

Abstract

HIV persistence in latently infected, resting CD4+ T cells is broadly considered a barrier to eradicate HIV. Activation of the provirus using latency-reversing agents (LRAs) followed by immune-mediated clearance to purge reservoirs has been touted as a promising therapeutic approach. Histone deacetylases (HDACs) and histone acetyltransferases (HATs) control the acetylation level of lysine residues in histones to regulate the gene transcription. Several clinical HDAC inhibitors had been examined as LRAs, which induced HIV activation in vitro and in vivo. Here we report the discovery of a series of selective and potent class I HDAC inhibitors based on aryl ketones as a zinc binding group, which reversed HIV latency using a Jurkat model of HIV latency in 2C4 cells. The SAR led to the discovery of a highly selective class I HDAC inhibitor 10 with excellent potency. HDACi 10 induces the HIV gag P24 protein in patient latent CD4+ T cells.
Keywords: HDAC, inhibitors, HIV, latency, LRA
Human Immunodeficiency virus (HIV) has been an epidemic for over 30 years. By 2019, 74.9 million people had been infected with HIV since the beginning of the epidemic and 37.9 million people globally were living with HIV.1 AIDS (acquired immune deficiency syndrome) caused by HIV used to be a universally fatal disease, but now can be managed as a chronic disease with the discovery and availability of combined antiretroviral therapy (cART). cART available in a single pill dosed once daily can control the virus in patients to below detectable levels, and various longer-acting combinations are in development. However, HIV is not curable with cART due to the establishment of a latent pool of HIV-infected resting memory CD4+ T cells that are transcriptionally silent.2 These HIV latent CD4+ T cells have an extremely long biological half-life and may be activated, resurrecting HIV gene transcription and causing HIV rebound when cART treatment is interrupted.3 Many HIV patients have difficulties continuing the cART treatment due to long-term toxicities, side effects, and the need for daily compliance to avoid viral resistance. Furthermore, life-long therapy presents a significant financial burden to global health care networks. It is also known that the low level of viremia in HIV patients contributes to chronic inflammation, immune dysfunction, and accelerated aging.4 Thus, there is a huge unmet medical need for an HIV cure treatment. One approach toward the HIV cure, often referred to as “shock and kill”,5 is to use pharmaceutical latency-reversing agents (LRAs) to reverse HIV-1 latency and turn on production of viral proteins in latently infected CD4+ T cells. This activation would expose these cells to killing by an immune-mediated mechanism or via cytopathic effects. LRAs that have been identified through mechanism-directed approaches include protein kinase C agonists, histone deacetylase inhibitors (HDACi), histone methylation inhibitors, DNA methyltransferase inhibitors, inhibitors of bromodomain and extra terminal domain proteins, and unclassified agents such as disulfiram.6
Histone deacetylases (HDACs) and histone acetyl transferases (HATs) together regulate the acetylation level of lysine residues of histones. Histone acetylation diminishes the charge–charge interaction between nucleic acids in DNA, and this relaxed conformation induces gene expression.7,8 Eighteen unique isoforms of HDACs have been discovered and are separated into four distinct classes.9 Class I (HDACs 1, 2, 3, and 8), Class II (HDACs 4, 5, 6, 7, 9, and 10), and Class IV (HDAC11) are Zn2+-dependent, and Class III are the NAD+-dependent Sirtuins (SirT1–7). HDAC inhibitors have been developed for oncology, and recently these HDACs have been broadly explored as LRAs. The clinical studies have shown that single and multiple administrations result in induced RNA transcription in CD4+ T cells from cART suppressed HIV+ subjects10−12 using vorinostat, panobinostat, and romidepsin. We recently reported that clinical administration of histone deacetylase inhibitors (vorinostat and panobinostat) induced HIV gag p24, and ex vivo stimulation produced sufficient viral antigen to elicit immune mediated cell killing using anti-gp120/CD3 bispecific antibody.13 However, these repurposed HDACis tested in clinical trials are pan-HDAC inhibitors without any subtype selectivity, which cause significant adverse effects (AEs) in clinical tests.14−16 The AEs may be acceptable for short-term cancer chemotherapies. However, the AEs would limit the doses for potential chronic treatment of HIV patients. Subtype selective HDAC inhibitors may be helpful to increase the therapeutic window for HIV activation.
Our collaborators at IRBM recently disclosed17 a series of selective HDAC inhibitors based on ethyl ketone, exemplified by HDACi 1 shown in Figure 1. HDACi 1 is a class I HDAC inhibitor with potent inhibitory activity for HDAC1–3 enzymes. We also profiled HDACi 1 for the ability to reverse HIV latency, using a Jurkat model of HIV latency (2C4 cells), which was produced in the same manner as a similar Jurkat T-cell line that utilized an eGFP reporter gene.18 In this cell line, reactivation of a quiescent HIV provirus is measured by quantification of a luciferase reporter gene in the HIV provirus. HDACi 1 shows moderate activation efficacy with EC50 at 198 nM in this cell assay. We obtained the X-ray crystal structure of a complex of HDACi 1 with the HDAC2 enzyme at high resolution of 1.6 Å. Several key interactions between HDACi 1 and the enzyme are observed. The ketone exists as the hydrate and forms bidentate chelation with the metal zinc ion. The linear alkane chain linker goes through the narrow hydrophobic tunnel and connects the surface binding group and the ketone. The amide NH and the imidazole form a bidentate chelation with the enzyme Asp104 side chain carboxylic acid. The imidazole and the amide carbonyl form hydrogen bonds with water molecules in the water network. The bicyclic heteroaromatic methoxy quinoline has van der Waals interactions with the protein surface. The ketone is presumed to become hydrated with the water bound to zinc in the HDAC enzyme after binding to the pocket. This X-ray crystal structure shows that there is a foot pocket under the zinc binding ketone, which has also been reported previously.19 Replacement with an aromatic ring for the ethyl group is possible to fit in the foot pocket to potentially improve the selectivity for class I HDACs, and at the same time enhance the potency and physicochemical properties. Here we report a series of potent and selective class I HDAC inhibitors based on aryl ketones as the zinc binding group for reversing HIV latency.
