

Abstract

Phosphoinositide 3-kinases (PI3Ks) mediate a series of events related to cell growth, proliferation, survival, and differentiation. Overexpression of PI3Ks can lead to the dysregulation of cell homeostasis and cause tumorigenesis. In this study, rationally designed compounds were investigated as PI3Kα-selective inhibitors. Our efforts culminated in the discovery of a series of quinazolin-4(3H)-one derivatives with 2-substituted-N-methylpropanamide substitutions as PI3Kα-selective inhibitors. The best compound, 10, has PI3Kα enzymatic and cellular IC50 values of 1.8 and 12.1 nM, respectively. It exhibits biochemical selectivities for PI3Kα over PI3Kβ/δ/γ of 150/7.72/7.67-fold and cellular selectivities of 115/15.1/>826-fold, respectively. Compound 10 is 59% orally bioavailable with a dose-normalized AUC of 3090 nM. These effects translated into in vivo conditions, as 10 significantly time- and dose-dependently inhibited phosphorylation of Akt in BT-474 subcutaneous xenograft mice and inhibited tumor growth.
Keywords: PI3Kα inhibitor, breast cancer, PIK3CA mutant, structure-based design, DMPK, PD study
The phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) signaling pathway plays a key role in a series of cellular functions related to cell growth, proliferation, survival, and differentiation.1 PI3Ks are a class of lipid kinases that catalyze the phosphorylation of phosphoinositides at the 3-hydroxyl position. PI3Ks are divided into three classes (I, II, and III) on the basis of substrate specificity, sequence homology, and structural features.2 Class I PI3Ks are heterodimers formed by a regulatory subunit and a catalytic (p110) subunit, including PI3Kα, PI3Kβ, PI3Kδ, and PI3Kγ.3
The clinical development of pan-PI3K and dual PI3K/mTOR inhibitors has been limited by off-target toxicities such as GI toxicities, hepatotoxicity, and mood alterations. Such toxicities have limited the dose escalation.4 Isoform-specific inhibitors could allow administration at higher, pharmacologically active doses with fewer off-target toxicities.5
One of the most attractive isoforms is the PI3Kα subtype. It is ubiquitously expressed and associated with angiogenesis and glucose homeostasis.6 The PI3K/mTOR pathway is frequently dysregulated in cancers, often because of activating mutations or amplification of PIK3CA. Gain of function mutations in PIK3CA, the gene encoding the p110α catalytic subunit of PI3K, are among the most common somatic alterations in solid tumors,7,8 such as 42% to 55% of endometrial,9 42% of cervical,10 27%–36% of breast,11,12 18% of colorectal,13 13% of head and neck,14 and 12% of ovarian cancers.15 BYL-719 is the first and only PI3Kα inhibitor that has been approved by the FDA (in May 2019) for the treatment of PIK3CA mutant HER2–/ER+ metastatic advanced breast cancer.16
On the basis of published binding interactions between the PI3Kα ATP binding pocket and small molecules such as BYL-719,17 residue Gln859 is unique for the PI3Kα isoform and contributes to its selectivity against other PI3K isoforms. The hydrogen-bond formation of PI3Kα-selective inhibitors with Gln859 is essential to the selectivity. Our starting point came from the design and synthesis of compound 1 through the hybridization between our previously reported pyrido[1,2-a]pyrimidine scaffold18 and l-prolinamide as seen in BYL-719, which was essential to the selectivity (Figure 1). To our delight, compound 1 showed very good PI3Kα activity in a biochemical assay (IC50 = 0.5 nM), and the selectivity for PI3Kα over PI3Kβ/δ/γ was 130/3.4/7.4-fold. However, it demonstrated modest cellular potency (IC50 = 188.0 nM in MCF-7 cells; Table 1), which might be due to poor permeability (A to B: 0.45 × 10–6 cm/s, MDCK assay) and was speculated to be related to high polar surface area.19
Figure 1.
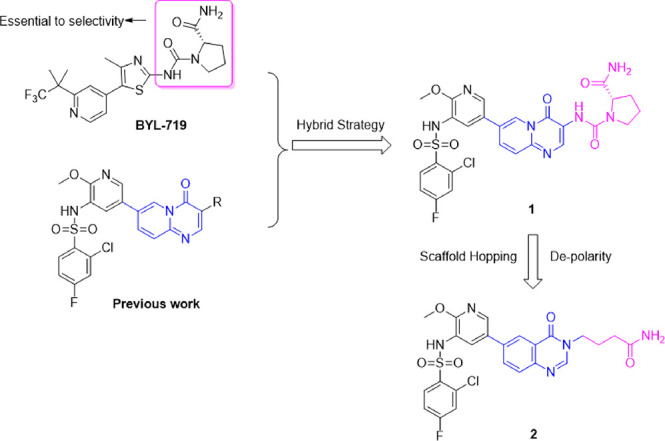
Our design strategy.
