

Abstract
A series of Mycobacterium tuberculosis TMPK (MtbTMPK) inhibitors based on a reported compound 3 were synthesized and evaluated for their capacity to inhibit MtbTMPK catalytic activity and the growth of a virulent M. tuberculosis strain (H37Rv). Modifications of the scaffold of 3 failed to afford substantial improvements in MtbTMPK inhibitory activity and antimycobacterial activity. Optimization of the substitution pattern of the D ring of 3 resulted in compound 21j with improved MtbTMPK inhibitory potency (three-fold) and H37Rv growth inhibitory activity (two-fold). Moving the 3-chloro substituent of 21j to the para-position afforded isomer 21h, which, despite a 10-fold increase in IC50-value, displayed promising whole cell activity (minimum inhibitory concentration (MIC) = 12.5 μM).
Keywords: tuberculosis, Mycobacterium tuberculosis, MtbTMPK, 1-(1-arylethylpiperidin-4-yl)thymine
1. Introduction
Tuberculosis (TB) is an airborne infectious disease caused by Mycobacterium tuberculosis (M. tuberculosis). Still belonging to the top ten causes of death worldwide, TB was responsible for claiming 1.5 million lives in 2018, thereby preceding AIDS [1]. The World Health Organization (WHO) launched the “END TB” strategy in 2014, aiming at reducing the incidence of TB by 90% and the number of deaths from TB by 95% by 2035 compared with 2015 levels [2]. Nevertheless, the progress towards this sustainable development goal is disappointing, and the continuing increase in drug-resistant TB cases makes the situation more challenging [1,3].
Patients with drug-sensitive TB are currently treated with a combination regimen, consisting of a two-month treatment with first-line agents rifampicin, isoniazid, pyrazinamide and ethambutol, followed by a four-month treatment with rifampicin and isoniazid. Although this schedule has decreased TB mortality, these gains are being threatened by the advent of coinfection with HIV/AIDS, poor patient adherence and a deficient health care system. Additionally, the emergence of multidrug-resistant TB (MDR-TB) and extensively drug-resistant TB (XDR-TB) further erodes the ambitions of the WHO program [1]. In the case of MDR/XDR TB, a wide palette of second- and third-line anti-TB drugs are used, e.g., fluoroquinolones, ethionamide, thioacetazone, clarithromycin and clofazimine [4,5]. However, this regimen not only requires more toxic and costly medications; it also requires a longer treatment duration (up to 24 months), resulting in a poor outcome. Moreover, soon after their introduction, resistance has already developed to the newly approved agents bedaquiline [6] and delamanid [7], with the use of pretomanid restricted to a limited and specific population of patients [8]. Thus, novel anti-TB agents are needed to effectively shorten the treatment regime and cure MDR-/XDR-TB.
Thymidylate kinase, a key enzyme for the synthesis of the DNA building block thymidine-5′-triphosphate, is indispensable for bacterial survival [9,10,11]. The availability of co-crystal structures of MtbTMPK [12,13,14,15,16] has aided the discovery of MtbTMPK inhibitors, featuring both nucleoside [14,17,18,19,20] and non-nucleoside [14,15,20,21] structures.
A previous work from our laboratory started from compound 1 (Figure 1), originally reported by AstraZeneca as an inhibitor of TMPK’s of Gram-positive bacteria [22]. After we found that it also potently inhibited MtbTMPK, a SAR investigation demonstrated that it could be converted to the achiral inhibitor 2 [14]. Further modifications led to the identification of 1-(1-arylethylpiperidin-4-yl)thymine analog 3, which, compared to 1, displayed a nine-fold lower minimum inhibitory concentration (MIC) value for H37Rv [14].
Figure 1.

Structures of reported MtbTMPK inhibitors 1, 2 and 3 and their TMPK inhibitory potency and antitubercular activity [14,20]. Compound 3 was resynthesized and re-evaluated herein as a reference. MIC: minimum inhibitory concentration.
In this manuscript, we describe our optimization efforts towards finding potent antimycobacterial agents based on 3 by introducing modifications on the A/B/C/D ring (Figure 2) and by altering the linker part. An overview of the synthesized analogs is presented in Figure 2.
Figure 2.

An overview of the synthesized compounds 3, 21a-21q, 26 and 27 in this study.
2. Results and Discussion
2.1. Chemistry
The envisioned analogs were synthesized according to a literature-reported procedure [14]. Briefly, the synthesis encompassed the reductive amination of N3-benzyloxymethyl (BOM)-protected piperidinyl thymine with the appropriate aldehyde, followed by trifluoroacetic acid (TFA)-mediated BOM deprotection to yield the desired analogs [14]. The required aldehyde intermediates (Scheme 1) were obtained through the reduction of commercially available substituted methyl phenylacetate esters with LiAlH4 [23] and subsequent re-oxidation by pyridinium chlorochromate (PCC) [24,25]. When the required methyl esters were not commercial available, substituted phenoxy- or benzyloxyaryl analogs were obtained through either Chan-Lam coupling of hydroxyphenylacetic methyl ester with a boronic acid [26] or alkylation of the hydroxyphenylacetic methyl ester with benzyl bromide under basic conditions [27].
Scheme 1.

Synthesis of crude aldehyde intermediates. Reagents and conditions: (a) LiAlH4 and tetrahydrofuran (THF); (b) pyridinium chlorochromate (PCC) and dichloromethane (DCM); (c) substituted phenylboronic acid, pyridine, 4Å molecular sieves, Cu(OAc)2 and 1,2-dichloroethane; (d) substituted benzyl bromide, K2CO3/Cs2CO3, NaI and dimethylformamide (DMF) and (e) substituted fluorobenzene, K2CO3, DMF and 90 °C.
Due to the low yield of 17a–17c in the Chan-Lam coupling reaction, a different synthetic route was applied for the synthesis of aldehydes 18a–18c, which was based on nucleophilic aromatic substitution of the phenol with the appropriate 4-subsituted fluorobenzene [28]. Then, a PCC-mediated oxidation furnished the desired aldehydes, which were used without further purification in the reductive amination step (Scheme 2).
Scheme 2.

Synthesis of the final compounds. Reagents and conditions: (a) 28, sodium triacetoxyborohydride and 1,2-dichloroethane; (b) trifluoroacetic acid (TFA), Et3SiH and 73 °C; (c) (±)-29, sodium triacetoxyborohydride and 1,2-dichloroethane; (d) (i) aq. NaOH, MeOH and 50 °C; (ii) 1N aq. HCl and H2O; (e) 28, N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC.HCl), 4-dimethylaminopyridine (DMAP) and DCM; (f) Pd/C (10%), H2, HCOOH (0.5%) and isopropanol/H2O (10/1 v/v) and (g) 4-phenylpiperidine, sodium triacetoxyborohydride and 1,2-dichloroethane.
Compounds 21a–21q and 23 were synthesized via reductive amination of N3-BOM-protected piperidinyl thymine with the appropriate aldehyde [14], followed by trifluoroacetic acid (TFA)-mediated BOM deprotection (Scheme 2) [29]. Potential dimerization during the TFA-mediated deprotection [14] was precluded by adding Et3SiH as a cation scavenger [29].
Amide 26 was obtained via N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide (EDC)-mediated coupling of 28 with 2-(3-phenoxyphenyl)acetic acid, which was obtained by hydrolysis of the corresponding methyl ester. The BOM group in the resulting amide intermediate was removed by catalytic hydrogenation with Pd/C [30].
A one-step reductive amination of 4-phenylpiperidine with aldehyde 12b afforded the phenyl analog 27.
2.2. Biological Activity
All synthesized compounds were evaluated for their capacity to inhibit MtbTMPK. Our previously reported analog 3, the starting point, was resynthesized and its reported inhibitory potency confirmed (Table 1, IC50 values of 11 and 17 µM, respectively) [14]. Replacement of the thymine ring by a phenyl (27) led to a complete loss of the inhibitory potency. Additionally, repositioning of the thymine ring from the para to the meta-position of the piperidine ring [20,22] (racemic compound 23) resulted in a substantial (>10-fold) loss in the inhibitory potency. In-line with earlier observations, with a one-carbon linker [14,21], amide analog 26 displayed a weak enzyme inhibition.
Table 1.
Mycobacterium tuberculosis TMPK (MtbTMPK) inhibitory potency of the compounds.
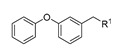 (3, 23, 26, 27) |
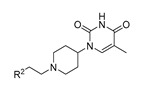 (21a-21c, 21e, 21n) |
||||
|---|---|---|---|---|---|
| Compound | R1 | IC50 (μM) a | Compound | R2 | IC50 (μM) |
| 3 |
|
17 ± 2 | 21a |
|
680 ± 90 |
| 27 |
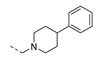
|
NI b | 21b |
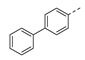
|
NI b |
| (±)-23 |
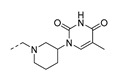
|
192 ± 53 | 21n |
|
95 ± 14 |
| 26 |
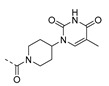
|
230 ± 63 | 21c |
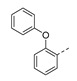
|
100 ± 15 |
| 21e |
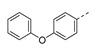
|
160 ± 22 | |||
a Values for which standard deviations are given are the calculated mean values of at least two measurements. b NI: no inhibition at a concentration of 0.2 mM.
Having established that 1-(1-arylethylpiperidin-4-yl)thymine is preferable for inhibitory potency, our efforts were then directed toward the exploration of the biphenyl ether tail. Deletion of the terminal phenoxy group (analog 21a) resulted in a 40-fold decrease in inhibitory potency. Docking studies based on the X-ray co-crystal structure of MtbTMPK with our previously reported 1-(piperidin-4-yl)thymine inhibitor (PDB 5NR7) [14] were performed to rationalize the observed SAR. Docking indicated that the loss of a hydrophobic interaction with Tyr39 accounts for the drop in inhibitory potency (Figure 3A). The addition of a methylene moiety between the terminal phenyl ring and C-phenoxy group (analog 21n) caused a five-fold decrease in the inhibitory potency. The docking pose of compound 21n in MtbTMPK (Figure 3B) showed that the elongated benzyloxy phenyl resulted in a weaker hydrophobic interaction with Tyr39 than the phenoxy moiety of 3. Omitting the oxygen atom between the two phenyl rings led to a complete loss of enzyme inhibitory potency (compound 21b). Moreover, moving the terminal phenoxy ring of 3 to the ortho or para-position of ring C (compounds 21c/21e) had a negative effect on the inhibitory activity.
Figure 3.

(A) Structure overlay of compound 3 (cyan) and 21a (yellow) docked in the MtbTMPK (PDB 5NR7) [14] active site. (B) Structure overlay of compound 3 (cyan) and 21n (green) docked in the MtbTMPK (PDB 5NR7) [14] active site. All residues interacting with the inhibitors, including the hydrophobic contact (gray wire) and hydrogen-bonding interaction (residues in orange wire, hydrogen bonds indicated in magenta), were calculated using LigPlus [31]. Illustration was created using Chimera [32].
Based on these results, further optimization efforts focused on the substitution pattern of the terminal phenyl ring of 3 and 21n. As shown in Table 2, most of the substituted analogs exhibited a small decrease in inhibitory activity compared to 3. Of note, the introduction of sterically demanding electron withdrawing substituents (21k/21l/21m) resulted in a significant drop in MtbTMPK inhibitory potency.
Table 2.
MtbTMPK inhibitory activity of the compounds.
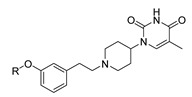
| Compound | R | IC50 (μM) a | Compound | R | IC50 (μM) |
|---|---|---|---|---|---|
| 3 |
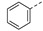
|
17 ± 2 | 21k |
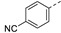
|
126 ± 10 |
| 21f |
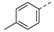
|
49 ± 7 | 21l |
|
91 ± 10 |
| 21g |
|
45 ± 2 | 21m |
|
135 ± 22 |
| 21h |
|
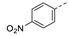
49 ± 8 | 21o |
|
70 ± 9 |
| 21i |
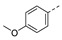
|
34 ± 5 | 21p |
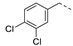
|
ND b |
| 21j |
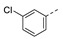
|
5.0 ± 0.7 | 21q |
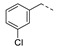
|
44 ± 2 |
a Values for which standard deviations are given are the calculated mean values of at least two measurements. b Solubility issue.
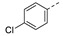
Interestingly, the introduction of a 3-chloro (21j) but not a 4-chloro substituent (21h) afforded a significant improvement in the inhibitory potency due to an edge-to-face π-stacking interaction between the 3-chlorobenzene ring and Tyr39, as found for compound 3 (Figure 4). Introduction of a 3-chloro substituent in the benzyl analog 21n also had a beneficial impact on the inhibitory activity.
Figure 4.

(A) Structure overlay of compound 3 (cyan), 21j (yellow) and 21h (purple) in the MtbTMPK (PDB 5NR7) [14] active site. (B) Detailed representation of the interactions of the D rings of 3, 21h and 21j, with the side chain of Tyr39. All residues interacting with the inhibitors, including the hydrophobic contact (gray wire) and hydrogen-bonding interaction (residues in orange wire, hydrogen bonds indicated in magenta), were calculated using LigPlus [31]. Illustration was created using Chimera [32].
