

Abstract
Drug delivery plays a vital role in medicine and health, but the on‐demand delivery of large‐sized drugs using stimuli‐triggered carriers is extremely challenging. Most present capsules consist of polymeric dense shells with nanosized pores (<10 nm), thus typically lack permeability for nano/microparticle drugs. Here, a pinecone‐inspired smart microcage with open network shells, assembled from cellulose nanofibrils (CNFs), is reported for nano/microparticle drug delivery. The approach allows the nanoarchitectured, functionalized CNFs to assemble into mechanically robust, haystack‐like network shells with tunable large‐through pores and polypeptide‐anchored points on a large scale. Such open network shells can intelligently open/close triggered by lesion stimuli, making the therapy “always on‐demand.” The resulting pinecone‐inspired microcages exhibit integrated properties of superior structural stability, superhydrophilicity, and pH‐triggered, smart across‐shell transport of emerging antimicrobial silver nanoparticles and bioactive silicate nanoplatelets (sizes of >100 nm), which enable both extraordinary anti‐infection and bone regeneration. This work provides new insights into the design and development of multifunctional encapsulation and delivery carriers for medical and environmental applications.
Keywords: antimicrobial activity, bone regeneration, drug delivery, microcage, self‐assembly
Pinecone‐inspired nanoarchitectured smart microcages with open network shells are assembled from nanoarchitectured, functionalized cellulose nanofibrils using a layer‐by‐layer self‐assembly technique. The resulting pinecone‐inspired microcages exhibit integrated properties of superior structural stability, superhydrophilicity, and pH‐triggered, smart across‐shell transport of emerging antimicrobial silver nanoparticles and bioactive silicate nanoplatelets (sizes of >100 nm), which enable both extraordinary anti‐infection and bone regeneration.
1. Introduction
Advanced encapsulation and delivery materials are attractive for their fascinating capability of on‐demand targeting and release of various cargos, like drugs, particles, flavors, and even cells; they have been widely studied for medical, electrical, environmental, and energy applications.[ 1 , 2 , 3 , 4 ] In particular, these delivery materials in forms of membranes, particles, liposomes, and hydrogels, as suitable vehicles for overcoming pharmacokinetic limitations associated with conventional drugs, are commonly used to achieve direct action along with low toxicity in drug delivery.[ 4 , 5 , 6 ] It is possible to use such drug delivery platforms to deliver a drug in spatial‐, temporal‐, and dosage‐controlled fashions.[ 3 , 7 , 8 ] Nowadays, postoperative complications driven by implant‐associated infection and delayed tissue regeneration have caused a growing health and financial burden worldwide. For instance, device‐associated infections account for 25.6% of all health‐care‐associated infections in the US,[ 9 ] and 5–10% of fractures are prone to delayed bony union, nonunion, and even the development of pseudarthrosis.[ 10 ] To reduce these two complications (i.e., infection and delayed healing), most current cutting‐edge measures focus on developing drug‐delivery materials at the implant/tissue interface,[ 11 , 12 ] and it remains challenging to create delivery materials tailored to patients' needs with the capability to accurately deliver various drugs to the cells/tissues over time.
Versatile capsules for drug delivery must possess selective encapsulation of different drugs with tailorable permeability, smart loading/release function, and robust mechanical properties (to protect cargos from external disturbances).[ 13 , 14 , 15 , 16 ] Due to the flexible choice of components and precise control of structure/properties, layer‐by‐layer (LbL) self‐assembly has proven powerful to construct various drug‐delivery capsules. However, most of its currently explored capsules are mainly made from synthetic polymers or biopolymers.[ 13 , 17 , 18 ] Such polymer microcapsules usually exhibit dense and random porous shells with limited pore size of <10 nm, thus typically lack the capability to deliver large‐sized drugs, such as nanoparticles and macromolecules of >106 Da.[ 19 , 20 , 21 , 22 ] Moreover, these microcapsules are usually composed of conventional polymers and have poor mechanical properties. Nowadays, emerging nanoparticle drug therapeutics offer a promising treatment modality for various diseases.[ 23 , 24 , 25 , 26 ] These drugs, such as nanoparticles, nanoplatelets, macromolecules, polymer‐drug conjugates, etc., have relatively large size and have proven advantages of striking efficacy, prolonged drug circulation lifetimes, and low drug resistance; some of them have been clinically approved.[ 27 , 28 ] However, overcoming the barriers for large‐sized drug delivery is complex and calls for exploration of new approaches. This challenge is due to the lack of suitable delivery vehicles that can across‐shell transport and encapsulate large‐sized drugs, then can control drug dose to reduce toxicity over time, and protect the drugs from varying loads. Nature offers a simple yet efficient strategy for material assembly: introducing a porous and fibrous structure to improve material utilization and to obtain unique properties.[ 29 , 30 , 31 ] For instance, spider webs, bird nests, and fish gills all possess a porous structure but are structurally robust and multifunctional. Inspired by such biostructures, assembling an innovative type of microcapsules with highly porous structures and high stability using anisotropic fibrous materials could be an effective strategy to deliver large‐sized drugs on‐demand; however, design of such materials is still a long‐standing challenge.
