

Abstract
Insulin-regulated aminopeptidase (IRAP) is a transmembrane zinc metallopeptidase with many important biological functions and an emerging pharmacological target. Although previous structural studies have given insight on how IRAP recognizes linear peptides, how it recognizes its physiological cyclic ligands remains elusive. Here, we report the first crystal structure of IRAP with the macrocyclic peptide inhibitor HA08 that combines structural elements from angiotensin IV and the physiological substrates oxytocin and vasopressin. The compound is found in the catalytic site in a near canonical substrate-like configuration and inhibits by a competitive mechanism. Comparison with previously solved structures of IRAP along with small-angle X-ray scattering experiments suggests that IRAP is in an open conformation in solution but undergoes a closing conformational change upon inhibitor binding. Stabilization of the closed conformation in combination with catalytic water exclusion by the tightly juxtaposed GAMEN loop is proposed as a mechanism of inhibition.
Keywords: Enzyme, inhibitor, X-ray crystallography, small-angle X-ray diffraction, aminopeptidase, mechanism
Insulin-regulated aminopeptidase (IRAP, EC 3.4.11.3) is a transmembrane zinc metalloprotease that belongs to the M1 family of aminopeptidases. IRAP has been reported to have several diverse biological functions including the regulation of trafficking of glucose transporter 4 and the generation of antigenic peptides for cross-presentation.1,2 A hallmark of IRAP is its ability to degrade cyclic bioactive peptides such as oxytocin and vasopressin in brain, and thus it plays roles in cognitive function.3,4 Furthermore, IRAP is localized in areas of the brain associated with learning and memory and is the receptor for angiotensin IV (AngIV), which inhibits its enzymatic activity.5,6 Given the role of AngIV in improving memory tasks in experimental animals, development of IRAP inhibitors has been pursued as potential therapeutics for cognitive disorders.7−9 The ability of IRAP to degrade cyclic peptides has inspired the development of macrocyclic analogues of AngIV, which act as potent IRAP inhibitors with significant selectivity and have the capacity to alter dendritic spine density and morphology in neuronal cultures.10−12
Despite the pharmacological interest in IRAP, the crystal structure of the extracellular enzymatic domain was only recently solved.13,14 It features a four-domain concave structure with a large internal cavity extending away from the Zn(II) containing catalytic site. Compared to structures of homologous enzymes, the conserved GAMEN loop at the catalytic site was found in a unique conformation and it was hypothesized that this conformation allows accommodation of the more bulky cyclic peptidic substrates.15 There is only a single structure of IRAP with an inhibitor reported, a transition-state analogue phosphinic pseudopeptide.16 In that structure, apparent ligand-induced conformational changes induce closing of the overall structure and formation of a large internal cavity with no access to the external solvent. Currently, no crystal structures exist with AngIV in complex with IRAP, and as a result the mode of binding and mechanism of inhibition remain controversial.
In an effort to understand the mechanism of action of macrocyclic inhibitors and gain insight on the ability of IRAP to trim cyclic peptides as well as the mechanism of inhibition by AngIV, we solved the crystal structure of the soluble extracellular domain of IRAP in complex with HA08, a potent macrocyclic inhibitor inspired from the structures of oxytocin and AngIV (Scheme 1).11 Since HA08 was previously characterized using membrane preparations from cells that express IRAP, we first characterized the inhibitor using high-purity recombinant soluble IRAP expressed in mammalian cells as described before.14 In accordance to previous studies,11 HA08 was found to be a potent IRAP inhibitor with a pIC50 of 7.8 and 8.3, for substrates Arg-AMC and Leu-AMC, respectively. Furthermore, it was found to be quite selective versus the highly homologous enzymes ERAP2 and ERAP117 (Figure 1A). Interestingly, the pattern of inhibition selectivity was similar for AngIV suggesting that HA08 and AngIV may use the same binding site and mode of action (Figure 1B). Michaelis–Menten analysis revealed a substrate-inhibition behavior, that has not been reported for IRAP before, but has been reported before for the homologous ERAP1.18 Addition of HA08 resulted in the increase of the calculated KM parameter while it did not affect the Vmax suggesting that the inhibitor acts through a competitive mechanism (Figure 1C, D).
Scheme 1. Chemical Structure of HA08.

Figure 1.

