

Abstract
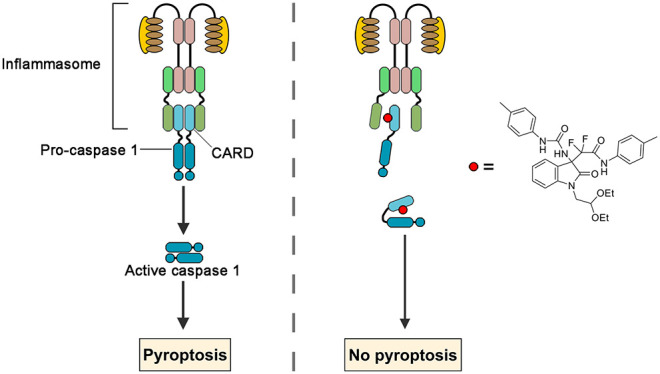
Dysregulation of the inflammatory response is a key driver of many debilitating and costly diseases including immune disorders, cancer, and infection. Pyroptosis is a highly inflammatory form of programmed cell death, triggered by various stimuli and meditated by the activation of inflammatory caspases. Pharmacologic agents that provide strategies to modulate pyroptosis for research and clinical practice are still very limited. In current study, we identify 3-difluoroalkyl quaternary oxindoles as chemical inhibitors of caspase-1, the pyroptosis driving caspase. Our results demonstrated compound 6 could directly bind to the CARD domain of pro-caspase-1 to inhibit its infammasome recruitment and pharmacologic inhibition of pyroptotic cell death by compound 6 is partially efficacious in sepsis models. Compound 6 is thus a potential therapeutic for inflammatory disorders and a tool for further study of the inflammation in human health and disease.
Keywords: Quaternary oxindoles, pyroptosis, macrophage, caspase-1
In the human body, the innate immune system mounts the initial response to pathogenic microorganisms and danger signals derived within human cells.1,2 Following the detection of immunological challenges, an inflammatory signal cascade is activated in innate immune cells and pyroptotic cell death is triggered.3 This lytic cell death results in the release of inflammatory molecules that recruit immune effector cells and thus effectively amplify the inflammatory response.4 Tight control of pyroptosis is crucial to allow the immune system to initiate antimicrobial and inflammatory processes while avoiding excessive tissue damage. The clinical importance of pyroptotic cell death reaches beyond infectious diseases, as dysregulated inflammation is a key driver of numerous hereditary and acquired inflammatory disorders.4−8
Recent studies have offered a glimpse into the sophisticated mechanisms by which pyroptosis is controlled and executed. Pyroptotic cell death is initiated by cytosolic danger signals that stimulate receptors such as NLR family members NLRP1, NLRP3, and NLRC4, as well as AIM2 and pyrin, resulting in protein oligomerization and the recruitment of a scaffolding protein known as ASC. ASC in turn engages with the inflammatory caspases, such as caspase-1, to form a high molecular weight cytosolic complex called the inflammasome.2 The inflammasome subsequently coordinates the cleavage and activation of caspases. Active caspases cleave the proinflammatory cytokines (IL-1β and IL-18) and pore-forming protein gasdermin D (GSDMD) to their active forms, ultimately resulting in robust cell lysis with rapid release of proinflammatory cytokines.3,9
As one prominent member of the pro-inflammatory class, caspase-1 has been found constitutively or inducibly expressed in immune response elements such as macrophages, neutrophils and microglia.10 Upon pyroptosis initiation, inactive pro-caspase-1 could be recruited to multiple types of inflammasomes via interactions between caspase recruitment domain (CARD). Within the complex, caspase-1 is converted to an active enzyme via dimerization, followed by an autocatalytic reaction that generates an active molecule composed of two large (P20) and two small (P10) subunits.11 Since caspase-1 serves as a primary effector for numerous external and internal stimuli, identification of small molecule inhibitors that target the caspase-1 is an important step toward developing effective therapeutics for the treatment of inflammation disorders.12 To date, at least three caspase-1 inhibitors have entered clinical evaluation.13 All these agents are active site inhibitors that act through covalent modification of the catalytic cysteine residue, it would be valuable for research and development of new therapeutics to identify news agents act through alternative mechanisms such as allosteric inhibition.