Finally, all compounds were evaluated for their in vitro antimycobacterial activity (Table 3). Consistent with the observations on the enzyme inhibitory activities, modifications of the scaffold of 3 did not yield analogs with superior antimycobacterial activity (21a-21c, 21e, 21n, 23 and 26). Remarkably, analog 27, in which the thymine moiety is replaced by a phenyl ring, showed potent antimycobacterial activity but lacked selectivity, as evidenced by its equipotent cytotoxicity. The introduction of substituents on the distal phenyl ring of 3 afforded several analogs with improved antimycobacterial activity (21f–21j). Substitution of the D-ring of the benzyloxy analog 21n also contributed to the growth inhibitory activity. However, the selectivity vs. MRC-5 fibroblasts was modest.
Table 3.
Antimycobacterial activity against H37Rv and cytotoxicity against MRC-5 fibroblasts of the compounds in this study.
| Compound | MIC a
(H37Rv, μM) |
IC50
(MRC-5, μM) |
SI b | Compound | MIC (H37Rv, μM) |
IC50
(MRC-5, μM) |
SI |
|---|---|---|---|---|---|---|---|
| 3 | 37 | >64 | >1.73 | 21k | ≥50 | >64 | - |
| 21a | >50 | >64 | - | 21l | 37 | >64 | >1.73 |
| 21b | >50 | 22.22 | - | 21m | 37 | >64 | >1.73 |
| 21c | 50 | 26.81 | 0.54 | 21n | 37 | 27.57 | 0.74 |
| 21e | >50 | >64 | - | 21o | 19 | 7.08 | 0.37 |
| 21f | 19 | 8.42 | 0.44 | 21p | 12.5 | 4.22 | 0.34 |
| 21g | 19 | 5.66 | 0.30 | 21q | 25 | 19.84 | 0.79 |
| 21h | 12.5 | 5.81 | 0.46 | 23 | >50 | 21.77 | - |
| 21i | 25 | >64 | >2.56 | 26 | >50 | >64 | - |
| 21j | 19 | 25.24 | 1.33 | 27 | 6.25 | 8.06 | 1.29 |
a Minimum inhibitory concentration (MIC) is the minimum concentration required to inhibit >99% growth of Mycobacterium tuberculosis H37Rv in the liquid culture. b Selectivity index (SI): IC50 (MRC-5)/MIC (H37Rv).
3. Materials and Methods
3.1. Enzymatic Assay
The enzymatic assay was performed as previously reported on the recombinant purified MtbTMPK [14,33]. As described by Blondin et al. [34], compounds were evaluated by a serial dilution method using the spectrophotometric assay at fixed concentrations of adenosine triphosphate (ATP) (0.5 mM) and thymidine monophosphate (dTMP; 0.05 mM). The reaction medium includes 50-mM Tris-HCl; pH 7.4; 50-mM KCl; 2-mM MgCl2; 0.2-mM nicotinamide adenine dinucleotide (NADH); 1-mM phosphoenol pyruvate and 2 units each of coupling enzymes (lactate dehydrogenase, pyruvate kinase and nucleoside diphosphate kinase). From the experimental data, the IC50 value was calculated using KaleidaGraph 4.5.3.
3.2. Computational Studies
For the molecular modeling, X-ray structure of the MtbTMPK (PDB entry 5NR7 [14]) was analyzed using AutoDock vina and AutodockTools-1.5.6. [35]. In ChemDraw 3D 16.0, the PDB files of all ligands were generated after the energy was minimized (minimum RMS gradient: 0.001). The PDBQT file of the ligands and receptors were prepared by AutodockTools-1.5.6, including atom types, atomic partial charges and the information on the ligand torsional degrees. Using a grid spacing of 0.375 and 60 × 60 × 60 numbers of grid points, the prepared PDBQT files of ligands and receptors were docked (centered on the MtbTMPK active site PHE70 CE2, the coordinates x, y and z were −0.997, 26.240 and −4.528, correspondingly) through the Lamarckian 4.2 method. Each ligand was docked in Autodock vina 3 times, with each time generating 20 possible conformations. Chimera in combination with LigPlus were used to analyze the results.
3.3. In Vitro Antituberculosis Assay
The MIC values of all compounds were determined as previously described [36]. In brief, M. tuberculosis H37Rv (ATCC 27294) was grown to optical density at 650 nm wavelength (OD650nm) 0.2 in Middlebrook 7H9 medium supplemented with 0.4% glucose, 0.03% Bacto casitone and 0.05% Tyloxapol (7H9/glucose/casitone/Tyloxapol) prior to further 1000-fold dilution in fresh medium. Drugs were 2-fold serially diluted in duplicate in 7H9/glucose/casitone/Tyloxapol (50 µL/well) in a concentration range spanning 100-0.049 µM in sterile 96-well U-bottom clear polystyrene microtiter plates. Isoniazid and DMSO as positive and negative controls, respectively. An equal volume (50 µL) of diluted cells was added to the plates with the serial drug dilution. Plates were sealed in Ziplock bags and incubated at 37 °C. After 7–14 days, plates were read with an enlarging inverted mirror plate reader. The MIC was recorded as the concentration that fully inhibited all visible growth.
3.4. In Vitro Cytotoxicity Assay
The cytotoxicity of compounds on MRC-5 fibroblasts was performed exactly as previously reported [14].
3.5. Chemistry
All reagents and solvents were purchased from standard commercial sources and were of analytical grade. All synthetic compounds described in this study were checked with analytical TLC (Macherey−Nagel precoated F254 aluminum plates, Düren, Germany), visualized under UV light at 254 nm and purified by column chromatography (CC) on a Reveleris X2 (Grace, BÜCHI, Flawil, Switzerland) automated flash unit. All final compounds and some intermediates were measured with Varian Mercury 300/75 MHz (Palo Alto, CA, USA) or a Bruker AVANCE (Fällanden, Zürich, Switzerland) Neo® 400/100 MHz spectrometer at 298.15 K using tetramethylsilane (TMS) as an internal standard. The analysis and confirmation of the final compounds were conducted with 1H, 13C, HSQC and HMBC NMR spectral data (Supplementary Materials). High-resolution mass spectrometry was performed on a Waters LCT Premier XETM (Waters, Zellik, Belgium) time-of-flight (TOF) mass spectrometer equipped with a standard electrospray ionization (ESI) and modular LockSprayTM interface (Waters, Zellik, Belgium). The purity of the tested compounds was determined by LC-MS analysis using a Waters AutoPurification system equipped with a Waters Cortecs C18 column (2.7 μm, 100 × 4.6 mm), as was a gradient system of formic acid in H2O (0.2%, v/v)/MeCN with a gradient of 95:5 to 0:100 in 6.5 min at a flow rate of 1.44 mL/min.
General procedure A: Synthesis of biphenyl ether aldehyde building blocks. According to a literature report [26], hydroxyphenylacetic ester derivatives (1.0 eq), phenylboronic acid derivatives (3.0 eq.), Cu(OAc)2 (2.0 eq.), 4Å molecular sieves (0.18 g/mmol ester) and pyridine (3.0 eq.) in 1,2-dichloroethane (6.0 mL/mmol ester) afforded the biphenyletheracetic ester intermediates. To a solution of the biphenyletheracetic ester intermediates (1.0 eq.) in dry tetrahydrofuran (THF) (6.0 mL/mmol ester intermediate) was added LiAlH4 (2.0 eq.) at 0 °C under N2 atmosphere, and the resulting mixture was then stirred at room temperature for 1 h [23]. After complete consumption of the starting material, the reaction mixture was quenched with aq. Na+/K+ tartrate solution (5.0 mL/mmol LiAlH4), and the mixture was stirred at room temperature overnight and then filtered. The collected filtrate was dried and concentrated to afford a crude alcohol intermediate, which was oxidized by PCC (2.0 eq.) for 2 h in dichloromethane (DCM) (5.0 mL/mmol PCC) [24,25]. The reaction mixture was filtered through a short silica column. The collected filtrate was concentrated in vacuo and used in the next step without any additional purification.
General procedure B: Synthesis of final compounds. To a solution of aldehyde intermediates (1.0 eq.) and 28 (1.0 eq.) in 1,2-dichloroethane (33 mL/mmol aldehyde) was added sodium triacetoxyborohydride (2.0 eq.). The resulting mixture was stirred at room temperature for overnight to afford the BOM-protected intermediate [14], which was deprotected with TFA (17 mL/mmol aldehyde) in the presence of triethylsilane [29] (17 mL/mmol aldehyde) at 73 °C for 4 h. After cooling down to room temperature, the reaction mixture was evaporated in vacuo to remove TFA, and the residue was adjusted to pH 6 with 1N aq. HCl. Purification of the resulting mixture by column chromatography gave the desired compounds.
2-([1,1’-Biphenyl]-4-yl)acetaldehyde (6). To a solution of methyl 2-([1,1’-biphenyl]-4-yl)acetate (0.3 g, 1.3 mmol) in dry THF (7.8 mL) was added LiAlH4 (0.10 g, 2.7 mmol) to give alcohol intermediate, which was oxidized with PCC (0.56 g, 2.6 mmol) in DCM (13.0 mL) to yield aldehyde 6 (C14H12O, 0.22 g, 1.1 mmol).
2-(2-Phenoxyphenyl)acetaldehyde (12a). Following the general procedure A, methyl 2-(2-hydroxyphenyl)acetate (1.1 g, 8.1 mmol), phenylboronic acid (2.9 g, 24 mmol), Cu(OAc)2 (2.9 g, 16 mmol), 4Å molecular sieves (1.5 g) and pyridine (1.9 mL, 24 mmol) in 1,2-dichloroethane (49 mL) afforded the ester intermediate methyl 2-(2-phenoxyphenyl)acetate 10a (eluent system: 10% ethylacetate in petroleum ether, C15H14O3, 0.40 g, 1.6 mmol, 21% yield). 1H NMR (300 MHz, CDCl3) δ ppm 3.63 (s, 3 H, OCH3), 3.72 (s, 2 H, CH2), 6.90 (dd, J = 8.1, 1.0 Hz, 1 H, Ph), 6.95–7.01 (m, 2 H, Ph), 7.06–7.15 (m, 2 H, Ph), 7.21–7.37 (m, 4 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 35.6 (1 C, CH2), 51.8 (1 C, OCH3), 118.3 (2 C, Ph), 118.8 (1 C, Ph), 123.0 (1 C, Ph), 123.6 (1 C, Ph), 125.8 (1 C, Ph), 128.6 (1 C, Ph), 129.6 (2 C, Ph), 131.4 (1 C, Ph), 155.0 (1 C, Ph), 157.2 (1 C, Ph), 171.7 (1 C, CO). Then, 10a (0.20 g, 0.83 mmol) was treated with LiAlH4 (63 mg, 1.7 mmol) in dry THF (5.0 mL) to give alcohol intermediate, which was oxidized with PCC (0.34 g, 1.6 mmol) in DCM (8.0 mL) to yield aldehyde 12a (C14H12O2, 0.15 g, 0.70 mmol).
2-(3-Phenoxyphenyl)acetaldehyde (12b). Following the general procedure A, methyl 2-(3-hydroxyphenyl)acetate (1.1 g, 8.1 mmol), phenylboronic acid (2.9 g, 24 mmol), Cu(OAc)2 (2.9 g, 16 mmol), 4Å molecular sieves (1.5 g) and pyridine (1.9 mL, 24 mmol) in 1,2-dichloroethane (49 mL) afforded the ester intermediate methyl 2-(3-phenoxyphenyl)acetate 10b (eluent system: 10% ethylacetate in petroleum ether, C15H14O3, 0.80 g, 3.3 mmol, 41% yield). 1H NMR (300 MHz, CDCl3) δ ppm 3.62 (s, 2 H, CH2), 3.71 (s, 3 H, OCH3), 6.90 - 6.95 (m, 1 H, Ph), 6.98 (t, J = 2.1 Hz, 1 H, Ph), 7.01–7.07 (m, 3 H, Ph), 7.13 (tt, J = 7.4, 1.1 Hz, 1 H, Ph), 7.26–7.40 (m, 3 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 40.9 (1 C, CH2), 52.0 (1 C, OCH3), 117.3 (1 C, Ph), 119.0 (2 C, Ph), 119.7 (1 C, Ph), 123.3 (1 C, Ph), 124.0 (1 C, Ph), 129.7 (3 C, Ph), 135.7 (1 C, Ph), 156.9 (1 C, Ph), 157.4 (1 C, Ph), 171.6 (1 C, CO). Then, 10b (0.20 g, 0.83 mmol) was treated with LiAlH4 (63 mg, 1.7 mmol) in dry THF (5.0 mL) to give alcohol intermediate, which was oxidized with PCC (0.35 g, 1.6 mmol) in DCM (8.0 mL) to yield aldehyde 12b (C14H12O2, 0.15 g, 0.71 mmol).