Herein, inspired by self‐adaptive open/close function of the pinecone, we present a facile methodology to create pinecone‐inspired microcages, which are assembled from functionalized cellulose nanofibrils (CNFs) and with biomimetic architectures, for large‐sized drug delivery. The premise of our design is that the structural size, surface chemistry, and packing structure of CNFs are controlled using the unique polypeptide‐anchored architecture to assemble haystack‐like shells with highly open‐through pores. The pinecone‐inspired microcages have shown the integrated properties of tunable porous structures, superhydrophilicity, robust mechanical stability, and pH‐triggered, on‐demand loading and release of silver nanoparticles (AgNPs, sizes up to 100 nm) and bioactive silicate nanoplatelets (SiNPs). These microcages may be used to reduce the two major complications (i.e., infection and delayed healing) associated with traumatic injuries.
2. Results and Discussion
2.1. Pinecone‐Inspired Design for Microcages
Natural pinecone displays special structures with a set of microfibrous scales constructed on pinecone core, which can anisotropically restrict the swelling/shrinkage of their organic matrices upon humidity stimuli, then help scales open and close to sow nuts (Figure 1a top). Inspired by this biostructure and its unique functional characteristics, a biomimetic microcage with pinecone‐like structure/function was developed (Figure 1a bottom) for pH‐responsive nanoparticle drug delivery. We prepared pinecone‐inspired microcages based on three criteria: i) the microcages can serve as vehicles for large‐sized (up to 100 nm) particle drugs via open network shells; ii) the microcages must be stimuli‐responsive and able to controllably load and release the drugs on demand; iii) the microcages are mechanically robust, readily incorporated onto any implants, and can be used at any time postpreparation. The first requirement was satisfied by forming fibrous shells with nano/micropore structures using LbL assembly that is capable of exerting nanometer control over shell structures; rigid CNFs were used as building blocks to construct this open network barrier allowing across‐shell transport of large‐sized drugs, such as solid nano/microparticles. To satisfy the other two criteria—effective drug delivery capability and assembly strength—our material design was based on a biocompatible polypeptide, i.e., poly‐l‐lysine (PL), which was chosen as a “binder” to build bonding points among haystack‐like CNFs to assemble a stable framework structure, and also as pH‐triggerable “active site” (net positive charge varies with pH) to load and release charged large‐sized drugs (e.g., AgNPs and SiNPs), and to reduce infection and improve bone regeneration simultaneously.
Figure 1.
