

Abstract
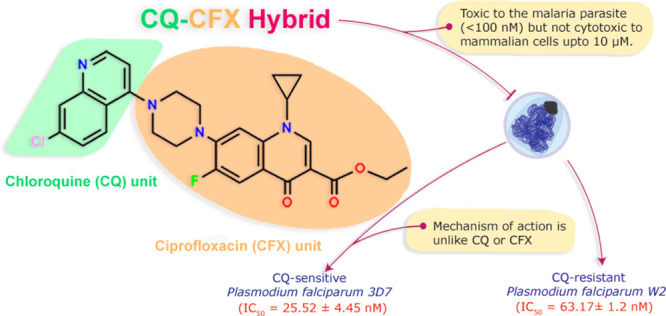
Antimalarial drug resistance is a serious obstacle in the persistent quest to eradicate malaria. There is a need for potent chemical agents that are able to act on drug-resistant Plasmodium falciparum populations at reasonable concentrations without any related toxicity to the host. By rational drug design, we envisaged to address this issue by generating a novel hybrid drug possessing two pharmacophores that can act on two unique and independent targets within the cell. We synthesized a new class of ciprofloxacin-based hybrid molecules, which have been integrated with acridine, quinolone, sulphonamide, and cinnamoyl pharmacophores (1–4). We realized a potent chloroquinolone-ciprofloxacin-based antimalarial hybrid (2, CQ-CFX) whose mechanism of action is unlike that of its parent molecules indicating a unique biological target. CQ-CFX is not only potent against CQ-resistant and susceptible strains of Plasmodium falciparum at low nanomolar concentrations (IC50 values are 63.17 ± 1.2 nM and 25.52 ± 4.45 nM, respectively) but is also not toxic to mammalian and bacterial systems up to 20 μM and 1 μM, respectively.
Keywords: Hybrid drug design, synthesis, structure activity relationship, antimalarial activity
Malaria, caused by the apicomplexan Plasmodium falciparum, is still a major threat to mankind. The most successful treatment strategy that is currently employed involves the use of an artemisinin derivative in combination with a partner drug. The combination of Artemether and lumefantrine has an overall treatment efficacy of about 93.4% with certain places showing greater than 10% failure rate.1 According to the WHO global database on antimalarial drug efficacy and resistance, the rate of Artemisinin combination Therapy (ACT) failure has increased to greater than 10% since 2010 in areas like Cambodia, Thailand, Vietnam, and Myanmar. The emergence and spread of these resistant strains over the coming years may prove detrimental to our goal to eradicate malaria. Therefore, there is a need for the discovery and innovation of a new class of antimalarial compounds that retain effective capabilities at low concentrations and possess hitherto unexploited mechanisms of action to curb the spread of resistant parasites.
In this context, the concept of hybrid drug design is at its infancy and can be promising as one can use existing antimalarial agents to generate combinations that possess two or more pharmacophores that can act on multiple distinct targets within the parasite.2−8 We conceptualized that a novel combination of such molecules would be more potent than the parent drugs and would also be highly demanding for the parasites to become resistant against. Fluoroquinolones, since their discovery in the 1970s, have been used as broad-spectrum antibacterial agents for the treatment of a wide variety of Gram-negative and Gram-positive bacterial infections.9 They target the bacterial Gyrase and Topoisomerase IV and exert their toxicity by forming a ternary complex of enzyme–drug–DNA and consequently block DNA replication.10 It has been seen that fluoroquinolones are active against the in vitro culture of P. falciparum.11 This is attributed to the presence of a prokaryotic organelle known as the Apicoplast within the parasite.12 Among all the fluoroquinolone-based antibiotics tested against P. falciparum, grepafloxacin, gatifloxacin, and moxifloxacin are among the most active. They have displayed IC50 values greater than 10 μM against different P. falciparum strains (3D7 and/or NF54-R).13 However, we were interested in working with Ciprofloxacin (CFX). It is a second generation fluoroquinolone, introduced in 1987, and is included in the WHO list of essential medicines for adults as well as children.14,15 CFX treatment blocks apicoplast DNA replication and causes the formation of abnormal apicoplast and exerts “delayed death” phenotype in P. falciparum as well as T. gondii.16,17 Our group has previously characterized the nuclear-encoded apicoplast targeted gyrase B and established it as a viable target in malaria monotherapy using CFX.18,19
The enhancement of antimalarial activity of CFX was achieved by Dubar et al.20 In this work, ethyl ester derivatives of CFX bearing a ferrocenyl substituent at N-1 or at the C-7 position of the quinolone ring were synthesized. The introduction of ferrocenyl substituents and ethyl esterification of the carboxylic group made these molecules more hydrophobic than their precursor drug CFX. In parallel to the enhancement of lipophilicity, the “prodrug” form of CFX was also achieved by esterification of the carboxylic group. These derivatives were 15-fold and 30-fold more active than their precursor drug CFX as antimalarials.