(Panel A) HA08 is a potent inhibitor of IRAP, but much less potent for ERAP2 and inactive for ERAP1. Titrations follow the fluorescent signal produced by trimming of the small substrates Leu-AMC (for IRAP and ERAP1) or Arg-AMC (for IRAP and ERAP2). pIC50 values calculated from mathematical fits are indicated. (Panel B) AngIV is a potent inhibitor of IRAP but a weaker inhibitor of ERAP2 and ERAP1. Titrations are as in Panel A but using AngIV instead of HA08 (substrate used was Leu-AMC). (Panel C) Michaelis–Menten analysis of IRAP using substrate Arg-AMC, in the presence of increasing concentrations of HA08. Solid lines represent computational fits to a substrate inhibition model. (Panel D) Calculated KM and Vmax parameters for each inhibitor concentration from the fits shown in panel C.
Recombinant IRAP was cocrystallized with HA08, and crystals yielded useful diffraction data to 3.20 Å. The structure was solved by molecular replacement using the closed structure of IRAP (PDB ID: 5MJ6) as a search model.16 Crystallographic data and refinement statistics are shown in Supporting Information Table 1. The overall useful resolution of the anisotropic diffraction data was fairly low and the Rfree of the structure after refinement convergence was rather high for structures of comparable resolution. Τhe overall B-factor (temperature factor) however is low, at 38.0 Å2, and the electron density in the binding site region, as well as the residual electron density assigned to the ligand, is well-defined allowing an unequivocal placement of protein side-chains and ligand. From a survey of individual atomic B-factors, the fit of model to electron density, and a general assessment of map quality, it appears that the most problematic regions of electron density correspond to Domain I of molecule A and to several external loops of both molecules in the asymmetric unit, whereas most other regions (including at the ligand) are surprisingly well-defined for the resolution (Supporting Information Figure 1).
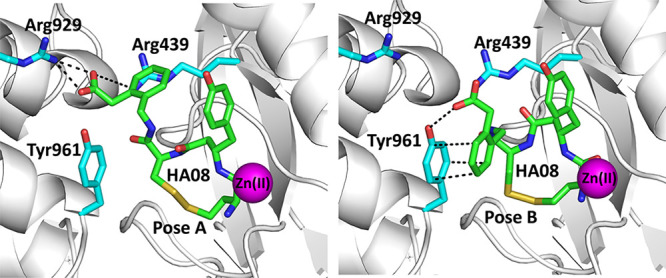
Overall, the structure of the enzyme was found to be identical to a previously solved structure of IRAP with a phosphinic pseudotripeptide transition-state analogue.16 As a result, it appears that the presence of HA08 stabilized the closed conformation of IRAP. Significant electron density was found in the catalytic site and interpreted to belong to the HA08 inhibitor (Supporting Information Figure 2). In both monomers of the asymmetric unit, the electron density seen in the binding site could not be completely accounted for by a single HA08 pose. Instead, it could be accounted for as a combination of two poses (Figure 2). Overall, the inhibitor was found in a conformation similar to what has been proposed by molecular dynamic simulations in recent studies.12,19 The amino-terminus and first peptide bond of the inhibitor are found in a canonical substrate-like orientation, similar to the transition-state analogue DG025.14 The side-chain of the β-tyrosine amino acid residue is located in the S1 pocket of the enzyme and the disulfide bond abuts to the S1′ pocket. The phenylacetic acid group in the C-terminal moiety of the inhibitor was found in two different orientations: in the first, the carboxyl group makes electrostatic interactions with Arg929 and Arg439. In the second, its phenyl moiety makes pi-stacking interactions with Tyr961. It should be noted that in the previously solved IRAP-inhibitor structure, interactions with Tyr961 were interpreted to constitute the S2′ pocket of the enzyme. Thus, it appears that the S2′ pocket of the enzyme is not unequivocally set and may be redefined depending on the chemical groups carried by the inhibitor.
Figure 2.
Schematic representation of HA08 in the active site of IRAP (shown in gray cartoon representation). HA08 is shown in green sticks, nearby IRAP residues in cyan sticks, and catalytic Zn(II) atom as a magenta sphere (oxygen atoms are in red, nitrogen in blue, and sulfur in yellow). Blue mesh indicates the 2Fo – Fc electron density map contoured at 1.5 sigma (carve = 1.8). (Panel A) Pose A of the inhibitor HA08. The C-terminal end makes electrostatic interactions with Arg929 of domain IV and Arg439. (Panel B) Pose B of the inhibitor HA08. The C-terminal end of the inhibitor makes pi-stacking interactions and possibly hydrogen bonding interactions with Tyr961 of the domain IV of the enzyme. Interactions are indicated by dotted lines.