In this work, we identified compound 6, a quaternary carbon stereocenter compound, as an inhibitor for pyroptotic cell death through high throughput screen. The results showed compound 6 blocks pyroptotic cell death and interleukin-1β (IL-1β) release in both murine and human macrophages. Studies on mechanism of action of compound 6 suggested it directly binds to procaspase-1 and nonselectively inhibits inflammasome-mediated caspase-1 cleavage. Furthermore, compound 6 treatment partially decreases inflammatory cytokine release, macrophage infiltration and substantially prolongs survival in sepsis models. Together, these findings demonstrated the proof of principle that caspase cleavage is a drugable process. The results also provided a basis for compound 6 as a tool for further study of the inflammation in human health and disease and a potential therapeutic for inflammatory disorders.
Results and Discussion
Compound 6 Inhibits Pyroptosis in Macrophage
Taking advantage of the robustness of the macrophage pyroptosis that is induced by combinatorial treatment with LPS and nigericin, through using the level of extracellular IL-1β as surrogate of pyroptosis, we screened a collection of 2787 chemicals featured with 467 compounds containing quaternary carbon stereocenter in immortalized adherent murine macrophage cell line J744A.1. Among the hits with a pronounced pyroptosis inhibition effect (Figure 1A), one group that drew our attention are the compounds sharing a common C3-difluoroalkyl oxindole backbone (Figure 1B). To generate potent pyroptosis inhibitors, we conducted structural modification studies to design and synthesize a number of molecules centered around the C3-difluoroalkyl oxindole core; the effort was concentrated on varying three aspects of the core structure and six representative analogues that are reported herein (Figure 1C). To identify structural analogues with the highest potency, we biologically assessed the compounds for their ability to inhibit macrophage pyroptosis, as the primary screen, using the level of extracellular interleukin-1β (IL-1β) as surrogate. To primarily analyze the acute toxicity of the compounds, we also analyzed their effects on live zebrafish embryos. Among those compounds, compound 6 demonstrated the highest inhibitory effect (EC50 = 5.5 μM) and low toxicity (no nonspecific toxicity was observed at 200 μM) (Figure 1C). The relative molecular mass of compound 6 is 580 with a lipid–water partition coefficient of 4.10. Thus, compound 6 is a promising antipyroptosis agent, therefore selected for further in-depth biological analysis.
Figure 1.

Screen and structural modification studies of pyroptosis-inhibiting compounds. (A) A graph showing results from the chemical screen in the J744A.1 cell line. Each point represents the calculated Z score of each compound on IL-1β releasing phenotype (a negative Z score represents active compounds). The dotted line marks the Z-score threshold of ≤ –2 that was applied to obtain primary hits in the hit window. Red data points indicated the primary hits which sharing the common backbone in B. (B) The C3-difluoroalkyl oxindole backbone for derivatization involving modifications at the 3 positions (red circles). (C) In vitro structure–activity relationship study and in vivo toxicity assessments of selected analogues. For anti-inflammation inhibition, the EC50 is the concentration of a compound that gives half-maximal response. The maximal response of the compound was presented as IL-1β concentration from immortalized murine macrophages. For nonspecific toxicity, the EC100 represents the concentration when 100% of the treated zebrafish embryos exhibit either early lethality, variable embryonic defects, or developmental delay. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. Compound concentrations and IL-1β data are means of ≥3 experimental replicates.
A set of assays was applied to confirm and characterize the pryoptosis inhibition potency of compound 6. Administrated the primary bone marrow-derived macrophages (BMDM) and J774.A1 cells with LPS and nigericin led to the formation of large bubbles, swelling, as well as the uptake of propidium iodide (PI) (visualized by epifluorescent microscopy, Figure 2A and Figure S1A). Cells treated under the same conditions in the presence of compound 6 displayed normal morphology and negative PI staining (Figure 2A and Figure S1A). Furthermore, compound 6 inhibited IL-1β release and cell death in a dose-dependent manner (Figure 2B and Figure S1B and C). To determine whether compound 6 is also effective at inhibiting pyroptosis in human cells, we next tested compound 6 for its pyroptosis-inhibiting ability in human THP-1 monocytes. Compound 6 inhibited formation of the pyroptotic pore and cell death in THP-1 cells as measured by PI uptake and LDH release (Figure 2C). During bacterial infection as well as other immunological challenges, adenosine triphosphate (ATP) is released by bacteria and host cells and acting as an inducer of pyroptosis.14 When using ATP as separate activator of the NLRP3 inflammasome, similar suppression of macrophage pyroptosis by compound 6 was observed (Figure S2). Together, these data suggest that compound 6 blocks pyroptotsis in macrophages across multiple scenarios.