However, despite the growing importance of CFX in clinical settings of bacterial infections, the lack of activity of CFX in the first life cycle of malaria at clinically relevant concentration prevents their use in monotherapy. All antibiotics, including CFX, are slow acting antimalarials.21 However, the prokaryotic apicoplast is a comparatively new target in malaria chemotherapy and extensively divergent from existing targets of known antimalarials. Therefore, in the search for the hybrid partner, we restricted ourselves to possible pharmacophores that are active in the first life cycle of the parasite.22,23 Additionally, such modifications could help to increase lipophilicity in hybrid molecules. This increase in lipophilicity might help the hybrid molecule to traverse the multiple membranes of the parasite and its plastid and reach the potential target sites as the biological membranes are lipophilic in nature.24
Structure–activity relationship (SAR) studies have demonstrated that structural modification at the C-7 position of the fluoroquinolone ring including alkylation and acylation at the secondary amine of the piperazine moiety results in the loss of antibacterial activity.25,26 Therefore, we chose to functionalize CFX at the terminal N atom of the piperazine moiety with various first life cycle active antimalarial scaffolds (acridine, quinoline, sulphonamide, and cinnamoyl) to prepare CFX-based hybrid antimalarials 1–4.
Synthesis
The detailed synthetic pathway for the targeted CFX-based hybrid molecules 1–4 (Figure 1) starting from commercially available 2,4,5-trifluorobenzoic acid (6) is depicted in Scheme 1. These molecules (1–4) were synthesized in seven steps with 32–80% overall yield. The synthesis of the ethyl ester of CFX 11 was adapted from the previous method reported elsewhere.27 For the building of key quinolone cycle (10), 2,4,5-trifluorobenzoic acid 6 was first reacted with Vilsmeier–Haack reagent (oxalyl chloride and DMF) in dichloromethane to afford acyl chloride (7) in quantitative yield, which was then condensed with ethyl 3-(diethylamino)acrylate (5) in a mixture of triethylamine/toluene to obtain N,N-diethyl enaminone intermediate (8) in 76% yield. Formation of enaminone intermediate (8) was confirmed by recording the 1H NMR spectrum. The ethyl 3-(diethylamino) acrylate (5) was synthesized by Michael addition of diethylamine to ethyl propionate in acetonitrile with 88% yield as previously reported elsewhere.28 In a one-pot process, transaminolysis of 8 with cyclopropylamine followed by cyclization with potassium carbonate in DMF afforded difluoroquinolone (10) in 68% yield. The cyclization of 8 to fluoroquinolone 10 was confirmed by the ∼0.9 ppm deshielding of H-2 proton of 10 with respect to the vinylic-H of 8 in the 1NMR spectra. The ethyl ester of CFX (11) was prepared by an aromatic nucleophilic substitution of the C-7 fluorine atom of difluoroquinolone 10 with piperazine in 98% yield. Finally, nucleophilic substitution reaction between secondary amine of piperazine moiety at the C-7 position of 11 with corresponding chloro substituted first life cycle active antimalarial scaffolds (ArCl) afforded the final ciprofloxacin-based hybrid compounds 1–4 in 30–40% yields. The structures of 1–4 were confirmed by 1H NMR, 13C NMR, FT-IR, and mass spectrometric analysis. In addition, compounds 2 and 3 were characterized by single crystal X-ray diffraction analysis. A detailed account of the synthesis and characterization has been given in the Supporting Information (Figures S6–S22).
Figure 1.

Molecular structures of CFX-based hybrid molecules.
Scheme 1. Synthetic Pathways to the Ciprofloxacin Derivatives 1–4.

X-ray Crystallography
The X-ray suitable single crystals of 2 and 3 were grown from MeCN solutions at room temperature. The crystal structure of 2 belongs to monoclinic space group P21/n with one molecule per asymmetric unit.29 The quinolone moiety assumes a planar structure and the C=O bond length of the quinolone moiety was found to be 1.239 Å, while the C=O bond of the ester group was found to be 1.208 Å. The propyl group at the quinolone N1 forms an obtuse geometry with an angle of 119.70° (N1—C23—C24) and 120.60° (N1—C23—C25) from the N1 atom which allow H-bonding between H atoms associated with C atoms of cyclopropyl and O and Cl atoms of the neighboring moiety (Figure 2a).
Figure 2.

ORTEP representations of (a) 2 and (b) 3. Thermal ellipsoids are shown at 50% probability.