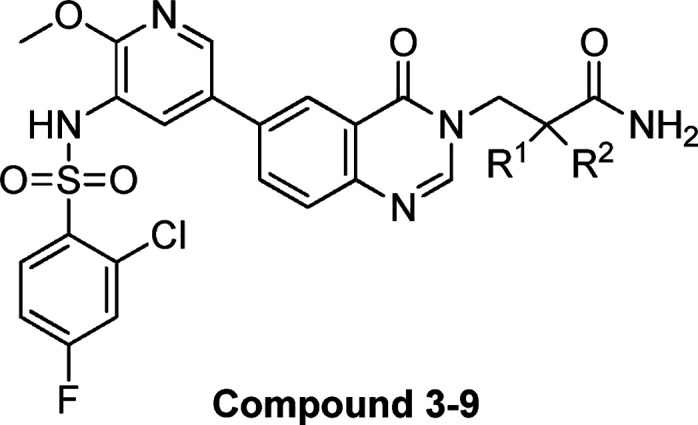
Table 1. Structures and PI3K Enzyme and Cell Inhibitory Potencies of Compounds 1–9.
| compd | R1 | R2 | PI3Kα IC50 (nM) | selectivity folds over PI3Kβ/δ/γ | cellulara IC50 (nM) |
|---|---|---|---|---|---|
| 1 | – | – | 0.5 | 130/3.4/7.4 | 188 |
| 2 | – | – | 1.1 | 30/1.91/– | 9.43 |
| 3 | –H | –Me | 2.0 | 83.2/5.03/7.38 | 17.1 |
| 4 | –F | –F | 2.7 | 46.6/2.69/3.15 | – |
| 5 (rac) | –H | –NH2 | 5.4 | 33.9/4.75/7.39 | 134 |
| 6 | –H | –H | 1.2 | 59.0/3.34/3.94 | 16.8 |
| 7 (rac) | –H | –Et | 6.9 | 19.3/–/– | 52.3 |
| 8 (rac) | –H | –iPr | 25.5 | 32.6/–/– | 125 |
| 9 | –CH2CH2– | 5.7 | 14.4/0.35/3.61 | 39.9 | |
MCF-7 pAktS473 inhibitory IC50 values.
We envisioned that reducing the polarity of 1 might improve its cellular potency. With compound 1 as a template, scaffold hopping20,21 together with modification of l-prolinamide were conducted, leading to the generation of compound 2 (Figure 1). As shown in Table 1, although the biochemical potency was maintained, the selectivity over PI3Kβ decreased significantly (30 vs 130). The linker between the hinge binding region motif and the solvent-accessible region motif of 2 was too flexible, which was unfavorable to the conformation needed to form hydrogen-bonding interactions with Gln859 in PI3Kα. Next, different side chains that could help restrict the conformation were introduced into the linker to explore the optimal conformation, such as methyl, gem-difluoro, amine, ethyl, isopropyl, and cyclopropyl (Table 1, entries 3–9). All of these compounds had good PI3Kα inhibition potency, but the selectivity over the other PI3K subtypes varied. Compound 3 with a single methyl substituent on the linker displayed the highest selectivity among all of these compounds. The pharmacokinetic profile of compound 3 was then evaluated in mice. Compound 3 seemed to have a moderate DMPK profile (see Table 3). However, when evaluated in a 28 day ovarian carcinoma SKOV-3 subcutaneous xenograft mouse model at doses of 10, 30, and 100 mg kg–1 day–1 delivered once daily by oral gavage, compound 3 did not show significant tumor growth inhibition. It is speculated that the low efficacy might be due to low drug concentration in tumor tissue resulting from the unfavorable structural feature of the primary amide. The primary amide has a high polar surface area, which may be detrimental to the distribution of compound 3 in the body.
Table 3. DMPK Profiles of Selected Compoundse.
| compd | Cmax (nM)a | DNAUC0–last (nM h)b | Cl (mL min–1 kg–1)c | F (%)d |
|---|---|---|---|---|
| 3 | 4453 | 1631 | 9.8 | 56.2 |
| 10 | 22167 | 3090 | 4.2 | 59.4 |
| 12 | 9133 | 723 | – | – |
| 18 | 4730 | 100 | – | – |
| 21 | 1441 | 302 | – | – |
| 40 | 10623 | 1021 | 5.5 | 18.4 |
| 42 | 5000 | 1372 | – | – |
| 43 | 24700 | 2018 | 9.0 | 9.0 |
| 44 | 24033 | 5704 | – | – |
| 45 | 37000 | 6260 | – | – |
Cmax: plasma maximum compound concentration.