2-(4-Phenoxyphenyl)acetaldehyde (12c). Following the general procedure A, methyl 2-(4-hydroxyphenyl)acetate (0.60 g, 4.4 mmol), phenylboronic acid (1.6 g, 13 mmol), Cu(OAc)2 (1.6 g, 8.8 mmol), 4Å molecular sieves (0.70 g) and pyridine (1.1 mL, 13 mmol) in 1,2-dichloroethane (26 mL) afforded the ester intermediate methyl 2-(4-phenoxyphenyl)acetate 10c (eluent system: 10% ethylacetate in petroleum ether, C15H14O3, 0.66 g, 2.7 mmol, 62% yield). 1H NMR (300 MHz, CDCl3) δ ppm 3.63 (s, 2 H, CH2), 3.72 (s, 3 H, OCH3), 6.95–7.06 (m, 4 H, Ph), 7.08–7.15 (m, 1 H, Ph), 7.23–7.29 (m, 2 H, Ph), 7.31–7.39 (m, 2 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 40.3 (1 C, CH2), 52.0 (1 C, OCH3), 118.8 (4 C, Ph), 123.2 (1 C, Ph), 128.7 (1 C, Ph), 129.6 (2 C, Ph), 130.5 (2 C, Ph), 156.3 (1 C, Ph), 157.0 (1 C, Ph), 172.0 (1 C, CO). Then, 10c (0.20 g, 0.83 mmol) was treated with LiAlH4 (63 mg, 1.6 mmol) in dry THF (5.0 mL) to give alcohol intermediate, which was oxidized with PCC (0.3 g, 1.4 mmol) in DCM (7.0 mL) to yield aldehyde 12c (C14H12O2, 0.13 g, 0.62 mmol).
2-(3-(p-Tolyloxy)phenyl)acetaldehyde (12d). Following the general procedure A, methyl 2-(3-hydroxyphenyl)acetate (0.60g, 4.4 mmol), p-tolylboronic acid (1.8 g, 13 mmol), Cu(OAc)2 (1.6 g, 8.8 mmol), 4Å molecular sieves (0.79 g) and pyridine (1.1 mL, 13 mmol) in 1,2-dichloroethane (26 mL) afforded the ester intermediate methyl 2-(3-(p-tolyloxy)phenyl)acetate 10d (eluent system: 10% ethylacetate in petroleum ether, C16H16O3, 0.34 g, 1.3 mmol, 30% yield). 1H NMR (300 MHz, CDCl3) δ ppm 2.37 (s, 3 H, CH3), 3.62 (s, 2 H, CH2), 3.72 (s, 3 H, OCH3), 6.89–6.99 (m, 4 H, Ph), 7.00–7.05 (m, 1 H, Ph), 7.14–7.21 (m, 2 H, Ph), 7.25–7.32 (m, 1 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 20.6 (1 C, CH3), 40.9 (1 C, CH2), 51.9 (1 C, OCH3), 116.7 (1 C, Ph), 119.1 (3 C, Ph), 123.5 (1 C, Ph), 129.6 (1 C, Ph), 130.1 (2 C, Ph), 132.9 (1 C, Ph), 135.6 (1 C, Ph), 154.4 (1 C, Ph), 157.9 (1 C, Ph), 171.6 (1 C, CO). Then, 10d (0.20 g, 0.78 mmol) was treated with LiAlH4 (59 mg, 1.6 mmol) in dry THF (4.7 mL) to give alcohol intermediate, which was oxidized with PCC (0.28 g, 1.3 mmol) in DCM (6.5 mL) to yield aldehyde 12d (C15H14O2, 0.13 g, 0.58 mmol).
2-(3-(3,4-Dichlorophenoxy)phenyl)acetaldehyde (12e). Following the general procedure A, methyl 2-(3-hydroxyphenyl)acetate (0.60 g, 4.4 mmol), (3,4-dichlorophenyl)boronic acid (2.5 g, 13 mmol), Cu(OAc)2 (1.6 g, 8.8 mmol), 4Å molecular sieves (0.79 g) and pyridine (1.1 mL, 13 mmol) in 1,2-dichloroethane (26 mL) afforded the ester intermediate methyl 2-(3-(3,4-dichlorophenoxy)phenyl)acetate 10e (eluent system: 10% ethylacetate in petroleum ether, C15H12Cl2O3, 0.73 g, 2.3 mmol, 53% yield). 1H NMR (300 MHz, CDCl3) δ ppm 3.63 (s, 2 H, CH2), 3.71 (s, 3 H, OCH3), 6.86 (dd, J = 8.9, 2.8 Hz, 1 H, Ph), 6.90–6.99 (m, 2 H, Ph), 7.06–7.12 (m, 2 H, Ph), 7.31 (d, J = 7.9 Hz, 1 H, Ph), 7.37 (d, J = 8.8 Hz, 1 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 40.8 (1 C, CH2), 52.1 (1 C, OCH3), 117.8 (1 C, Ph), 118.0 (1 C, Ph), 120.2 (1 C, Ph), 120.3 (1 C, Ph), 125.1 (2 C, Ph), 130.0 (1 C, Ph), 130.9 (1 C, Ph), 133.1 (1 C, Ph), 136.1 (1 C, Ph), 156.1 (1 C, Ph), 156.3 (1 C, Ph), 171.5 (1 C, CO). Then, 10e (0.20 g, 0.64 mmol) was treated with LiAlH4 (49 mg, 1.3 mmol) in dry THF (3.8 mL) to give alcohol intermediate, which was oxidized with PCC (0.23 g, 1.1 mmol) in DCM (5.5 mL) to yield aldehyde 12e (C14H10Cl2O2, 0.10 g, 0.36 mmol).
2-(3-(4-Chlorophenoxy)phenyl)acetaldehyde (12f). Following the general procedure A, methyl 2-(3-hydroxyphenyl)acetate (0.60 g, 4.4 mmol), (4-chlorophenyl)boronic acid (2.1 g, 13 mmol), Cu(OAc)2 (1.6 g, 8.8 mmol), 4Å molecular sieves (0.79 g) and pyridine (1.1 mL, 13 mmol) in 1,2-dichloroethane (26 mL) afforded the ester intermediate methyl 2-(3-(4-chlorophenoxy)phenyl)acetate 10f (eluent system: 10% ethylacetate in petroleum ether, C15H13ClO3, 0.40 g, 1.4 mmol, 33% yield). 1H NMR (300 MHz, CDCl3) δ ppm 3.62 (s, 2 H, CH2), 3.71 (s, 3 H, OCH3), 6.87–6.99 (m, 4 H, Ph), 7.05 (d, J = 7.3 Hz, 1 H, Ph), 7.25–7.34 (m, 3 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 40.9 (1 C, CH2), 52.1 (1 C, OCH3), 117.4 (1 C, Ph), 119.8 (1 C, Ph), 120.2 (2 C, Ph), 124.5 (1 C, Ph), 128.4 (1 C, Ph), 129.8 (2 C, Ph), 129.9 (1 C, Ph), 136.0 (1 C, Ph), 155.7 (1 C, Ph), 157.1 (1 C, Ph), 171.6 (1 C, CO). Then, 10f (0.20 g, 0.72 mmol) was treated with LiAlH4 (55 mg, 1.5 mmol) in dry THF (4.3 mL) to give alcohol intermediate, which was oxidized with PCC (0.31 g, 1.5 mmol) in DCM (7.5 mL) to yield aldehyde 12f (C14H11ClO2, 0.13 g, 0.53 mmol).
2-(3-(4-Methoxyphenoxy)phenyl)acetaldehyde (12g). Following the general procedure A, methyl 2-(3-hydroxyphenyl)acetate (0.60 g, 4.4 mmol), (4-methoxyphenyl)boronic acid (2.0 g, 13 mmol), Cu(OAc)2 (1.6 g, 8.8 mmol), 4Å molecular sieves (0.79 g) and pyridine (1.1 mL, 13 mmol) in 1,2-dichloroethane (26 mL) afforded the ester intermediate methyl 2-(3-(4-methoxyphenoxy)phenyl)acetate 10g (eluent system: 10% ethylacetate in petroleum ether, C16H16O4, 0.33 g, 1.2 mmol, 28% yield). 1H NMR (300 MHz, CDCl3) δ ppm 3.60 (s, 2 H, CH2), 3.70 (s, 3 H, OCH3), 3.82 (s, 3 H, (Ph)OCH3), 6.82–6.87 (m, 1 H, Ph), 6.88–6.94 (m, 3 H, Ph), 6.95–7.03 (m, 3 H, Ph), 7.22–7.29 (m, 1 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 41.0 (1 C, CH2), 52.0 (1 C, OCH3), 55.6 (1 C, (Ph)OCH3), 114.8 (2 C, Ph), 116.0 (1 C, Ph), 118.4 (1 C, Ph), 120.9 (2 C, Ph), 123.2 (1 C, Ph), 129.6 (1 C, Ph), 135.6 (1 C, Ph), 149.8 (1 C, Ph), 155.9 (1 C, Ph), 158.6 (1 C, Ph), 171.7 (1 C, CO). Then, 10g (0.20 g, 0.74 mmol) was treated with LiAlH4 (56 mg, 1.5 mmol) in dry THF (4.4 mL) to give alcohol intermediate, which was oxidized with PCC (0.32 g, 1.5 mmol) in DCM (7.5 mL) to yield aldehyde 12g (C15H14O3, 0.14 g, 0.58 mmol).
2-(3-(3-Chlorophenoxy)phenyl)acetaldehyde (12h). Following the general procedure A, methyl 2-(3-hydroxyphenyl)acetate (0.40 g, 2.9 mmol), 3-chlorophenylboronic acid (1.4 g, 8.8 mmol), Cu(OAc)2 (1.1 g, 5.9 mmol), 4Å molecular sieves (0.50 g) and pyridine (0.71 mL, 8.8 mmol) in 1,2-dichloroethane (17 mL) afforded the ester intermediate methyl 2-(3-(3-chlorophenoxy)phenyl)acetate 10h (eluent system: 10% ethylacetate in petroleum ether, C15H13ClO3, 0.28 g, 1.0 mmol, 34% yield). 1H NMR (300 MHz, CDCl3) δ ppm 3.63 (s, 2 H, CH2), 3.71 (s, 3 H, OCH3), 6.88–6.96 (m, 2 H, Ph), 6.98 (t, J = 1.9 Hz, 1 H, Ph), 7.00 (t, J = 2.2 Hz, 1 H, Ph), 7.04–7.14 (m, 2 H, Ph), 7.17–7.36 (m, 2 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 40.9 (1 C, CH2), 52.1 (1 C, OCH3), 116.8 (1 C, Ph), 117.9 (1 C, Ph), 118.9 (1 C, Ph), 120.2 (1 C, Ph), 123.3 (1 C, Ph), 124.8 (1 C, Ph), 130.0 (1 C, Ph), 130.5 (1 C, Ph), 135.0 (1 C, Ph), 136.0 (1 C, Ph), 156.5 (1 C, Ph), 158.1 (1 C, Ph), 171.5 (1 C, CO). Then, 10h (0.20 g, 0.72 mmol) was treated with LiAlH4 (55 mg, 1.4 mmol) in dry THF (4.3 mL) to give alcohol intermediate, which was oxidized with PCC (0.19 g, 0.87 mmol) in DCM (4.3 mL) to yield aldehyde 12h (C14H11ClO2, 0.10 g, 0.41 mmol).
2-(3-(Benzyloxy)phenyl)acetaldehyde (15a). According to a literature procedure [27] with minor changes, methyl 2-(3-hydroxyphenyl)acetate (0.30 g, 2.2 mmol), benzyl bromide (0.26 mL, 2.2 mmol), K2CO3 (0.61 g, 4.4 mmol), sodium iodide (33 mg, 0.22 mmol) in dimethylformamide (DMF) (10 mL) at room temperature for overnight afforded the ester intermediate methyl 2-(3-(benzyloxy)phenyl)acetate 13a (eluent system: 10% ethylacetate in petroleum ether, C16H16O3, 0.33 g, 1.3 mmol, 58% yield). 1H NMR (300 MHz, CDCl3) δ ppm 3.65 (s, 2 H, CH2), 3.73 (s, 3 H, OCH3), 5.10 (s, 2 H, (Ph)CH2O), 6.83–7.05 (m, 3 H, Ph), 7.21–7.52 (m, 6 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 41.1 (1 C, CH2), 52.0 (1 C, OCH3), 69.8 (1 C, (Ph)CH2O), 113.4 (1 C, Ph), 115.8 (1 C, Ph), 121.8 (1 C, Ph), 127.5 (2 C, Ph), 127.9 (1 C, Ph), 128.5 (2 C, Ph), 129.5 (1 C, Ph), 135.4 (1 C, Ph), 136.9 (1 C, Ph), 158.9 (1 C, Ph), 171.8 (1 C, CO). Then, 13a (0.20 g, 0.78 mmol) was treated with LiAlH4 (59 mg, 1.6 mmol) in dry THF (4.7 mL) to give alcohol intermediate, which was oxidized with PCC (0.34 g, 1.6 mmol) in DCM (8.0 mL) to yield aldehyde 15a (C15H14O2, 0.14 g, 0.62 mmol).
2-(3-((2-Chlorobenzyl)oxy)phenyl)acetaldehyde (15b). Following the procedure as described for 15a, methyl 2-(3-hydroxyphenyl)acetate (0.40 g, 2.9 mmol), 1-(bromomethyl)-2-chlorobenzene (0.60 g, 2.9 mmol), K2CO3 (0.81 g, 5.9 mmol), sodium iodide (44 mg, 0.29 mmol) in DMF (13 mL) afforded the ester intermediate methyl 2-(3-((2-chlorobenzyl)oxy)phenyl)acetate 13b (eluent system: 10% ethylacetate in petroleum ether, C16H15ClO3, 0.26 g, 0.89 mmol, 30% yield). 1H NMR (300 MHz, CDCl3) δ ppm 3.63 (s, 2 H, CH2), 3.71 (s, 3 H, OCH3), 5.17 (s, 2 H, (Ph)CH2O), 6.86–7.01 (m, 3 H, Ph), 7.23–7.36 (m, 3 H, Ph), 7.41 (dd, J = 7.0, 2.1 Hz, 1 H, Ph), 7.53–7.63 (m, 1 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 41.2 (1 C, CH2), 52.1 (1 C, OCH3), 67.1 (1 C, (Ph)CH2O), 113.4 (1 C, Ph), 116.0 (1 C, Ph), 122.1 (1 C, Ph), 126.9 (1 C, Ph), 128.8 (1 C, Ph), 129.0 (1 C, Ph), 129.3 (1 C, Ph), 129.6 (1 C, Ph), 132.6 (1 C, Ph), 134.7 (1 C, Ph), 135.5 (1 C, Ph), 158.7 (1 C, Ph), 171.8 (1 C, CO). Then, 13b (0.20 g, 0.69 mmol) was treated with LiAlH4 (52 mg, 1.4 mmol) in dry THF (4.1 mL) to give alcohol intermediate, which was oxidized with PCC (0.25 g, 1.2 mmol) in DCM (6.0 mL) to yield aldehyde 15b (C15H13ClO2, 0.14 g, 0.53 mmol).