One of the characteristics of HA08 is its high selectivity versus homologous enzymes. To understand this selectivity, we superimposed the IRAP/HA08 structure with the homologous enzymes ERAP1 and ERAP2 (Figure 3A and B). In IRAP, HA08 makes close van der Waals interactions with Ala427 adjacent to the GAMEN motif. In ERAP1 the equivalent residue is a serine, Ser316, whose hydroxyl group would clash with the macrocyclic body of HA08, making the observed pose impossible in ERAP1 and thus explaining the inability of HA08 to inhibit ERAP1 (Figure 1A). This would not be a problem for ERAP2, which also has an alanine residue in the same position. However, two sets of interactions between the phenylacetic group of HA08 and IRAP residues Arg929 and Tyr961 would not be possible for ERAP2 since the equivalent location residues (His860 and Ser869) are not capable of making similar interactions with HA08 (Figure 3A and B). Thus, while this pose of HA08 is also possible in ERAP2, lack of key interactions would be expected to reduce the affinity of the inhibitor, as observed experimentally (Figure 1A).
Figure 3.
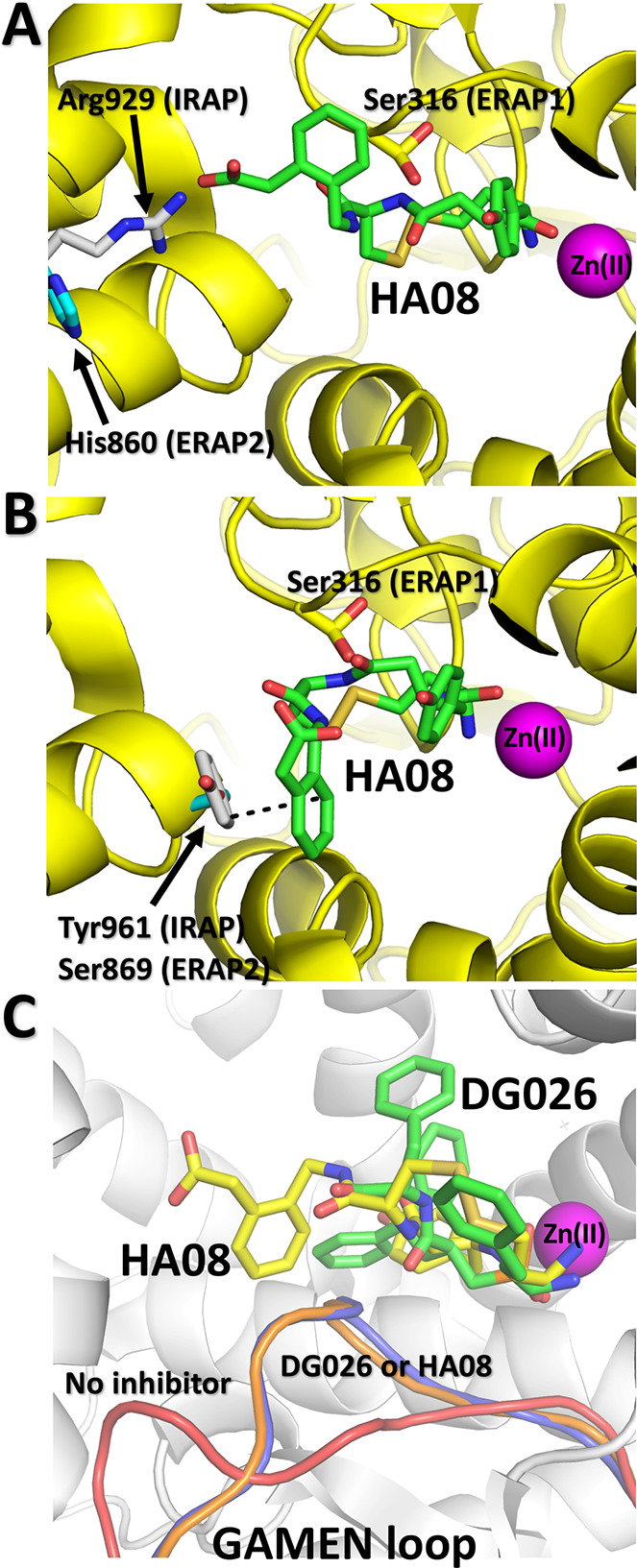
(Panel A) Alignment of the HA08 pose A (green sticks) in the active site of ERAP1 (PDB code 6Q4R) and ERAP2 (PDB code 5AB0). IRAP residues are shown in gray sticks, ERAP1 residues are shown in yellow sticks, and ERAP2 residues are shown in cyan sticks. (Panel B) As panel A but for pose B of HA08. (Panel C) Comparison of the conformation of the GAMEN loop in the IRAP/HA08 structure (blue ribbon) versus the empty IRAP (red ribbon, PDB code 5C97) and IRAP with bound transition state-analogue DG026 (orange ribbon, PDB code 5MJ6).