DNAUC0–last: dose-normalized AUC0–last.
Cl: clearance.
F: biovailability.
po vehicle: 0.5% MC + 0.2% Tween 80. iv vehicle: 1:1 10% HP-βCD/10% solutol, pH 8.5.
To further improve the pharmacokinetic profile of compound 3, we then focused on the exploration of substitutions at the amide nitrogen. A series of compounds containing secondary and tertiary amide moieties were synthesized and tested. As shown in Table 2, most of the secondary amides could maintain both the potency and selectivity (compounds 10 and 12–23), and several molecules displayed good cellular activities (e.g., 10, 12 and 13). However, the enzymatic potency of the compounds with a tertiary amide against PI3Kα decreased significantly, along with the selectivity (Table 2, 24–26). This may be attributed to the loss of hydrogen-bond formation with Gln859, which is unique in PI3Kα. It was consistent with Novartis’s conclusion that the interactions with Gln859 and Ser854 were critical to the selectivity. The PI3Kα inhibitory potency of the compounds decreased with the replacement of methyl by more bulky groups, while the selectivity was maintained (Table 2, 10 vs 14, 15, and 19).
Table 2. Structures and PI3K Enzyme and Cell Inhibitory Potencies of Compounds 10–26.
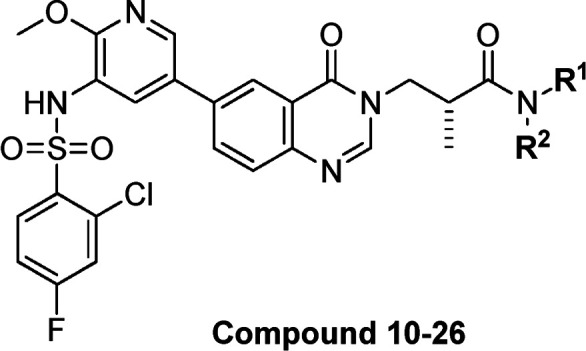

MCF-7 pAktS473 inhibitory IC50 values.
To better understand the absolute configuration of compound 10, the binding modes in PI3Kα ATP-binding pocket were simulated. According to the binding modes, only the R-configured compound 10 could form an extra hydrogen bond with Ser854 contributing to the crucial PI3Kα selectivity, while compound 11 could not (Figure 2). This was in accordance with the biochemical selectivity (Table 2, 10 vs 11).
Figure 2.
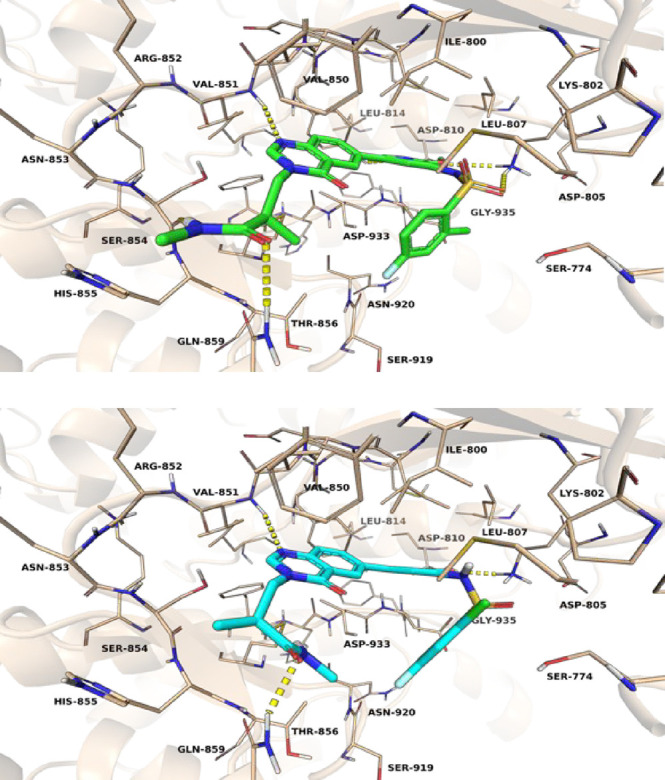
Simulated binding modes of compounds 10 (top) or 11 (bottom) with PI3Kα.