2-(3-((3,4-Dichlorobenzyl)oxy)phenyl)acetaldehyde (15c). Following the procedure as described for 15a, methyl 2-(3-hydroxyphenyl)acetate (0.40 g, 2.9 mmol), 3,4-dichlorobenzyl bromide (0.70 g, 2.9 mmol), K2CO3 (0.81 g, 5.9 mmol), sodium iodide (44 mg, 0.29 mmol) in DMF (13 mL) afforded the ester intermediate methyl 2-(3-((3,4-dichlorobenzyl)oxy)phenyl)acetate 13c (eluent system: 10% ethylacetate in petroleum ether, C16H14Cl2O3, 0.75 g, 2.3 mmol, 84% yield). 1H NMR (300 MHz, CDCl3) δ ppm 3.62 (s, 2 H, CH2), 3.70 (s, 3 H, OCH3), 5.01 (s, 2 H, (Ph)CH2O), 6.87 (d, J = 8.2 Hz, 1 H, Ph), 6.93 (br. s., 2 H, Ph), 7.21–7.30 (m, 2 H, Ph), 7.41–7.48 (m, 1 H, Ph), 7.54 (s, 1 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 41.1 (1 C, CH2), 52.1 (1 C, OCH3), 68.4 (1 C, (Ph)CH2O), 113.4 (1 C, Ph), 115.8 (1 C, Ph), 122.3 (1 C, Ph), 126.5 (1 C, Ph), 129.2 (1 C, Ph), 129.7 (1 C, Ph), 130.5 (1 C, Ph), 131.8 (1 C, Ph), 132.6 (1 C, Ph), 135.6 (1 C, Ph), 137.3 (1 C, Ph), 158.4 (1 C, Ph), 171.7 (1 C, CO). Then, 13c (0.20 g, 0.62 mmol) was treated with LiAlH4 (47 mg, 1.2 mmol) in dry THF (3.7 mL) to give alcohol intermediate, which was oxidized with PCC (0.26 g, 1.2 mmol) in DCM (6.0 mL) to yield aldehyde 15c (C15H12Cl2O2, 0.18 g, 0.60 mmol).
2-(3-((3-Chlorobenzyl)oxy)phenyl)acetaldehyde (15d). Following the procedure as described for 15a, methyl 2-(3-hydroxyphenyl)acetate (0.40 g, 2.9 mmol), m-chlorobenzyl bromide (0.60 g, 2.9 mmol), K2CO3 (0.81 g, 5.9 mmol), sodium iodide (44 mg, 0.29 mmol) in DMF (13 mL) afforded the ester intermediate methyl 2-(3-((3-chlorobenzyl)oxy)phenyl)acetate 13d (eluent system: 10% ethylacetate in petroleum ether, C16H15ClO3, 0.62 g, 2.1 mmol, 73% yield). 1H NMR (400 MHz, CDCl3) δ ppm 3.61 (s, 2 H, CH2), 3.70 (s, 3 H, OCH3), 5.03 (s, 2 H, (Ph)CH2O), 6.84–6.94 (m, 3 H, Ph), 7.23–7.28 (m, 1 H, Ph), 7.29–7.32 (m, 3 H, Ph), 7.35–7.48 (m, 1 H, Ph). 13C NMR (101 MHz, CDCl3) δ ppm 41.2 (1 C, CH2), 52.1 (1 C, OCH3), 69.1 (1 C, (Ph)CH2O), 113.4 (1 C, Ph), 115.8 (1 C, Ph), 122.1 (1 C, Ph), 125.3 (1 C, Ph), 127.4 (1 C, Ph), 128.0 (1 C, Ph), 129.6 (1 C, Ph), 129.8 (1 C, Ph), 134.5 (1 C, Ph), 135.5 (1 C, Ph), 139.0 (1 C, Ph), 158.6 (1 C, Ph), 171.9 (1 C, CO). Then, 13d (0.20 g, 0.69 mmol) was treated with LiAlH4 (52 mg, 1.4 mmol) in dry THF (4.1 mL) to give alcohol intermediate, which was oxidized with PCC (0.26 g, 1.2 mmol) in DCM (6.0 mL) to yield aldehyde 15d (C15H13ClO2, 0.14 g, 0.52 mmol).
2-(3-(4-Cyanophenoxy)phenyl)acetaldehyde (18a). To a solution of methyl 2-(3-hydroxyphenyl)acetate (0.35 g, 2.1 mmol) in THF (13 mL) was added LiAlH4 (0.16 g, 4.2 mmol) afforded 3-(2-hydroxyethyl)phenol (0.24 g, 1.4 mmol), which was reacted with 4-fluorobenzonitrile (0.21 g, 1.7 mmol), K2CO3 (0.40 g, 2.9 mmol) in DMF (10 mL) at 90 °C gave 4-(3-(2-hydroxyethyl)phenoxy)benzonitrile [28] 17a (eluent system: 25% ethylacetate in petroleum ether, C15H13NO2, 0.14 g, 0.59 mmol, 40% yield). 1H NMR (300 MHz, CDCl3) δ ppm 2.89 (t, J = 6.6 Hz, 2 H, CH2), 3.89 (t, J = 6.4 Hz, 2 H, CH2(OH)), 6.89–7.06 (m, 4 H, Ph), 7.11 (dd, J = 7.6, 0.59 Hz, 1 H, Ph), 7.36 (t, J = 7.4 Hz, 1 H, Ph), 7.49–7.69 (m, 2 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 38.9 (1 C, CH2), 63.3 (1 C, CH2(OH)), 105.8 (1 C, Ph), 110.0 (1 C, Ph), 118.0 (2 C, Ph), 118.3 (1 C, CN), 120.9 (1 C, Ph), 125.7 (1 C, Ph), 130.2 (1 C, Ph), 134.1 (2 C, Ph), 141.4 (1 C, Ph), 154.9 (1 C, Ph), 161.5 (1 C, Ph). Then, 17a was oxidized by PCC (0.25 g, 1.2 mmol) in DCM (6.0 mL) to give aldehyde 18a (C15H11NO2, 0.11 g, 0.44 mmol).
2-(3-(4-Nitrophenoxy)phenyl)acetaldehyde (18b). Following the procedure described for 18a, 3-(2-hydroxyethyl)phenol (0.30 g, 2.2 mmol), 1-fluoro-4-nitrobenzene (0.37 g, 2.6 mmol) and K2CO3 (0.60 g, 4.3 mmol) in DMF (16 mL) afforded 2-(3-(4-nitrophenoxy)phenyl)ethan-1-ol 17b (eluent system: 25% ethylacetate in petroleum ether, C14H13NO4, 0.52 g, 2.0 mmol, 92% yield). 1H NMR (300 MHz, CDCl3) δ ppm 2.90 (t, J = 6.4 Hz, 2 H, CH2), 3.89 (t, J = 6.4 Hz, 2 H, CH2(OH)), 6.93–7.07 (m, 4 H, Ph), 7.13 (d, J = 7.6 Hz, 1 H, Ph), 7.37 (t, J = 7.7 Hz, 1 H, Ph), 8.13–8.24 (m, 2 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 38.8 (1 C, CH2), 63.2 (1 C, CH2(OH)), 117.1 (2 C, Ph), 118.4 (1 C, Ph), 121.0 (1 C, Ph), 125.9 (2 C, Ph), 126.0 (1 C, Ph), 130.3 (1 C, Ph), 136.5 (1 C, Ph), 141.5 (1 C, Ph), 154.9 (1 C, Ph), 163.2 (1 C, Ph). Then, 17b was oxidized by PCC (0.86 g, 4.0 mmol) in DCM (20 mL) to give aldehyde 18b (C14H11NO4, 0.36 g, 1.4 mmol).
2-(3-(4-(Trifluoromethyl)phenoxy)phenyl)acetaldehyde (18c). Following the procedure described for 18a, 3-(2-hydroxyethyl)phenol (0.30 g, 2.2 mmol), 1-fluoro-4-(trifluoromethyl)benzene (0.43 g, 2.6 mmol) and CsCO3 (0.85 g, 2.6 mmol) in DMF (16 mL) at 90 °C gave 2-(3-(4-(trifluoromethyl)phenoxy)phenyl)ethan-1-ol 17c (eluent system: 25% ethylacetate in petroleum ether, C15H13F3O2, 0.13 g, 0.46 mmol, 21% yield). 1H NMR (300 MHz, CDCl3) δ ppm 2.87 (t, J = 6.6 Hz, 2 H, CH2), 3.86 (t, J = 6.6 Hz, 2 H, CH2(OH)), 6.89–6.98 (m, 2 H, Ph), 7.01–7.12 (m, 3 H, Ph), 7.33 (t, J = 6.6 Hz, 1 H, Ph), 7.58 (d, J = 8.8 Hz, 2 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 38.9 (1 C, CH2), 63.3 (1 C, CH2(OH)), 117.7 (1 C, Ph), 117.8 (2 C, Ph), 120.4 (2 C, Ph), 124.2 (q, J = 271.5 Hz, 1 C, CF3), 125.1 (1 C, Ph), 127.0 (2 C, Ph), 130.0 (1 C, Ph), 141.1 (1 C, Ph), 155.9 (1 C, Ph), 160.3 (1 C, Ph). Then, 17c was oxidized by PCC (0.20 g, 0.92 mmol) in DCM (4.6 mL) to give aldehyde 18c (C15H11F3O2, 0.12 g, 0.43 mmol).
5-Methyl-1-(1-phenethylpiperidin-4-yl)pyrimidine-2,4(1H,3H)-dione (21a). Following the general procedure B, phenylacetaldehyde (36 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21a (eluent system: 5% MeOH in DCM, 53 mg, 0.17 mmol, 56% yield). 1H NMR (300 MHz, DMSO-d6) δ ppm 1.53–1.93 (m, 7 H, 5-CH3, piperdyl-3-yl, piperidyl-5-yl), 2.00–2.16 (m, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 2.57 (d, J = 8.2 Hz, 2 H, CH2N), 2.66–2.82 (m, 2 H, PhCH2), 3.06 (d, J = 10.8 Hz, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.16–4.36 (m, 1 H, piperidyl-4-yl), 7.10–7.37 (m, 5 H, Ph), 7.63 (s, 1 H, H-6), 11.19 (s, 1 H, NH). 13C NMR (75 MHz, DMSO-d6) δ ppm 12.0 (1 C, 5-CH3), 29.9 (2 C, piperdyl-3-yl, piperidyl-5-yl), 33.0 (1 C, PhCH2), 52.4 (2 C, piperidyl-2-yl, piperidyl-6-yl), 52.6 (1 C, piperidyl-4-yl), 59.3 (1 C, CH2N), 108.9 (1 C, C-5), 125.8 (1 C, Ph), 128.2 (2 C, Ph), 128.6 (2 C, Ph), 137.7 (1 C, C-6), 140.4 (1 C, Ph), 150.8 (1 C, C-2), 163.7 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C18H23N3O2 + H]+ 314.1863, found 314.1855.
1-(1-(2-([1,1’-Biphenyl]-4-yl)ethyl)piperidin-4-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (21b). Following the general procedure B, 6 (60 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21b (eluent system: 5% MeOH in DCM, 39 mg, 0.10 mmol, 33% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.72–1.93 (m, 5 H, piperdyl-3a-yl, piperidyl-5a-yl, 5-CH3), 1.98–2.20 (m, 2 H, piperdyl-3b-yl, piperidyl-5b-yl), 2.89–3.04 (m, 2 H, PhCH2), 3.26–3.61 (m, 4 H, piperidyl-2-yl, piperidyl-6-yl), 4.39–4.53 (m, 1 H, piperidyl-4-yl), 7.33–7.40 (m, 3 H, Ph), 7.46 (t, J = 7.6 Hz, 2 H, Ph), 7.59–7.71 (m, 5 H, H-6, Ph), 11.30 (s, 1 H, NH), 2 H (CH2N) could not be observed. 13C NMR (101 MHz, DMSO-d6) δ ppm 12.1 (1 C, 5-CH3), 27.7 (2 C, piperdyl-3-yl, piperidyl-5-yl), 29.9 (1 C, PhCH2), 51.4 (3 C, piperidyl-2-yl, piperidyl-6-yl, piperidyl-4-yl), 109.1 (1 C, C-5), 126.6 (2 C, Ph), 126.8 (2 C, Ph), 127.4 (1 C, Ph), 128.9 (2 C, Ph), 129.3 (2 C, Ph), 137.5 (3 C, C-6, Ph), 139.9 (1 C, Ph), 150.8 (1 C, C-2), 163.7 (1 C, C-4), C (CH2N) could not be observed. HRMS (ESI): m/z [M + H]+ Calcd. for [C24H27N3O2 + H]+ 390.2176, found 390.2186.