The crystal structure of CFX-based hybrid molecule 3 belongs to the triclinic space group P1 with one molecule per asymmetric unit (Figure 2b). The quinolone moiety forms a planar structure and the C=O bond length of the quinolone moiety was found to be at a distance of 1.236 Å, while the C=O bond of the ester was found to be 1.206 Å. The propyl group at the quinolone N1 forms an obtuse geometry with an angle of 118.74° from the N1 atom allowing H-bonding between H atoms associated with the C22 atom of cyclopropyl and the O atoms of the neighboring moiety. The piperazine moiety adopts the chair conformation. The trifluoromethylbenzenesulfonyl group forms slightly obtuse right angles with the N3 atom of the piperazine moiety (N3—S1—C14 = 105.33°).
The Hybrid Compounds Are Potent In Vitro Antimalarials
Ciprofloxacin derivatives 1–4 were tested for their in vitro antimalarial activity against Chloroquine (CQ) sensitive (Pf 3D7) as well as CQ resistant (Pf W2) strains. Ciprofloxacin and Chloroquine were taken as controls and their IC50 values were determined (Table 1). All the dose–response curves are presented in the Supporting Information, Figures S1–S4.
Table 1. In Vitro Antimalarial Activity against CQ-Susceptible (3D7) and CQ-Resistant (W2) P. falciparum Strains after the 1st (48 h) and 2nd (96 h) Life Cycle.
| IC50 (nM)a 1st life Cycle (48 h) |
IC50 (nM)a 2nd life Cycle (96 h) |
|||||
|---|---|---|---|---|---|---|
| 3D7 | W2 | 3D7 | W2 | RI | Log P | |
| 1 | 359.40 ± 1.15 | nd | 311.30 ± 1.07 | nd | 6.31 | |
| 2 | 25.52 ± 4.45 | 63.17 ± 1.2 | 13.52 ± 0.2 | 30.64 ± 1.07 | 2.47 | 5.02 |
| 3 | 37.63 ± 1.18 | 146.2 ± 1.19 | 14.83 ± 1.09 | 33.39 ± 1.11 | 3.88 | 4.29 |
| 4 | 9109.00 ± 1.18 | nd | 4786.00 ± 1.05 | nd | 3.81 | |
| CFX | 45350.00 ± 1.18 | 37080.00 ± 1.04 | 26260.00 ± 0.99 | 22850.00 ± 1.04 | 1.62 | 1.32 |
| CQ | 12.56 ± 0.66 | 430.60 ± 1.09 | nd | nd | 34.2 | 3.93 |
IC50 values are from a single experiment with the duplicate sample. CQ = chloroquine, CFX = ciprofloxacin: Resistance Indices (RI) = IC50 (W2)/IC50(3D7) after first life cycle were 0calculated using ChemDraw ultra 7.0; nd = not determined.
The synthesized hybrids 1–4 exhibited significant antimalarial activity. Hybrids 1–3 exhibited IC50 values in nanomolar ranges at both cycles. Hybrid 4 bearing cinnamoyl moiety exhibited IC50 value in the micromolar μM range. The activity of hybrids 1–4 was improved in comparison to that of CFX. A remarkable increase in the antimalarial activities (∼103-fold) at both the life cycles was observed especially for hybrids 2–3 in comparison to CFX. The higher activities of hybrids 1–4 compared to CFX can be attributed to the increased lipophilicity of hybrid molecules (log P values in Table 1), as well as to the presence of two pharmacophores in the hybrids. In the hybrids 2–4, the antiplasmodial activity (1st cycle and second cycle against Pf3D7 and PfW2) increased in the order 2 > 3 > 4. This is consistent with the increasing order of lipophilicity [logP: 2 (5.02) > 3 (4.09) > 4 (3.81)]. However, Hybrid 1 bearing the acridine scaffold does not follow the order. The most potent hybrids 2–3 were tested for their antimalarial activity against the Pf W2 strain. Both the hybrids exhibited IC50 values in nanomolar ranges at first as well as second life cycle against PfW2 strain. The antimalarial activities of hybrids 2–3 at both the life cycle were 2 × 102 to 7 × 102-fold higher in comparison to that of CFX. Importantly, hybrids 2–3 were also more potent than CQ against the CQ-resistant PfW2 strain. These hybrids exhibited 3- and 7-fold higher activity, respectively, in the first cycle against PfW2 strain than CQ. In addition, these hybrids also displayed 8- and 17-fold better resistance indices, respectively, than CQ.
Cytotoxicity Study of Most Potent Molecule, CQ-CFX
The cytotoxicity of the most potent compound 2 (CQ-CFX) was determined against mammalian fibroblast NIH3T3 cells using concentrations ranging from 0.3125 to 20 μM by MTT assay. The studies revealed that CQ-CFX did not produce any significant cytotoxicity up to 10 μM even when incubated for up to 72 h (Figure 3). Further, it is noteworthy that molecules 1–4 did not show any hemolysis of infected RBCs or uninfected RBCs at their highest tested concentrations (10–45 μM). Therefore, these molecules may display minimal toxic effects in vivo.