Compounds 10, 12, 18, and 21, which demonstrated good biochemical and cellular potencies together with reasonable selectivities, were chosen for evaluation of their DMPK profiles in animals (Table 3). Compound 10 with a terminal methyl amide showed the best overall PK behavior, achieving the highest plasma exposure, low clearance, and good bioavailability in mice. Hence, SAR exploration was continued based on compound 10.
With the optimal methyl amide side chain in the solvent-accessible region in hand, the left hydrophobic pocket substituents were then explored. Replacement of the methoxy group on the pyridine ring was first tested. As shown in Table 4, only compounds 31 and 32 with trifluoromethyl and methyl substituents showed PI3Kα potencies and selectivities comparable to those of 10 at the enzymatic level. Most of the compounds exhibited decreased or lost cellular potency. We postulated that the oxygen atom of methoxy group in compound 10 can form an internal hydrogen bond with the hydrogen atom of the sulfonamide, which decreased the acidity and polarity of the sulfonamide and led to better permeability. When more bulky substituents were introduced at this position, the PI3Kα potency decreased (Table 4, compounds 31 and 32 vs 27–30 and 33–37). As a result, the tolerability at the pyridine 2-position was limited. The 2-methoxy substituent in compound 10 seemed to be the optimal choice.
Table 4. Structures and PI3K Enzyme and Cell Inhibitory Potencies of Compounds 27–37.
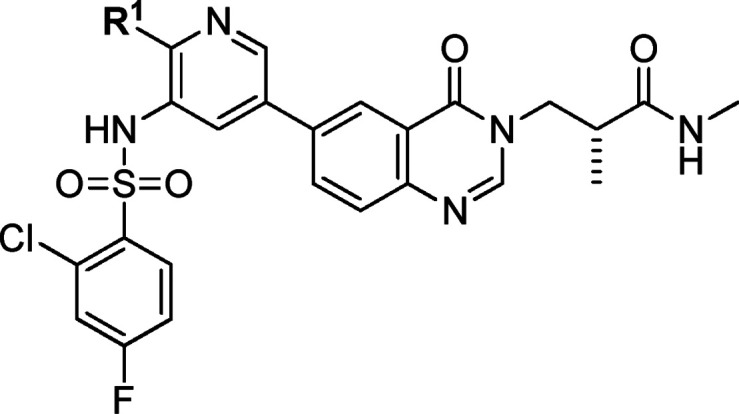
| compd | R1 | PI3Kα IC50 (nM) | selectivity folds over PI3Kβ/δ/γ | cellulara IC50 (nM) |
|---|---|---|---|---|
| 27 | –OCH2CH3 | 13.7 | 153/–/– | 116 |
| 28 | –OCH(CH3)2 | – | –/–/– | 387 |
| 29 | – OCH(CH2)2 | – | –/–/– | 55.3 |
| 30 (rac) | –OCF3 | 22.8 | 94.9/–/– | >1000 |
| 31 (rac) | –CF3 | 0.6 | 196/–/– | 972 |
| 32 | –CH3 | 3.0 | 406/17/21 | 397 |
| 33 | –CH2CH3 | 6.0 | 309/13.8/14.4 | 347 |
| 34 | –CH2OH | 30.0 | >334/17.9/12.5 | >1000 |
| 35 | –CH2OCH3 | >1000 | –/–/– | >1000 |
| 36 | –NH2 | 19.3 | 283/–/– | >1000 |
| 37 | –N(CH3)2 | 8.4 | 59.5/–/– | – |
MCF-7 pAktS473 inhibitory IC50 values.
Different substituted heterocycles or benzenesulfonamides were then explored, which were buried deep in the hydrophobic region of the binding pocket. As shown in Table 5, among several heterocyclic sulfonamides, only 40 demonstrated good in vitro potency together with reasonable selectivity, although with high PI3Kδ activity. The two monosubstituted phenyl sulfonamides 42 and 43 gave good results. Also, 2,3- and 2,4-disubstitution on the phenyl ring were favorable for the potency, and 2,3-dichloro-substituted 44 showed the best in vitro profile in terms of potency and selectivity. However, the potencies of 3,5- and 3,4-disubstituted phenyl sulfonamides were decreased significantly. Especially, 3,4-dichloro analogue 47 showed the lowest PI3Kβ selectivity.
Table 5. Structures and PI3K Enzyme and Cell Inhibitory Potencies of Compound 38-47.
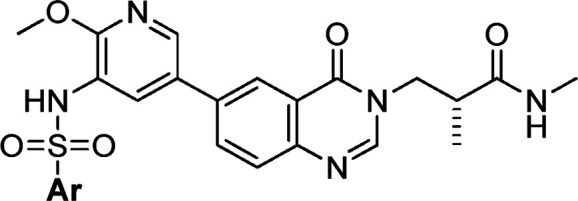

MCF-7 pAktS473 inhibitory IC50 values.