Figure 1.
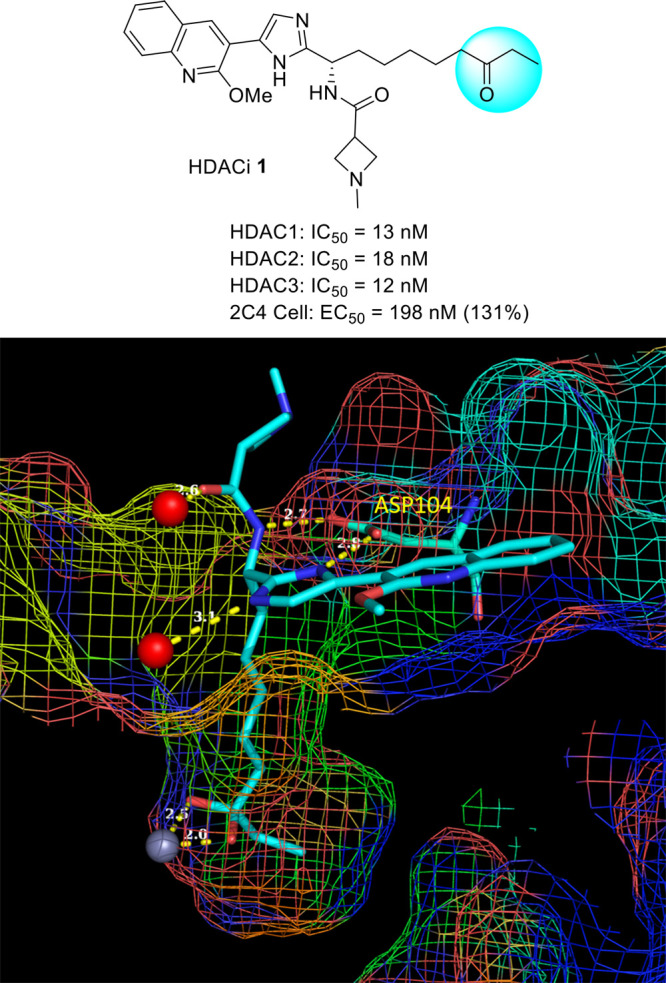
X-ray crystal structure of HDACi 1 bound to HDAC2 enzyme.
A synthetic route was designed for the quick exploration of the different aryl ketones as zinc binding groups, depicted in Scheme 1. We prepared two building blocks with terminal double bonds for cross metathesis to quickly explore the structure activity relationships (SAR) of different heteroaryl ketones as the zinc binding group and different substitutions on the amide, the imidazole replacements, and the aryl substitutions on the imidazole. This synthesis is exemplified by the preparation of compound 10. (S)-2-Boc-amino-4-pentenoic acid 2 was converted to an ester quantitively by 2-fluoro-2′-bromoacetylphenone 3 in DMF with cesium carbonate as the base. This ester then condensed with ammonium acetate in toluene under heating to form the imidazole ring, which was then protected with Boc anhydride catalyzed with dimethyl aminopyridine to afford compound 4 in excellent yield. The isoxazole-3-carboxylic acid 5 was converted to the Weinreb amide 6, which later reacted with but-3-en-1-yl magnesium bromide in THF at elevated temperature to provide the aryl ketone compound 7 in good yield. The cross-metathesis reaction between compounds 4 and 7 was successfully carried out in toluene catalyzed by Umicore M71 SIPr ruthenium catalyst20 to form the coupled product 8 with good yield. We experienced much poorer yield for the cross metathesis if the imidazole in compound 4 is not protected by Boc. The Boc protection on both the imidazole and the amino groups in compound 8 was then removed by treatment of trifluoroacetic acid in methylene chloride, followed by double bond reduction on the resulting product using palladium on carbon catalyzed hydrogenation to give compound 9. The free amine was finally coupled with 1-methylazetidine-3-carboxylic acid using (1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate in DMF/DCM with N-methylmorpholine as base to generate compound 10 in good yield.21
Scheme 1.

Reagents and conditions: (i) Cs2CO3, DMF, 2 h, 100%; (ii) NH4OAc, toluene, 110 °C, 3 h; (iii) Boc2O, DMAP, CH2Cl2, rt, 3 h, 92% two steps; (iv) EDCI, HOBT, MMP, MeONHMe·HCl, DMF/DCM, overnight, 72%; (v) CH2=CHCH2CH2MgBr, THF, 60 °C, 3 h, 100%; (vi) UMICOREM71 SIPR, toluene, 55 °C, 3 h, 78%; (vii) TFA/CH2Cl2, rt, 92%; (viii) Pd/C, H2, MeOH, 92%; (ix) 1-methylazetidine-3-carboxylic acid, MMP, COMU, CH2Cl2/DMF, 79%.
In the similar synthetic sequence depicted in Scheme 1, the compounds in Table 1 were prepared applying different starting materials for the substitutions Ar1, Ar2, and R in the general structures. These compounds were evaluated in the inhibitory activity and selectivity for HDAC1, 2, 3, 6, and 8, as well as in a cellular assay for activation of HIV latency. For this initial SAR study searching for a heteroaryl ketone, several five-membered hetero aryls and the pyridinyl were prepared. The 3-isoxazolyl ketone in compound 10 shows the highest potency with subnanomolar inhibition to HDAC1, 2, and 3. This inhibition translated to exceptionally high potency in the cellular HIV latency activation assay with EC50 of 4.7 nM. Compound 10 also has excellent selectivity over HDAC6 and moderate selectivity over HDAC8 (37 fold to HDAC1). The 2-oxazolyl (Compound 11) and 2-thiazolyl (Compound 12) are less potent by at least 10-fold in the inhibitory assay compare to 10. 4-Methyl substitution on the oxazolyl (Compound 13) causes further loss of inhibitory activity, while the 5-methyl substitution on the 3-isoxazolyl (Compound 14) is more tolerated with only less than 2-fold loss of potency. On the other hand, six-membered aryl group such as 2-pyridinyl (Compound 15) is much weaker in the inhibitory assays, possibly due to the larger aromatic ring size and its unfit shape.
