

Abstract
Background
Germline DNA damage repair gene mutations (gDDRm) have been found in approximately 12% of patients with metastatic prostate cancer (mPCa). Previous studies of the clinical impact of gDDRm have mainly been in the setting of metastatic castration‐resistant prostate cancer (mCRPC). This study aimed to determine the prognostic value of gDDRm in de novo metastatic and castration‐sensitive prostate cancer (mCSPC).
Materials and Methods
We retrospectively collected the records of 139 consecutive men with de novo mCSPC who initially received systemic therapies following guidelines. This included 128 patients who underwent genetic testing at our center and 11 patients referred to our center after being identified as gDDRm carriers. Time to mCRPC was collected. Kaplan‐Meier and log‐rank analysis were used to analyze the association between gDDRm and clinical outcomes. Survival outcomes were adjusted using multivariable Cox regression models.
Results
Of the 139 patients with de novo mCSPC, 28 gDDRm carriers were identified. Median time progressing to mCRPC was significantly shorter in patients carrying gDDRm than in those without mutations (8.3 vs 13.2 months; hazard ratio [HR], 2.37; p < .001). Moreover, median progression time was almost halved in BRCA2 carriers (6.3 vs. 13.2 months; HR, 3.73; p < .001). Subgroup analysis revealed that the presence of gDDRm indicated poor therapy response regardless of disease volume and prostate‐specific antigen nadir within the first 7 months. Presence of gDDRm remained independently associated with increased risk of progression to mCRPC in multivariate analysis (adjusted HR, 1.98; p = .006).
Conclusion
Our study suggested that positive gDDRm status predicted rapid progression to castration resistance in patients with de novo mCSPC. We propose identifying gDDRm status at the time of diagnosis for mCSPC patients, considering it is the first step of tailoring individualized treatment. In addition, DNA repair genes were a good therapeutic target for poly (ADP‐ribose) polymerase inhibitors, and our results call for more frontline targeted therapy trials in gDDRm carriers to prolong the progression time.
Implications for Practice
Results of this study suggested that positive germline DNA damage repair gene mutation (gDDRm) status predicted earlier progression to castration resistance in patients with de novo metastatic and castration‐sensitive prostate cancer (mCSPC). These findings indicated the importance of intense therapy for some subgroups of mCSPC, especially for mCSPC harboring gDDRm with low‐volume disease. Moreover, gDDRm was a good therapeutic target for poly (ADP‐ribose) polymerase inhibitors, and these findings call for more molecular marker driven trials moving to the mTNPC setting.
Keywords: De novo metastatic prostate cancer, Germline mutations, DNA repair genes, BRCA2, Prognostic value
Short abstract
There is increasing interest in defining the role of germline DNA damage repair gene mutations (gDDRm) in de novo metastatic castration sensitive prostate cancer (mCSPC) cases to potentially guide therapy choices. This article focuses on the association between gDDRm status and time to castration resistance to determine the prognostic value of gDDRm in mCSPC cases receiving standard androgen deprivation treatment‐based therapies.
Introduction
Germline mutations in several DNA repair genes (DRGs), especially BRCA2 alterations, have been reported to be associated with increased risk of prostate cancer (PCa) 1, 2. Recently, two landmark publications revealed that patients harboring germline DNA damage repair gene mutations (gDDRm) accounted for 8%–12% of men with metastatic prostate cancer (mPCa) 3, 4, which was significantly higher than that in localized PCa (5%) and the general population (3%) 3, 5. Our previous study also confirmed a similar mutation prevalence in Chinese patients with PCa, although there is a large difference in risk of PCa between China and the West 6. Moreover, gDDRm has been identified to be associated with aggressive disease and poor survival 7, 8, indicating that patients with DNA repair deficiency may have an inferior response to standard of care systemic therapies. To elucidate the role of gDDRm in response to systemic therapy, many case series have been reported 9, 10, 11, 12, 13. However, most previous studies on the prognostic value of gDDRm have focused on patients with metastatic and castration‐resistant prostate cancer (mCRPC), with few data reported in patients with metastatic castration‐sensitive prostate cancer (mCSPC). Owing to insufficient data and conflicting results, the consensus on the prognostic value of gDDRm in response to systemic therapy in patients with mCSPC has not yet been reached.
De novo mPCa represents the more aggressive disease compared with recurrent mPCa and is associated with almost 50% of PCa‐related death 14, 15, 16. Most patients with de novo mPCa missed the opportunity to receive surgical treatment and were initially treated with androgen deprivation treatment (ADT), ADT plus abiraterone, or ADT plus docetaxel. Patients with mCSPC will inevitably progress to mCRPC, although the progression time varies. Moreover, few biomarkers estimating time to castration resistance makes it difficult for individual management. Recent studies indicated that the use of poly (ADP‐ribose) polymerase (PARP) inhibitors or platinum‐based chemotherapy might be of benefit for patients with gDDRm 17, 18. Thus, there is an increasing interest in defining the role of gDDRm in de novo mCSPC cases to potentially guide therapy choices.
In this study, we focused on the association between gDDRm status and time to castration resistance to determine the prognostic value of gDDRm in mCSPC cases receiving standard ADT based therapies.
Subjects, Materials, and Methods
Patient Cohort
This study included 139 consecutive patients with de novo mPCa who received treatment at Fudan University Shanghai Cancer Center. All patients had been previously tested for gDDRm between January 2018 and March 2019. This cohort included 128 patients who underwent genetic testing at our center, which has been reported in our previous study 6, and 11 patients who were referred to our center after identified as gDDRm carriers. Importantly, patients were selected regardless of family history, age of diagnosis, or any other known genetic background.