Comparison of the overall structure of IRAP in the absence or presence of the HA08 inhibitor suggests a conformational change similar to the one observed for a transition-state analogue peptide.16 To investigate if this conformational change occurs also in solution and it is not a result of crystallization, we utilized small-angle X-ray scattering (SAXS). This experimental approach has been successfully used before to probe the conformations of the homologous enzyme ERAP1.20 Guinier analysis of SAXS data was used to derive radii-of-gyration (Rg). Soluble IRAP was calculated to have an Rg value of 54.7 ± 1.1 Å whereas in the presence of HA08 the Rg value was 51.6 ± 1.1 Å (Figure 4A). This indicates that the scattering particles in solution become more compact upon addition of HA08. It should be noted that this Rg value is consistent with a dimer structure in solution, as indicated by size-exclusion chromatographic analysis (Figure 4B) and as suggested previously.16,21 It should be noted that the dimer topology suggested by X-ray crystallography should allow dimer formation even for the membrane-bound IRAP form, since dimerization is mediated by domain IV interactions which are furthest away from the transmembrane region. Furthermore, this topology would not preclude ligand-induced conformational changes in membrane IRAP, although this needs to be confirmed experimentally. Scattering data were also analyzed by pairwise distribution to evaluate the distribution of interatomic distances within the scattering particle. This analysis allowed us to calculate the parameter Dmax which corresponds to the maximum distance of any two atoms in the scattering particle. Similar to the Rg analysis, the presence of HA08 reduced the Dmax of IRAP from 190 to 164 Å (Figure 4A). Both results are consistent with a model wherein IRAP is an open dimer in solution and assumes a more compact conformation upon HA08 binding. Thus, the conformational change that became evident by comparing the crystal structure of empty and inhibitor-bound IRAP appears to also apply in solution.
Figure 4.

(Panel A) Calculated Rg and Dmax parameters from small-angle X-ray scattering experiments of IRAP in solution in the presence or absence of inhibitor HA08. (Panel B) Size-exclusion chromatogram of IRAP analyzed on a Sephacryl 200 16/60 HR column run at 0.5 mL/min. MW standards are indicated.
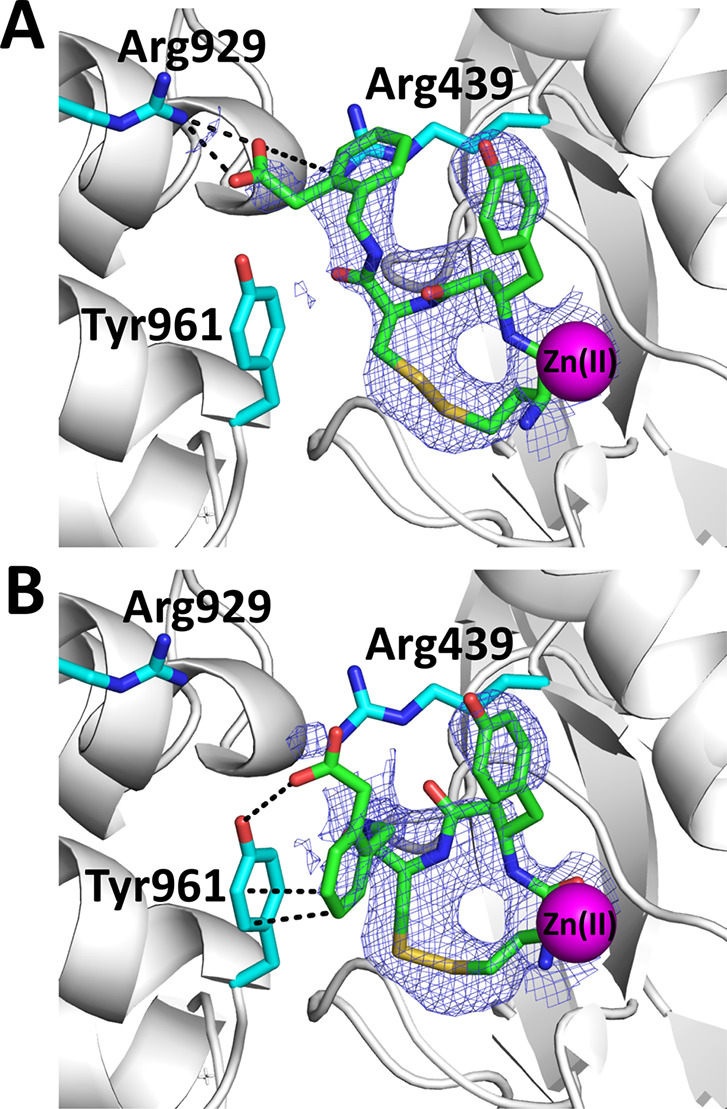
The structure presented here can provide insight on open questions regarding IRAP specificity and mechanism of inhibition by macrocyclic peptide analogues. In previous studies, the unusual configuration of the GAMEN loop in the empty IRAP structure was proposed to be responsible for IRAP’s ability to trim cyclic peptides,13,14 in the context that it allows for additional space in the catalytic site to accommodate the more bulky cyclic peptides. However, in both inhibitor-bound structures available, the GAMEN loop moves to make tight interactions with the inhibitor (Figure 3C). Accordingly, in the IRAP/HA08 structure, the macrocyclic nature of the inhibitor does not appear to require IRAP to be in the open