Table 1. HDAC Inhibitors with Aryl Ketones as the Zinc Binding Groups.

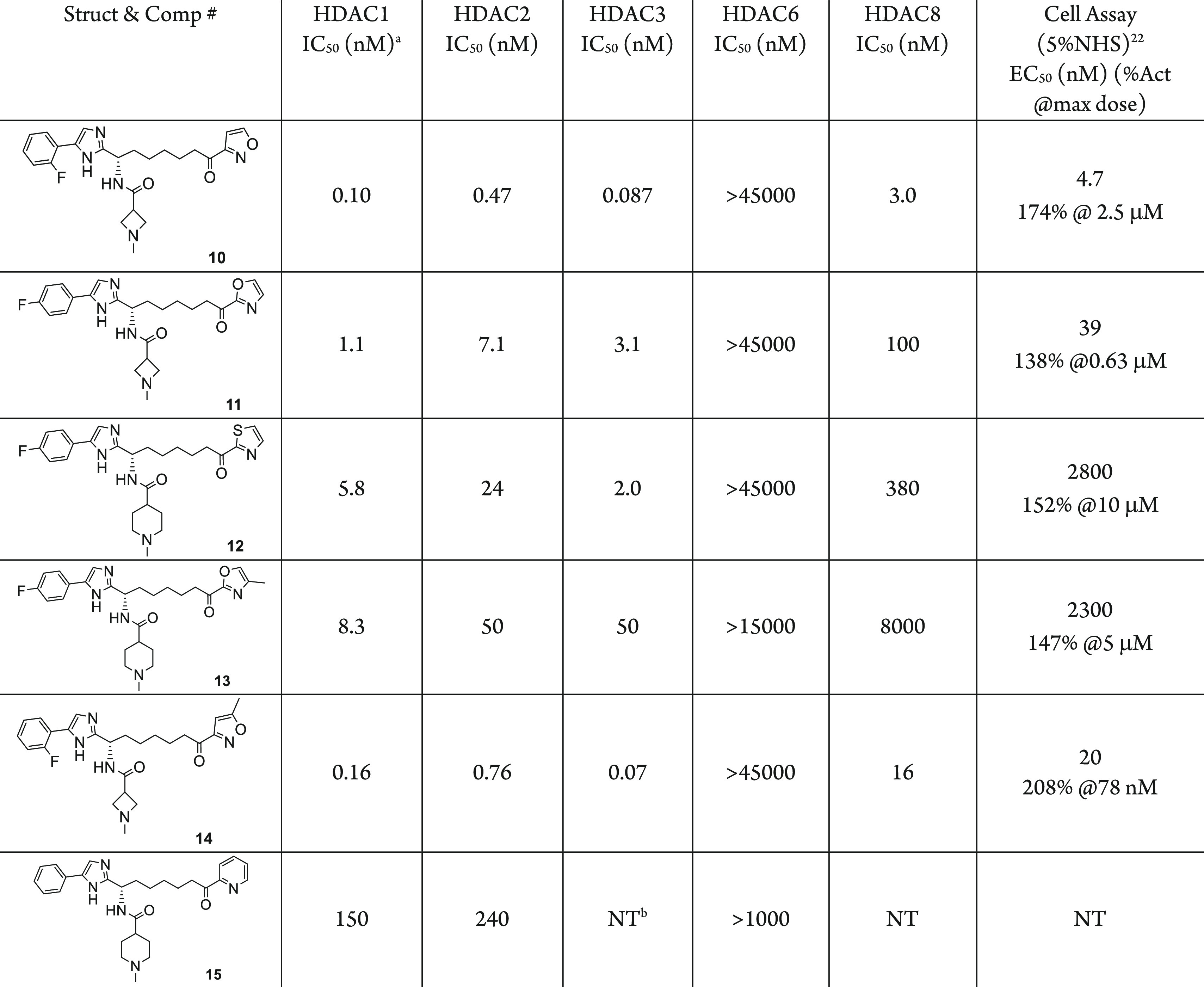
Reported values are the average of ≥2 independent measurements with standard deviation less than 3-fold of the reported mean.
NT = not tested.
To investigate the interaction of compound 10 with the HDAC enzyme, we obtained an X-ray crystal structure of HDACi 10 bound to the HDAC2 enzyme with a resolution of 1.6 Å (Figure 2).23 HDACi 10 and HDACi 1 have similar interactions with the HDAC2 protein at the surface, the hydrophobic tunnel, the zinc, and the catalytic residues His141, His142, and Tyr304. The key interactions include the bidentate hydrogen bond of the amide NH and imidazole with the side chain of Asp104 and the van der Waal’s interaction between the aromatic substitution on the imidazole and the HDAC2 surface (Figure 2A). The amide carbonyl oxygen and the imidazole 3-position nitrogen each have a hydrogen bond interaction with one water molecule in the water network, which was also observed in the crystal structures of HDACi 1 with the HDAC2 enzyme. The ketone hydrate makes bidentate inner coordination sphere contacts to the catalytic zinc ion at the bottom of the tunnel. The enhanced binding potency is introduced by the exceptional fit of the isoxazole ring in the foot pocket where it contacts Met31, Gly301, Gly302, Tyr304, Leu140, His141, and Cys152 (Figure 2B). The structure suggests that substitutions on the isoxazole, or replacement of the isoxazole with larger aryl ring systems, would likely disrupt these optimal contacts and result in steric clashes with the HDAC2 foot pocket.