Figure 2.

Compound 6 inhibits pyroptotic cell death in macrophages. (A) Confocal microscopy images of murine primary macrophage, stained with propidium iodide (PI). (B) In a dose-dependent manner, compound 6 inhibited Niggericin induced IL-β release and cell death (measured as LDH release and Alamar blue assay) in murine primary macrophage. (C) Pyroptotic cell death in human THP cells was assessed through LDH release and PI uptake. *P < 0.05, **P < 0.01, ***P < 0.001, and ****P < 0.0001. LDH, Alamar blue assay and PI data are means ± SE and are representative of ≥3 experimental replicates.
Antipyroptotic Effects of Compound 6 Are Independent of the CCKB Pathway
During chemoinformatic analysis on compound 6 molecular structure, we noticed it showed structural similarity with AG-041R, a previous reported gastrin/cholecystokinin-2 (CCKB) receptor antagonist.15 To study the possibility that blocking the CCK pathway could alleviate macrophage pyropotosis, netazepide and proglumide, both selective CCKB receptor antagonists, were administrated onto immortalized macrophages when pyroptosis was induced by nigericin. Cell morphology and cell death assays showed no inhibition of pyroptotic cell death compared to control (Figure S3). These data suggest compound 6 inhibits pryroptosis through CCK pathway independent mechanisms.
Compound 6 Represses Caspase-1 Activation during Macrophage Pyroptosis
The execution of pyroptosis relies on a tightly regulated signaling cascade;2,3 to clarify the mechanism of the antipyroptotic effect of compound 6, we aimed to determine the target following a “downstream-to-upstream” order.
After being cleaved by inflammatory caspases, gasdermin D (GSDMD) executes pyroptosis by generating pores on the plasma membrane,16 so we first asked whether compound 6 affects the cleavage and translocation of GSDMD. Immunofluorescence experiments with anti-GSDMD antibody demonstrated, in both immortalized and primary macrophages, compound 6 administration could effectively repress the cytosol-plasma membrane translocation (Figure 3A). It is interesting to test whether compound 6 inhibits GSDMD-mediated cell death downstream of GSDMD cleavage, so we generated a human embryonic kidney (HEK)-293T cell line in which the expression of a cleaved p30 fragment of GSDMD (gasdermin-N domain) is driven by doxycycline (Dox) treatment. Taking advantage of a lack of expression of other pyroptosis-associated proteins, we can analyze the effect of compound 6 on gasdermin-N domain inducing osmotic lysis. In this experiment, no inhibition of pyroptotic pore formation and cell death was seen when compound 6 was administrated (Figure S4), again suggesting that compound 6 inhibits GSDMD indirectly. Because compound 6 appeared to be working on the step(s) upstream to gasdermin-N domain, the ability of compound 6 to inhibit GSDMD cleavage was tested. Cleavage of GSDMD was significantly reduced in compound 6-treated immortalized macrophages (Figure 3B), implying that compound 6 inhibits caspase-1 activity at these concentrations. To get a direct measure of the amount of active caspase-1 in a living cell, the fluorescent FAM-FLICA caspase-1 reagent was applied to nigericin-treated macrophages. The fluoromicroscopy results indicated clearly attenuated caspase-1 activity in the presence of compound 6 (Figure 3C and D); this observation was corroborated when the caspase-1 activity was determined with caspase-1 substrate Ac-YVAD-pNA (Figure 3E). To study the mechanism that compound 6 inhibits caspase-1 activity, we tested the effect of this drug on negericin-induced caspase-1 cleavage in immortalized macrophages. As shown in Figure 3F, compound 6 substantially abrogated caspase-1 cleavage. To determine whether the effect of compound 6 on caspase-1 cleavage is specific or whether this drug could inhibit activation of other inflammatory caspases, we also analyzed the cleavage of caspase-8 and no inhibition was observed (Figure 3F).
Figure 3.