Moreover, patients had to have histologically confirmed prostate adenocarcinoma and received ADT only or combination therapy (ADT plus abiraterone or docetaxel). Patients receiving additional concurrent anticancer therapies were excluded. The clinical characteristics of the study population were retrospectively reviewed via medical records and telephone interview. Baseline clinical information, including age at diagnosis, baseline prostate‐specific antigen (PSA), Gleason score, disease volume, PSA nadir within the first 7 months of initial treatment, and family history of any cancer, was collected. High‐volume disease (defined as the presence of visceral metastasis or four or more bone lesions with at least one outside of the vertebral column and pelvis according to the CHAARTED trial) was annotated 19. For all the 139 patients, 100 (72%) patients were initially treated with ADT, with 18 (13%) patients and 21 (15%) patients receiving ADT plus abiraterone and ADT plus docetaxel, respectively. Finally, 99 (71%) patients have progressed to mCRPC at last follow‐up. Median follow‐up was 9.2 months. Written informed consents were obtained from all participants prior to enrollment.
Sequencing and Detection of gDDRm
For 128 patients who were included in the cohort we previously analyzed, sequencing and bioinformatics methodology have been reported in our previous study 6. Different platforms were used as the sequencing platform evolved during the study. Next‐generation sequencing panel, including 63 PCa‐related genes for 58 patients, 508 genes for 1 patient, 618 genes for 11 patients, and whole‐exome for 58 patients, was used to sequence the germline DNA extracted from patients’ peripheral blood mononuclear cell. Annotations were defined with ANNOVAR (http://annovar.openbioinformatics.org/en/latest). Then, we retrieved information of variants from the Exome Aggregation Consortium ExAC Browser (http://exac.broadinstitute.org/), 1000 Genomes (www.1000genomes.org), the single‐nucleotide polymorphism database of the National Center for Biotechnology Information (dbSNP) version 139 (www.ncbi.nlm.nih.gov/projects/SNP), and ClinVar. For this analysis, we focused on DNA repair genes: ATM, ATR, BRCA1, BRCA2, BRIP1, CHEK2, ERCC3, FAM175A, FANCA, GEN1, MLH1, MRE11, MSH2, MSH6, NBN, PALB2, PMS2, RAD51C, and RAD51D. Based on the American College of Medical Genetics and Genomics criteria, the pathogenic and likely pathogenic mutations are defined as (a) all truncating mutations unless their allele frequency is 1% or higher in population databases or is identified as benign or likely benign in the ClinVar; (b) nonsynonymous mutations if their allele frequency is less than 1% and identified as pathogenic and likely pathogenic mutations in the ClinVar; and (c) in‐frameshift mutations, which affect more than three amino acids.
Outcome Measures and Statistical Analysis
The primary endpoint was progression to mCRPC, which represented the time from initial treatment to castration resistance. Castration resistance was defined according to the European Association of Urology (EAU) guidelines (2019 edition): biochemical progression (three consecutive rises in PSA 1 week apart resulting in two 50% increases over the nadir, and a PSA >2 ng/mL) or radiographic progression (the appearance of new lesions: 1 or ≥2 bone lesions on bone scan, or a soft tissue lesion as defined by the RECIST) and castrate serum testosterone <50 ng/dL or 1.7 nmol/L.
The study population was divided into two groups based on gDDRm status. Baseline characteristics, including Gleason score, PSA nadir within the first 7 months, disease volume, family history, and progression to mCRPC, were compared between two groups using Pearson's chi‐squared test or Fisher's exact test. Continuous variable, such as age at diagnosis and baseline PSA, were compared using Student's t test. Then, we used Kaplan‐Meier method to calculate survival (time to castration resistance) and used univariate and multivariate Cox proportional hazards model to analyze prognostic factors.
Finally, we assessed the prognostic value of germline BRCA2 mutations, using the Kaplan‐Meier method and Cox regression analysis. All p values were two‐sided and p < .05 was defined as statistically significant. Statistical analysis was performed using SPSS 20.0 and R 3.3.0.
Results
Patients’ Characteristics and Germline DNA Repair Gene Mutation Status
There were 139 patients with de novo mPCa enrolled in this study. Deleterious gDDRm were identified in 28 patients. The distribution of pathogenic mutation genes was as follows: BRCA2, 17 patients; ATM, 2 patients; MSH6, 2 patients; CHEK2, 1 patient; ERCC3, 1 patient; FANCA, 1 patient; GEN1, 2 patients; MSH2, 1 patient; PALB2, 2 patients (1 patient had both BRCA2 and MSH6 mutations). The detailed gDDRm types and locations in this cohort were shown in Table 1.
Table 1.