Figure 2.

X-ray crystal structure of HDACi 10 bound to HDAC2 enzyme. (A) Top view; (B) Foot pocket. Yellow dashes = vdW contacts; Red dashes = Hydrogen bonds; Green dashes = Hydrogen bond donor−π interactions; Orange dashes = π–π interactions.
The favorable interactions of HDACi 10 in the foot pocket with the enzymes led to very potent HDAC 1/2/3 inhibition and the most potent activity in the HIV latency assay. We thus evaluated the intermediate compound 9 in these in vitro assays. Even without the amide, compound 9 still shows excellent HDAC inhibitory activity with an IC50 of 8.5 nM against HDAC2 and cellular potency in the HIV latency activation with EC50 of 178 nM. This 18-fold loss in potency compared to compound 10 presumably is caused by the loss of a hydrogen bond interaction between the amide carbonyl oxygen and the water network, and the weaker interaction of the amine nitrogen to Asp-104 side chain acid. These results encouraged us to continue the SAR exploration using the amine analogs. HDAC inhibitors (Table 2) using additional five membered heterocycle replacements were synthesized applying the similar chemistry as in Scheme 1 and evaluated in the in vitro assays. The 4-oxazolyl (16), 3-pyrazolyl (17), and 2-imidazolyl (18) all resulted in over 100-fold loss of inhibitory potency compared to compound 9, with no cellular activity. The methyl substitution on the 4-isoxazolyl and 5-hydroxymethyl-2-oxazolyl also led to over 138-fold loss of inhibitory activity. Based on this SAR result, we selected the 3-isoxazolyl ketone as the zinc binding group for further SAR investigation.
Table 2. HDAC Inhibitors with Aryl Ketones as the Zinc Binding Groups.

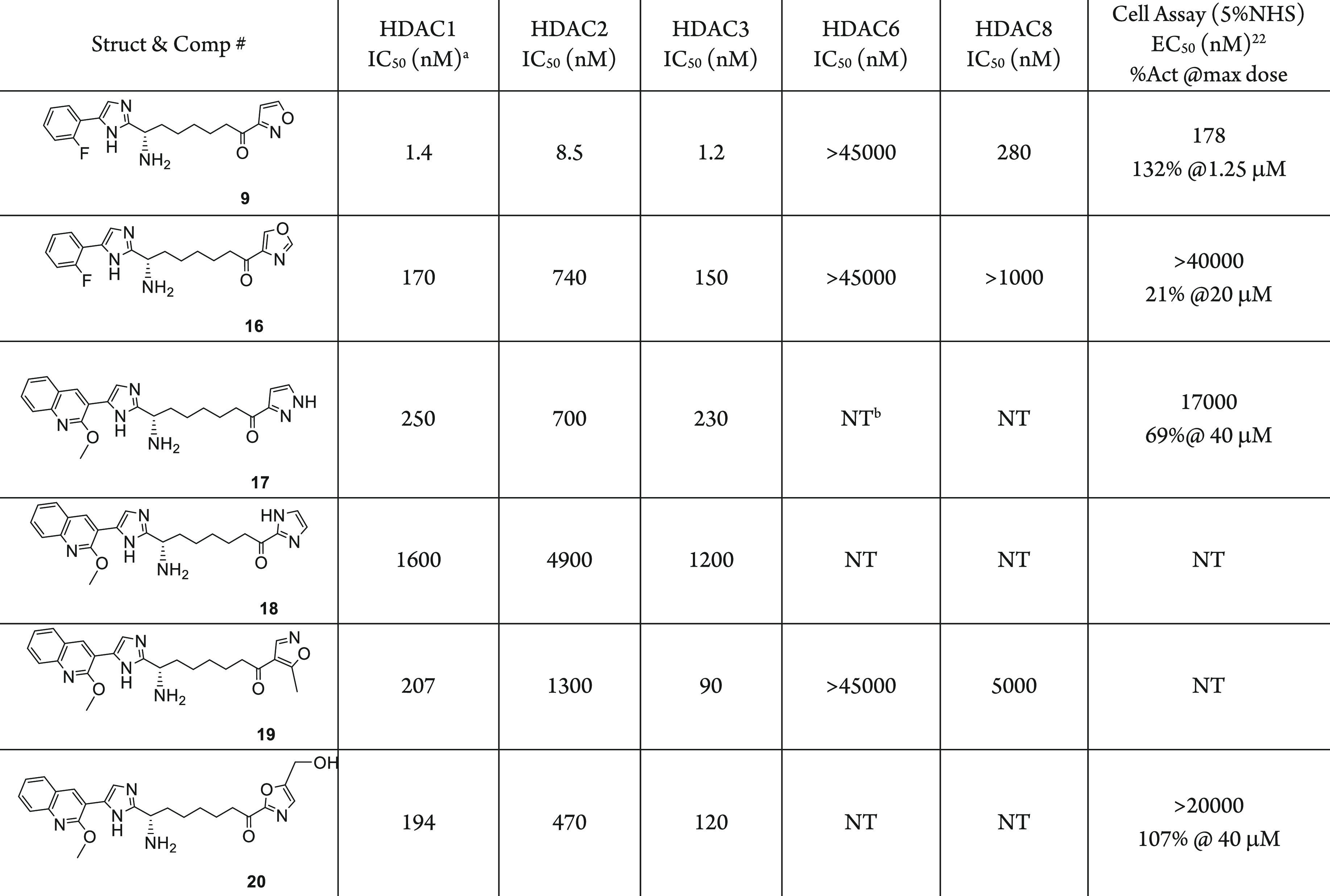
Reported values are the average of ≥2 independent measurements with standard deviation less than 3-fold of the reported mean.