5-Methyl-1-(1-(2-phenoxyphenethyl)piperidin-4-yl)pyrimidine-2,4(1H,3H)-dione (21c). Following the general procedure B, 12a (64 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21c (eluent system: 5% MeOH in DCM, 40 mg, 0.098 mmol, 32% yield). 1H NMR (300 MHz, DMSO-d6) δ ppm 1.52–1.62 (m, 2 H, piperdyl-3a-yl, piperidyl-5a-yl), 1.68–1.84 (m, 5 H, 5-CH3, piperdyl-3b-yl, piperidyl-5b-yl), 1.92–2.04 (m, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 2.50–2.55 (m, 2 H, CH2N), 2.64–2.75 (m, 2 H, PhCH2), 2.92 (d, J = 11.1 Hz, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.12–4.26 (m, 1 H, piperidyl-4-yl), 6.85–6.92 (m, 3 H, Ph), 7.02–7.15 (m, 2 H, Ph), 7.17–7.26 (m, 1 H, Ph), 7.29–7.38 (m, 3 H, Ph), 7.57 (d, J = 1.2 Hz, 1 H, H-6), 11.16 (s, 1 H, NH). 13C NMR (75 MHz, DMSO-d6) δ ppm 12.0 (1 C, 5-CH3), 27.3 (1 C, PhCH2), 29.9 (2 C, piperdyl-3-yl, piperidyl-5-yl), 52.3 (2 C, piperidyl-2-yl, piperidyl-6-yl), 52.3 (1 C, piperidyl-4-yl), 57.9 (1 C, CH2N), 108.9 (1 C, C-5), 117.1 (2 C, Ph), 119.8 (1 C, Ph), 122.6 (1 C, Ph), 124.2 (1 C, Ph), 127.7 (1 C, Ph), 129.9 (2 C, Ph), 131.1 (1 C, Ph), 131.8 (1 C, Ph), 137.6 (1 C, C-6), 150.8 (1 C, C-2), 153.8 (1 C, Ph), 157.6 (1 C, Ph), 163.6 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C24H27N3O3 + H]+ 406.2125, found 406.2133.
5-Methyl-1-(1-(4-phenoxyphenethyl)piperidin-4-yl)pyrimidine-2,4(1H,3H)-dione (21e). Following the general procedure B, 12c (64 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21e (eluent system: 5% MeOH in DCM, 65 mg, 0.16 mmol, 53% yield). 1H NMR (300 MHz, DMSO-d6) δ ppm 1.57–1.93 (m, 7 H, 5-CH3, piperdyl-3-yl, piperidyl-5-yl), 1.97–2.16 (m, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 2.54 (d, J = 7.3 Hz, 2 H, CH2N), 2.64–2.77 (m, 2 H, PhCH2), 3.04 (d, J = 11.1 Hz, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.19–4.29 (m, 1 H, piperidyl-4-yl), 6.87–6.98 (m, 4 H, Ph), 7.04–7.13 (m, 1 H, Ph), 7.18–7.26 (m, 2 H, Ph), 7.29–7.41 (m, 2 H, Ph), 7.60 (d, J = 0.9 Hz, 1 H, H-6), 11.17 (s, 1 H, NH). 13C NMR (75 MHz, DMSO-d6) δ ppm 12.0 (1 C, 5-CH3), 29.9 (2 C, piperdyl-3-yl, piperidyl-5-yl), 32.2 (1 C, PhCH2), 52.4 (2 C, piperidyl-2-yl, piperidyl-6-yl), 52.6 (1 C, piperidyl-4-yl), 59.3 (1 C, CH2N), 108.9 (1 C, C-5), 118.2 (2 C, Ph), 118.7 (2 C, Ph), 123.1 (1 C, Ph), 130.0 (2 C, Ph), 130.1 (2 C, Ph), 135.6 (1 C, Ph), 137.7 (1 C, C-6), 150.8 (1 C, C-2), 154.6 (1 C, Ph), 157.0 (1 C, Ph), 163.7 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C24H27N3O3 + H]+ 406.2125, found 406.2124.
5-Methyl-1-(1-(3-(p-tolyloxy)phenethyl)piperidin-4-yl)pyrimidine-2,4(1H,3H)-dione (21f). Following the general procedure B, 12d (69 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21f (eluent system: 5% MeOH in DCM, 40 mg, 0.095 mmol, 31% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.64 (d, J = 10.6 Hz, 2 H, piperdyl-3a-yl, piperidyl-5a-yl), 1.73 - 1.87 (m, 5 H, 5-CH3, piperdyl-3b-yl, piperidyl-5b-yl), 2.05 (t, J = 11.1 Hz, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 2.28 (s, 3 H, PhCH3), 2.54 (d, J = 8.4 Hz, 2 H, CH2N), 2.72 (t, J = 7.5 Hz, 2 H, PhCH2), 3.02 (d, J = 11.5 Hz, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.19–4.29 (m, 1 H, piperidyl-4-yl), 6.77 (d, J = 8.1 Hz, 1 H, Ph), 6.87 (s, 1 H, Ph), 6.90 (d, J = 8.3 Hz, 2 H, Ph), 6.98 (d, J = 7.6 Hz, 1 H, Ph), 7.19 (d, J = 8.4 Hz, 2 H, Ph), 7.26 (t, J = 7.8 Hz, 1 H, Ph), 7.61 (s, 1 H, H-6), 11.20 (s, 1 H, NH). 13C NMR (101 MHz, DMSO-d6) δ ppm 12.0 (1 C, 5-CH3), 20.2 (1 C, PhCH3), 30.0 (2 C, piperdyl-3-yl, piperidyl-5-yl), 32.8 (1 C, PhCH2), 52.4 (2 C, piperidyl-2-yl, piperidyl-6-yl), 52.5 (1 C, piperidyl-4-yl), 59.0 (1 C, CH2N), 108.9 (1 C, C-5), 115.5 (1 C, Ph), 118.4 (1 C, Ph), 118.8 (2 C, Ph), 123.4 (1 C, Ph), 129.7 (1 C, Ph), 130.3 (2 C, Ph), 132.5 (1 C, Ph), 137.7 (1 C, C-6), 142.7 (1 C, Ph), 150.8 (1 C, C-2), 154.2 (1 C, Ph), 157.1 (1 C, Ph), 163.7 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C25H29N3O3 + H]+ 420.2282, found 420.2286.
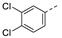
1-(1-(3-(3,4-Dichlorophenoxy)phenethyl)piperidin-4-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (21g). Following the general procedure B, 12e (85 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21g (eluent system: 5% MeOH in DCM, 56 mg, 0.12 mmol, 39% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.79 (s, 3 H, 5-CH3), 1.91–2.06 (m, 2 H, piperdyl-3a-yl, piperidyl-5a-yl), 2.10–2.19 (m, 2 H, piperdyl-3b-yl, piperidyl-5b-yl), 2.96–3.08 (m, 2 H, PhCH2), 3.09–3.22 (m, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 3.25–3.37 (m, 2 H, CH2N), 3.56–3.73 (m, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.46–4.59 (m, 1 H, piperidyl-4-yl), 7.02 (dd, J = 8.9, 2.9 Hz, 2 H, Ph), 7.06 (br. s., 1 H, Ph), 7.15 (d, J = 7.3 Hz, 1 H, Ph), 7.30 (d, J = 2.8 Hz, 1 H, Ph), 7.34–7.46 (m, 2 H, H-6, Ph), 7.65 (d, J = 8.9 Hz, 1 H, Ph), 11.34 (s, 1 H, NH). 13C NMR (101 MHz, DMSO-d6) δ ppm 12.2 (1 C, 5-CH3), 27.1 (2 C, piperdyl-3-yl, piperidyl-5-yl), 29.4 (1 C, PhCH2), 50.5 (1 C, piperidyl-4-yl), 51.2 (2 C, piperidyl-2-yl, piperidyl-6-yl), 56.3 (1 C, CH2N), 109.3 (1 C, C-5), 117.8 (1 C, Ph), 118.7 (1 C, Ph), 119.5 (1 C, Ph), 120.2 (1 C, Ph), 125.16 (d, J = 43.5 Hz, 1 C, Ph), 130.6 (1 C, Ph), 131.6 (2 C, Ph), 132.0 (1 C, Ph), 137.4 (1 C, C-6), 139.6 (1 C, Ph), 150.7 (1 C, C-2), 155.8 (1 C, Ph), 156.4 (1 C, Ph), 163.7 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C24H25Cl2N3O3 + H]+ 474.1346, found 474.1333.
1-(1-(3-(4-Chlorophenoxy)phenethyl)piperidin-4-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (21h). Following the general procedure B, 12f (75 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21h (eluent system: 5% MeOH in DCM, 40 mg, 0.091 mmol, 30% yield). 1H NMR (300 MHz, DMSO-d6) δ ppm 1.57–1.67 (m, 2 H, piperdyl-3a-yl, piperidyl-5a-yl), 1.68–1.87 (m, 5 H, 5-CH3, piperdyl-3b-yl, piperidyl-5b-yl), 2.03 (t, J = 12.3 Hz, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 2.54 (d, J = 8.5 Hz, 2 H, CH2N), 2.72 (t, J = 7.4 Hz, 2 H, PhCH2), 3.00 (d, J = 12.0 Hz, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.22 (tt, J = 12.1, 4.5 Hz, 1 H, piperidyl-4-yl), 6.83 (dd, J = 7.8, 1.9 Hz, 1 H, Ph), 6.92 (s, 1 H, Ph), 6.96–7.06 (m, 3 H, Ph), 7.24–7.33 (m, 1 H, Ph), 7.38–7.43 (m, 2 H, Ph), 7.59 (s, 1 H, H-6), 11.17 (s, 1 H, NH). 13C NMR (75 MHz, DMSO-d6) δ ppm 12.0 (1 C, 5-CH3), 30.0 (2 C, piperdyl-3-yl, piperidyl-5-yl), 32.7 (1 C, PhCH2), 52.4 (2 C, piperidyl-2-yl, piperidyl-6-yl), 52.5 (1 C, piperidyl-4-yl), 59.0 (1 C, CH2N), 108.9 (1 C, C-5), 116.3 (1 C, Ph), 119.2 (1 C, Ph), 120.0 (2 C, Ph), 124.3 (1 C, Ph), 126.9 (1 C, Ph), 129.8 (2 C, Ph), 129.9 (1 C, Ph), 137.6 (1 C, C-6), 143.0 (1 C, Ph), 150.8 (1 C, C-2), 155.8 (1 C, Ph), 156.1 (1 C, Ph), 163.7 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C24H26ClN3O3 + H]+ 440.1736, found 440.1750.
1-(1-(3-(4-Methoxyphenoxy)phenethyl)piperidin-4-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (21i). Following the general procedure B, 12g (74 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21i (eluent system: 5% MeOH in DCM, 40 mg, 0.092 mmol, 30% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.65 (d, J = 10.6 Hz, 2 H, piperdyl-3a-yl, piperidyl-5a-yl), 1.74–1.87 (m, 5 H, piperdyl-3b-yl, piperidyl-5b-yl, 5-CH3), 2.05 (t, J = 11.3 Hz, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 2.52–2.56 (m, 2 H, CH2N), 2.70 (t, J = 8.3 Hz, 2 H, PhCH2), 3.02 (d, J = 11.4 Hz, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 3.74 (s, 3 H, OCH3), 4.19–4.29 (m, 1 H, piperidyl-4-yl), 6.72 (d, J = 8.1 Hz, 1 H, Ph), 6.83 (s, 1 H, Ph), 6.92–7.01 (m, 5 H, Ph), 7.24 (t, J = 7.9 Hz, 1 H, Ph), 7.61 (s, 1 H, H-6), 11.20 (s, 1 H, NH). 13C NMR (101 MHz, DMSO-d6) δ ppm 12.0 (1 C, 5-CH3), 30.0 (2 C, piperdyl-3-yl, piperidyl-5-yl), 32.8 (1 C, PhCH2), 52.4 (2 C, piperidyl-2-yl, piperidyl-6-yl), 52.5 (1 C, piperidyl-4-yl), 55.4 (1 C, OCH3), 59.0 (1 C, CH2N), 108.9 (1 C, C-5), 114.7 (1 C, Ph), 115.0 (2 C, Ph), 117.6 (1 C, Ph), 120.6 (2 C, Ph), 123.0 (1 C, Ph), 129.6 (1 C, Ph), 137.7 (1 C, C-6), 142.6 (1 C, Ph), 149.4 (1 C, Ph), 150.8 (1 C, C-2), 155.5 (1 C, Ph), 157.9 (1 C, Ph), 163.7 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C25H29N3O4 + H]+ 436.2231, found 436.2234.