Pathogenic and likely pathogenic germline mutations in our cohort (n = 28)
| ID | Gene | Chr | Start | End | Ref. | Alt. | NC change | AA change | Mutation type |
|---|---|---|---|---|---|---|---|---|---|
| 1 | BRCA2 | 13 | 32944609 |
32944610 |
AAAA | c.8402_8403insAAAA |
p.F2801Lfs* |
Frameshift ins | |
| 2 | BRCA2 | 13 |
32914137 |
32914137 |
C | A | c.5645C>A |
p.S1882* |
nonsense |
| 3 | BRCA2 | 13 |
32971034 |
32971034 |
G | T | c.9502‐1G>T |
p.X3168_splice |
Splice |
| 4 | BRCA2 | 13 |
32914174 |
32914174 |
C | G | c.5682C>G |
p.Y1894* |
nonsense |
| 5 | BRCA2 | 13 |
32900690 |
32900691 |
AT | c.571_572insAT |
p.M192Ifs* |
Frameshift ins | |
| 6 | BRCA2 | 13 |
32914174 |
32914174 |
C | G | c.5682C>G |
p.Y1894* |
nonsense |
| 7 | BRCA2 | 13 |
32944692 |
32944692 |
C | T | c.8485C>T |
p.Q2829* |
nonsense |
| 8 | BRCA2 | 13 | 32912902 |
32912905 |
AAGA | c.4410_4413delAAGA | p. K1472Tfs*6 | Frameshift del | |
| 9 | BRCA2 | 13 | 32914066 | 32914069 | AATT | c.5574_5577delAATT | p. I1859Kfs*3 | Frameshift del | |
| 10 | BRCA2 | 13 | 32911145 | 32911148 | GACA | c.2653_2656delGACA | p. D885Mfs*9 | Frameshift del | |
| 11 | BRCA2 | 13 | 32911337 | 32911337 | T | c.2845delT |
p.Y949Mfs*11 |
Frameshift del | |
| 12 | ATM | 11 | 108236104 | 108236104 | C | T | c.9040C>T | p.Q3014* | nonsense |
| 13 | ATM | 11 | 108121756 | 108121756 | G | T | c.1564G>T | p.E522* | nonsense |
| 7 | MSH6 | 2 | 48033792 | 48033795 | TAAC | c.4001+2delTAAC | p.X1334_splice | Splice | |
| 14 | MSH6 | 2 |
48033792 |
48033795 |
TAAC | c.4001+2delTAAC | p.X1334_splice | Splice | |
| 15 | BRCA2 | 13 | 32930649 | 32930652 | CAGG | c.7520_7523delCAGG | p.G2508Vfs*15 | Frameshift del | |
| 16 | BRCA2 | 13 | 32914356 | 32914356 | C | G | c.C5864G | p.S1955* | nonsense |
| 17 | CHEK2 | 22 | 29130450 | 29130464 |
TCCTCAGG TTCTTGG |
c.246_260delTCCT CAGGTTCTTGG |
p.D82_E86del | Inframeshift del | |
| 18 | ERCC3 | 2 | 128030510 | 128030511 | CT | c.1757_1758delCT | p.Q586Rfs*17 | Frameshift del | |
| 19 | FANCA | 16 | 89874714 | 89874715 |
CACAATGCCT TGCAGGCTAC |
c.583_584insCACAAT GCCTTGCAGGCTAC |
E195Gfs*57 | Frameshift ins | |
| 20 | GEN1 | 2 | 17955667 | 17955667 | C | T | c.C1201T | p.R401* | nonsense |
| 21 | GEN1 | 2 | 17955667 | 17955667 | C | T | c.C1201T | p.R401* | nonsense |
| 22 | MSH2 | 2 | 47702205 | 47702205 | C | T | c.C1801T | p.Q601* | nonsense |
| 23 | BRCA2 | 13 | 32914174 | 32914174 | C | G | c.5682C>G | p.Y1894* | nonsense |
| 24 | BRCA2 | 13 | 32914127 | 32914130 | GAGA | c.5638_5641delGAGA | p.E1879Ifs*29 | Frameshift del | |
| 25 | BRCA2 | 13 | 32968969 | 32968969 | G | c.9401delG | p.G3134Afs*29 | Frameshift del | |
| 26 | BRCA2 | 13 | 32911681 | 32911684 | GTCA | c.3192_3195delGTCA | p.S1064Lfs*12 | Frameshift del | |
| 27 | PALB2 | 16 | 23646807 | 23646807 | A | c.1059delA | p.S354Lfs*2 | Frameshift del | |
| 28 | PALB2 | 16 | 23652432 | 23652432 | T | c.47delT | p.K16Sfs*2 | Frameshift del |
Abbreviations: AA, amino acid; Alt., alternative base(s); Chr, chromosome; ID, identifier; NC, nucleotide; Ref = reference base(s).
Overall, patients with gDDRm had a younger age of onset than patients without mutations (median, 63 vs. 66 years, p = .025). The frequency of patients receiving ADT alone after diagnosis was similar between gDDRm carriers and noncarriers (71% vs. 72%). At the last follow‐up, 71% of patients had progressed to mCRPC in our cohort. Eighty‐six percent of gDDRm carriers progressed to castration resistance whereas 68% of gDDR wild‐type cases progressed. Detailed clinical and pathological characteristics of patients were summarized in Table 2. Groups by gDDRm status appeared comparable with regard to clinical prognostic variables.
Table 2.