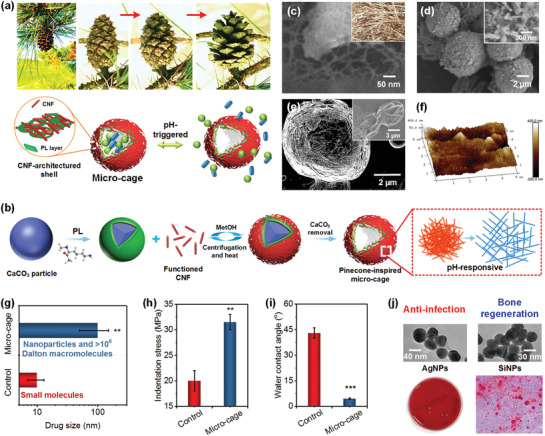
a) Top: A set of optical photographs showing the close–open process of a pinecone due to humidity stimuli. Bottom: schematic illustration of the pinecone‐inspired microcages for pH‐responsive nanoparticle drug delivery. b) Schematic showing the formation process of CNF architectured microcage for large‐sized drug delivery using LbL assembly technology. SEM images of c) freeze‐dried CNFs and d) PL/CNF5 shelled CaCO3 particles. The inset in (c) represents a snapshot image of haystack of Bermuda grass. The inset in (d) shows haystack‐like CNF stacked surface. e) SEM image of the free‐standing (PL/CNF5)2/PL microcage obtained after freeze‐drying. The inset in (e) presents collapsed capsules from polymers only, like PL/poly‐l‐glutamic acid capsules (PL/PG). f) AFM images of PL/CNF5 shell of the microcage. Comparison of g) drug size range, h) indentation stress at 30% strain, and i) surface wettability between the control polymeric capsules (PL/PG) and microcages. j) Top: TEM images of the loaded AgNPs and SiNPs. Bottom: The schematics of their applications for anti‐infection and bone regeneration.
The scheme to synthesize the pinecone‐inspired microcages is shown in Figure 1b. Figure 1c and Figure S1a in the Supporting Information describe the random packing of CNFs, indicating a stacking structural feature like “haystack.” A prelayer of PL was deposited onto porous CaCO3 particles (Figure S1b, Supporting Information) to facilitate the LbL assembly of CNFs. After CNF deposition, besides electrostatic interaction, a thermal treatment (85 °C, 3 Torr, 15 min) was introduced to promote the formation of a hydrogen bonding network among the individual CNFs.[ 13 , 32 ] Then, removal of CaCO3 template resulted in the formation of anisotropic CNF‐based microcapsules with highly porous networked, haystack‐like shells, which we called “microcages.” Scanning electron microscope (SEM) imaging of PL/CNF shells demonstrated that, with increasing number of CNF layers, their surface roughness increased (Figure 1d; Figure S1c, Supporting Information). In contrast to the dense shells of pure polymeric capsules, our microcages exhibited unique CNF‐architectured porous shells (Figure S1d, Supporting Information). The pinecone‐like assembly enabled the construction of a highly open porous network shell, which is only partially filled due to the repulsive interactions of like‐charged CNFs. This was a major feature in terms of structure design that differentiated pinecone‐inspired microcages from other microcapsules, which often have limited molecular‐level permeability.[ 20 , 33 ] We built one PL bonding layer after the assembly of every five CNF layers to enhance the stability of the microcages while maintaining their porous structures. Based on this unique design, we obtained innovative free‐standing PL/CNF microcages after freeze‐drying that were distinctly different from typical capsules made from polymers alone which may collapse in dry and wet conditions, such as PL/poly‐l‐glutamic acid capsules (Figure 1e, inset).[ 11 ] Further atomic force microscopy (AFM) analyses indicated that, in contrast to the smooth PL layer (Figure S1e, Supporting Information), the incorporation of CNFs led to an obvious increase in surface roughness from 2–5 to 10–50 nm (Figure 1f), showing a topography that can enhance cell–material interactions for clinical applications.[ 34 , 35 ] Our unique pinecone‐inspired design allowed the microcages to possess remarkable loading capability with drug size >100 nm, more than ten times higher than that of the control polymer capsules (Figure 1g). Meanwhile, both their mechanical property and hydrophilicity were greatly enhanced compared to the control capsules (>150% improved compressive strength and from hydrophilic to superhydrophilic; Figure 1h,i). Based on such fascinating properties, our pinecone‐inspired microcages achieved smart delivery of AgNPs and SiNPs, to reduce the infection and delayed healing of traumatic injuries (Figure 1j).