NT = not tested.
We also carried out the SAR to search for heterocyclic replacements to the imidazole ring using the same synthetic strategy as depicted in Scheme 1 for the synthesis of compounds 9 and 10. We recently reported our work on HDAC inhibitors using ethyl ketone as the zinc binding group, in which we described the SAR for the replacement of the surface imidazole.24,25 We aimed to reduce a hydrogen bond donor in an attempt to improve the permeability of the compound. The isoxazole in place of imidazole (21) is well tolerated with only a 2-fold loss of inhibitory activity. However, the N-aryl triazole (22) and the two N-aryl pyrazole (23, 24) all cause significant loss of HDAC inhibitory potency (Table 3). We used oxazole and oxadiazole in the ethylketone HDAC inhibitors, and a similar impact on the inhibitory potency was observed.24 Compound 25 with an oxazole replacement for imidazole showed about 2-fold loss of activity compared to compound 10. By contrast, the oxadiazole replacement compound 26 had over 500-fold loss of inhibitory activity.
Table 3. HDAC Inhibitors on SAR for Imidazole Replacements.

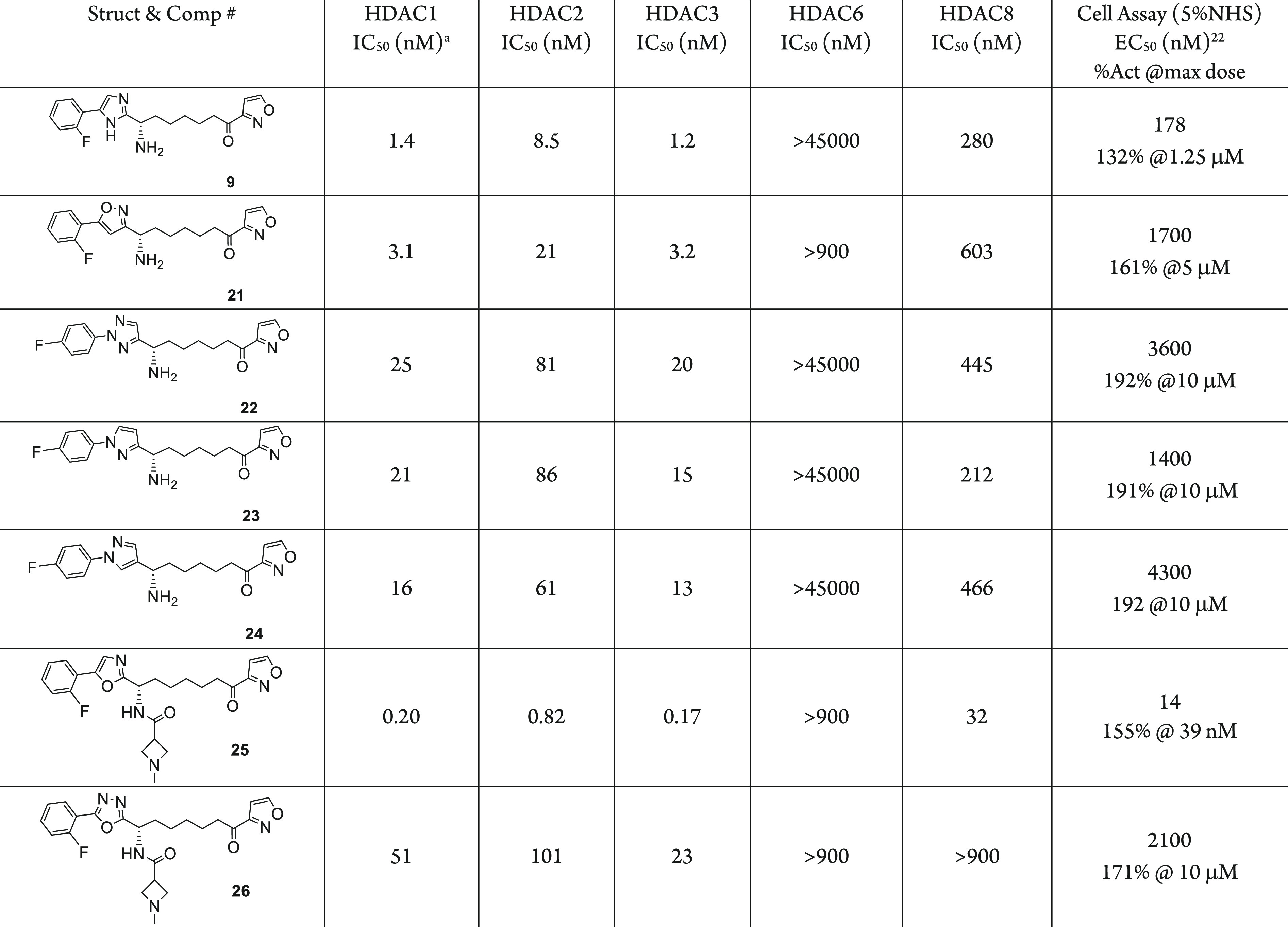
Reported values are the average of ≥2 independent measurements with standard deviation less than 3-fold of the reported mean.
NT = not tested.
We also investigated the binding inhibitory kinetic profile of HDACi 1 and 10, to understand the enormous improvement of inhibitory potency of the 3-isoxazolyl ketone compared to the ethyl ketone. We tested the kinetics of the binding and inhibitory activity to the HDAC3 enzyme applying a global progress curve analysis (GPCA, see the Supporting Information for details). The ethyl ketone HDACi 1 is a fast on (kon = 1.70 × 106 /nM/min) and fast off inhibitor with a residence time of only 5.6 min. The 3-isoxazolyl ketone HDACi 10 is a relatively slower on (kon = 1.82 × 10–2 /nM/min) inhibitor, which still reaches full inhibition within an hour. However, HDACi 10 is an inhibitor with very slow off rate having a residence time of 35 h. This long residence time is presumed to be caused by the tight fit of the isoxazole in the foot pocket. We hypothesize that this long residence time of HDACi 10 may provide prolonged efficacy.