Compound 6 prevents translocation of GSDMD and activation of caspase-1 during macrophage pyroptosis. (A) Left panels, immunofluorescence images and line intensity histograms demonstrate the localization of GSDMD in the cytosol in LPS primed macrophages and its translocation to the plasma membrane after stimulation by Nigericin, while compound 6 inhibits the translocation. For line intensity histograms, each color represents a single cell. White lines show representative cross sections of macrophage for data sampling. Right panels, percentage of cell with cytomembrane-localized GSDMD. (B) Representative Western blot analysis and relative bar graph quantification of full-length and cleaved N-terminal of GSDMD in J774A.1 cells after treatment with compound 6. Tubulin was used as a loading control. (C) Confocal microscopy images of J774A.1 cell which were stained with FAM-FLICA caspase-1 activity probe (green) and Hochest (blue). (D) Quantification of FLICA fluorescence intensity; at least 20 cells from each group were analyzed. (E) Caspase-1 activity in macrophage pyroptosis with or without compound 6 treatment, assayed with YVAD-afc fluorogenic substrate. (F) Representative Western blot analysis and relative bar graph quantification of Caspase-1 and Caspase-8 in J774A.1 cells after treatment with compound 6. Tubulin was used as a loading control. The positions of the full-length and cleaved caspase proteins are indicated. A, scale bar = 25 μm. C, scale bar = 5 μm. **P < 0.01, ***P < 0.001, and ****P < 0.0001. A, D, and E, data expressed as mean ± SEM from n ≥ 3 independent experiments. B and C, representative gels from n ≥ 3 independent experiments.
Previous data suggested compound 6 could inhibit caspase-1 cleavage; since inflammasomes serve as the platform for caspase-1 activation, we examined the effect of compound 6 on the recruitment of caspase-1 to inflammasomes. The immunostaining results indicated with the presence of compound 6, significantly less caspase-1 protein colocalized with ASC, one of the key components of inflammasome (Figure 4A). We next further confirmed the effect of compound 6 on NLRP3-mediated ASC oligomerization, a key upstream event for caspase-1 cleavage. To visualize ASC oligomerization lively, eGFP-tagged ASC was expressed in marine primary macrophages. Upon NLRP3 activation by nigericin, ASC-eGFP condensed into a single speck in each cell; treatment with compound 6 has no effect on speck formation (Figure 4B). As we had observed that compound 6 could not prevent NLRP3-induced ASC oligomerization, we next tested whether compound 6 could prevent direct NLRP3-ASC interactions. In immortalized macrophages, interaction between endogenous ASC and NLRP3 was analyzed with a coimmunoprecipitation assay; the results indicated compound 6 did not alter the interaction (Figure 4C). Recent studies suggested that ROS production is induced by many inflammasome stimulators, and elevated ROS is essential for inflammasome activation.17,18 To examine the effect of compound 6 on ROS level, a set of flow cytometry assays was deployed. As expected, nigericin administration resulted in ROS generation, as demonstrated by a marked shift in DCFH-DA fluorescence, while compound 6 administration resulted in a supershift in fluorescence (Figure 4D), the higher levels of ROS in compound 6-treated samples likely due to diminished cell death. In all, these results suggest that compound 6 does not inhibit inflammasome assembly and activation, further suggesting that caspase-1 is a direct target of compound 6.
Figure 4.

Compound 6 does not impair inflammasome assembly and activation. (A) Confocal microscopy images of localization of ASC and caspase-1 revealed by immunostaining. Bar graph, colocalization coefficients between the ASC proteins and caspase-1. Pearson’s correlation coefficient values of the two signals in 20 to 25 randomly selected cells were calculated and plotted. (B) Confocal microscopy images of primary macrophages were transfected with ASC-GFP plasmid and treated with LPS and nigericin to induce ASC speck formation. Arrowheads highlight the ASC specks. Bar graph, the percentage of cell with ASC speck. (C) Co-IP experiments in J774A.1 cells showed compound 6 does not block interaction between components of the NLRP3 inflammasome. (D) Representative FACS plot of intracellular ROS production in primary macrophages. (E) Effects of compound 6 on release of IL-1β induced by lethal factor, poly(dA:dT) or Flagellin in J774A.1 cells as measured by ELISA assay. (F) Effects of compound 6 on cell death induced by cytoplasmic LPS in J774A.1 cells as measured by LDH release. *P < 0.05, **P < 0.01, and ***P < 0.001. Data expressed as mean ± SEM from n ≥ 3 independent experiments, Data expressed as mean ± SEM from n ≥ 3 independent experiments, representative gels from n ≥ 3 independent experiments.