conformation so that it can bind, but to rather induce the closing conformational change. Thus, the ability of IRAP to process cyclic peptides may not be due to the more open nature of the active site but rather to the conformational flexibility of the GAMEN loop that can provide sufficient active-site plasticity to accommodate different shapes of substrates and by extension, inhibitors. Interestingly, similar analysis with the homologous enzyme aminopeptidase N (Supporting Information Figure 3) crystallized in both empty and AngIV-bound forms, indicated no change in the GAMEN loop configuration upon ligand binding.22 Therefore, either the conformational transition of the GAMEN loop is a unique property of IRAP or there are additional conformations of aminopeptidase N not yet observed.
Unlike the nonhydrolyzable pseudopeptide inhibitor cocrystallized with IRAP before, HA08 contains a putatively scissile peptide bond that is juxtaposed against the catalytic Zn(II) in a position that should, in principle, allow catalysis. In this context, the structure of IRAP with HA08 opens questions regarding the mechanism of inhibition of HA08 and by extrapolation of AngIV. Close examination of the structure, however, can provide some insight toward answering that question. Unlike the empty IRAP, the GAMEN loop moves to close juxtaposition with the inhibitor (Figure 3C). The catalytic cycle of M1 aminopeptidases necessitates a nucleophilic attack on the carbon atom of the scissile bond by a water molecule assisted by the ionized carboxylate of the active site residue Glu (Glu465 in the case of IRAP).23,24 However, the close juxtaposition of the GAMEN loop on the bound inhibitor does not allow space for motion of water molecules to interact with Glu465 and no ordered water molecule is found in the structure at that location. This observation suggests that for catalysis to take place, motions of the GAMEN loop during the catalytic cycle would be necessary. Therefore, stabilization of the GAMEN loop by the inhibitor could act as a bottleneck to catalysis.
An additional insight into the inhibitory properties of HA08 comes from its interactions with domain IV of IRAP (Figure 2). These interactions are found with both poses of the inhibitor and are consistent with structure–activity analyses performed previously.10 The conformational closing of IRAP upon inhibitor binding revealed by SAXS measurements is likely due to those interactions that “pull” domains II and IV closer together. Thus, HA08 appears to stabilize the closed form of IRAP. This, however, should be unproductive for the catalytic cycle, both by limiting GAMEN loop plasticity and by not allowing substrate–product exchange since in the closed structure the active site is completely excluded from the outside solvent. Synergy of the two mechanisms proposed here, i) exclusion of catalytic water molecule and ii) stabilization of a conformation that does not allow product−substrate exchange, may be sufficient to explain the inhibitory properties of HA08. This is however a nontypical mechanism of inhibition that may require additional studies to confirm or disprove.