The HDACi 10 was then selected for counter screen against other subtype HDAC enzymes. It is a potent inhibitor to class I HDAC enzymes HDAC1, 2, 3, and has excellent selectivity against other HDACs, except moderate selectivity over HDAC8 (38 fold over HDAC1) (Table 4). These selectivities are the results of the specific interactions described before between compound 10 and the HDAC enzymes.
Table 4. HDACi 10 Selectivity over HDAC Enzymes.
| Comp # | HDAC1 IC50 (nM) | HDAC2 IC50 (nM) | HDAC3 IC50 (nM) | HDAC4 IC50 (μM) | HDAC5 IC50 (μM) | HDAC6 IC50 (μM) | HDAC7 IC50 (μM) | HDAC8 IC50 (nM) | HDAC11 IC50 (μM) |
|---|---|---|---|---|---|---|---|---|---|
| 10 | 0.08 | 0.47 | 0.09 | >50 | >50 | >45 | >50 | 3.0 | >50 |
The HDACi 10 was evaluated in pharmacokinetic tests in several preclinical species for predicting the dose of in vivo assays. This compound has quite high total clearance in all the tested preclinical species mouse, rat, and dog, however with reasonable half-life because of its quite high volume of distribution (Table 5). The oral bioavailabilities in mouse, rat, and dog are moderate with F% as 18, 16, and 7.2, respectively. The plasma protein binding fractions of compound 10 are also listed in Table 5 for these preclinical species. The plasma protein binding fraction in human plasma is 91.5%. Thus, compound 9 has reasonable free fraction in plasma from all species. HDACi 10 demonstrated its in vivo efficacy by elevating the H4 acetylation level and the H1F0 gene expression when dosed orally to rats, which will be reported in detail elsewhere.26
Table 5. Pharmacokinetic Parameters of Compound 10.
| Mouse |
Rat |
Dog |
||||
|---|---|---|---|---|---|---|
| PK Parameters | IV dose | PO dose | IV dose | PO dose | IV dose | PO dose |
| Dose (mg/kg) | 2 | 10 | 0.5 | 5 | 1 | 2 |
| Cl (mL/min/kg) | 54 | 156 | 61 | |||
| Vss (L/kg) | 3.9 | 37 | 1.5 | |||
| AUCN (μM·h) | 1.38 | 0.118 | 0.188 | 0.61 | ||
| T1/2 (h) | 5.2 | 5.5 | 2.3 | |||
| Cmax (μM) | 0.48 | 0.050 | 0.071 | |||
| Tmax (h) | 0.25 | 0.38 | 0.75 | |||
| F (%) | 18 | 16 | 7.2 | |||
| Plasma protein binding % | 96.2 | 88.6 | 91.2 | |||
Plasma protein binding % in human plasma is 91.5.
We recently reported a robust method to detect HIV gag p24 in CD4+ T cell lysates from aviremic HIV+ individuals following viral reactivation with LRAs.13 Using this method, we evaluated HDACi 10 in reversing HIV latency from aviremic patient CD4+ T cells and inducing HIV gag p24 proteins (Figure 3). CD4+ T cells isolated from the blood of HIV+ individuals were treated with vehicle (DMSO), HDACi 10 (50 nM, about 1xEC50 free), and vorinostat (400 nM) for 72 h. CD4+ T cells were lysed, and gag p24 protein was quantified. HDACi 10 significantly induces HIV gag p24 protein at 0.06 pg/mL with significant difference (n = 3, p < 0.05) from vehicle (DMSO) treatment (Figure 3A).This induction is to a greater extent than observed following treatment with 400 nM vorinostat, which is a much weaker HDAC inhibitor with EC50 of 1600 nM in the 2C4 cell activation assay. The higher concentration of vorinostat (2000 nM) caused significant cell death due to its cytotoxicity. Furthermore, dose titration of HDACi 10 from 15 nM to 500 nM resulted in increased levels of viral protein (Figure 3B). For the dose increase to 150 nM and 500 nM, the HIV gag p24 protein expression decreased, presumably due to cytotoxicity of HDACi 10 at high concentration for extended duration in culture. This class-I HDACi 10 was evaluated in the safety test in the rodent model which showed a limited safety margin; however, the report is out of the scope for this letter and would be published separately.
Figure 3.

HDACi 10 activates cART-suppressed HIV patient CD4+ T cells with detectable HIV gag p24 proteins. (a) HDACi 10 activates aviremic patient’s CD4+ T cell HIV latency and induces HIV gag p24 proteins significantly higher than the vehicle, vorinostat. (n = 3, p < 0.05). (b) Dose titration of HDACi 10 on activation of aviremic patient’s CD4+ T cell HIV latency and induces HIV gag p24 proteins.
In summary, we described the identification of a series of class-I HDAC inhibitors based on an aryl ketone. SAR of the aryl ketone led to the discovery of the 3-isoxazolyl ketone as the best zinc binding group for the HDAC inhibitors, which have excellent HDAC1, 2, 3 inhibitory activity and are highly selective against other subtype HDACs. The X-ray crystal structure of HDACi 10 bound to the HDAC2 enzyme demonstrates that favorable interactions of the isoxazole group in the foot pocket of HDAC2 lead to the high potency. The lead HDACi 10 can activate HIV latency in a Jurkat model using 2C4 cells, with exceptional single digit nanomolar potency. HDACi 10 has an acceptable pharmacokinetic profile in preclinical species with moderate oral exposure. The binding inhibitory kinetic assays demonstrated that the 3-isoxazolyl ketone HDACi 10 has a very slow off rate from the HDAC3 enzyme. Furthermore, HDACi 10 activates ART-suppressed HIV+ patient latent CD4+ T cells and induces the expression of HIV gag p24 proteins. Overall, we have identified a series of class-I HDAC inhibitors with excellent inhibitory potency and selectivity, in which the lead HDACi 10 demonstrates efficacy to activate HIV latency and induces protein p24 for potential cellular kill of HIV latent CD4+ T cells when combined with other killing methods.