2.2. Structural Control of Pinecone‐Inspired Microcages
To tailor the network shells of microcages, various building blocks of functionalized CNFs were used. As illustrated in Figure 2a, the CNFs were negatively charged; the increase of hydrolysis time caused significantly more negative zeta‐potentials ranging from −18 to −56 mV. These results could be attributed to the presence of sulfonic acid ester groups on increasing CNF surfaces.[ 13 , 36 ] Surface roughness of CNF layers confirmed the size change of CNFs (Figure S2, Supporting Information). The AFM scans directly revealed the haystack morphology of the randomly packed CNFs (Figure 2b). Incomplete hydrolysis (<1 h) of cellulose led to a large particle‐like roughness with signs of local aggregation and bundling of nanofibrils (Figure 2b1,b2), while 3 h hydrolyzing resulted in a rough surface (Figure 2b3). Further increasing hydrolysis time (6 and 12 h) caused the CNF layer to be smoother. Unlike the conventional polymer multilayers with a characteristic growth rate of 2–4 nm per bilayer, the CNF multilayers showed a much lower increment of 1–1.5 nm per layer (Figure S3, Supporting Information).[ 37 , 38 , 39 ] As shown in Figure 2c, after 3 cycles of PL/CNF5 assembly, the resultant LC3 films achieved a thickness of 29 nm without disassembly, confirming the strong hydrogen bonding of CNF networks and physical bonding (i.e., electrostatic interaction and molecular entanglement) of PL layers.[ 40 ] The surface roughness of these coatings varied due to their different components and structures (Figure 2d). In contrast to the smooth PL prelayer (Ra of 1.3 nm), the construction of CNF layers led to significantly higher surface roughness (LC3 Ra of 10.5 nm).
Figure 2.
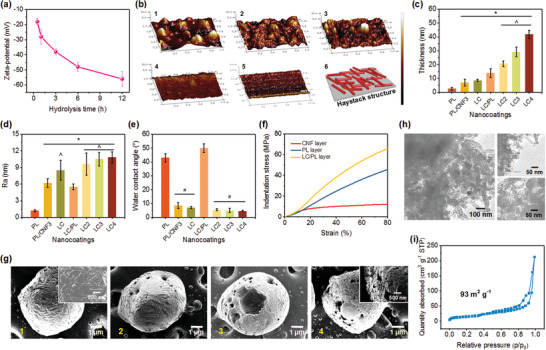
a) Zeta‐potential and b) microstructure of the films assembled using CNFs obtained under different hydrolysis times. b1–b5) AFM images of CNF films obtained with hydrolysis times of 0.5, 1, 3, 6, and 12 h, and b6) a schematic description of a haystack‐like assembled structure. c) Thickness, d) surface roughness, and e) surface wettability of various PL/CNF assembled shells. f) Microindentation–strain curve of the CNF, PL, and LC/PL shells. g) SEM images showing the template removal process of the LC3 microcages. g1–g4) Microcages after immersing in EDTA for 0, 0.5, 1, and 1.5 h, respectively. h) TEM images and i) N2 adsorption–desorption isotherm of porous LC3 microcages. The top right and bottom right images in (h) show the shell structure and internal structure of the microcages, respectively.
Besides the microstructures, two other properties (i.e., surface wettability and mechanical property) for drug delivery vehicles were also evaluated. Figure 2e presents the water contact angles (WCAs) of pinecone‐inspired microcage shells using 3 µL droplets. In contrast to shells with PL as the outermost layer (WCAs of 40°–50°), the change to CNF outermost layer allowed for greatly decreased WCA to 4.5°, which was superhydrophilic. The WCAs of LC shells decreased slightly with increasing layer numbers, due to the enhanced surface roughness according to the Cassie model.[ 41 , 42 ] Using a single‐nanofiber mechanical tester equipped with a 40 µm spherical‐tipped indenter, we performed dynamic microindentation measurement of the shells (Figure 2f; Figure S4, Supporting Information). Under the same indentation strain, the LC/PL shells exhibited higher indentation stress compared to the controls (PL and CNF layers). Our unique PL‐anchored CNF shells showed a remarkable rigidity (Young's modulus of 12.4 GPa; Figure S4, Supporting Information) and resilient behavior, and could provide the mechanical capacity to protect entrapped drugs by their rigid–flexible coupling networks of soft PL polypeptide and rigid CNFs. We also examined the template removal process of the pinecone‐inspired microcages using SEM (Figure 2g). After CaCO3 removal, many large‐through pores formed on the microcage shell due to the fence‐style stacking of CNFs, indicating the pinecone‐like stacking structures (Figure 2g4 inset). This open network shell is also confirmed by transmission electron microscope (TEM) (Figure 2h). Obviously, both the external and internal portions of the microcages show a mesh structure with abundant large‐through mesopores and macropores. In addition, N2 adsorption–desorption isotherm (Figure 2i) demonstrated that LC3 microcages possessed abundant open mesopores with a few macropores, and achieved Brunauer–Emmet–Teller (BET) surface area of 93 m2 g−1 and porosity of 0.23 cm3 g−1 with 72.5% mesopores.