1-(1-(3-(3-Chlorophenoxy)phenethyl)piperidin-4-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (21j). Following the general procedure B, 12h (75 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21j (eluent system: 5% MeOH in DCM, 63 mg, 0.14 mmol, 47% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.65 (d, J = 9.8 Hz, 2 H, piperdyl-3a-yl, piperidyl-5a-yl), 1.71–1.89 (m, 5 H, piperdyl-3b-yl, piperidyl-5b-yl, 5-CH3), 2.06 (t, J = 11.0 Hz, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 2.53–2.62 (m, 2 H, CH2N), 2.69–2.79 (m, 2 H, PhCH2), 3.03 (d, J = 11.6 Hz, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.17–4.31 (m, 1 H, piperidyl-4-yl), 6.88 (dd, J = 8.1, 1.8 Hz, 1 H, Ph), 6.92–7.00 (m, 2 H, Ph), 7.02 (t, J = 2.2 Hz, 1 H, Ph), 7.08 (d, J = 7.8 Hz, 1 H, Ph), 7.15–7.21 (m, 1 H, Ph), 7.33 (t, J = 7.8 Hz, 1 H, Ph), 7.37–7.43 (m, 1 H, Ph), 7.60 (d, J = 0.9 Hz, 1 H, H-6), 11.20 (s, 1 H, NH). 13C NMR (101 MHz, DMSO-d6) δ ppm 12.0 (1 C, 5-CH3), 30.0 (2 C, piperdyl-3-yl, piperidyl-5-yl), 32.7 (1 C, PhCH2), 52.4 (3 C, piperidyl-2-yl, piperidyl-6-yl, piperidyl-4-yl), 58.9 (1 C, CH2N), 108.9 (1 C, C-5), 116.7 (1 C, Ph), 116.8 (1 C, Ph), 118.0 (1 C, Ph), 119.6 (1 C, Ph), 123.0 (1 C, Ph), 124.7 (1 C, Ph), 130.0 (1 C, Ph), 131.4 (1 C, Ph), 133.9 (1 C, Ph), 137.6 (1 C, C-6), 143.1 (1 C, Ph), 150.8 (1 C, C-2), 155.5 (1 C, Ph), 158.1 (1 C, Ph), 163.7 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C24H26ClN3O3 + H]+ 440.1736, found 440.1740.
4-(3-(2-(4-(5-Methyl-2,4-dioxo-3,4-dihydropyrimidin-1(2H)-yl)piperidin-1- yl)ethyl)phenoxy)benzonitrile (21k). Following the general procedure B, 18a (72 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21k (eluent system: 5% MeOH in DCM, 50 mg, 0.12 mmol, 43% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.79 (s, 3 H, 5-CH3), 2.00 (d, J = 10.8 Hz, 2 H, piperdyl-3a-yl, piperidyl-5a-yl), 2.21 (d, J = 11.6 Hz, 2 H, piperdyl-3b-yl, piperidyl-5b-yl), 3.05 (br. s., 2 H, PhCH2), 3.15 (br. s., 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 3.32 (s, 2 H, CH2N), 3.66 (br. s., 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.44–4.63 (m, 1 H, piperidyl-4-yl), 7.03–7.16 (m, 4 H, Ph), 7.21 (d, J = 6.6 Hz, 1 H, Ph), 7.35–7.53 (m, 2 H, Ph, H-6), 7.86 (d, J = 8.4 Hz, 2 H, Ph), 11.32 (br. s., 1 H, NH). 13C NMR (101 MHz, DMSO-d6) δ ppm 12.2 (1 C, 5-CH3), 27.1 (2 C, piperdyl-3-yl, piperidyl-5-yl), 29.3 (1 C, PhCH2), 50.3 (1 C, piperidyl-4-yl), 51.2 (2 C, piperidyl-2-yl, piperidyl-6-yl), 56.2 (1 C, CH2N), 105.2 (1 C, Ph), 109.3 (1 C, C-5), 118.1 (2 C, Ph), 118.7 (2 C, Ph, CN), 120.5 (1 C, Ph), 125.6 (1 C, Ph), 130.7 (1 C, Ph), 134.7 (2 C, Ph), 137.3 (1 C, C-6), 139.7 (1 C, Ph), 150.7 (1 C, C-2), 154.7 (1 C, Ph), 161.0 (1 C, Ph), 163.6 (1 C. C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C25H26N4O3 + H]+ 431.2078, found 431.2085.
5-Methyl-1-(1-(3-(4-nitrophenoxy)phenethyl)piperidin-4-yl)pyrimidine-2,4(1H,3H)-dione (21l). Following the general procedure B, 18b (78 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21l (eluent system: 5% MeOH in DCM, 67 mg, 0.15 mmol, 40% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.79 (s, 3 H, 5-CH3), 1.93–2.06 (m, 2 H, piperdyl-3a-yl, piperidyl-5a-yl), 2.20 (d, J = 11.6 Hz, 2 H, piperdyl-3b-yl, piperidyl-5b-yl), 2.98–3.08 (m, 2 H, PhCH2), 3.09–3.24 (m, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 3.41–3.55 (m, 2 H, CH2N), 3.67 (d, J = 10.6 Hz, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.44–4.62 (m, 1 H, piperidyl-4-yl), 7.09–7.17 (m, 4 H, Ph), 7.24 (d, J = 7.5 Hz, 1 H, Ph), 7.38 (s, 1 H, H-6), 7.49 (t, J = 7.8 Hz, 1 H, Ph), 8.27 (d, J = 9.1 Hz, 2 H, Ph), 11.32 (s, 1 H, NH). 13C NMR (101 MHz, DMSO-d6) δ ppm 12.2 (1 C, 5-CH3), 27.0 (2 C, piperdyl-3-yl, piperidyl-5-yl), 29.3 (1 C, PhCH2), 50.5 (1 C, piperidyl-4-yl), 51.2 (2 C, piperidyl-2-yl, piperidyl-6-yl), 56.2 (1 C, CH2N), 109.3 (1 C, C-5), 117.5 (2 C, Ph), 118.9 (1 C, Ph), 120.7 (1 C, Ph), 125.9 (1 C, Ph), 126.2 (2 C, Ph), 130.8 (1 C, Ph), 137.3 (1 C, C-6), 139.4 (1 C, Ph), 142.3 (1 C, Ph), 150.7 (1 C, C-2), 154.5 (1 C, Ph), 162.7 (1 C, Ph), 163.6 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C24H26N4O5 + H]+ 451.1976, found 451.1971.
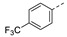
5-Methyl-1-(1-(3-(4-(trifluoromethyl)phenoxy)phenethyl)piperidin-4-yl)pyrimidine-2,4(1H,3H)-dione (21m). Following the general procedure B, 18c (85 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21m (eluent system: 5% MeOH in DCM, 64 mg, 0.14 mmol, 44% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.79 (s, 3 H, 5-CH3), 2.00 (d, J = 12.0 Hz, 2 H, piperdyl-3a-yl, piperidyl-5a-yl), 2.20 (d, J = 11.4 Hz, 2 H, piperdyl-3b-yl, piperidyl-5b-yl), 2.99 - 3.08 (m, 2 H, PhCH2), 3.16 (d, J = 9.5 Hz, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 3.39–3.55 (m, 2 H, CH2N), 3.67 (d, J = 10.6 Hz, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.47–4.59 (m, 1 H, piperidyl-4-yl), 7.05 (d, J = 7.9 Hz, 1 H, Ph), 7.10–7.20 (m, 4 H, Ph), 7.39 (br. s., 1 H, H-6), 7.44 (t, J = 7.8 Hz, 1 H, Ph), 7.75 (d, J = 8.5 Hz, 2 H, Ph), 11.32 (s, 1 H, NH). 19F NMR (377 MHz, DMSO-d6) δ ppm -60.16 (s, 3 F). 13C NMR (101 MHz, DMSO-d6) δ ppm 12.2 (1 C, 5-CH3), 27.1 (2 C, piperdyl-3-yl, piperidyl-5-yl), 29.3 (1 C, PhCH2), 50.4 (1 C, piperidyl-4-yl), 51.1 (2 C, piperidyl-2-yl, piperidyl-6-yl), 56.3 (1 C, CH2N), 109.3 (1 C, C-5), 118.0 (2 C, Ph), 118.4 (1 C, Ph), 120.2 (1 C, Ph), 123.20 (q, J = 32.6 Hz, 1 C, Ph), 124.27 (q, J = 271.3 Hz, 1 C, CF3), 125.2 (1 C, Ph), 127.5 (2 C, Ph), 130.6 (1 C, Ph), 137.3 (1 C, C-6), 139.6 (1 C, Ph), 150.7 (1 C, C-2), 155.2 (1 C, Ph), 160.3 (1 C, Ph), 163.6 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C25H26F3N3O3 + H]+ 474.1999, found 474.1989.
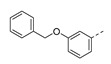
1-(1-(3-(Benzyloxy)phenethyl)piperidin-4-yl)-5-methylpyridine-2,4(1H,3H)-dione (21n). Following the general procedure B, 15a (69 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21n (eluent system: 5% MeOH in DCM, 58 mg, 0.14 mmol, 46% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.66 (d, J = 10.8 Hz, 2 H, piperdyl-3a-yl, piperidyl-5a-yl), 1.74–1.90 (m, 5 H, 5-CH3, piperdyl-3b-yl, piperidyl-5b-yl), 2.07 (t, J = 11.3 Hz, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 2.55 (t, J = 9.0 Hz, 2 H, CH2N), 2.70 (t, J = 7.4 Hz, 2 H, PhCH2), 3.04 (d, J = 11.3 Hz, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.20–4.31 (m, 1 H, piperidyl-4-yl), 5.08 (s, 2 H, (Ph)CH2O), 6.82 (t, J = 7.8 Hz, 2 H, Ph), 6.90 (s, 1 H, Ph), 7.19 (t, J = 7.9 Hz, 1 H, Ph), 7.30–7.35 (m, 1 H, Ph), 7.39 (t, J = 7.4 Hz, 2 H, Ph), 7.43–7.48 (m, 2 H, Ph), 7.63 (s, 1 H, H-6), 11.20 (s, 1 H, NH).13C NMR (101 MHz, DMSO-d6) δ ppm 12.0 (1 C, 5-CH3), 30.0 (2 C, piperdyl-3-yl, piperidyl-5-yl), 33.0 (1 C, PhCH2), 52.4 (2 C, piperidyl-2-yl, piperidyl-6-yl), 52.4 (1 C, piperidyl-4-yl), 59.2 (1 C, CH2N), 69.0 (1 C, (Ph)CH2O), 108.9 (1 C, C-5), 112.1 (1 C, Ph), 115.2 (1 C, Ph), 121.1 (1 C, Ph), 127.7 (2 C, Ph), 127.8 (1 C, Ph), 128.4 (2 C, Ph), 129.2 (1 C, Ph), 137.2 (1 C, Ph), 137.7 (1 C, C-6), 142.0 (1 C, Ph), 150.8 (1 C, C-2), 158.4 (1 C, Ph), 163.7 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C25H29N3O3 + H]+ 420.2282, found 420.2277.
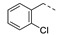
1-(1-(3-((2-Chlorobenzyl)oxy)phenethyl)piperidin-4-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (21o). Following the general procedure B, 15b (79 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21o (eluent system: 5% MeOH in DCM, 90 mg, 0.20 mmol, 65% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.73–2.23 (m, 7 H, piperdyl-3-yl, piperidyl-5-yl, 5-CH3), 2.92 (br. s., 2 H, PhCH2), 3.21–3.67(m, 4 H, piperidyl-2-yl, piperidyl-6-yl), 4.35–4.68 (m, 1 H, piperidyl-4-yl), 5.12 (s, 2 H, (Ph)CH2O), 6.85–6.93 (m, 2 H, Ph), 6.97 (br. s., 1 H, Ph), 7.26 (t, J = 7.8 Hz, 1 H, Ph), 7.36–7.49 (m, 4 H, Ph), 7.52 (s, 1 H, H-6), 11.31 (br. s., 1 H, NH), 2 H (CH2N) could not be observed. 13C NMR (101 MHz, DMSO-d6) δ ppm 12.2 (1 C, 5-CH3), 27.0 (2 C, piperdyl-3-yl, piperidyl-5-yl), 30.2 (1 C, PhCH2), 50.4 (1 C, piperidyl-4-yl), 51.5 (2 C, piperidyl-2-yl, piperidyl-6-yl), 68.2 (1 C, (Ph)CH2O), 109.2 (1 C, C-5), 112.7 (1 C, Ph), 115.6 (1 C, Ph), 121.4 (1 C, Ph), 126.2 (2 C, Ph), 127.3 (1 C, Ph), 127.8 (1 C, Ph), 129.7 (1 C, Ph), 130.4 (1 C, Ph), 133.1 (1 C, Ph), 137.5 (1 C, C-6), 139.7 (1 C, Ph), 150,8 (1 C, C-2), 158.3 (1 C, Ph), 163.7 (1 C, C-4), C (CH2N) could not be observed. HRMS (ESI): m/z [M + H]+ Calcd. for [C25H28ClN3O3 + H]+ 454.1892, found 454.1902.