Glossary
Abreviations
- HDAC
histone deacetylase
- HDACi
histone deacetylase inhibitor
- HIV
human immunodefiency virus
- LRA
latency reversing agency
- AIDS
acquired immune deficiency syndrome
- cART
combined antiretroviral therapy
- AEs
adverse effects
- SAR
structure activity relation
- DMF
dimethylformaldehyde
- THF
tetrahydrofuran
- DCM
dichloromethane
- DMAP
4-dimethylaminopyridine
- EDCI
1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride
- HOBT
1-hydroxybenzotriazole hydrate
- MMP
4-methylmorpholine
- TFA
trifluoroacetic acid
- MeOH
methanol
- COMU
(1-cyano-2-ethoxy-2-oxoethylidenaminooxy)dimethylamino-morpholino-carbenium hexafluorophosphate
- GPCA
global progress curve analysis
- PK
pharmacokinetic.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00302.
Synthetic methods, characterization of key compounds, biology assay protocols, and data (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- UNAIDS website, Global HIV & AIDS statistics - 2019 fact sheet. URL (accessed Jun 12, 2020): https://www.unaids.org/en/resources/fact-sheet.
- Barton K.; Burch B.; Soriano-Sarabia N.; Margolis D. M. Prospects for treatment of latent HIV. Clin. Pharmacol. Ther. 2013, 93 (1), 46–56. 10.1038/clpt.2012.202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siliciano J. D.; Kajdas J.; Finzi D.; Quinn T. C.; Chadwick K.; Margolick J. B.; Kovacs C.; Gange S. J.; Siliciano R. F. Long-term follow-up studies confirm the stability of the latent reservoir for HIV-1 in resting CD4+ T cells. Nat. Med. 2003, 9, 727. 10.1038/nm880. [DOI] [PubMed] [Google Scholar]
- Nasi M.; Pinti M.; De Biasi S.; Gibellini L.; Ferraro D.; Mussini C.; Cossarizza A. Aging with HIV infection: A journey to the center of inflammAIDS, immunosenescence and neuro HIV. Immunol. Lett. 2014, 162, 329. 10.1016/j.imlet.2014.06.012. [DOI] [PubMed] [Google Scholar]
- Deeks S. Shock and Kill. Nature 2012, 487, 439. 10.1038/487439a. [DOI] [PubMed] [Google Scholar]
- For review:Darcis G.; Driessche B. V.; Lint G. V. HIV Latency: Should We Shock or Lock. Trends Immunol. 2017, 38, 217–228. 10.1016/j.it.2016.12.003. [DOI] [PubMed] [Google Scholar]
- Grunstein M. Histone acetylation in chromatin structure and transcription. Nature 1997, 389, 349–352. 10.1038/38664. [DOI] [PubMed] [Google Scholar]
- Pazin M. J.; Kadonaga J. T. What’s Up and Down with Histone Deacetylation and Transcription?. Cell 1997, 89, 325–328. 10.1016/S0092-8674(00)80211-1. [DOI] [PubMed] [Google Scholar]
- Gregoretti L.; Lee Y.; Goodson H. V. Molecular Evolution of the Histone Deacetylase Family: Functional Implications of Phylogenetic Analysis. J. Mol. Biol. 2004, 338, 17. 10.1016/j.jmb.2004.02.006. [DOI] [PubMed] [Google Scholar]
- Archin N. M.; Liberty A. L.; Kashuba A. D.; Choudhary S. K.; Kuruc J. D.; Crooks A. M.; Parker D. C.; Anderson E. M.; Kearney M. F.; Strain M. C.; Richman D. D.; Hudgens M. G.; Bosch R. J.; Coffin J. M.; Eron J. J.; Hazuda D. J.; Margolis D. M. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487 (7408), 482–5. 10.1038/nature11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen T. A.; Tolstrup M.; Brinkmann C. R.; Olesen R.; Erikstrup C.; Solomon A.; Winckelmann A.; Palmer S.; Dinarello C.; Buzon M.; Lichterfeld M.; Lewin S. R.; Ostergaard L.; Sogaard O. S. Panobinostat, a histone deacetylase inhibitor, for latent-virus reactivation in HIV-infected patients on suppressive antiretroviral therapy: a phase 1/2, single group clinical trial. Lancet HIV 2014, 1 (1), e13–21. 10.1016/S2352-3018(14)70014-1. [DOI] [PubMed] [Google Scholar]
- Tapia G.; Højen J. F.; Ökvist M.; Olesen R.; Leth S.; Nissen S. K.; VanBelzen D. J.; O’Doherty U.; Mørk A.; Krogsgaard K.; Søgaard O. S.; Østergaard L.; Tolstrup M.; Pantaleo G.; Sommerfelt M. A. Sequential Vacc-4x and romidepsin during combination antiretroviral therapy (cART): Immune responses to Vacc-4x regions on p24 and changes in HIV reservoirs. J. Infect. 2017, 75, 555–571. 10.1016/j.jinf.2017.09.004. [DOI] [PubMed] [Google Scholar]