In summary, we report here the first structure of IRAP in complex with a macrocyclic inhibitor. Analysis of the structure along with biochemical and SAXS data provide insight on the inhibitor selectivity and mechanism of action as well as the enzymatic mechanism of IRAP and its selectivity for cyclic peptides. Detailed knowledge of how this macrocyclic inhibitor interacts with IRAP can serve as a starting point for the optimization of this compound as a lead for the preclinical development of IRAP inhibitors for therapeutic applications.
Acknowledgments
We thank the beamline scientists of EMBL-Hamburg for their assistance during data collection at the P13 beamline of PETRA III, Hamburg, Germany.
Glossary
Abbreviations
- IRAP
insulin-regulated aminopeptidase
- ERAP1
endoplasmic reticulum aminopeptidase 1
- ERAP2
endoplasmic reticulum aminopeptidase 2
- SAXS
small-angle X-ray scattering
- Leu-AMC
l-leucine-7-amido-4-methylcoumarin
- Arg-AMC
l-arginine-7-amido-4-methylcoumarin
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.0c00172.
Figure showing the unbiased omit map, table showing data collection and refinement statistics, description of methods used for protein expression purification, enzymatic assays, crystallization, data collection, structure determination, and small-angle X-ray scattering experiments (PDF)
Author Present Address
⊥ (P.G.) Department of Neurobiology, Hellenic Pasteur Institute, Athens 11521, Greece.
Author Contributions
A.M. prepared recombinant protein, performed crystallization experiments, solved the structure, and performed enzymatic assays. Em.S. processed diffraction data and helped solve the crystal structure. P.G. assisted with crystallization trials, performed diffraction experiments, and helped solve the crystal structure. Z.M. and L.J.S. designed, performed, and analyzed SAXS experiments. M.L. and M.H. synthesized and were engaged in the design of the inhibitor. E.S. supervised the study, helped solve the crystal structure, processed data, and wrote the paper with contributions from all coauthors. All authors have given approval to the final version of the manuscript.
We acknowledge support of this work by the project “INSPIRED-The National Research Infrastructures on Integrated Structural Biology, Drug Screening Efforts and Drug Target Functional Characterization” (Grant MIS 5002550), which is implemented under the Action “Reinforcement of the Research and Innovation Infra- structure”, funded by the Operational Programme “Competitiveness, Entrepreneurship and Innovation” (Grant NSRF 2014–2020) and cofinanced by Greece and the European Union (European Regional Development Fund). Synchrotron access was funded by iNEXT (Proposal ID 5589) and by EMBL-Hamburg (Proposal MX-619). We also acknowledge support from the Kjell and Märta Beijer Foundation and King Gustaf V and Queen Victoria’s Foundation of Freemasons. Research support was also provided by the National Institutes of Health (grant no. AI-038996 to L.J.S.). This research used beamline 16-ID LiX of the National Synchrotron Light Source II, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Brookhaven National Laboratory under Contract No. DE-SC0012704. The Life Science Biomedical Technology Research resource is primarily supported by the National Institute of Health, National Institute of General Medical Sciences (NIGMS) through a Center Core P30 Grant (P30GM133893), and by the DOE Office of Biological and Environmental Research (KP1605010).
The authors declare no competing financial interest.
Notes
Atomic coordinates and structure factors have been deposited in the Protein Data Bank (www.pdb.org), with PDB code 6YDX.