Figure 3.
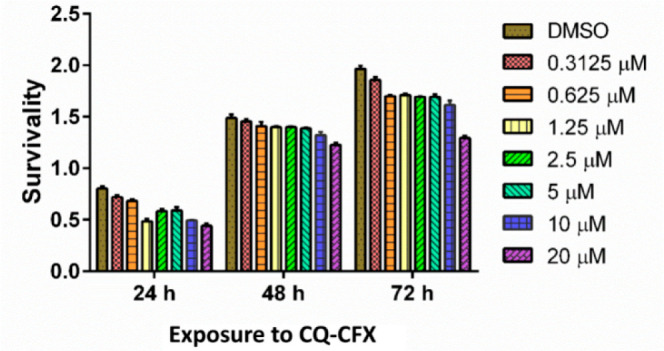
Survivability of NIH3T3 cells in the presence of 2 (CQ-CFX).
CQ-CFX Shows Stage Specificity in Inhibition
The life cycle of Plasmodium is organized into discrete stages termed as Ring, Trophozoite, and Schizont. Each stage is morphologically distinct and corresponds to a particular phase in the cell cycle wherein various proteins are differentially expressed.30 We found that CQ-CFX shows stage specificity in its action. The compound was more effective during the Ring and the Schizont stages compared to the Trophozoite stage (Figure 4). The biological target/s of CQ-CFX may be maximally expressed during these stages.
Figure 4.

Stage-specific effects of CQ-CFX treatment. The drug-treated groups were plotted as percentages calculated against the untreated control group.
CQ-CFX Does Not Retain CFX-like Behavior
Ciprofloxacin exhibits a characteristic delayed-death phenotype in Plasmodium falciparum.31 This is due to the fact that Ciprofloxacin targets the prokaryotic PfGyrase localized in a plastidlike organelle known as the apicoplast. Since there is an adequate pool of apicoplast derived metabolites within the parasite, damage to the apicoplast does not manifest any cellular toxicity in the first life cycle of the parasite. Therefore, the parasitemia in the first life cycle remains unaffected. It is however evident in the second life cycle, where the now dysfunctional apicoplasts are unable to maintain the metabolic needs of the parasite. Thus, the parasitemia in the second life cycle drops sharply compared to the first life cycle. We wanted to check whether CQ-CFX shows a similar pattern of inhibition.
We looked at the inhibition in the first and second life cycle parasitemia across a range of concentrations below the IC50 value of CFX and CQ-CFX. In the CFX treated parasites a stark statistically significant difference in parasitemia was seen. This pattern was absent in the CQ-CFX treated parasites (Figure 5A). In order to corroborate the absence of delayed death on a protein level, we looked at the accumulation of an unprocessed band of the single-stranded DNA binding protein (PfSSB). The PfSSB gene is located within the P. falciparum nucleus. Therefore, the protein is synthesized outside the apicoplast and is targeted to the organelle via a signal peptide.
Figure 5.

(A) Synchronized ring stage parasites were treated with various concentrations of CFX and CQ-CFX (as indicated at the bottom), and the parasitemia was recorded at 48 h and 96 h and plotted accordingly. (B) The cell lysate from the 96 h drug-treated parasites was probed with polyclonal antibodies specific for PfSSB (single-stranded binding protein). PfActin was used to ensure equal loading of the untreated control and the vehicle (DMSO) treated lysates. The arrowhead marks the unprocessed PfSSB specific band that builds in the CFX treated lysate. (C) Antibacterial activity of CFX compared with CQ-CFX. An actively growing culture of the DH10β strain of E. coli was aliquoted at OD600 of 0.2 into respective treatment groups as indicated and treated with the corresponding drug for 6 h. The OD600 values were logged, and the data were plotted as a percentage of the untreated control group.
We have previously observed that a nuclear-encoded apicoplast targeted (NEAT) protein like PfSSB cannot successfully translocate into a dysfunctional apicoplast and therefore accumulates in the cytosol with the apicoplast targeting signal peptide intact.32 This unprocessed protein can be seen as a higher molecular weight band in an immunoblot probed with antibodies against PfSSB. CFX treatment generated this unprocessed form of PfSSB in the second life cycle by virtue of its mechanism of action. This was not observed in CQ-CFX treated parasites (Figure 5B). We failed to see the accumulation of this unprocessed form of PfSSB in the parasites treated across an effective range of concentrations of CQ-CFX (Supporting Information, Figure S5). This suggests that the mechanism of CQ-CFX action is unlike that of CFX.