Several compounds were selected for profiling of the pharmacokinetic properties in mice (Table 3). Compounds 43, 44, and 45 displayed reasonable plasma exposure. The efficacies of these three compounds together with compound 10 were then evaluated in PIK3CA mutant human breast ductal carcinoma BT-474 mouse subcutaneous xenograft models.
When these compounds were orally dosed at 30 mg/kg once daily, compounds 44 and 45 demonstrated severe toxicity toward Balb/c nude mice, while the animals in groups dosed with compounds 10 and 43 could tolerate these compounds. After dosing at 30 mg/kg once daily for 21 days, compound 10 achieved the best efficacy, inhibiting tumor growth by 73.0% (Figure 3). Compounds 44 and 45 were tested further at lower dosages (10 and 20 mg/kg once daily), but neither showed significant efficacy (data not shown). 10 was then selected for further studies.
Figure 3.
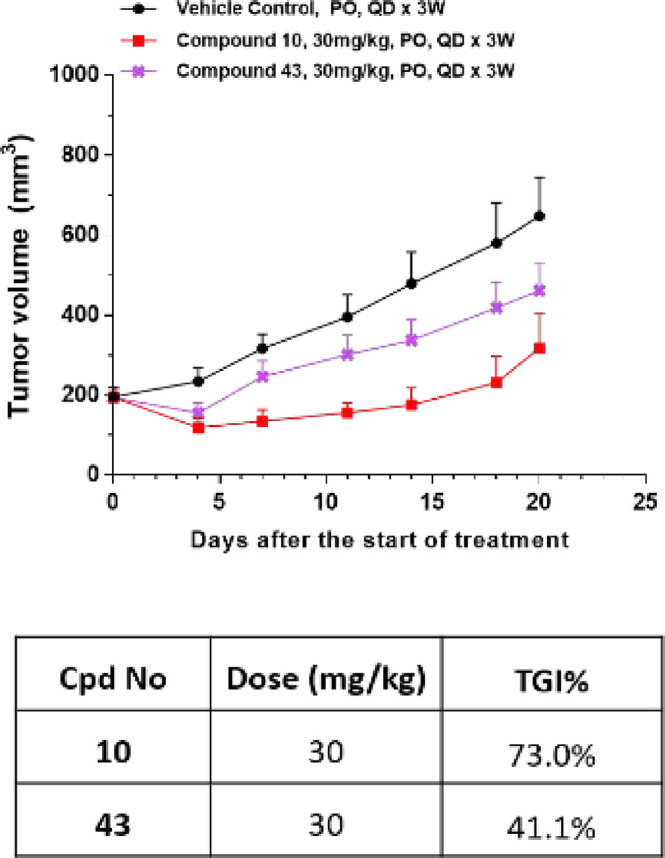
In vivo efficacies of compounds 10 and 43 in PIK3CA mutant human breast ductal carcinoma BT-474 mouse subcutaneous xenograft models (vehicle: 0.5% MC + 0.2% Tween 80).
The selectivity profile for compound 10 among the class I PI3K isoforms was also evaluated in cell-based assays.22 In cell-based assays assessing the ability to inhibit Akt phosphorylation, compound 10 was a potent inhibitor in cells sensitive to PI3Kα inhibition (IC50 = 12.1 nM in the PIK3CA mutant human breast ductal carcinoma BT-474 cell line) but not in cells sensitive to PI3Kβ inhibition (IC50 = 1393 nM in PTEN null breast adenocarcinoma MDA-MB-468 cells), PI3Kδ inhibition (IC50 = 183 nM in the Jeko-1 B cell line), or PI3Kγ inhibition (IC50 > 10 μM in the monocytic RAW264 cell line).
The multidose tumor inhibition of compound 10 was validated in a BT-474 mouse subcutaneous xenograft model. Compound 10 could dose-dependently suppress tumor growth by 62.5% (15 mpk), 86.0% (30 mpk), and 90.7% (40 mpk), respectively (Figure 4a), which was attributed to inhibition of the PI3Ks downstream critical node of Akt phosphorylation. Western blot studies confirmed that compound 10 inhibited the phosphorylation of Akt in a dose- and time-dependent manner (Figure 4b).
Figure 4.