1-(1-(3-((3,4-Dichlorobenzyl)oxy)phenethyl)piperidin-4-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (21p). Following the general procedure B, 15c (90 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21p (eluent system: 5% MeOH in DCM, 55 mg, 0.11 mmol, 37% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.79 (s, 3 H, 5-CH3), 1.92–2.06 (m, 2 H, piperdyl-3a-yl, piperidyl-5a-yl), 2.08–2.24 (m, 2 H, piperdyl-3b-yl, piperidyl-5b-yl), 2.85–3.02 (m, 2 H, PhCH2), 3.06–3.22 (m, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 3.24–3.34 (m, 2 H, CH2N), 3.65 (br. s., 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 4.45–4.59 (m, 1 H, piperidyl-4-yl), 5.13 (s, 2 H, (Ph)CH2O), 6.85–6.95 (m, 2 H, Ph), 6.98 (br. s., 1 H, Ph), 7.28 (t, J = 7.9 Hz, 1 H, Ph), 7.38–7.48 (m, 2 H, H-6, Ph), 7.68 (d, J = 8.3 Hz, 1 H, Ph), 7.73 (d, J = 1.5 Hz, 1 H, Ph), 11.33 (s, 1 H, NH). 13C NMR (101 MHz, DMSO-d6) δ ppm 12.2 (1 C, 5-CH3), 27.2 (2 C, piperdyl-3-yl, piperidyl-5-yl), 29.7 (1 C, PhCH2), 50.6 (1 C, piperidyl-4-yl), 51.2 (2 C, piperidyl-2-yl, piperidyl-6-yl), 56.5 (1 C, CH2N), 67.5 (1 C, (Ph)CH2O), 109.3 (1 C, C-5), 112.8 (1 C, Ph), 115.5 (1 C, Ph), 121.5 (1 C, Ph), 127.8 (1 C, Ph), 129.4 (1 C, Ph), 129.8 (1 C, Ph), 130.4 (1 C, Ph), 130.7 (1 C, Ph), 131.1 (1 C, Ph), 137.4 (1 C, C-6), 138.3 (2 C, Ph), 150.7 (1 C, C-2), 158.2 (1 C, Ph), 163.7 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C25H27Cl2N3O3 + H]+ 488.1502, found 488.1518.
1-(1-(3-((3-Chlorobenzyl)oxy)phenethyl)piperidin-4-yl)-5-methylpyrimidine-2,4(1H,3H)-dione (21q). Following the general procedure B, 15d (79 mg, 0.30 mmol), 28 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 21q (eluent system: 5% MeOH in DCM, 38 mg, 0.084 mmol, 28% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.78 (s, 5 H, 5-CH3, piperdyl-3a-yl, piperidyl-5a-yl), 2.00 (br. s., 2 H, piperdyl-3b-yl, piperidyl-5b-yl), 2.84 (br. s., 2 H, PhCH2), 4.31–4.46 (m, 1 H, piperidyl-4-yl), 5.12 (s, 2 H, (Ph)CH2O), 6.87 (t, J = 8.3 Hz, 2 H, Ph), 6.94 (br.s., 1 H, Ph), 7.23 (t, J = 7.8 Hz, 1 H, Ph), 7.37–7.47 (m, 3 H, Ph), 7.50–7.59 (m, 2 H, Ph, H-6), 11.26 (s, 1 H, NH), 2 H (CH2N) and 4 H (piperidyl-2-yl, piperidyl-6-yl) could not be observed. 13C NMR (101 MHz, DMSO-d6) δ ppm 12.1 (1 C, 5-CH3), 28.6 (2 C, piperdyl-3-yl, piperidyl-5-yl), 31.3 (1 C, PhCH2), 51.5 (1 C, piperidyl-4-yl), 51.7 (2 C, piperidyl-2-yl, piperidyl-6-yl), 68.1 (1 C, (Ph)CH2O), 109.0 (1 C, C-5), 112.5 (1 C, Ph), 115.4 (1 C, Ph), 121.3 (1 C, Ph), 126.1 (1 C, Ph), 127.3 (1 C, Ph), 127.7 (1 C, Ph), 129.5 (1 C, Ph), 130.4 (1 C, Ph), 133.1 (1 C, Ph), 137.5 (1 C, C-6), 139.7 (1 C, Ph), 150.8 (1 C, C-2), 158.2 (1 C, Ph), 163.7 (1 C, C-4), C (CH2N) and C (Ph) could not be observed. HRMS (ESI): m/z [M + H]+ Calcd. for [C25H28ClN3O3 + H]+ 454.1892, found 454.1899.
5-Methyl-1-(1-(3-phenoxyphenethyl)piperidin-3-yl)pyrimidine-2,4(1H,3H)-dione (23). Following the general procedure B, 12b (65 mg, 0.30 mmol), 29 (0.10 g, 0.30 mmol) and sodium triacetoxyborohydride (0.13 g, 0.61 mmol) in dichloroethane (10 mL) afforded the BOM-protected intermediate, which was deprotected with TFA (5.0 mL) in the presence of triethylsilane (5.0 mL) at 73 °C for 4 h to give the 23 (eluent system: 5% MeOH in DCM, 57 mg, 0.14 mmol, 46% yield). 1H NMR (400 MHz, DMSO-d6) δ ppm 1.43–1.56 (m, 1 H, piperdyl-5a-yl), 1.59–1.80 (m, 6 H, 5-CH3, piperdyl-4-yl, piperdyl-5b-yl), 2.07 (t, J = 12.9 Hz, 1 H, piperdyl-6a-yl), 2.22 (t, J = 10.3 Hz, 1 H, piperdyl-2a-yl), 2.56 (t, J = 4.6 Hz, 2 H, CH2N), 2.68–2.81 (m, 3 H, piperdyl-6b-yl, PhCH2), 2.82–2.90 (m, 1 H, piperdyl-2b-yl), 4.31–4.43 (m, 1 H, piperdyl-3-yl), 6.81 (dd, J = 8.1, 1.8 Hz, 1 H, Ph), 6.90 (s, 1 H, Ph), 6.95–7.04 (m, 3 H, Ph), 7.12 (t, J = 7.6 Hz, 1 H, Ph), 7.28 (t, J = 7.8 Hz, 1 H, Ph), 7.38 (t, J = 7.9 Hz, 2 H, Ph), 7.68 (s, 1 H, H-6), 11.23 (s, 1 H, NH). 13C NMR (101 MHz, DMSO-d6) δ ppm 12.0 (1 C, 5-CH3), 23.9 (1 C, piperdyl-5-yl), 28.1 (1 C, piperdyl-4-yl), 32.3 (1 C, PhCH2), 51.0 (1 C, piperdyl-3-yl), 52.2 (1 C, piperdyl-6-yl), 56.5 (1 C, piperdyl-2-yl), 59.2 (1 C, CH2N), 108.6 (1 C, C-5), 116.1 (1 C, Ph), 118.5 (2 C, Ph), 119.0 (1 C, Ph), 123.3 (1 C, Ph), 123.9 (1 C, Ph), 129.7 (1 C, Ph), 130.0 (2 C, Ph), 138.0 (1 C, C-6), 142.7 (1 C, Ph), 150.8 (1 C, C-2), 156.5 (1 C, Ph), 156.7 (1 C, Ph), 163.7 (1 C, C-4). HRMS (ESI): m/z [M + H]+ Calcd. for [C24H27N3O3 + H]+ 406.2125, found 406.2125.
5-Methyl-1-(1-(2-(3-phenoxyphenyl)acetyl)piperidin-4-yl)pyrimidine-2,4(1H,3H)-dione (26). To a solution of 10b (0.2 g, 0.82 mmol) in MeOH (5.0 mL) was added 1M NaOH (5.0 mL), the resulting reaction mixture was stirred at 50 °C for 2 h. After cooling to room temperature, the reaction mixture was treated with 1N aq. HCl to pH 2–3. The generated white precipitate was collected through filtration and dried in vacuo. The intermediate (0.17 g, 0.74 mmol), 28 (0.26 g, 0.79 mmol), EDC HCl (0.29 g, 1.5 mmol) and 4-dimethylaminopyridine (DMAP) (9.1 mg, 7.4 μmol) were dissolved in DCM (30 mL), and the reaction mixture was stirred at room temperature for overnight to afford BOM protected intermediate, which was deprotected with Pd/C (10%), H2 and HCOOH (0.5%) in i-propanol/H2O (10/1 mL) [30] to give 26 (eluent system: 5% MeOH in DCM, 45 mg, 0.11 mmol, 13% yield). 1H NMR (300 MHz, DMSO-d6) δ ppm 1.54–1.83 (m, 7 H, 5-CH3, piperdyl-3-yl, piperidyl-5-yl), 2.53–2.67 (m, 1 H, piperidyl-2/6-yl), 3.07 (t, J = 10.8 Hz, 1 H, piperidyl-2/6-yl), 3.59–3.85 (m, 2 H, COCH2), 4.05 (d, J = 13.5 Hz, 1 H, piperidyl-2/6-yl), 4.43–4.56 (m, 2 H, piperidyl-4-yl, piperidyl-2/6-yl), 6.82–6.91 (m, 2 H, Ph), 6.95–7.03 (m, 3 H, Ph), 7.08–7.15 (m, 1 H, Ph), 7.31 (t, J = 7.8 Hz, 1 H, Ph), 7.34–7.41 (m, 2 H, Ph), 7.53 (d, J = 0.9 Hz, 1 H, H-6), 11.20 (s, 1 H, NH). 13C NMR (101 MHz, DMSO-d6) δ ppm 12.1 (1 C, 5-CH3), 29.7 (1 C, piperidyl-3/5-yl), 30.5 (1 C, piperidyl-3/5-yl), 38.9 (1 C, (CO)CH2), 40.8 (1 C, piperidyl-2/6-yl), 44.7 (1 C, piperidyl-2/6-yl), 52.1 (1 C, piperidyl-4-yl), 109.1 (1 C, C-5), 116.7 (1 C, Ph), 118.7 (2 C, Ph), 119.5 (1 C, Ph), 123.5 (1 C, Ph), 124.4 (1 C, Ph), 129.9 (1 C, Ph), 130.1 (2 C, Ph), 137.7 (1 C, Ph), 138.2 (1 C, C-6), 150.8 (1 C, C-2), 156.6 (1 C, Ph), 156.7 (1 C, Ph), 163.8 (1 C, C-4), 168.5 (1 C, CO(CH2)). HRMS (ESI): m/z [M + H]+ Calcd. for [C24H25N3O4 + H]+ 420.1918, found 420.1925.
1-(3-Phenoxyphenethyl)-4-phenylpiperidine (27). To a solution of 12b (0.12 g, 0.56 mmol) and 4-phenylpiperidine (0.14 g, 0.87 mmol) in dichloroethane (10 mL) was added sodium triacetoxyborohydride (0.24 g, 1.1 mmol), the resulting mixture was stirred at room temperature for overnight. The reaction mixture was diluted with DCM, and extracted with sat. NaHCO3 and brine. The collected organic layer was dried, concentrated and purified to give 27 (58 mg, 0.16 mmol, 29% yield). 1H NMR (300 MHz, CDCl3) δ ppm 1.91 (br. s., 4 H, piperdyl-3-yl, piperidyl-5-yl), 2.12–2.31 (m, 2 H, piperidyl-2a-yl, piperidyl-6a-yl), 2.46–2.62 (m, 1 H, piperidyl-4-yl), 2.65–2.77 (m, 2 H, CH2N), 2.83–2.97 (m, 2 H, PhCH2), 3.20 (d, J = 10.8 Hz, 2 H, piperidyl-2b-yl, piperidyl-6b-yl), 6.82 - 6.95 (m, 2 H, Ph), 6.96–7.07 (m, 3 H, Ph), 7.12 (t, J = 7.3 Hz, 1 H, Ph), 7.17–7.46 (m, 8 H, Ph). 13C NMR (75 MHz, CDCl3) δ ppm 33.0 (2 C, piperdyl-3-yl, piperidyl-5-l), 33.2 (1 C, PhCH2), 42.4 (1 C, piperidyl-4-yl), 54.1 (2 C, piperidyl-2-yl, piperidyl-6-yl), 60.3 (1 C, CH2N), 116.5 (1 C, Ph), 118.8 (1 C, Ph), 119.1 (1 C, Ph), 123.2 (1 C, Ph), 123.6 (1 C, Ph), 126.2 (1 C, Ph), 126.8 (3 C, Ph), 128.4 (2 C, Ph), 129.6 (1 C, Ph), 129.7 (2 C, Ph), 142.1 (1 C, Ph), 145.9 (1 C, Ph), 157.1 (1 C, Ph), 157.8 (1 C, Ph). HRMS (ESI): m/z [M + H]+ Calcd. for [C25H27NO + H]+ 358.2166, found 358.2164.
4. Conclusions
Starting from the earlier reported compound 3, 19 analogs were synthesized and evaluated for the inhibitory potencies of both MtbTMPK and M. tuberculosis (H37Rv) in vitro growths. Selected substituents on the terminal phenyl ring slightly improved the inhibitory potency. Surprisingly, the 3-Cl analog (21j) was three-fold more potent than compound 3 as the MtbTMPK inhibitor. Substitution of the distal phenyl ring of 3 and 21n afforded analogs with superior whole cell antimycobacterial activity compared to 3. These results provide possible directions for further investigations of MtbTMPK inhibitors as antituberculosis agents.
Supplementary Materials
The following are available online at https://www.mdpi.com/1420-3049/25/12/2805/s1, Figures S1–S43: NMR spectra of final compounds.
Author Contributions
Chemistry and writing—original draft preparation, Y.J.; conceptualization, S.V.C.; methodology, H.E.F., G.C., H.M.-L. and H.I.M.B.; writing—review and editing, F.H., H.M.-L., H.I.M.B. and S.V.C. and supervision, S.V.C. and M.D.P.R. All authors discussed the results and contributed to the final manuscript. All authors have read and agreed to the published version of the manuscript.
Funding
This research was funded by the China Scholarship Council (grant number 201607060021) and, in part, by the Intramural Research Program of the NIAID, NIH. S.V.C. thanks the Hercules Foundation (project AUGE/17/22 “Pharm-NMR”).
Conflicts of Interest
The authors declare no conflict of interest.