Supplementary Material
References
- Keller S. R. The insulin-regulated aminopeptidase: a companion and regulator of GLUT4. Front. Biosci., Landmark Ed. 2003, 8, s410–20 10.2741/1078. [DOI] [PubMed] [Google Scholar]
- Rowntree L. C.; Nguyen T. H. O.; Halim H.; Purcell A. W.; Rossjohn J.; Gras S.; Kotsimbos T. C.; Mifsud N. A. Inability To Detect Cross-Reactive Memory T Cells Challenges the Frequency of Heterologous Immunity among Common Viruses. J. Immunol. 2018, 200 (12), 3993–4003. 10.4049/jimmunol.1800010. [DOI] [PubMed] [Google Scholar]
- Herbst J. J.; Ross S. A.; Scott H. M.; Bobin S. A.; Morris N. J.; Lienhard G. E.; Keller S. R. Insulin stimulates cell surface aminopeptidase activity toward vasopressin in adipocytes. Am. J. Physiol. 1997, 272 (4 Pt 1), E600–6. 10.1152/ajpendo.1997.272.4.E600. [DOI] [PubMed] [Google Scholar]
- Albiston A. L.; Diwakarla S.; Fernando R. N.; Mountford S. J.; Yeatman H. R.; Morgan B.; Pham V.; Holien J. K.; Parker M. W.; Thompson P. E.; Chai S. Y. Identification and development of specific inhibitors for insulin-regulated aminopeptidase as a new class of cognitive enhancers. Br. J. Pharmacol. 2011, 164 (1), 37–47. 10.1111/j.1476-5381.2011.01402.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lew R. A.; Mustafa T.; Ye S.; McDowall S. G.; Chai S. Y.; Albiston A. L. Angiotensin AT4 ligands are potent, competitive inhibitors of insulin regulated aminopeptidase (IRAP). J. Neurochem. 2003, 86 (2), 344–50. 10.1046/j.1471-4159.2003.01852.x. [DOI] [PubMed] [Google Scholar]
- Fernando R. N.; Larm J.; Albiston A. L.; Chai S. Y. Distribution and cellular localization of insulin-regulated aminopeptidase in the rat central nervous system. J. Comp. Neurol. 2005, 487 (4), 372–90. 10.1002/cne.20585. [DOI] [PubMed] [Google Scholar]
- Lee J.; Albiston A. L.; Allen A. M.; Mendelsohn F. A.; Ping S. E.; Barrett G. L.; Murphy M.; Morris M. J.; McDowall S. G.; Chai S. Y. Effect of I.C.V. injection of AT4 receptor ligands, NLE1-angiotensin IV and LVV-hemorphin 7, on spatial learning in rats. Neuroscience 2004, 124 (2), 341–9. 10.1016/j.neuroscience.2003.12.006. [DOI] [PubMed] [Google Scholar]
- Chai S. Y.; Yeatman H. R.; Parker M. W.; Ascher D. B.; Thompson P. E.; Mulvey H. T.; Albiston A. L. Development of cognitive enhancers based on inhibition of insulin-regulated aminopeptidase. BMC Neurosci. 2008, 9 (Suppl 2), S14. 10.1186/1471-2202-9-S2-S14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson H.; Hallberg M. Discovery of inhibitors of insulin-regulated aminopeptidase as cognitive enhancers. Int. J. Hypertens. 2012, 2012, 789671. 10.1155/2012/789671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersson H.; Demaegdt H.; Vauquelin G.; Lindeberg G.; Karlen A.; Hallberg M.; Erdelyi M.; Hallberg A. Disulfide cyclized tripeptide analogues of angiotensin IV as potent and selective inhibitors of insulin-regulated aminopeptidase (IRAP). J. Med. Chem. 2010, 53 (22), 8059–71. 10.1021/jm100793t. [DOI] [PubMed] [Google Scholar]
- Andersson H.; Demaegdt H.; Johnsson A.; Vauquelin G.; Lindeberg G.; Hallberg M.; Erdelyi M.; Karlen A.; Hallberg A. Potent macrocyclic inhibitors of insulin-regulated aminopeptidase (IRAP) by olefin ring-closing metathesis. J. Med. Chem. 2011, 54 (11), 3779–92. 10.1021/jm200036n. [DOI] [PubMed] [Google Scholar]
- Diwakarla S.; Nylander E.; Gronbladh A.; Vanga S. R.; Khan Y. S.; Gutierrez-De-Teran H.; Ng L.; Pham V.; Savmarker J.; Lundback T.; Jenmalm-Jensen A.; Andersson H.; Engen K.; Rosenstrom U.; Larhed M.; Aqvist J.; Chai S. Y.; Hallberg M. Binding to and Inhibition of Insulin-Regulated Aminopeptidase by Macrocyclic Disulfides Enhances Spine Density. Mol. Pharmacol. 2016, 89 (4), 413–24. 10.1124/mol.115.102533. [DOI] [PubMed] [Google Scholar]