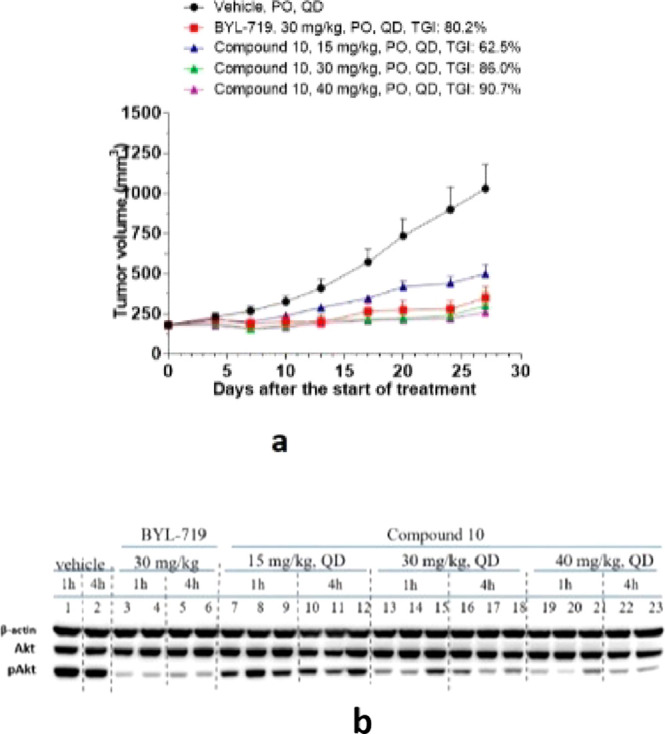
(a) In vivo efficacy of compound 10 with various doses in PIK3CA mutant human breast ductal carcinoma BT-474 mouse subcutaneous xenograft models. (b) Compound 10 inhibited the phosphorylation of Akt in a time- and dose-dependent manner. (Vehicle: 0.5% MC + 0.2% Tween 80.)
According to our structure–activity exploration, we discovered a series of novel selective PI3Kα inhibitors with a 2-substituted N-methylpropanamide side chain to contribute to the formation of hydrogen bonds with Gln859 and Ser854, which are unique in the PI3Kα isoform. To find the optimal conformation of compounds to form these interactions, several different substituents were introduced into the compounds. Among the substituents, (R)-2-methyl was the best one. This chiral R substituent could force the compound to adopt a conformation enabling interactions with Gln859 and Ser854, while the compound with the S substituent could interact only with Gln859. In conclusion, the (R)-2-methyl group could offer a conformation that the compound needed to improve the PI3Kα selectivity.
We can see significant pharmacokinetics profile improvement when compound 10 is compared with 3. The only structural difference of these two compounds is the terminal N-methyl substituent. The introduction of a methyl group into the terminal amide could contribute a lot to mouse in vivo plasma exposure, and the maximum concentration was also increased by nearly 5-fold. We concluded that methylation of the amide nitrogen enhanced the oral pharmacokinetics properties of this series of compounds.
To summarize, we have identified compound 10, a quinazolin-4(3H)-one derivative with a 2-substituted N-methylpropanamide substituent, as a potent and selective PI3Kα inhibitor. Additionally, 10 has excellent human metabolic stability in microsomal and hepatocyte incubations (Table 6). It demonstrated good in vitro potencies in enzymatic and cellular levels, decent drug-like properties, significant in vivo tumor suppression, and reasonable selectivities. The data support 10 to be a potential preclinical candidate, and further optimization and evaluation work will be reported in due course.
Table 6. Profiles of Compound 10a.
| compound 10 profile | results | |
|---|---|---|
| in vitro activity | enzymatic PI3Kα/β/δ/γ IC50 (nM) | 1.8/271.0/13.9/13.8 |
| PI3Kα/β/δ/γ cellular pAktS473 IC50 (nM)b | 12.1/1393/183/>10000 | |
| DMPK | mouse iv 1 mpk (t1/2, Vd, Cl) | 0.966 h, 0.179 L/kg, 4.16 mL min–1 kg–1 |
| mouse oral 10 mpk (Cmax, DNAUC0–last, F) | 22167 nM, 3090 nM h mg–1 kg–1, 59.4% | |
| rat iv 1 mpk (t1/2, Vd, Cl) | 1.22 h, 0.321 L/kg, 5.28 mL min–1 kg–1 | |
| rat oral 3 mpk (Cmax, DNAUC0–last, F) | 2327 nM, 2711 nM h mg–1 kg–1, 46.9% | |
| ADMET | PPB (human, mouse) | 98.6%, 98.6% |
| permeability 10–6 cm/s (A to B, B to A, efflux ratio) | 2.2, 28.5, 13.1 | |
| MMS remaining (T = 60 min) | 44.6% (h), 32.0% (r), 53.9% (d), 32.7% (m), 13.7% (monkey) | |
| HMS in vivo Clint (mL min–1 kg–1) | 641.9 (m), 58.9 (r), <44 (d), <17.8 (h), <23 (monkey) | |
| hERG IC50 (μM) | 27.5 | |
| CYP (1A2, 2C9, 2C19, 2D6, 3A4) IC50 (μM) | >50, 6.1, 21.6, >50, 23.3 | |
Abbreviations: Vd, volume distribution; Cmax, maximum compound concentration in plasma; F, bioavailability; DNAUC0–last, dose-normalized AUC0–last; Cl, clearance; PPB, plasma protein binding; MMS, microsome metabolism stability; HMS, hepatocyte metabolism stability.