To date, varieties of canonical inflammasome have been identified;2,6,19 to determine whether the effect of compound 6 on caspase-1 activation is specific for the NLRP3 inflammasome, we stimulated macrophages with different pyroptosis triggers, in the presence or absence of compound 6. Release of IL-1β was decreased by compound 6 from immortalized macrophages stimulated with the NLRP1 ligand,20 anthrax lethal toxin (Figure 4E). Similar attenuation of pyroptosis was observed when microbial Flagellin was delivered into J774A.1 with electroporation to activate NAIP/NLRC4 inflammasome21 (Figure 4E). The effect of compound 6 on the non-NLR AIM2 inflammasome was examined by transfecting primary macrophages with the dsDNA analog Poly(dA: dT).22 Compound 6 treatment also repressed the IL-1β secretion in this scenario (Figure 4E). To examine whether compound 6 could inhibit noncanonical caspases, we analyzed the effect of compound 6 on macrophage pyroptotic death induced by cytosolic LPS. The data suggested the presence of compound 6 could partially inhibit other pyroptosis-inducing caspases such as caspase-11 (Figure 4F). These results suggested that compound 6 inhibits the activity of pyroptosis-inducing caspases in a nonselective fashion, further supporting the hypothesis that compound 6 binds to caspase-1 directly and inhibited its recruitment to inflammasomes.
Compound 6 Likely Binds to the CARD Domain on Caspase-1 to Block Its Cleavage
To explore whether compound 6 directly inhibits caspase-1 cleavage, we incubated purified procaspase-1 with preformed ASC pyroptosomes in the presence of compound 6. As shown in Figure 5A, incubation of proteins at 37 °C resulted in caspase-1 cleavage, which was inhibited by compound 6. To provide additional support for the direct effect of compound 6 on caspase-1, we performed a drug affinity-responsive target stability (DARTS) assay which is a well-established target identification method.23 It is on the basis of the principle that the increasing of proteolysis resistance of the target protein is generated by the interaction with a small molecular ligand. We utilized immunoblotting to examine the stability of caspase-1 in cell lysates digested by different concentrations of Pronase. Representative data shown in Figure 5B clearly indicated that the protease susceptibility of caspase-1 is significantly reduced in cell lysates pretreated with compound 6, compared with the Histone 3 control group. To confirm the specificity of this protective effect, one inactive analog of compound 6 (compound 3) was added and no change on protease susceptibility was observed (Figure 5C). This observation suggested a possible direct interaction between compound 6 and caspase-1 that will be confirmed by X-ray crystallography in the future.
Figure 5.

Compound 6 interacts with pro-Caspase-1 CARD to block its cleavage. (A) Pyroptosome assay showed compound 6 blocks ASC mediated caspase-1 cleavage. (B and C) DARTS assay revealed compound 6 protects the caspase-1 from proteolysis and this protection is relying on the CARD domain. (A, B, and C) Representative gels from n ≥ 3 independent experiments. (B and C) Histone 3 protein served as a loading indicator. (D) Surface docking model of compound 6–CARD complex; compound 6 is green. Enlarged image showing the details of interaction. The yellow and blue dashed lines indicate the hydrogen bonds. (C and D) human caspase-1 CARD, PDB ID: 5FNA. (E) Predicted molecular bonds and distance of residues between compound 6 and caspase-1. (F) Protein sequence alignment of the N-terminal of caspase proteins. Asterisks (*) indicates the amino acids potentially interacting with compound 6. Gray shadowed sequences are CARD.
In order to gain deeper insight into the mechanism by which compound 6 prevents caspase-1 cleavage, binding models were generated with an automated molecular docking platform Glide24 based on the negative binding affinity values and the model with the lowest free energy (Glide score = −3.72) was shown as Figure 5C. Examination of this proposed binding pose clearly suggested that compound 6 occupies a hydrophobic groove on the CARD domain of caspase-1 by interacting with key residues Ala2 and Lys8 (Figure 5C, D, and E). Inactive analogs, compound 4 and 5, were used in the modeling, the results indicating the benzyl group at the C3-substituent of oxindole could be critical for compound 6 to fit into the hydrophobic groove and form necessary aromatic hydrogen bonds with Ala2 and Lys8. To test this model, we removed the CARD domain on caspase-1 and tested the protease susceptibility; the results indicated the protective effect of compound 6 is relying on the CARD domain (Figure 5B, lower panel). Further modeling study indicated compound 6 could not bind to other pyrotosis-inducing caspases (human caspase-4, caspase-5, and mouse caspase-11) through bonding with the conserved amino acids on the N-terminal of the proteins (Figure 5F).