2.3. Nanoparticle Drug Loading and Release
Our pinecone‐inspired microcages were intended to be used for local drug delivery at injury sites, thus we anchored them on a precoated implant. Fluorescent isothiocyanate (FITC) imaging (Figure 3a) revealed the uniformly distributed particles with intact shell structures. By tailoring the incubation (8 × 106 particles mL−1 suspension), the capsule density was facilely regulated (Figure 3b). Within 150 min, the capsule density on implants achieved 16 800 cm−2, suggesting a rapid assembly process. Before template removal, the shells consisting of PL−FITC presented an inhomogeneous fluorescent morphology with abundant dark sites, further confirming the porous and haystack‐like structure (Figure 3c). Moreover, we observed that the structure of cage‐like capsules remained after CaCO3 removal, although the random packing of CNFs left abundant open spaces serving as large‐through pores (Figures 2h and 3c; Figure S5, Supporting Information). In addition, the long‐term stability of PL/CNF shells was examined (Figure 3d; Figure S6, Supporting Information). No obvious changes were observed in both film thickness and surface roughness of the microcage shells after immersing in PBS for up to 1 week. Besides the physical bonding from the PL layer, this result could be ascribed to van der Waals interactions that could stabilize the haystack structure owing to the hydrogen bonding network.[ 40 , 43 ] Such an assembly manner is distinct from conventional LbL polymeric capsules with dense shells typically based on electrostatic interactions.
Figure 3.
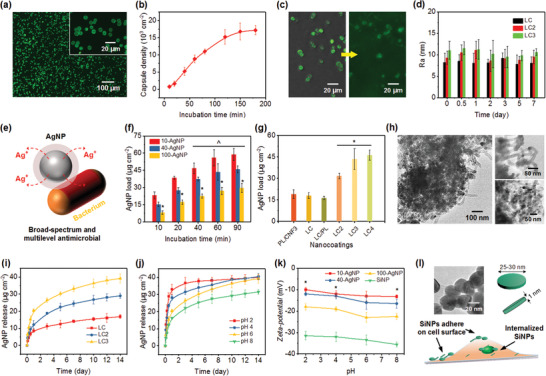
a) CLSM images of LC3 shelled CaCO3 particles with incubation time of 120 min in particle suspension. PL used in the shells is labeled with FITC. b) Assembly of microcages as a function of incubation time in particle suspension (8 × 106 particles mL−1). c) CLSM images of LC3 microcages (left) before and (right) after CaCO3 decomposition. d) Surface roughness of the PL/CNF films after immersion in PBS with different durations. e) Schematic showing the general mechanism for the antimicrobial action of AgNPs via the release of Ag+ ions. f) Loading of AgNPs with various sizes using LC3 microcages with increasing incubation time. g) Loading capability of various PL/CNF assembled carriers using 40 nm AgNPs for 60 min. *p < 0.01 compared to PL/CNF3, LC, and LC/PL coatings. h) TEM images showing the LC3 microcages after 40‐AgNP loading. The top right and bottom right images show the shell structure and internal structure of the microcages, respectively. Cumulative release profiles of 40 nm AgNPs i) from various microcages under pH of 6 and j) from LC3 microcages under various pH values in PBS solution. k) Zeta‐potential of AgNPs and SiNPs under different pH values. l) Top left: TEM image of SiNPs. Top right: The shape and dimension of SiNPs. Bottom: Schematic showing the approach of SiNPs to promote osteogenic differentiation.