Additionally, since Ciprofloxacin is an antibacterial agent, we hypothesized that the compound could be toxic to prokaryotic systems that are completely dependent on their DNA Gyrase for replication. However, we saw that CQ-CFX fails to inhibit the growth of E. coli at all concentrations tested (Figure 5C), indicating that the CFX moiety in CQ-CFX is not functional. On a separate note, the lack of any antibacterial activity makes CQ-CFX a better candidate than CFX for in vivo test as there would be minimal damage to the gut flora while maintaining an enhanced antimalarial effect.
CQ-CFX Does Not Retain CQ-like Behavior
During the intraerythrocytic stages of the Plasmodium falciparum life cycle, the parasite relies on the host erythrocyte’s hemoglobin as a nutrient source.33 In the acidic food vacuole, dedicated proteases break down hemoglobin into free amino acids. The heme component, Fe(II)-protoporphyrin IX (FP), in hemoglobin is released as a byproduct in these reactions. Free heme is toxic, as it can generate free radicals via Fenton reactions and damage the lipid bilayers in the parasite and the host erythrocyte. Therefore, it is biocrystallized to an inert crystalline lattice known as hemozoin.34 A vast array of protein complexes including Falcipain-2 and Heme Detoxification Protein (HDP) are involved in this process of biomineralization.35 Chloroquine (CQ) has been shown to disrupt the association of hemoglobin with Falcipain 2 and therefore inhibits hemozoin polymerization.36 Therefore, one of the hallmarks of Chloroquine mediated toxicity is the reduction in the size of the hemozoin crystal compared to the untreated parasites. We decided to use this phenotype to screen whether the CQ moiety in CQ-CFX is still functional. The parasites were treated with CQ and CQ-CFX separately and observed for a duration of 48 h (Figure 6A). The CQ treated parasites showed a statistically significant reduction in the size of the hemozoin (Figure 6B). However, CQ-CFX treated parasites did not show any impairment in their ability to form hemozoin in the food vacuole. This suggests that perhaps the CQ related function is absent or minimal in the hybrid molecule. In order to corroborate this assumption, we looked at another facet of Chloroquine’s physical chemistry. In addition to disrupting Falcipain 2 and hemoglobin interaction, CQ also binds to Fe(II)-protoporphyrin IX and forms a complex that sterically inhibits its crystallization.37 We observed the ability of the hybrid to bind to Fe(II)-protoporphyrin IX using UV–vis spectroscopy. The concentration of Hemin was kept constant at 2.4 μM and Chloroquine, Ciprofloxacin, and CQ-CFX were added in small increments. Chloroquine showed a clear concentration-dependent association with Hemin denoted by the reduction in the characteristic Soret band of Hemin with increasing amounts of Chloroquine. This pattern was absent across a range of concentrations of CFX and CQ-CFX tested, ranging from 100 nM to 4.5 μM (Figure 6C). This suggests that the CQ moiety in CQ-CFX does not retain its function.
Figure 6.

(A) Panel shows the comparison of the reduction in hemozoin crystal size upon CQ and CQ-CFX treatment. (B) The ratio of hemozoin crystal to the cell size derived from images of parasites treated with the indicated drugs for 24 h. Image analysis and estimation were done using ImageJ. (C) Spectrophotometric titration of monomeric heme (blue dotted line) with increasing concentrations of (i) Ciprofloxacin (CFX), (ii) Chloroquine (CQ), and (iii) CQ-CFX. The characteristic Soret band of heme ranges between 380 and 420 nm. The decreasing band between 380 and 420 nm in the Chloroquine titration is distinctive of the heme binding.
Conclusion
In this study, we present a novel class of CFX-based hybrid molecules whose antimalarial activities were found to be remarkably higher than the precursor drug CFX; due to their unique structural design. Hybrids (2–3) were found to be much more potent than CQ against CQ-resistant Plasmodium falciparum strains. It is intriguing that the CQ-CFX hybrid compound is an effective antimalarial agent that works at low nanomolar concentrations. The hybrid is also not toxic to mammalian cells in its antimalarial range of concentrations, in vitro. Presently, the limitation of this study is the lack of in vivo validation of the efficacy of CQ-CFX. Also, it is possible that the ester prodrugs may not survive in vivo; hence, the efficacy of the acid forms of the compounds need to be tested further for their antimalarial activity. It is intriguing that CQ-CFX works against CQ resistant parasites. It will be interesting to study the stage specific effect of CQ-CFX that may allow us to explore the mechanism of action of the molecule. To the best of our knowledge, this is the first example of CFX-based hybrid molecules that exhibit nanomolar antimalarial activity in the first as well as second life cycle of the malaria parasite, Plasmodium falciparum.