Compound 6 Treatment Increases Survival in Sepsis Models
To test the potential of compound 6 as a therapy agent for immunological disorders, we examined its effect on inflammatory response in murine sepsis models.
In the murine sepsis shock model, a single dose of 20 mg/kg compound 6 or saline was administered onto animals 30 min before a lethal dose of LPS (25 mg/kg) was given. Histological images of lung tissue showed decreased levels of alveolar congestion, exudate, and damage of epithelial architecture, in compound 6-treated mice as compared to saline-treated mice (Figure 6 A and B). Serum taken from these animals demonstrated a significant decrease in the release of IL-1β in mice treated with compound 6 (Figure 6C). Survival curves in mice further demonstrated the therapeutic effect of compound 6, wherein it increased median survival by 8 h (P = 0.05, Figure 6D). To observe the macrophage response to compound 6 treatment directly, a zebrafish sepsis model25 was deployed. Macrophages (labeled with mepg1:eGFP transgenic) were quiescent in an untreated larva, located primarily in the dorsal aorta (DA). Exposure to LPS resulted in migration of macrophages from the DA into the interstitial space, in compound 6 treated larvae, the density of macrophages in the interstitial space was significantly lower (Figure 6E). On the basis of these data, it is promising that optimized derivatives of compound 6 might be of use in the treatment of acute immunological disorders.
Figure 6.

Compound 6 partially attenuatesin vivoinflammatory response. (A) Effects of compound 6 on histopathological changes of lung tissues in LPS-induced acute lung injury mice. Lungs (n = 5 from each experimental group) were processed for histological evaluation at 12 h after LPS challenge. The representative histological section of the lungs was stained by hematoxylin and eosin (H & E staining, magnification ×100). (B) Acute lung injury score (expressed using arbitrary units.). (C) ELISA analysis indicated compound 6 treatment decreased the IL-1β level in serum. (D) Survival curves were analyzed by log-rank (Mantel-Cox) test demonstrating an increase in median survival of 8 h in the pyrooxindole treatment group. (E) Upper panel, schematic cartoon of zebrafish larvae; the box showed the region imaged. Lower panels, confocal microscopy images of larvae intestine at 7 days post fertilization (pdf); macrophages were labeled with Tg(mpeg1:eGFP) transgenic. Compound 6 treatment (5 μM) reduces macrophage infiltration into intestine. The scatter plot, quantification of macrophages in the imaged regions. **P < 0.01. (B and D) Data are presented as means ± SEM, n > 12 for each group.
We describe here compound 6, a potent pyroptosis inhibitor that is active in both of murine and human macrophages. This inhibitory ability translates to animal models of sepsis as well, where compound 6 inhibits macrophage infiltration and increases median survival. In the context of our current understanding of pyropotosis, we characterized the mechanism of action of compound 6. The data demonstrate compound 6 does not block assembly of inflammasome and generation of reactive oxygen species, but possibly inhibits caspases activation through binding to the caspase recruitment domain.
Caspases-1, the crucial effector molecules in pyroptosis, are among the promising targets for pharmacological modulation of inflammatory cell death. The search programs focusing on caspase-1 inhibitors are pursued by a number of researchers, and several synthetic caspase-1 inhibitors have entered clinical research. Unlike most of the reported caspase-1 inhibitors, which block the active site as pseudosubstrates, compound 6 provides the proof of principle that caspase cleavage can be specifically inhibited. This innovative mechanism of action opened new research avenues for discovery of new anti-inflammatory medicines. In addition, as a chemical probe, compound 6 allows researchers to better understand the biology of caspase-1.
The therapeutic efficacy of inhibiting the inflammasome-pyroptosis axis has already been demonstrated clinically with the efficacy of IL-1β receptor antagonists to treat inflammatory diseases,26 while biologics such as canakinumab and rilonacept have been shown to increase the risk of infections.27,28 IL-1β maturation can be mediated by a number of different enzymes besides caspase-1;29 thus, specific targeting of caspase-1 will not result in the complete blockade of the IL-1β signal in vivo, and immune responses may remain at a certain extent. Small molecule Caspase-1 inhibitors like compound 6 may therefore have fewer immunosuppressive effects and also be more cost-effective than biologic agents. The future clinical development of compound 6 and its derivatives may result in the development of new therapeutics for inflammatory disorders.
Acknowledgments
We thank Dr. Dingyu Wang and Mr. Yufang Wang from Xiang Gao’s lab at Nanjing University for help during this project.