- Hermans S. J.; Ascher D. B.; Hancock N. C.; Holien J. K.; Michell B. J.; Chai S. Y.; Morton C. J.; Parker M. W. Crystal structure of human insulin-regulated aminopeptidase with specificity for cyclic peptides. Protein Sci. 2015, 24 (2), 190–9. 10.1002/pro.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mpakali A.; Saridakis E.; Harlos K.; Zhao Y.; Papakyriakou A.; Kokkala P.; Georgiadis D.; Stratikos E. Crystal Structure of Insulin-Regulated Aminopeptidase with Bound Substrate Analogue Provides Insight on Antigenic Epitope Precursor Recognition and Processing. J. Immunol. 2015, 195 (6), 2842–2851. 10.4049/jimmunol.1501103. [DOI] [PubMed] [Google Scholar]
- Mpakali A.; Giastas P.; Mathioudakis N.; Mavridis I. M.; Saridakis E.; Stratikos E. Structural Basis for Antigenic Peptide Recognition and Processing by Endoplasmic Reticulum (ER) Aminopeptidase 2. J. Biol. Chem. 2015, 290 (43), 26021–32. 10.1074/jbc.M115.685909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mpakali A.; Saridakis E.; Harlos K.; Zhao Y.; Kokkala P.; Georgiadis D.; Giastas P.; Papakyriakou A.; Stratikos E. Ligand-Induced Conformational Change of Insulin-Regulated Aminopeptidase: Insights on Catalytic Mechanism and Active Site Plasticity. J. Med. Chem. 2017, 60 (7), 2963–2972. 10.1021/acs.jmedchem.6b01890. [DOI] [PubMed] [Google Scholar]
- Georgiadis D.; Mpakali A.; Koumantou D.; Stratikos E. Inhibitors of ER Aminopeptidase 1 and 2: From Design to Clinical Application. Curr. Med. Chem. 2019, 26 (15), 2715–2729. 10.2174/0929867325666180214111849. [DOI] [PubMed] [Google Scholar]
- Evnouchidou I.; Kamal R. P.; Seregin S. S.; Goto Y.; Tsujimoto M.; Hattori A.; Voulgari P. V.; Drosos A. A.; Amalfitano A.; York I. A.; Stratikos E. Coding single nucleotide polymorphisms of endoplasmic reticulum aminopeptidase 1 can affect antigenic peptide generation in vitro by influencing basic enzymatic properties of the enzyme. J. Immunol. 2011, 186 (4), 1909–13. 10.4049/jimmunol.1003337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barlow N.; Vanga S. R.; Sävmarker J.; Sandström A.; Burns P.; Hallberg A.; Åqvist J.; Gutiérrez-De-Terán H.; Hallberg M.; Larhed M.; Chai S. Y.; Thompson P. E. Macrocyclic peptidomimetics as inhibitors of insulin-regulated aminopeptidase (IRAP). RSC Medicinal Chemistry 2020, 11 (2), 234–244. 10.1039/C9MD00485H. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stamogiannos A.; Maben Z.; Papakyriakou A.; Mpakali A.; Kokkala P.; Georgiadis D.; Stern L. J.; Stratikos E. Critical Role of Interdomain Interactions in the Conformational Change and Catalytic Mechanism of Endoplasmic Reticulum Aminopeptidase 1. Biochemistry 2017, 56 (10), 1546–1558. 10.1021/acs.biochem.6b01170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ascher D. B.; Cromer B. A.; Morton C. J.; Volitakis I.; Cherny R. A.; Albiston A. L.; Chai S. Y.; Parker M. W. Regulation of insulin-regulated membrane aminopeptidase activity by its C-terminal domain. Biochemistry 2011, 50 (13), 2611–22. 10.1021/bi101893w. [DOI] [PubMed] [Google Scholar]
- Wong A. H.; Zhou D.; Rini J. M. The X-ray crystal structure of human aminopeptidase N reveals a novel dimer and the basis for peptide processing. J. Biol. Chem. 2012, 287 (44), 36804–13. 10.1074/jbc.M112.398842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tholander F.; Muroya A.; Roques B. P.; Fournie-Zaluski M. C.; Thunnissen M. M.; Haeggstrom J. Z. Structure-based dissection of the active site chemistry of leukotriene A4 hydrolase: implications for M1 aminopeptidases and inhibitor design. Chem. Biol. 2008, 15 (9), 920–9. 10.1016/j.chembiol.2008.07.018. [DOI] [PubMed] [Google Scholar]
- Kochan G.; Krojer T.; Harvey D.; Fischer R.; Chen L.; Vollmar M.; von Delft F.; Kavanagh K. L.; Brown M. A.; Bowness P.; Wordsworth P.; Kessler B. M.; Oppermann U. Crystal structures of the endoplasmic reticulum aminopeptidase-1 (ERAP1) reveal the molecular basis for N-terminal peptide trimming. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (19), 7745–50. 10.1073/pnas.1101262108. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.