PI3Kα/β/δ/γ specific cellular acitivities used the BT-474/MDA-MB-468/Jeko-1/RAW264 cell lines.
Acknowledgments
We thank the LTD Department, RSD Biology Department, and OIU Department of WuXi AppTec Co., Ltd. for pharmacokinetics studies, enzymatic potency and MCF-7 cellular studies, and mouse in vivo efficacy studies. We thank the Membrane Biology Division of HD Biosciences for BT-474/MDA-MB468/RAW264.7/Jeko-1 cellular studies.
Glossary
Abbreviations
- PI3K
phosphatidylinositol 3-kinase
- mTOR
mammalian target of rapamycin
- GI
gastrointestinal
- DMPK
pharmacokinetics
- Cl
clearance
- F%
bioavailability
- DNAUC
dose-normalized area under the curve
- SAR
structure–activity relationship
- mpk
milligrams per kilogram
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00239.
Experimental procedures and analytical data, chemistry schemes, and spectral data of compounds 1–47 (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All of the authors approved the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Zhang J.-Q.; Luo Y.-J.; Xiong Y.-S.; Yu Y.; Tu Z.-C.; Long Z.-J.; Lai X.-J.; Chen H.-X.; Luo Y.; Weng J.; Lu G. Design, synthesis, and biological evaluation of substituted pyrimidines as potential phosphatidylinositol 3-kinase (PI3K) inhibitors. J. Med. Chem. 2016, 59, 7268–7274. 10.1021/acs.jmedchem.6b00235. [DOI] [PubMed] [Google Scholar]
- Curigliano G.; Shah R. R. Safety and Tolerability of Phosphatidylinositol-3-Kinase (PI3K) Inhibitors in Oncology. Drug Saf. 2019, 42, 247–262. 10.1007/s40264-018-0778-4. [DOI] [PubMed] [Google Scholar]
- Heffron T. P.; Wei B.; Olivero A.; Staben S. T.; Tsui V.; Do S.; Dotson J.; Folkes A. J.; Goldsmith P.; Goldsmith R.; Gunzner J.; Lesnick J.; Lewis C.; Mathieu S.; Nonomiya J.; Shuttleworth S.; Sutherlin D. P.; Wan N. C.; Wang S.; Wiesmann C.; Zhu B.-Y. Rational Design of Phosphoinositide 3-Kinase α Inhibitors That Exhibit Selectivity over the Phosphoinositide 3-Kinase β Isoform. J. Med. Chem. 2011, 54, 7815–7833. 10.1021/jm2007084. [DOI] [PubMed] [Google Scholar]
- Thorpe L. M.; Yuzugullu H.; Zhao J. J. PI3K in cancer: divergent roles of isoforms, modes of activation and therapeutic targeting. Nat. Rev. Cancer 2015, 15, 7–24. 10.1038/nrc3860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis H.; Ma C. X. PI3K Inhibitors in Breast Cancer Therapy. Curr. Oncol. Rep. 2019, 21, 110. 10.1007/s11912-019-0846-7. [DOI] [PubMed] [Google Scholar]
- Hanker A. B.; Kaklamani V.; Arteaga C. L. Challenges for the Clinical Development of PI3K Inhibitors: Strategies to Improve Their Impact in Solid Tumors. Cancer Discovery 2019, 9, 482–491. 10.1158/2159-8290.CD-18-1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juric D.; Rodon J.; Tabernero J.; Janku F.; Burris H. A.; Schellens J. H. M.; Middleton M. R.; Berlin J.; Schuler M.; Gil-Martin M.; Rugo H. S.; Seggewiss-Bernhardt R.; Huang A.; Bootle D.; Demanse D.; Blumenstein L.; Coughlin C.; Quadt Q.; Baselga J. Phosphatidylinositol 3-Kinase α-Selective Inhibition With Alpelisib (BYL719) in PIK3CA-Altered Solid Tumors: Results From the First-in-Human Study. J. Clin. Oncol. 2018, 36, 1291–1299. 10.1200/JCO.2017.72.7107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janku F.; Yap T. A.; Meric-Bernstam F. Targeting the PI3K pathway in cancer: are we making headway?. Nat. Rev. Clin. Oncol. 2018, 15, 273–291. 10.1038/nrclinonc.2018.28. [DOI] [PubMed] [Google Scholar]