Increasing drug resistance of microorganisms has resulted in a growing interest in exploring novel antimicrobial agents; AgNPs are of particular interest owing to their robust bactericidal activities.[ 44 , 45 ] Compared to antibiotics, AgNPs are less likely to induce microbial resistance. This is due to their multilevel antimicrobial modes (Figure 3e), like binding bacterial cell structural elements, and interfering with bacterial integrity, energy production and conservation.[ 46 , 47 ] In contrast to the direct administration of AgNPs, we aimed to deliver AgNPs using pinecone‐inspired microcages to prevent postoperative infection in a sustained and controlled manner that may achieve long‐term effects, less toxicity, and minimal side effects. AgNPs with different sizes were utilized (Figure S7a, Supporting Information), and over 80% of the 10‐AgNPs, 40‐AgNPs, and 100‐AgNPs were in the diameter range of 8–16, 36–55, and 88–110 nm, respectively. Unlike most microcapsules that are only permeable to small molecules (<106 Da), our microcages allowed for across‐shell transport of these large AgNPs (up to 100 nm). Loading of various AgNPs using pinecone‐inspired microcages was performed under solution pH of 2, as illustrated in Figure 3f. Loading of 10‐, 40‐, and 100‐AgNPs within 60 min achieved 56, 43, and 27 µg cm−2, respectively, with a loading efficiency range of 27–45% (Figure S8, Supporting Information). This feature of loading after vehicle preparation enables the capability to load drugs into the microcages before surgical implantation thereby avoiding the need to store drug‐loaded implants, which could be challenging due to concerns with drug stability. Moreover, different stacking structures led to different AgNP loading (Figure 3g). In contrast to PL/CNF3 microcages (19 µg cm−2), LC/PL microcages exhibited a slightly lower 40‐AgNP loading of 16.3 µg cm−2, suggesting the desirable binding effect of PL layer without compromising the shell permeability. The structures of the whole microcages remained stable; no obvious size changes were found after drug loading (Figure S9, Supporting Information). By increasing the shell thickness, 40‐AgNP loading was enhanced although the shell pore size decreased, for instance, from 31.6 µg cm−2 for LC2 microcages to 43.5 µg cm−2 for LC3 microcages. This result could be due to the increase of loading site and space while having adequate permeability for AgNPs. In addition, no obvious changes were observed in the loading capabilities of 10‐ and 40‐AgNP after storing the microcages in PBS for 1 week (Figure S10, Supporting Information). TEM images (Figure 3h) revealed the detailed loading location of AgNPs in the microcages. Obviously, the LC3 microcage shells are permeable to AgNPs with diameters of 40 nm; abundant AgNPs can penetrate into the interior and be encapsulated (Figure 3h bottom right).
Similar to the humidity stimuli for pinecones, the loading and release properties of pinecone‐inspired microcages can be controlled by external pH. Controlled release works effectively for 40‐AgNPs, using smart microcages, due to their large‐through and pH‐triggered “open/close” pores. As shown in Figure 3i, burst release within one day was triggered due to pH variation, followed by sustained release until 14 days. For instance, the cumulative release of AgNPs from LC3 microcages was 20 µg cm−2 within the first day and achieved 39.5 µg cm−2 after 2 weeks. In contrast, relatively fewer AgNPs (17 µg cm−2) were released from LC microcages. More strikingly, the “burst effect” of AgNP release was effectively weakened, which could be due to the combined effect of shielding of interspersed PL layers and tortuous channels of haystack‐like CNF layers (Figure 3h). The comparison of release profiles of pinecone‐inspired microcages triggered by different pH levels from 2 to 8 (Figure 3j) revealed that, at the lower acidic pH (i.e., pH 2), the CNF shells turned to “open” and allowed the across‐shell transport of large particles. This smart function could be due to the changes in external pH that led to changes in surface charges of CNFs having abundant carboxylic and hydroxylic surface groups.