Glossary
Abbreviations
- ATP
adenosine triphosphate
- BMDM
bone marrow-derived macrophages
- CARD
caspase recruitment domain
- CCKB
cholecystokinin-2
- DA
dorsal aorta
- DARTS
drug affinity-responsive target stability
- Dox
doxycycline
- EC50
half maximal effective concentration
- FLICA
fluorochrome-labeled inhibitors of caspases
- GSDMD
gasdermin D
- HEK
human embryonic kidney
- IL-1β
interleukin-1β
- LDH
lactate dehydrogenase
- IP
immunoprecipitation
- LPS
lipopolysaccharides
- NLR
NOD-like receptor
- PI
propidium iodide
- ROS
reactive oxygen species
- SEM
standard errors of the mean
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00070.
Author Contributions
X.L., D.M., and J.Z. conceived and supervised the project. J.-S.Y. synthesized the chemicals and performed purity and structure analysis. Q.X. and Y.W. performed the bioactivities assays and ligand-protein docking. All authors analyzed and discussed the data. The manuscript was written by Q.X., J.Y., and X.L.
Author Contributions
∇ Q.X., J.-S.Y., and Y.W. contributed equally to this work.
This research was undertaken, in part, thanks to grant funding from National Natural Science Foundation of China (NSFC 31970765 and NSFC 31671505 to X.L., NSFC 21725203 to J.Z., and NSFC 21901074 to J.-S.Y.)
The authors declare no competing financial interest.
Supplementary Material
References
- Brubaker S. W.; Bonham K. S.; Zanoni I.; Kagan J. C. Innate immune pattern recognition: a cell biological perspective. Annu. Rev. Immunol. 2015, 33, 257–90. 10.1146/annurev-immunol-032414-112240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broz P.; Dixit V. M. Inflammasomes: mechanism of assembly, regulation and signalling. Nat. Rev. Immunol. 2016, 16 (7), 407–20. 10.1038/nri.2016.58. [DOI] [PubMed] [Google Scholar]
- Rathinam V. A. K.; Zhao Y.; Shao F. Innate immunity to intracellular LPS. Nat. Immunol. 2019, 20 (5), 527–533. 10.1038/s41590-019-0368-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J.; Gao W.; Shao F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem. Sci. 2017, 42 (4), 245–254. 10.1016/j.tibs.2016.10.004. [DOI] [PubMed] [Google Scholar]
- Voet S.; Srinivasan S.; Lamkanfi M.; van Loo G.. Inflammasomes in neuroinflammatory and neurodegenerative diseases. EMBO Mol. Med. 2019, 11 ( (6), ), 10.15252/emmm.201810248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochnicki T.; Latz E. Inflammasomes on the Crossroads of Innate Immune Recognition and Metabolic Control. Cell Metab. 2017, 26 (1), 71–93. 10.1016/j.cmet.2017.06.018. [DOI] [PubMed] [Google Scholar]
- Hotamisligil G. S. Inflammation, metaflammation and immunometabolic disorders. Nature 2017, 542 (7640), 177–185. 10.1038/nature21363. [DOI] [PubMed] [Google Scholar]
- Van Opdenbosch N.; Lamkanfi M. Caspases in Cell Death, Inflammation, and Disease. Immunity 2019, 50 (6), 1352–1364. 10.1016/j.immuni.2019.05.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man S. M.; Kanneganti T. D. Converging roles of caspases in inflammasome activation, cell death and innate immunity. Nat. Rev. Immunol. 2016, 16 (1), 7–21. 10.1038/nri.2015.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franchi L.; Eigenbrod T.; Munoz-Planillo R.; Nunez G. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat. Immunol. 2009, 10 (3), 241–7. 10.1038/ni.1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sollberger G.; Strittmatter G. E.; Garstkiewicz M.; Sand J.; Beer H. D. Caspase-1: the inflammasome and beyond. Innate Immun. 2014, 20 (2), 115–25. 10.1177/1753425913484374. [DOI] [PubMed] [Google Scholar]
- Denes A.; Lopez-Castejon G.; Brough D. Caspase-1: is IL-1 just the tip of the ICEberg?. Cell Death Dis. 2012, 3, e338 10.1038/cddis.2012.86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudelova J.; Fleischmannova J.; Adamova E.; Matalova E. Pharmacological caspase inhibitors: research towards therapeutic perspectives. J. Physiol Pharmacol 2015, 66 (4), 473–82. [PubMed] [Google Scholar]