Baseline clinical and pathological characteristics, by germline mutation status
| Characteristics | Patients with germline mutations (n = 28) | Patients without germline mutations (n = 111) | p value |
|---|---|---|---|
| Median age at diagnosis (IQR), yr | 63 (58–67) | 66 (60–71) | .025 |
| Median baseline PSA (IQR), ng/mL | 238 (98–407) | 100 (47–252) | .9 |
| Gleason grade group, n (%) | .99 | ||
| 1 | 0 (0) | 0 (0) | |
| 2 | 0 (0) | 3 (2.7) | |
| 3 | 3 (11) | 9 (8.1) | |
| 4 | 8 (29) | 38 (34) | |
| 5 | 17 (60) | 59 (53) | |
| Unknown | 0 (0) | 2 (1.8) | |
| Metastases volume,a n (%) | .11 | ||
| High volume | 23 (82) | 70 (63) | |
| Low volume | 5 (18) | 39 (35) | |
| Unknown | 0 (0) | 2 (1.8) | |
| PSA nadir within the first 7 mo of initial treatment, ng/mL | .7 | ||
| <0.2 | 5 (18) | 28 (25) | |
| 0.2–4 | 13 (46) | 47 (43) | |
| >4 | 10 (36) | 36 (32) | |
| Initial therapy regimen after diagnosis, n (%) | .4 | ||
| ADT only | 20 (71) | 80 (72) | |
| ADT + docetaxel | 6 (21) | 15 (14) | |
| ADT + abiraterone | 2 (7.1) | 16 (14) | |
| Family history of cancers, n (%) | 10 (36) | 31 (28) | .4 |
| Progression to mCRPC at the last follow‐up, n (%) | 24 (86) | 75 (68) | .058 |
High‐volume disease was defined as the presence of visceral metastases or ≥ 4 bone lesions with at least one outside of the vertebral column and pelvis according to the CHAARTED trial.
Abbreviations: ADT, androgen deprivation therapy; IQR, interquartile range; mCRPC, metastatic and castration‐resistant prostate cancer; PSA, prostate‐specific antigen.
Germline DNA Repair Gene Mutations and Time to Castration Resistance
Analysis of progression to mCRPC included all 139 patients (Fig. 1). Patients with gDDRm had a median time to castration resistance of 8.3 versus 13.2 months in patients without gDDRm (hazard ratio [HR], 2.37; 95% confidence interval [CI], 1.48–3.80; p < .001; Fig. 2A). After adjusting treatment type, age of onset, baseline PSA, Gleason score, metastases volume, and PSA nadir within first 7 months, multivariate Cox regression model revealed that gDDRm was independently associated with higher risk of progression to mCRPC (adjusted HR, 1.98; 95% CI, 1.22–3.23; p = .006; Table 3). Moreover, high‐volume disease (adjusted HR, 1.74; 95% CI; 1.07–2.82; p = .024; Table 3) and PSA nadir >4 ng/mL within the first 7 months (adjusted HR, 1.57; 95% CI, 1.02–2.41; p = .042; Table 3) were also significantly associated with higher risk of progression to mCRPC.
Figure 1.
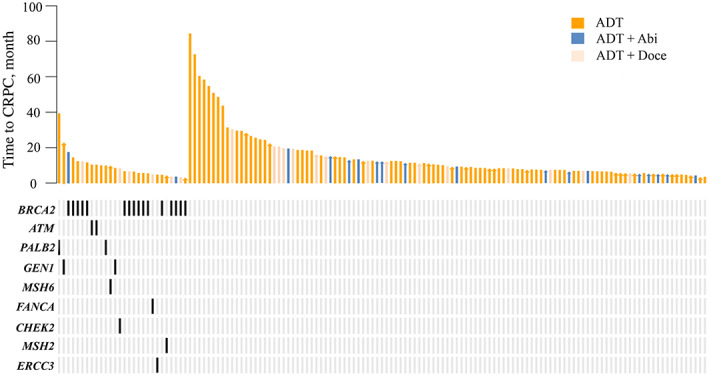
Swimmers plot of time to metastatic CRPC from initial treatment, stratified by germline DNA damage repair gene mutation status.Abbreviations: Abi, abiraterone; ADT, androgen deprivation therapy; CRPC, castration resistance prostate cancer; Doce, docetaxel chemotherapy.
Figure 2.
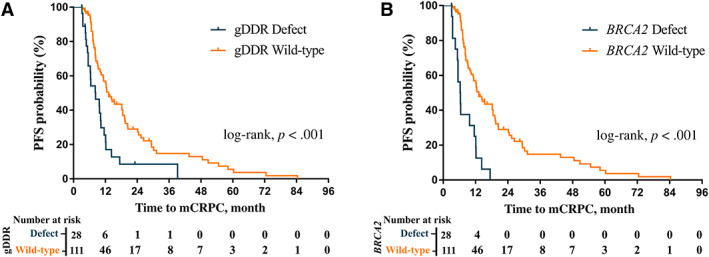
Kaplan‐Meier plots of time to mCRPC from initial treatment in patients. (A): Time to mCRPC from initial treatment in patients by gDDR gene mutation. (B): Time to mCRPC from initial treatment in patients with and without BRCA2 mutation.Abbreviations: gDDR, germline DNA damage repair; mCRPC, metastatic castration resistance prostate cancer
Table 3.
