, Martin G, Bolufer P et al. Incidence and prognostic value of FLT3 internal tandem duplication and D835 mutations in acute myeloid leukemia. Haematologica 2003;88:19–24. [PubMed] [Google Scholar]
- 10. Perl AE, Martinelli G, Cortes JE et al. Gilteritinib significantly prolongs overall survival in patients with relapsed/refractory (R/R) acute myeloid leukemia (AML): Results from the phase III ADMIRAL trial. Cancer Res 2019;79(suppl 13):CT184a. [Google Scholar]
- 11. Swerdlow SH, Campo E, Harris NL et al. WHO Classification of Tumours of Haematopoietic and Lymphoid Tissues. 4th ed. Lyon, France: International Agency for Research on Cancer, 2008. [Google Scholar]
- 12. Cheson BD, Bennett JM, Kopecky KJ et al. Revised recommendations of the International Working Group for Diagnosis, Standardization of Response Criteria, Treatment Outcomes, and Reporting Standards for Therapeutic Trials in Acute Myeloid Leukemia. J Clin Oncol 2003;21:4642–4649. [DOI] [PubMed] [Google Scholar]
- 13. Perl AE, Altman JK, Cortes J et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: A multicentre, first‐in‐human, open‐label, phase 1‐2 study. Lancet Oncol 2017;18:1061–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Usuki K, Sakura T, Kobayashi Y et al. Clinical profile of gilteritinib in Japanese patients with relapsed/refractory acute myeloid leukemia: An open‐label phase 1 study. Cancer Sci 2018;109:3235–3244. [DOI] [PMC free article] [PubMed] [Google Scholar]