Acknowledgments
P.M. acknowledges financial support under the Swarna Jayanti Fellowship (No. DST/SJF-02/CSA-02/2013-14), DST-PURSE II, and DST-FIST for funding the single crystal X-ray crystallography facility at SPS, JNU. The authors thank AIRF, JNU for the NMR and the Mass instrumentation facilities. Financial assistance from Department of Biotechnology grant (BT/PR21569/NNT/28/1234/2017) and DPRP, Department of Science and Technology, Government of India is acknowledged. S.K.D. acknowledges University Potential of Excellence 2 (University Grant Commission), Department of Science and Technology (DST-PURSE II), and UGC-SAP for funding. P.V. acknowledges UGC for a fellowship. The authors acknowledge Agam Prasad Singh, NII, New Delhi for his help and input during this work. The authors also acknowledge the help extended by Ms. Sonam Chorol, SPS, JNU in the synthesis and chromatographic purification of one of the batches of the molecule 2.
Glossary
Abbreviations
- CFX
ciprofloxacin
- CQ
chloroquine
- CQ-CFX
chloroquine–ciprofloxacin hybrid compound
- HDP
heme detoxification protein
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00196.
Details of biological assay protocols, synthetic procedures, and in vitro data (PDF)
Author Contributions
∥ The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript. S.D. and P.V. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- WHO, World Health Organization , World malaria report 2018, Page no. 56, http://apps.who.int/iris/bitstream/handle/10665/275867/9789241565653-eng.pdf, Last accessed 2020-05-07.
- Muregi F. W.; Ishih A. Next-Generation Antimalarial Drugs: Hybrid Molecules as a New Strategy in Drug Design. Drug Dev. Res. 2009, 71 (1), 20–32. 10.1002/ddr.20345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepe D. A.; Toumpa D.; André-Barrès C.; Menendez C.; Mouray E.; Baltas M.; Grellier P.; Papaioannou D.; Athanassopoulos C. M.. Synthesis of Novel G Factor or Chloroquine-Artemisinin Hybrids and Conjugates with Potent Antiplasmodial Activity. ACS Med. Chem. Lett. 2020, 11, 921. 10.1021/acsmedchemlett.9b00669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fröhlich T.; Reiter C.; Saeed M. E. M.; Hutterer C.; Hahn F.; Leidenberger M.; Friedrich O.; Kappes B.; Marschall M.; Efferth T.; Tsogoeva S. B. Synthesis of Thymoquinone-Artemisinin Hybrids: New Potent Antileukemia, Antiviral, and Antimalarial Agents. ACS Med. Chem. Lett. 2018, 9 (6), 534–539. 10.1021/acsmedchemlett.7b00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda D.; Capela R.; Albuquerque I. S.; Meireles P.; Paiva I.; Nogueira F.; Amewu R.; Gut J.; Rosenthal P. J.; Oliveira R.; Mota M. M.; Moreira R.; Marti F.; Prudêncio M.; O’Neill P. M.; Lopes F. Novel Endoperoxide-Based Transmission-Blocking Antimalarials with Liver- and Blood-Schizontocidal Activities. ACS Med. Chem. Lett. 2014, 5 (2), 108–112. 10.1021/ml4002985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira R.; Guedes R. C.; Meireles P.; Albuquerque I. S.; Gonçalves L. M.; Pires E.; Bronze M. R.; Gut J.; Rosenthal P. J.; Prudêncio M.; Moreira R.; O’Neill P. M.; Lopes F. Tetraoxane-Pyrimidine Nitrile Hybrids as Dual Stage Antimalarials. J. Med. Chem. 2014, 57 (11), 4916–4923. 10.1021/jm5004528. [DOI] [PubMed] [Google Scholar]
- Njogu P. M.; Gut J.; Rosenthal P. J.; Chibale K. Design, Synthesis, and Antiplasmodial Activity of Hybrid Compounds Based on (2R,3S)-N-Benzoyl-3-phenylisoserine. ACS Med. Chem. Lett. 2013, 4 (7), 637–641. 10.1021/ml400164t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellot F.; Coslédan F.; Vendier L.; Brocard J.; Meunier B.; Robert A. Trioxaferroquines as New Hybrid Antimalarial Drugs. J. Med. Chem. 2010, 53 (10), 4103–4109. 10.1021/jm100117e. [DOI] [PubMed] [Google Scholar]
- Wolfson J. S.; Hooper D. C. Fluoroquinolone antimicrobial agents. Clin. Microbiol. Rev. 1989, 2 (4), 378–424. 10.1128/CMR.2.4.378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper D. C. Mode of action of fluoroquinolones. Drugs 1999, 58 (Suppl 2), 6–10. 10.2165/00003495-199958002-00002. [DOI] [PubMed] [Google Scholar]
- Divo A. A.; Sartorelli A. C.; Patton C. L.; Bia F. J. Activity of fluoroquinolone antibiotics against Plasmodium falciparum in vitro. Antimicrob. Agents Chemother. 1988, 32 (8), 1182–1186. 10.1128/AAC.32.8.1182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFadden G. I.; Reith M. E.; Munholland J.; Lang-Unnasch N. Plastid in human parasites. Nature 1996, 381 (6582), 482. 10.1038/381482a0. [DOI] [PubMed] [Google Scholar]
- Anquetin G.; Greiner J.; Vierling P. Quinolone-based drugs against Toxoplasma gondii and Plasmodium spp. Curr. Drug Targets: Infect. Disord. 2005, 5 (3), 227–45. 10.2174/1568005054880172. [DOI] [PubMed] [Google Scholar]
- WHO, World Health Organization , WHO Model List of Essential Medicines, 19th List, Page no. 8, https://www.who.int/medicines/publications/essentialmedicines/EML_2015_FINAL_amended_NOV2015.pdf, Last accessed 2020. 05-07.