Cox regression analyses of time to mCRPC
| Univariate analysis | Multivariate analysis | |||
|---|---|---|---|---|
| Time to mCRPC | Hazard ratio (95% CI) | p value | Hazard ratio (95% CI) | p value |
| Model 1 | ||||
| Any gDDRm | 2.37 (1.48–3.80) | <.001 | 1.98 (1.22–3.23) | .006 |
| Treatment (combination therapy vs. ADT alone) | 1.00 (0.62–1.63) | .9 | 0.86 (0.51–1.43) | .6 |
| Age of onset, yr | 0.99 (0.96–1.01) | .4 | 0.99 (0.97–1.02) | .5 |
| Baseline PSA (log), ng/mL | 1.38 (0.99–1.91) | .058 | 1.14 (0.79–1.64) | .5 |
| Gleason grade group (4–5 vs. 1–3) | 1.63 (0.84–3.16) | .15 | 1.85 (0.95–3.61) | .070 |
| High‐volume disease (present vs. absent) | 1.86 (1.17–2.94) | .008 | 1.74 (1.07–2.82) | .024 |
| PSA nadir within the first 7 mo (>4 vs. ≤4), ng/mL | 1.36 (0.91–2.04) | .13 | 1.57 (1.02–2.41) | .042 |
| Model 2 | ||||
| mBRCA2 mutation | 3.73 (2.12–6.58) | <.001 | 3.14 (1.76–5.60) | <.001 |
| Treatment (combination therapy vs. ADT alone) | 0.94 (0.57–1.55) | .8 | 0.67 (0.39–1.14) | .14 |
| Age of onset, yr | 0.99 (0.97–1.01) | .4 | 0.99 (0.97–1.02) | .8 |
| Baseline PSA (log), ng/mL | 1.32 (0.94–1.86) | .11 | 1.05 (0.72–1.53) | .8 |
| Gleason grade group (4–5 vs. 1–3) | 1.48 (0.74–2.98) | .3 | 1.25 (0.61–2.55) | .5 |
| High‐volume disease (present vs absent) | 1.93 (1.20–3.11) | .007 | 1.97 (1.21–3.22) | .007 |
| PSA nadir within the first 7 mo (>4 vs ≤4), ng/mL | 1.42 (0.93–2.16) | .10 | 1.55 (0.98–2.44) | .059 |
Model 1 evaluated the effect of any gDDRm versus wild‐type. Model 2 evaluated the effect of BRCA2 mutation versus wild‐type.
High‐volume disease was defined as the presence of visceral metastases or ≥ 4 bone lesions with at least one outside of the vertebral column and pelvis according to the CHAARTED trial.
Abbreviations: ADT, androgen deprivation therapy; CI, confidence interval; gDDRm, germline DNA repair gene mutations; PSA, prostate‐specific antigen.
We stratified patients into two groups according to disease volume and PSA nadir within 7 months for further investigation, respectively. Interestingly, in both high‐ and low‐ volume metastatic disease groups, the shorter median time to mCRPC were observed in patients carrying gDDRm (8.2 vs. 11.3 months; p = .032; Fig. 3A; 10.2 vs. 24.2 months; p = .004; Fig. 3B). Furthermore, in group with PSA nadir ≤4 ng/mL within 7 months, the prognostic value of gDDRm remained significant and the median time to mCRPC was 9.7 versus 18.2 months for patients with and without gDDRm (p < .001; Fig. 3C). Besides, patients with the presence of gDDRm had a numerically shorter progression time than noncarriers for patients with PSA nadir >4 ng/mL, although no statistical difference was observed (6.5 vs. 11.3 months, p = .15; Fig. 3D).
Figure 3.
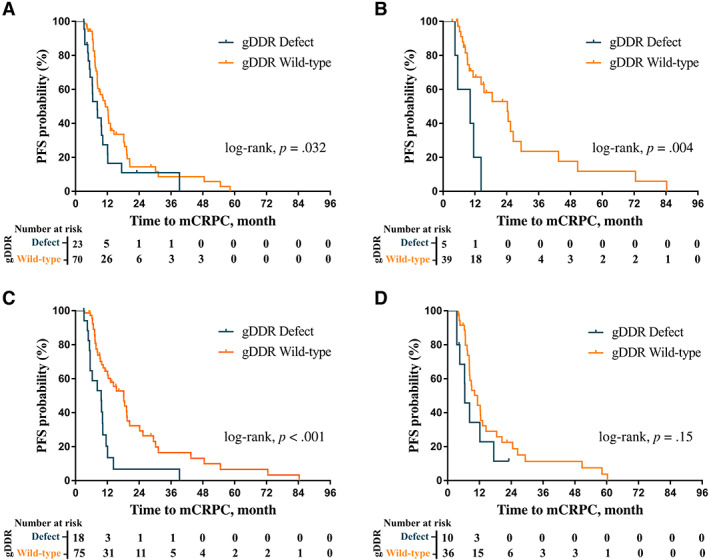
Kaplan‐Meier plots of time to mCRPC in patient subgroups. (A): Kaplan‐Meier plot of time to mCRPC from initial treatment in high‐volume disease patient subgroup by gDDR gene mutation (gDDRm). (B): Kaplan‐Meier plot of time to mCRPC from initial treatment in low‐volume disease patient subgroup by gDDRm. (C): Kaplan‐Meier plot of time to mCRPC from initial treatment in patient subgroup with prostate‐specific antigen nadir ≤4 ng/mL by gDDRm. (D): Kaplan‐Meier plot of time to mCRPC from initial treatment in patient subgroup with PSA nadir >4 ng/mL by gDDRmAbbreviations: gDDR, germline DNA damage repair; mCRPC, metastatic and castration resistance prostate cancer; PFS, progression‐free survival.
BRCA2 Mutations and Time to Castration Resistance
Similar to the role of gDDRm, Kaplan‐Meier analysis showed a shorter progression time in BRCA2 mutation carriers compared with noncarriers (6.3 vs. 13.2 months; HR, 3.73; 95% CI, 2.12–6.58; p < .001; Fig. 2B). This association remained significant after adjusting for other potential prognostic variables in a multivariate Cox regression model (adjusted HR, 3.14; 95% CI, 1.76–5.60; p < .001; Table 3). Additional factor such as high‐volume disease was also independently associated with higher hazard of progression to mCRPC in multivariate analysis (adjusted HR, 1.97; 95% CI, 1.21–3.22; p = .007; Table 3), whereas PSA nadir within first 7 months was nearly significantly associated with higher hazard of progression (adjusted HR, 1.55; 95% CI, 0.98–2.44; p = .059; Table 3).