- Wu G.; Swanson M.; Talla A.; Graham D.; Strizki J.; Gorman D.; Barnard R. J. O.; Blair W.; Søgaard O. S.; Tolstrup M.; Østergaard L.; Rasmussen T. A.; Sekaly R.-P.; Archin N. M.; Margolis D. M.; Hazuda D. J.; Howell B. J. HDAC inhibition induces HIV-1 protein and enables immune-based clearance following latency reversal. JCI Insight 2017, 2, e92901 10.1172/jci.insight.92901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvic M.; Talpur R.; Ni X.; Zhang C.; Hazarika P.; Kelly C.; Chiao J. H.; Reilly J. F.; Ricker J. L.; Richon V. M.; Frankel S. R. Phase 2 trial of oral vorinostat (suberoylanilide hydroxamic acid, SAHA) for refractory cutaneous T-cell lymphoma (CTCL). Blood 2007, 109 (1), 31. 10.1182/blood-2006-06-025999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis L.; Pan Y.; Smyth G. K.; George D. J.; McCormack C.; Williams-Truax R.; Mita M.; Beck J.; Burris H.; Ryan G.; Atadja P.; Butterfoss D.; Dugan M.; Culver K.; Johnstone R. W.; Prince H. M. Histone Deacetylase Inhibitor Panobinostat Induces Clinical Responses with Associated Alterations in Gene Expression Profiles in CutaneousT-Cell Lymphoma. Clin. Cancer Res. 2008, 14 (14), 4500. 10.1158/1078-0432.CCR-07-4262. [DOI] [PubMed] [Google Scholar]
- Søgaard O. S.; Graversen M. E.; Leth S.; Olesen R.; Brinkmann C. R.; Nissen S. K.; Kjaer A. S.; Schleimann M. H.; Denton P. W.; Hey-Cunningham W. J.; Koelsch K. K.; Pantaleo G.; Krogsgaard K.; Sommerfelt M.; Fromentin R.; Chomont N.; Rasmussen T. A.; Østergaard L.; Tolstrup M. The Depsipeptide Romidepsin Reverses HIV-1 Latency In Vivo. PLoS Pathog. 2015, 11 (9), e1005142 10.1371/journal.ppat.1005142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresciani A.; Ontoria J. M.; Biancofiore I.; Cellucci A.; Ciammaichella A.; Di Marco A.; Ferrigno F.; Francone A.; Malancona S.; Monteagudo E.; Nizi E.; Pace P.; Ponzi S.; Rossetti I.; Veneziano M.; Summa V.; Harper S. Improved Selective Class I HDAC and Novel Selective HDAC3 Inhibitors: Beyond Hydroxamic Acids and Benzamides. ACS Med. Chem. Lett. 2019, 10 (4), 481–486. 10.1021/acsmedchemlett.8b00517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson R.; Kim Y. K.; Hokello J.; Lassen K.; Friedman J.; Tyagi M.; Karn J. Epigenetic Silencing of Human Immunodeficiency Virus (HIV) Transcription by Formation of Restrictive Chromatin Structures at the Viral Long Terminal Repeat Drives the Progressive Entry of HIV into Latency. J. Virol. 2008, 82, 12291–12303. 10.1128/JVI.01383-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bressi J. C.; Jennings A. J.; Skene R.; Wu Y.; Melkus R.; de Jong R.; O’Connell S.; Grimshaw C. E.; Navre M.; Gangloff A. R. Exploration of the HDAC2 foot pocket: Synthesis and SAR of substituted N-(2-aminophenyl) benzamides. Bioorg. Med. Chem. Lett. 2010, 20, 3142–3145. 10.1016/j.bmcl.2010.03.091. [DOI] [PubMed] [Google Scholar]
- Clavier H.; Caijo F.; Borré E.; Rix D.; Boeda F.; Nolan S. P.; Mauduit M. Towards Long-Living Metathesis Catalysts by Tuning the N-Heterocyclic Carbene (NHC) Ligand on Trifluoroacetamide-Activated Boomerang Ru Complexes. Eur. J. Org. Chem. 2009, 2009, 4254. 10.1002/ejoc.200900407. [DOI] [Google Scholar]
- The detailed synthetic procedure can be found in the supplementary document, and also in patent application: Liu J.; Clausen D. J.; Yu W.; Kelly J. M.; Kim H. M.; Kozlowski J. A.. Preparation of imidazole derivatives as inhibitors of histone deacetylase useful for the treatment or prevention of HIV infection. PCT Int. Appl. (2020), WO 2020028150.
- The cell assay was carried out with addition of 0.1% and 5% of human normal serum (NHS). Only data with 5% NHS is reported here which included the serum shift caused by the impact of protein binding to the potency. The percent activation is relative to the activation from 2000 nM of vorinostat, which did not cause Jurkat 2C4 cell death with ∼20 h treatment.
- The data of X-ray crystal structures for HDACi 1 and 10 is uploaded to the Protein Data Bank (www.pdb.org) with code 6WBW and 6XDM, respectively.
- Yu W.; Liu J.; Yu Y.; Zhang V.; Clausen C.; Kelly J.; Wolkenberg S.; Beshore D.; Duffy J.; Chang C. C.; Myers R. W.; Klein D. J.; Fells J.; Holloway K.; Wu J.; Wu G.; Howell B. J.; Barnard R. J. O.; Kozlowski J. Discovery of ethyl ketone-based HDAC-1,2,3 selective inhibitors for the treatment of HIV latency. Bioorg. Med. Chem. Lett. 2020, 30, 127197. 10.1016/j.bmcl.2020.127197. [DOI] [PubMed] [Google Scholar]
- Clausen D. J.; Liu J.; Yu W.; Duffy J.; Chang C. C.; Myers R. W.; Klein D. J.; Fells J.; Holloway K.; Wu J.; Wu G.; Howell B. J.; Barnard R. J. O.; Kozlowski J.. Development of a selective HDAC inhibitor aimed at reactivating the HIV latent reservoir. Bioorg. Med. Chem. Lett. Accepted. [DOI] [PubMed] [Google Scholar]
- Maxwell J. W.; Barnard R. J. O. et al. Manuscript in preparation.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.