- Man S. M.; Karki R.; Kanneganti T. D. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev. 2017, 277 (1), 61–75. 10.1111/imr.12534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiba T.; Kinoshita Y.; Sawada M.; Kishi K.; Baba A.; Hoshino E. The role of endogenous gastrin in the development of enterochromaffin-like cell carcinoid tumors in Mastomys natalensis: a study with the specific gastrin receptor antagonist AG-041R. Yale J. Biol. Med. 1998, 71 (3–4), 247–55. [PMC free article] [PubMed] [Google Scholar]
- Shi J.; Zhao Y.; Wang K.; Shi X.; Wang Y.; Huang H.; Zhuang Y.; Cai T.; Wang F.; Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526 (7575), 660–5. 10.1038/nature15514. [DOI] [PubMed] [Google Scholar]
- Wang Y.; Shi P.; Chen Q.; Huang Z.; Zou D.; Zhang J.; Gao X.; Lin Z. Mitochondrial ROS promote macrophage pyroptosis by inducing GSDMD oxidation. J. Mol. Cell Biol. 2019, 11 (12), 1069–1082. 10.1093/jmcb/mjz020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harijith A.; Ebenezer D. L.; Natarajan V. Reactive oxygen species at the crossroads of inflammasome and inflammation. Front. Physiol. 2014, 5, 352. 10.3389/fphys.2014.00352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vance R. E. The NAIP/NLRC4 inflammasomes. Curr. Opin. Immunol. 2015, 32, 84–9. 10.1016/j.coi.2015.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyden E. D.; Dietrich W. F. Nalp1b controls mouse macrophage susceptibility to anthrax lethal toxin. Nat. Genet. 2006, 38 (2), 240–4. 10.1038/ng1724. [DOI] [PubMed] [Google Scholar]
- Matusiak M.; Van Opdenbosch N.; Vande Walle L.; Sirard J. C.; Kanneganti T. D.; Lamkanfi M. Flagellin-induced NLRC4 phosphorylation primes the inflammasome for activation by NAIP5. Proc. Natl. Acad. Sci. U. S. A. 2015, 112 (5), 1541–6. 10.1073/pnas.1417945112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugrin J.; Martinon F. The AIM2 inflammasome: Sensor of pathogens and cellular perturbations. Immunol Rev. 2018, 281 (1), 99–114. 10.1111/imr.12618. [DOI] [PubMed] [Google Scholar]
- Pai M. Y.; Lomenick B.; Hwang H.; Schiestl R.; McBride W.; Loo J. A.; Huang J. Drug affinity responsive target stability (DARTS) for small-molecule target identification. Methods Mol. Biol. 2015, 1263, 287–98. 10.1007/978-1-4939-2269-7_22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47 (7), 1739–49. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Philip A. M.; Wang Y.; Mauro A.; El-Rass S.; Marshall J. C.; Lee W. L.; Slutsky A. S.; dosSantos C. C.; Wen X. Y. Development of a zebrafish sepsis model for high-throughput drug discovery. Mol. Med. 2017, 23, 134–148. 10.2119/molmed.2016.00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinarello C. A.; Simon A.; van der Meer J. W. Treating inflammation by blocking interleukin-1 in a broad spectrum of diseases. Nat. Rev. Drug Discovery 2012, 11 (8), 633–52. 10.1038/nrd3800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard C.; Noe A.; Skerjanec A.; Holzhauer B.; Wernsing M.; Ligueros-Saylan M.; Thuren T. Safety and tolerability of canakinumab, an IL-1beta inhibitor, in type 2 diabetes mellitus patients: a pooled analysis of three randomised double-blind studies. Cardiovasc. Diabetol. 2014, 13, 94. 10.1186/1475-2840-13-94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapur S.; Bonk M. E. Rilonacept (arcalyst), an interleukin-1 trap for the treatment of cryopyrin-associated periodic syndromes. P T 2009, 34 (3), 138–41. [PMC free article] [PubMed] [Google Scholar]
- Netea M. G.; Simon A.; van de Veerdonk F.; Kullberg B. J.; Van der Meer J. W.; Joosten L. A. IL-1beta processing in host defense: beyond the inflammasomes. PLoS Pathog. 2010, 6 (2), e1000661 10.1371/journal.ppat.1000661. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.