Discussion
To shed a new light on the prognostic role of gDDRm in mCSPC, we retrospectively analyzed clinical outcome of patients with mCSPC by gDDRm status. Using a large number of patients and well‐adjusted analysis, we confirmed that the presence of gDDRm was an independent prognostic factor for ADT‐based therapies in patients with mCSPC. This finding is of great interest because not only prognostication improvement but also early identification of gDDRm may lead to precise application of targeted therapy such as PARP inhibitors or platinum‐based chemotherapy in the setting of mCSPC 17, 20, 21. Moreover, this strategy is gradually attracting attention because moving effective therapies earlier has produced great success in mCSPC 22, 23.
Our data echoed the biological finding from a recent paper suggesting that patients with CSPC with gDDRm presented elevated genomic instability and a mutational profile closely resembling mCRPC 24. For example, MED12L/MED12, a modulator of WNT/b‐catenin signaling, is preferentially amplified in both BRCA2‐driven primary tumors and sporadic mCRPC, which may help explain risk of early castration resistance for patients with germline BRCA2 mutations 24.
Although biologically reasonable, the evidence reported to data about the role of gDDRm in response to systemic therapies in PCa remained conflicting, and almost all prior studies focused on mCRPC 9, 10, 11, 12, 13. A retrospective data of 319 patients, including 22 gDDRm carriers, reported a worse outcome from abiraterone plus enzalutamide treatment for patients with mCRPC with gDDRm (3.3 vs. 6.2 months) 11. The data from the PROREPAIR‐B trial showed that germline BRCA2 mutations have a deleterious impact on mCRPC outcomes but not for non‐BRCA2 germline mutations 9. Conversely, results from the NCI9012 trial (NCT01576172) of abiraterone and the PARP inhibitor veliparib suggested that patients with PCa with DNA damage repair defects (including somatic and germline mutations) appear to be benefiting from abiraterone treatment 13. Similar results were observed in the data reported by Antonarakis et al., in which nine carriers of BRCA/ATM experienced a more prolonged progression‐free survival (PFS) compared with noncarriers (15.2 vs. 10.8 months) 12. Finally, in a retrospective series with 330 noncarriers and 60 gDDR carriers, there was no association between the presence of gDDRm and the response to abiraterone plus enzalutamide or docetaxel 10. The contradictory conclusions from prior studies is multifactorial. Retrospective design and a relatively limited number of patients with gDDRm would introduce bias in analysis. Also, heterogeneity of disease burden and prior treatments could play a role in disparate results. Moreover, different variants in DNA repair genes, even in the same genes, may lead to different responsiveness to specific therapies, and all prior studies did not control for other potential prognostic factors such as genomic aberrations in AR, TP53, and RB1 25, 26, which could also contribute to explain the conflicting data.
However, in the setting of mCSPC, the data about impact of gDDRm remained sparse. A recent study exploring the genomic alterations in patients who developed mCRPC in a short amount of time (median time to mCRPC, 1.17 years) found that BRCA2 and CDK12 mutations were significantly more common than described in The Cancer Genome Atlas cohort, and patients with germline or somatic BRCA2 alterations had the lower time to ADT progression compared with noncarriers, but the difference was not statistically significant, possibly owing to a limited number of patients carrying mutations and only two patients with germline mutations 27. To overcome these disadvantages, we enrolled 139 patients with mCSPC with full adjustment of well‐known risk factors. In addition to confirming that independent of other clinical prognostic factors, gDDRm indicated earlier progression to castration resistance in mCSPC, our subgroup analysis also revealed that gDDRm was associated with poor therapy response, regardless of disease volume. Currently, with multiple treatment options available for patients with mCSPC 28, 29, 30, treatment selection to optimize patients outcome has become increasingly difficult. Only disease volume has been validated as a reliable prognostic factor to guide treatment decisions 16, which impede the implement of individualized treatment. Our study provided another usable genomic biomarker to help identify patients who may benefit from intensified therapy and highlighted the necessity of intensified treatment in low‐volume mPCa carrying gDDRm. Besides, National Comprehensive Cancer Network Asia Consensus still recommends ADT alone as first‐line therapy for patients with mCSPC 31, and some patients preferred to receive ADT because of economic cost or intolerable adverse effect induced by combination therapy. Thus, patients receiving ADT constituted a considerable proportion of entire group in Asia, which also emphasizes the value of our study.
In addition to pinpointing individuals progressing rapidly to mCRPC, the presence of gDDRm is also a druggable biomarker indicating the benefit of PARP inhibitors or platinum‐based chemotherapy 17, 18. The National Comprehensive Cancer Network Prostate Cancer guideline now recommends germline genetic testing for all patients with mPCa, regardless of family history. Thus, increasingly more gDDRm carriers have been identified in the stage of mCSPC. However, the recommendation of use of PARP inhibitors remained confined to patients with mCRPC who have failed multiple lines of therapy, based on the evidence from PROfound study. Identifying the germline mutation status did not translate into clinical benefit for patients with mCSPC with gDDRm. Results from the SOLO1 trial revealed that median PFS was significantly longer when olaparib was used as a first‐line maintenance therapy in ovarian cancer (∼50 vs. 13.8 months) 32. The outstanding results achieved in ovarian cancer suggested that early engagement of PARP inhibitors for patients with gDDRm could also be considered in mPC, and more prospective trials were warranted to confirm this treatment strategy.