Footnotes
Sample Availability: Samples of the compounds 21a–21q, 23, 26 and 27 are available from the authors.
References
- 1.WHO . Global Tuberculosis Report 2019. World Health Organization; Geneva, Switzerland: 2019. [Google Scholar]
- 2.Uplekar M., Weil D., Lonnroth K., Jaramillo E., Lienhardt C., Dias H.M., Falzon D., Floyd K., Gargioni G., Getahun H., et al. WHO’s new end TB strategy. Lancet. 2015;385:1799–1801. doi: 10.1016/S0140-6736(15)60570-0. [DOI] [PubMed] [Google Scholar]
- 3.Lange C., Dheda K., Chesov D., Mandalakas A.M., Udwadia Z., Horsburgh C.R., Jr. Management of drug-resistant tuberculosis. Lancet. 2019;394:953–966. doi: 10.1016/S0140-6736(19)31882-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Karabanovich G., Dusek J., Savkova K., Pavlis O., Pavkova I., Korabecny J., Kucera T., Kocova Vlckova H., Huszar S., Konyarikova Z., et al. Development of 3,5-Dinitrophenyl-Containing 1,2,4-Triazoles and Their Trifluoromethyl Analogues as Highly Efficient Antitubercular Agents Inhibiting Decaprenylphosphoryl-beta-d-ribofuranose 2’-Oxidase. J. Med. Chem. 2019;62:8115–8139. doi: 10.1021/acs.jmedchem.9b00912. [DOI] [PubMed] [Google Scholar]
- 5.WHO . WHO Consolidated Guidelines on Drug-Resistant Tuberculosis Treatment. World Health Organization; Geneva, Switzerland: 2019. [PubMed] [Google Scholar]
- 6.Hoffmann H., Kohl T.A., Hofmann-Thiel S., Merker M., Beckert P., Jaton K., Nedialkova L., Sahalchyk E., Rothe T., Keller P.M., et al. Delamanid and Bedaquiline Resistance in Mycobacterium tuberculosis Ancestral Beijing Genotype Causing Extensively Drug-Resistant Tuberculosis in a Tibetan Refugee. Am. J. Respir. Crit. Care Med. 2016;193:337–340. doi: 10.1164/rccm.201502-0372LE. [DOI] [PubMed] [Google Scholar]
- 7.Haver H.L., Chua A., Ghode P., Lakshminarayana S.B., Singhal A., Mathema B., Wintjens R., Bifani P. Mutations in genes for the F420 biosynthetic pathway and a nitroreductase enzyme are the primary resistance determinants in spontaneous in vitro-selected PA-824-resistant mutants of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2015;59:5316–5323. doi: 10.1128/AAC.00308-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Food and Drug Administration FDA Approved Drug Products. [(accessed on 14 August 2019)]; Available online: https://www.accessdata.fda.gov/scripts/cder/daf/index.cfm?event=overview.process&ApplNo=212862.
- 9.Jong A.Y., Kuo C.L., Campbell J.L. The CDC8 gene of yeast encodes thymidylate kinase. J. Biol. Chem. 1984;259:11052–11059. [PubMed] [Google Scholar]
- 10.Cole S.T., Brosch R., Parkhill J., Garnier T., Churcher C., Harris D., Gordon S.V., Eiglmeier K., Gas S., Barry C.E., et al. Deciphering the biology of Mycobacterium tuberculosis from the complete genome sequence. Nature. 1998;393:537–544. doi: 10.1038/31159. [DOI] [PubMed] [Google Scholar]
- 11.Fioravanti E., Haouz A., Ursby T., Munier-Lehmann H., Delarue M., Bourgeois D. Mycobacterium tuberculosis thymidylate kinase: Structural studies of intermediates along the reaction pathway. J. Mol. Biol. 2003;327:1077–1092. doi: 10.1016/S0022-2836(03)00202-X. [DOI] [PubMed] [Google Scholar]
- 12.Li de la Sierra I., Munier-Lehmann H., Gilles A.M., Barzu O., Delarue M. X-ray structure of TMP kinase from Mycobacterium tuberculosis complexed with TMP at 1.95 A resolution. J. Mol. Biol. 2001;311:87–100. doi: 10.1006/jmbi.2001.4843. [DOI] [PubMed] [Google Scholar]
- 13.Fioravanti E., Adam V., Munier-Lehmann H., Bourgeois D. The crystal structure of Mycobacterium tuberculosis thymidylate kinase in complex with 3’-azidodeoxythymidine monophosphate suggests a mechanism for competitive inhibition. Biochemistry. 2005;44:130–137. doi: 10.1021/bi0484163. [DOI] [PubMed] [Google Scholar]
- 14.Song L., Merceron R., Gracia B., Quintana A.L., Risseeuw M.D.P., Hulpia F., Cos P., Ainsa J.A., Munier-Lehmann H., Savvides S.N., et al. Structure Guided Lead Generation toward Nonchiral, M. tuberculosis Thymidylate Kinase Inhibitors. J. Med. Chem. 2018;61:2753–2775. doi: 10.1021/acs.jmedchem.7b01570. [DOI] [PubMed] [Google Scholar]
- 15.Naik M., Raichurkar A., Bandodkar B.S., Varun B.V., Bhat S., Kalkhambkar R., Murugan K., Menon R., Bhat J., Paul B., et al. Structure guided lead generation for M. tuberculosis thymidylate kinase (Mtb TMK): Discovery of 3-cyanopyridone and 1,6-naphthyridin-2-one as potent inhibitors. J. Med. Chem. 2015;58:753–766. doi: 10.1021/jm5012947. [DOI] [PubMed] [Google Scholar]
- 16.Haouz A., Vanheusden V., Munier-Lehmann H., Froeyen M., Herdewijn P., Van Calenbergh S., Delarue M. Enzymatic and structural analysis of inhibitors designed against Mycobacterium tuberculosis thymidylate kinase. New insights into the phosphoryl transfer mechanism. J. Biol. Chem. 2003;278:4963–4971. doi: 10.1074/jbc.M209630200. [DOI] [PubMed] [Google Scholar]
- 17.Vanheusden V., Van Rompaey P., Munier-Lehmann H., Pochet S., Herdewijn P., Van Calenbergh S. Thymidine and thymidine-5’-O-monophosphate analogues as inhibitors of Mycobacterium tuberculosis thymidylate kinase. Bioorg. Med. Chem. Lett. 2003;13:3045–3048. doi: 10.1016/S0960-894X(03)00643-7. [DOI] [PubMed] [Google Scholar]
- 18.Vanheusden V., Munier-Lehmann H., Froeyen M., Dugue L., Heyerick A., De Keukeleire D., Pochet S., Busson R., Herdewijn P., Van Calenbergh S. 3’-C-branched-chain-substituted nucleosides and nucleotides as potent inhibitors of Mycobacterium tuberculosis thymidine monophosphate kinase. J. Med. Chem. 2003;46:3811–3821. doi: 10.1021/jm021108n. [DOI] [PubMed] [Google Scholar]
- 19.Van Poecke S., Munier-Lehmann H., Helynck O., Froeyen M., Van Calenbergh S. Synthesis and inhibitory activity of thymidine analogues targeting Mycobacterium tuberculosis thymidine monophosphate kinase. Bioorg. Med. Chem. 2011;19:7603–7611. doi: 10.1016/j.bmc.2011.10.021. [DOI] [PubMed] [Google Scholar]
- 20.Song L., Risseeuw M.D.P., Froeyen M., Karalic I., Goeman J., Cappoen D., Van der Eycken J., Cos P., Munier-Lehmann H., Van Calenbergh S. Elaboration of a proprietary thymidylate kinase inhibitor motif towards anti-tuberculosis agents. Bioorg. Med. Chem. 2016;24:5172–5182. doi: 10.1016/j.bmc.2016.08.041. [DOI] [PubMed] [Google Scholar]
- 21.Jian Y., Risseeuw M.D.P., Froeyen M., Song L., Cappoen D., Cos P., Munier-Lehmann H., van Calenbergh S. 1-(Piperidin-3-yl)thymine amides as inhibitors of M. tuberculosis thymidylate kinase. J. Enzym. Inhib. Med. Chem. 2019;34:1730–1739. doi: 10.1080/14756366.2019.1662790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Martinez-Botella G., Breen J.N., Duffy J.E., Dumas J., Geng B., Gowers I.K., Green O.M., Guler S., Hentemann M.F., Hernandez-Juan F.A., et al. Discovery of selective and potent inhibitors of gram-positive bacterial thymidylate kinase (TMK) J. Med. Chem. 2012;55:10010–10021. doi: 10.1021/jm3011806. [DOI] [PubMed] [Google Scholar]
- 23.Zhu Z., Chen H.-G., Goel O.P., Chan O.H., Stilgenbauer L.A., Stewart B.H. Phosphate prodrugs of PD154075. Bioorg. Med. Chem. Lett. 2000;10:1121–1124. doi: 10.1016/S0960-894X(00)00183-9. [DOI] [PubMed] [Google Scholar]
- 24.Elgaher W.A.M., Fruth M., Groh M., Haupenthal J., Hartmann R.W. Expanding the scaffold for bacterial RNA polymerase inhibitors: Design, synthesis and structure-activity relationships of ureido-heterocyclic-carboxylic acids. RSC Adv. 2014;4:2177–2194. doi: 10.1039/C3RA45820B. [DOI] [Google Scholar]
- 25.Elgaher W.A.M., Sharma K.K., Haupenthal J., Saladini F., Pires M., Real E., Mely Y., Hartmann R.W. Discovery and Structure-Based Optimization of 2-Ureidothiophene-3-carboxylic Acids as Dual Bacterial RNA Polymerase and Viral Reverse Transcriptase Inhibitors. J. Med. Chem. 2016;59:7212–7222. doi: 10.1021/acs.jmedchem.6b00730. [DOI] [PubMed] [Google Scholar]
- 26.Meanwell N.A., Krystal M.R., Nowicka-Sans B., Langley D.R., Conlon D.A., Eastgate M.D., Grasela D.M., Timmins P., Wang T., Kadow J.F. Inhibitors of HIV-1 Attachment: The Discovery and Development of Temsavir and its Prodrug Fostemsavir. J. Med. Chem. 2018;61:62–80. doi: 10.1021/acs.jmedchem.7b01337. [DOI] [PubMed] [Google Scholar]
- 27.Hrdlicka P.J., Jepsen J.S., Wengel J. Synthesis and Biological Evaluation of Conformationally Restricted and Nucleobase Modified Analogs of the Anticancer Compound 3’-C-Ethynylcytidine (ECyd) Nucleosides Nucleotides Nucleic Acids. 2005;24:397–400. doi: 10.1081/NCN-200059821. [DOI] [PubMed] [Google Scholar]
- 28.Jean-Yves M., Benoit J. Synthesis and Reactivity of 7-Azaindoles (1H-Pyrrolo(2,3-b)pyridine) Curr. Org. Chem. 2001;5:471–506. [Google Scholar]
- 29.Mehta A., Jaouhari R., Benson T.J., Douglas K.T. Improved Efficiency and Selectivity in Peptide-Synthesis-Use of Triethylsilane as a Carbocation Scavenger in Deprotection of Tert-Butyl Esters and Tert-Butoxycarbonyl-Protected Sites. Tetrahedron Lett. 1992;33:5441–5444. doi: 10.1016/S0040-4039(00)79116-7. [DOI] [Google Scholar]
- 30.Aleiwi B.A., Kurosu M. A reliable Pd-mediated hydrogenolytic deprotection of BOM group of uridine ureido nitrogen. Tetrahedron Lett. 2012;53:3758–3762. doi: 10.1016/j.tetlet.2012.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Laskowski R.A., Swindells M.B. LigPlot+: Multiple Ligand-Protein Interaction Diagrams for Drug Discovery. J. Chem. Inf. Model. 2011;51:2778–2786. doi: 10.1021/ci200227u. [DOI] [PubMed] [Google Scholar]
- 32.Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. UCSF chimera - A visualization system for exploratory research and analysis. J. Comput. Chem. 2004;25:1605–1612. doi: 10.1002/jcc.20084. [DOI] [PubMed] [Google Scholar]
- 33.Munier-Lehmann H., Chaffotte A., Pochet S., Labesse G. Thymidylate kinase of Mycobacterium tuberculosis: A chimera sharing properties common to eukaryotic and bacterial enzymes. Protein Sci. 2001;10:1195–1205. doi: 10.1110/ps.45701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blondin C., Serina L., Wiesmuller L., Gilles A.M., Barzu O. Improved spectrophotometric assay of nucleoside monophosphate kinase activity using the pyruvate kinase/lactate dehydrogenase coupling system. Anal. Biochem. 1994;220:219–221. doi: 10.1006/abio.1994.1326. [DOI] [PubMed] [Google Scholar]
- 35.Morris G.M., Huey R., Lindstrom W., Sanner M.F., Belew R.K., Goodsell D.S., Olson A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009;30:2785–2791. doi: 10.1002/jcc.21256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park Y., Pacitto A., Bayliss T., Cleghorn L.A., Wang Z., Hartman T., Arora K., Ioerger T.R., Sacchettini J., Rizzi M., et al. Essential but Not Vulnerable: Indazole Sulfonamides Targeting Inosine Monophosphate Dehydrogenase as Potential Leads against Mycobacterium tuberculosis. ACS Infect. Dis. 2017;3:18–33. doi: 10.1021/acsinfecdis.6b00103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.