- WHO, World Health Organization , WHO Model List of Essential Medicines for Children, 7th List, Page no. 11, https://apps.who.int/iris/bitstream/handle/10665/325772/WHO-MVP-EMP-IAU-2019.07-eng.pdf, Last accessed 2020-05-07.
- Wiesner J.; Reichenberg A.; Heinrich S.; Schlitzer M.; Jomaa H. The plastid-like organelle of apicomplexan parasites as drug target. Curr. Pharm. Des. 2008, 14 (9), 855–71. 10.2174/138161208784041105. [DOI] [PubMed] [Google Scholar]
- Goodman C. D.; Su V.; McFadden G. I. The effects of anti-bacterials on the malaria parasite Plasmodium falciparum. Mol. Biochem. Parasitol. 2007, 152 (2), 181–191. 10.1016/j.molbiopara.2007.01.005. [DOI] [PubMed] [Google Scholar]
- Dar A.; Prusty D.; Mondal N.; Dhar S. K. A unique 45-amino-acid region in the toprim domain of Plasmodium falciparum gyrase B is essential for its activity. Eukaryotic Cell 2009, 8 (11), 1759–1769. 10.1128/EC.00149-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar M. A.; Sharma A.; Mondal N.; Dhar S. K. Molecular cloning of apicoplast-targeted Plasmodium falciparum DNA gyrase genes: unique intrinsic ATPase activity and ATP-independent dimerization of PfGyrB subunit. Eukaryotic Cell 2007, 6 (3), 398–412. 10.1128/EC.00357-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubar F.; Anquetin G.; Pradines B.; Dive D.; Khalife J.; Biot C. Enhancement of the Antimalarial Activity of Ciprofloxacin Using a Double Prodrug/Bioorganometallic Approach. J. Med. Chem. 2009, 52 (24), 7954–7957. 10.1021/jm901357n. [DOI] [PubMed] [Google Scholar]
- Dahl E. L.; Rosenthal P. J. Multiple antibiotics exert delayed effects against the Plasmodium falciparum apicoplast. Antimicrob. Agents Chemother. 2007, 51 (10), 3485–3490. 10.1128/AAC.00527-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meunier B. Hybrid molecules with a dual mode of action: dream or reality?. Acc. Chem. Res. 2008, 41 (1), 69–77. 10.1021/ar7000843. [DOI] [PubMed] [Google Scholar]
- Walsh J. J.; Bell A. Hybrid drugs for malaria. Curr. Pharm. Des. 2009, 15 (25), 2970–85. 10.2174/138161209789058183. [DOI] [PubMed] [Google Scholar]
- Dive D.; Biot C. Ferrocene conjugates of chloroquine and other antimalarials: the development of ferroquine, a new antimalarial. ChemMedChem 2008, 3 (3), 383–91. 10.1002/cmdc.200700127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhi C.; Long Z. Y.; Manikowski A.; Comstock J.; Xu W. C.; Brown N. C.; Tarantino P. M. Jr.; Holm K. A.; Dix E. J.; Wright G. E.; Barnes M. H.; Butler M. M.; Foster K. A.; LaMarr W. A.; Bachand B.; Bethell R.; Cadilhac C.; Charron S.; Lamothe S.; Motorina I.; Storer R. Hybrid antibacterials. DNA polymerase-topoisomerase inhibitors. J. Med. Chem. 2006, 49 (4), 1455–65. 10.1021/jm0510023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubschwerlen C.; Specklin J. L.; Sigwalt C.; Schroeder S.; Locher H. H. Design, synthesis and biological evaluation of oxazolidinone-quinolone hybrids. Bioorg. Med. Chem. 2003, 11 (10), 2313–9. 10.1016/S0968-0896(03)00083-X. [DOI] [PubMed] [Google Scholar]
- Anquetin G.; Greiner J.; Mahmoudi N.; Santillana-Hayat M.; Gozalbes R.; Farhati K.; Derouin F.; Aubry A.; Cambau E.; Vierling P. Design, synthesis and activity against Toxoplasma gondii, Plasmodium spp. and Mycobacterium tuberculosis of new 6-fluoroquinolones. Eur. J. Med. Chem. 2006, 41 (12), 1478–93. 10.1016/j.ejmech.2006.07.003. [DOI] [PubMed] [Google Scholar]