Our study is still relevant in the treatment of recurrent mPCa. In this subgroup, the benefit of intensified therapy is less convincing and even more debatable in the era of new imaging. Thus, ADT alone was the preferred treatment for recurrent mPCa. Our results suggested that patients with gDDRm were prone to resistant to conventional ADT. Thus, there is an unmet need to prospectively test the hypothesis that patients with recurrent mPCa with gDDRm have worse outcome to ADT and therefore might benefit more from intensified therapy.
Considering that BRCA2 was identified as the core functional components in homologous recombination repair pathway and the most commonly mutated DNA repair gene in our cohort 33, we further separately examined the effect of BRCA2 and found median progression time was almost halved in BRCA2‐mutant cases compared with those without mutations (6.3 vs 13.2 months). The rapid progression to castration resistance were further enhanced when considering BRCA2 alone. Our data indicated discerning the different prognostic role in the full spectrum of DRGs is needed.
There are several limitations in our study. Although we achieved standard of care following guidelines in a single center, retrospective design remained a significant limitation. Second, we incorporated clinical prognostic factors such as high‐volume disease and PSA nadir after initial treatment, which were recommended in the EAU guidelines to perform a multivariate analysis. However, emerging evidence showed genomic alterations in TP53, PTEN, RB1, and Wnt‐pathway may also impact response to systemic therapy 25, 34, 35, which were not adjusted in our Cox regression model. Finally, that 72% of patients in our cohort were treated with ADT alone is also a limitation. Therefore, our conclusions were mainly based on the fact that gDDRm was associated with poor response to ADT in mPCa. Further studies were needed to compare the outcome of intensified therapy in patients with mCSPC carrying gDDRm with those without mutations, giving the era of combination therapy is coming.
Conclusion
Our results confirmed that the presence of gDDRm indicated earlier progression to castration resistance in mCSPC. We propose identifying gDDRm status at the diagnosis for patients with mCSPC, considering it is the first step of tailoring individualized treatment. In addition, gDDRm was a good therapeutic target for PARP inhibitors. Our results call for more frontline targeted therapy trials for patients with mCSPC with gDDRm to prolong the progression time.
Author Contributions
Conception/design: Junlong Wu, Dingwei Ye, Yao Zhu
Provision of study material or patients: Yu Wei, Junlong Wu, Hualei Gan
Collection and/or assembly of data: Yu Wei, Junlong Wu, Weijie Gu, Xiaojian Qin
Data analysis and interpretation: Yu Wei, Junlong Wu, Jun Wang, Guowen Lin, Bo Dai, Dingwei Ye
Manuscript writing: Yu Wei, Junlong Wu, Yao Zhu
Final approval of manuscript: Yu Wei, Junlong Wu, Weijie Gu, Jun Wang, Guowen Lin, Xiaojian Qin, Bo Dai, Hualei Gan, Dingwei Ye, Yao Zhu
Disclosures
The authors indicated no financial relationships.
Supporting information
See http://www.TheOncologist.com for supplemental material available online.
Supplemental Appendices
Acknowledgments
We wish to thank all our colleagues in the Department of Urology, Shanghai Cancer Center.
Contributed equally.
Disclosures of potential conflicts of interest may be found at the end of this article.
Contributor Information
Yu Wei, Email: dwyeli@163.com.
Dingwei Ye, Email: dwyeli@163.com.
Yao Zhu, Email: yaozhu09@fudan.edu.cn.
References
- 1. Kote‐Jarai Z, Leongamornlert D, Saunders E et al. BRCA2 is a moderate penetrance gene contributing to young‐onset prostate cancer: Implications for genetic testing in prostate cancer patients. Br J Cancer 2011;105:1230–1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Leongamornlert D, Mahmud N, Tymrakiewicz M et al. Germline BRCA1 mutations increase prostate cancer risk. Br J Cancer 2012;106:1697–1701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Pritchard CC, Mateo J, Walsh MF, et al. Inherited DNA‐repair gene mutations in men with metastatic prostate cancer. N Engl J Med 2016;375:443–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Robinson D, Van Allen Eliezer M, Wu Y‐M, et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015;161:1215–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Abeshouse A, Ahn J, Akbani R, et al. The molecular taxonomy of primary prostate cancer. Cell 2015;163:1011–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wei Y, Wu J, Gu W, et al. Germline DNA repair gene mutation landscape in chinese prostate cancer patients. Eur Urol 2019;76:280–283. [DOI] [PubMed] [Google Scholar]
- 7. Castro E, Goh C, Olmos D, et al. Germline brca mutations are associated with higher risk of nodal involvement, distant metastasis, and poor survival outcomes in prostate cancer. J Clin Oncol 2013;31:1748–1757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Na R, Zheng SL, Han M, et al. Germline mutations in atm and brca1/2 distinguish risk for lethal and indolent prostate cancer and are associated with early age at death. European Urology 2017;71:740–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Castro E, Romero‐Laorden N, Del Pozo A, et al. Prorepair‐b: A prospective cohort study of the impact of germline DNA repair mutations on the outcomes of patients with metastatic castration‐resistant prostate cancer. J Clin Oncol 2019;37:490–503. [DOI] [PubMed] [Google Scholar]