- Levine D. A.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. 10.1038/nature12113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- et al. Integrated genomic and molecular characterization of cervical cancer. Nature 2017, 543, 378–384. 10.1038/nature21386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comprehensive molecular portraits of human breast tumors. Nature 2012, 490, 61–70. 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerji S.; Cibulskis K.; Rangel-Escareno C.; et al. Sequence analysis of mutations and translocations across breast cancer subtypes. Nature 2012, 486, 405–409. 10.1038/nature11154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui V. W.; Hedberg M. L.; Li H.; et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discovery 2013, 3, 761–769. 10.1158/2159-8290.CD-13-0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine D. A.; Bogomolniy F.; Yee C. J.; et al. Frequent mutation of the PIK3CA gene in ovarian and breast cancers. Clin. Cancer Res. 2005, 11, 2875–2878. 10.1158/1078-0432.CCR-04-2142. [DOI] [PubMed] [Google Scholar]
- André F.; Ciruelos E.; Rubovszky G.; Campone M.; Loibl S.; Rugo H. S.; Iwata H.; Conte P.; Mayer I. A.; Kaufman B.; Yamashita T.; Lu Y.-S.; Inoue K.; Takahashi M.; Pápai Z.; Longin A.-S.; Mills D.; Wilke C.; Hirawat S.; Juric D. Alpelisib for PIKC3A-mutated, hormone receptor-positive advanced breast cancer. N. Engl. J. Med. 2019, 380, 1929–1940. 10.1056/NEJMoa1813904. [DOI] [PubMed] [Google Scholar]
- Furet P.; Guagnano V.; Fairhurst R. A.; Imbach-Weese P.; Bruce I.; Knapp M.; Fritsch C.; Blasco F.; Blanz J.; Aichholz R.; Hamon J.; Fabbro D.; Caravatti G. Discovery of NVP-BYL-719 a potent and selective phosphatidylinositol-3 kinase alpha inhibitor selected for clinical evaluation. Bioorg. Med. Chem. Lett. 2013, 23, 3741–3748. 10.1016/j.bmcl.2013.05.007. [DOI] [PubMed] [Google Scholar]
- Hui G.; Cheng W.; et al. Pyridino[1,2-a]pyrimidone analogue used as mTOR/PI3K inhibitor. WO 2015192761, 2015.
- Matsson P.; Kihlberg J. How Big Is Too Big for Cell Permeability?. J. Med. Chem. 2017, 60, 1662–1664. 10.1021/acs.jmedchem.7b00237. [DOI] [PubMed] [Google Scholar]
- Noji S.; Seki N.; Maeba T.; Sakai T.; Watanabe E.; Maeda K.; Fukushima K.; Noguchi T.; Ogawa K.; Toyonaga Y.; Negoro T.; Kawasaki H.; Shiozaki M. Concise SAR exploration based on the “Head-to-Tail” approach: discovery of PI4KIIIα inhibitors bearing diverse scaffolds. ACS Med. Chem. Lett. 2016, 7, 919–923. 10.1021/acsmedchemlett.6b00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H.; Xin M.-H.; Xie X.-X.; Mao S.; Zuo S.-J.; Lu S.-M.; Zhang S.-Q. Bioorg. Med. Chem. 2015, 23, 7765–7776. 10.1016/j.bmc.2015.11.027. [DOI] [PubMed] [Google Scholar]
- Barlaam B.; Cosulich S.; Delouvrié B.; Ellston R.; Fitzek M.; Germain H.; Green S.; Hancox U.; Harris C. S.; Hudson K.; Lambert-van der Brempt C.; Lebraud H.; Magnien F.; Lamorlette M.; Le Griffon A.; Morgentin R.; Ouvry G.; Page K.; Pasquet G.; Polanska U.; Ruston L.; Saleh T.; Vautier M.; Ward L. Discovery of 1-(4-(5-(5-amino-6-(5-tert-butyl-1,3,4-oxadiazol-2-yl)pyrazine-2-yl)-1-ethyl-1,2,4-triazol-3-yl)piperidin-1-yl)-3-hydroxypropan-1-one (AZD8835): A potent and selective inhibitor of PI3Kα and PI3Kδ for the treatment of cancers. Bioorg. Med. Chem. Lett. 2015, 25, 5155–5162. 10.1016/j.bmcl.2015.10.002. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.