- Truce W. E.; Brady D. G. Stereochemistry of Amine Additions to Acetylenic Sulfones and Carboxylic Esters1. J. Org. Chem. 1966, 31 (11), 3543–3550. 10.1021/jo01349a018. [DOI] [Google Scholar]
- CCDC 1899484 (for 2) and CCDC 1899485 (for 3) contain the supplementary crystallographic data for this paper. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via www.ccdc.cam.ac.uk/data_request/cif. (a) Crystal data for 2: C28 H26N4O3FCl, M = 520.16, monoclinic, P21/n, a = 13.9624(8) Å, b = 14.4936(8) Å, c = 14.0972(9) Å, α = 90°, β = 101.451(4)°, γ = 90°, V = 2796.0(3) Å3, Z = 4, Mo Kα radiation (λ = 0.71073 Å), T = 104.0(2) K, μ = 0.187 mm–1, F(000) = 1212, crystal size 0.15 mm × 0.10 mm × 0.08 mm, Bruker APEX-II CMOS diffractometer, 82979 reflections, 6957 independent reflections (Rint = 0.1600) and 4304 observed reflections [I ≥ 2σ(I)], 375 refined parameters, R = 0.0871, wR2 = 0.1965. (b) Crystal data for 3: C26H25F4N3O5S, M = 567.55, triclinic, P1, a = 8.1047(4)Å, b = 10.0759(5) Å, c = 16.0199(8) Å, α = 106.568(2)°, β = 96.388(2)°, γ = 96.915(2)°, V = 1230.12(11) Å3, Z = 2, Mo Kα radiation (λ = 0.71073 Å), T = 104.0(2) K, μ = 0.207 mm–1, F(000) = 588, crystal size 0.16 mm × 0.09mm × 0.06 mm, Bruker APEX-II CMOS diffractometer, 57339 reflections, 6129 independent reflections (Rint = 0.0319) and 5470 observed reflections [I ≥ 2σ(I)], 354 refined parameters, R = 0.0352, wR2 = 0.1170.
- Geary T. G.; Divo A. A.; Jensen J. B. Stage specific actions of antimalarial drugs on Plasmodium falciparum in culture. Am. J. Trop. Med. Hyg. 1989, 40 (3), 240–4. 10.4269/ajtmh.1989.40.240. [DOI] [PubMed] [Google Scholar]
- Fichera M. E.; Roos D. S. A plastid organelle as a drug target in apicomplexan parasites. Nature 1997, 390 (6658), 407–9. 10.1038/37132. [DOI] [PubMed] [Google Scholar]
- Prusty D.; Dar A.; Priya R.; Sharma A.; Dana S.; Choudhury N. R.; Rao N. S.; Dhar S. K. Single-stranded DNA binding protein from human malarial parasite Plasmodium falciparum is encoded in the nucleus and targeted to the apicoplast. Nucleic Acids Res. 2010, 38 (20), 7037–7053. 10.1093/nar/gkq565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francis S. E.; Sullivan D. J. Jr.; Goldberg D. E. Hemoglobin metabolism in the malaria parasite Plasmodium falciparum. Annu. Rev. Microbiol. 1997, 51, 97–123. 10.1146/annurev.micro.51.1.97. [DOI] [PubMed] [Google Scholar]
- Goldberg D. E.; Slater A. F.; Cerami A.; Henderson G. B. Hemoglobin degradation in the malaria parasite Plasmodium falciparum: an ordered process in a unique organelle. Proc. Natl. Acad. Sci. U. S. A. 1990, 87 (8), 2931–2935. 10.1073/pnas.87.8.2931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jani D.; Nagarkatti R.; Beatty W.; Angel R.; Slebodnick C.; Andersen J.; Kumar S.; Rathore D. HDP-a novel heme detoxification protein from the malaria parasite. PLoS Pathog. 2008, 4 (4), e1000053 10.1371/journal.ppat.1000053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chugh M.; Sundararaman V.; Kumar S.; Reddy V. S.; Siddiqui W. A.; Stuart K. D.; Malhotra P. Protein complex directs hemoglobin-to-hemozoin formation in Plasmodium falciparum. Proc. Natl. Acad. Sci. U. S. A. 2013, 110 (14), 5392–7. 10.1073/pnas.1218412110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray P. G.; Janneh O.; Raynes K. J.; Mungthin M.; Ginsburg H.; Ward S. A. Cellular uptake of chloroquine is dependent on binding to ferriprotoporphyrin IX and is independent of NHE activity in Plasmodium falciparum. J. Cell Biol. 1999, 145 (2), 363–76. 10.1083/jcb.145.2.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.