- 10. Mateo J, Cheng HH, Beltran H et al. Clinical outcome of prostate cancer patients with germline DNA repair mutations: Retrospective analysis from an international study. European urology 2018;73:687–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Annala M, Struss WJ, Warner EW et al. Treatment outcomes and tumor loss of heterozygosity in germline DNA repair‐deficient prostate cancer. Eur Urol 2017;72:34–42. [DOI] [PubMed] [Google Scholar]
- 12. Antonarakis ES, Lu C, Luber B et al. Germline DNA‐repair gene mutations and outcomes in men with metastatic castration‐resistant prostate cancer receiving first‐line abiraterone and enzalutamide. Eur Urol 2018;74:218–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Hussain M, Daignault‐Newton S, Twardowski PW et al. Targeting androgen receptor and DNA repair in metastatic castration‐resistant prostate cancer: Results from NCI 9012. J Clin Oncol 2018;36:991–999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Buzzoni C, Auvinen A, Roobol MJ et al. Metastatic prostate cancer incidence and prostate‐specific antigen testing: New insights from the european randomized study of screening for prostate cancer. Eur Urol 2015;68:885–890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Helgstrand JT, Røder MA, Klemann N et al. Trends in incidence and 5‐year mortality in men with newly diagnosed, metastatic prostate cancer‐a population‐based analysis of 2 national cohorts: Cancer 2018;124:2931–2938. [DOI] [PubMed] [Google Scholar]
- 16. Gravis G, Boher JM, Chen YH, et al. Burden of metastatic castrate naive prostate cancer patients, to identify men more likely to benefit from early docetaxel: Further analyses of CHAARTED and GETUG‐AFU15 studies. Eur Urol 2018;73:847–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Mateo J, Carreira S, Sandhu S et al. DNA‐repair defects and olaparib in metastatic prostate cancer. N Engl J Med 2015;373:1697–1708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pomerantz MM, Spisák S, Jia L et al. The association between germline BRCA2 variants and sensitivity to platinum‐based chemotherapy among men with metastatic prostate cancer. Cancer 2017;123:3532–3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Kyriakopoulos CE, Chen YH, Carducci MA et al. Chemohormonal therapy in metastatic hormone‐sensitive prostate cancer: Long‐term survival analysis of the randomized phase III E3805 CHAARTED trial. J Clin Oncol 2018;36:1080–1087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cheng HH, Pritchard CC, Boyd T et al. Biallelic inactivation of BRCA2 in platinum‐sensitive metastatic castration‐resistant prostate cancer. Eur Urol 2016;69:992–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Mateo J, Porta N, McGovern UB, et al. TOPARP‐B: A phase II randomized trial of the poly(ADP)‐ribose polymerase (PARP) inhibitor olaparib for metastatic castration resistant prostate cancers (MCRPC) with DNA damage repair (DDR) alterations. J Clin Oncol 2019;37:5005a. [Google Scholar]
- 22. James ND, de Bono JS, Spears MR et al. Abiraterone for prostate cancer not previously treated with hormone therapy. N Engl J Med 2017;377:338–351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. James ND, Sydes MR, Clarke NW et al. Addition of docetaxel, zoledronic acid, or both to first‐line long‐term hormone therapy in prostate cancer (STAMPEDE): Survival results from an adaptive, multiarm, multistage, platform randomised controlled trial. Lancet 2016;387:1163–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Taylor RA, Fraser M, Livingstone J et al. Germline BRCA2 mutations drive prostate cancers with distinct evolutionary trajectories. Nat Commun 2017;8:13671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hamid AA, Gray KP, Shaw G et al. Compound genomic alterations of TP53, PTEN, and RB1 tumor suppressors in localized and metastatic prostate cancer. Eur Urol 2019;76:89–97. [DOI] [PubMed] [Google Scholar]
- 26. Lord CJ, Ashworth A. BRCAness revisited. Nat Rev Cancer 2016;16:110–120. [DOI] [PubMed] [Google Scholar]
- 27. Mateo J, Seed G, Bertan C et al. Genomics of lethal prostate cancer at diagnosis and castration‐resistance. J Clin Invest 2020. [Epub ahead of print]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Teo MY, Rathkopf DE, Kantoff P. Treatment of advanced prostate cancer. Annu Rev Med 2019;70:479–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fizazi K, Tran N, Fein L et al. Abiraterone plus prednisone in metastatic, castration‐sensitive prostate cancer. N Engl J Med 2017;377:352–360. [DOI] [PubMed] [Google Scholar]
- 30. Sweeney CJ, Chen YH, Carducci M et al. Chemohormonal therapy in metastatic hormone‐sensitive prostate cancer. N Engl J Med 2015;373:737–746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hinotsu S, Namiki M, Ozono S et al. NCCN Asia Consensus Statement prostate cancer. Jpn J Clin Oncol 2018;48:964–965. [DOI] [PubMed] [Google Scholar]
- 32. Moore K, Colombo N, Scambia G et al. Maintenance olaparib in patients with newly diagnosed advanced ovarian cancer. N Engl J Med 2018;379:2495–2505. [DOI] [PubMed] [Google Scholar]
- 33. Wood RD, Mitchell M, Sgouros J et al. Human DNA repair genes. Science 2001;291:1284–1289. [DOI] [PubMed] [Google Scholar]
- 34. Zhang Z, Cheng L, Li J et al. Inhibition of the wnt/β‐catenin pathway overcomes resistance to enzalutamide in castration‐resistant prostate cancer. Cancer Res 2018;78:3147–3162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Isaacsson Velho P, Fu W, Wang H et al. Wnt‐pathway activating mutations are associated with resistance to first‐line abiraterone and enzalutamide in castration‐resistant prostate cancer. Eur Urol 2019;77:14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
See http://www.TheOncologist.com for supplemental material available online.
Supplemental Appendices