[ 13 , 40 , 48 ] At lower pH (2 and 4), the negative charge of CNFs decreases, which weakens electrostatic interaction with PL layer, and might affect the hydrogen bonding network of CNFs, thus resulting in the loosely packed porous shells. SEM observation reveals that the stacking CNFs in microcage shells became gradually loose and their formed pore sizes increased accordingly with increasing time after immersing the microcages in PBS at pH 4 (Figure S11, Supporting Information). At intermediate pH (6 and 8), strong van der Waals interactions derived from the denser hydrogen bonding network may form to stabilize the haystack structure, forming a compacted shell and preventing the transport of large‐sized particles. Considering the typical acidic environment of injury sites, the release of AgNPs under lower pH makes the pinecone‐inspired microcages a highly promising candidate for clinical applications. In our open fracture rat model, we found that the pH of tissue samples around uninfected and infected femurs on 10 days after surgery was around 7.1 and 6.3, respectively (Figure S12, Supporting Information). This was probably because bacteria, such as Staphylococcus aureus, produce acidic substances and may lead to massive infiltration of neutrophils and macrophages. Moreover, the zeta‐potential of AgNPs increased with increasing pH and particle diameters (Figure 3k). This change also contributed to the rapid release of AgNPs at lower pH. In addition, for bone healing, the bioactive SiNPs (Figure 3l), which were capable of promoting osteogenic differentiation of mesenchymal stem cells,[ 25 , 49 , 50 ] were investigated. The negatively charged (−31 to 35 mV) SiNPs of Na+ 0.7[(Mg5.5Li0.3)Si8O20(OH)4]− 0.7 show a disc‐shaped morphology with 40−70 nm in diameter (Figure 3l; Figure S7b, Supporting Information) and achieved a loading efficiency of ≈41% in the microcages (Figure S8, Supporting Information).
2.4. Anti‐Infection and Bone Regeneration
To evaluate the antibacterial activity of our AgNP‐loaded pinecone‐inspired microcages, we challenged the nanocoatings with S. aureus via in vitro bacterial assays (Figure 4a). LC, LC2, and LC3 microcages loaded with 40‐AgNPs exhibited an increasing killing efficacy of 32.5%, 65.2%, and 83.3%, respectively. Loading with 10‐AgNPs resulted in enhanced S. aureus killing compared to 40‐AgNPs (Figure 4a; Figure S13, Supporting Information). In contrast to the control (50 × 10−6 m AgNP suspension), the AgNP‐loaded LC3 microcages exhibited higher antibacterial activity against S. aureus of 87.6% killing using 10‐AgNPs and 83.3% killing using 40‐AgNPs. This result confirmed the unique capability of pinecone‐inspired microcages to deliver large‐sized drugs, nano/microparticles (Figure 3h), in a postpreparation loading manner. Moreover, the viabilities of both human osteoblast and human mesenchymal stem cell (hMSC) were examined using Trypan blue assay; no significant toxicity of the slowly released AgNPs from microcages was observed (Figure 4b; Figure S14, Supporting Information). The control AgNP suspensions showed viabilities of 74% and 71% for osteoblast and hMSC, respectively, while significantly higher viabilities of 84% and 85% were observed with 40‐AgNP‐loaded microcages. Obviously, the pinecone‐inspired microcages showed significantly reduced toxicity toward cells yet with strong S. aureus killing efficacy. This result could be ascribed to the on‐demand and sustained release manner rather than a one‐time and high‐concentration dosage. The dose‐dependent and time‐dependent studies of killing efficacy toward S. aureus were also conducted (Figure 4c; Figure S13, Supporting Information). With increasing AgNP loading dose (15–50 µg cm−2), the microcages exhibited an increasing killing efficacy from <30% to >80%. The kinetics suggested that both 10‐AgNP and 40‐AgNP‐loaded microcages displayed relatively fast bactericidal activity, exhibiting >66% killing within 3 h, and maintaining a high efficacy of >80% after 6 h.
Figure 4.
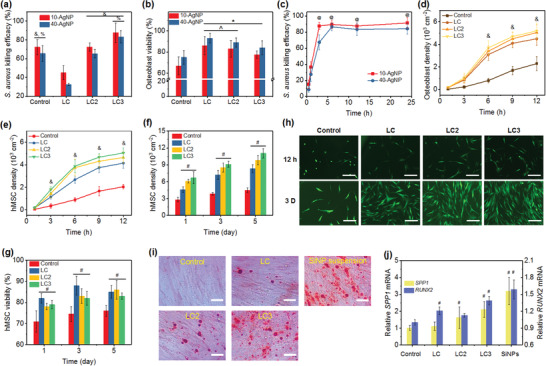
a) S. aureus killing efficacy and b) osteoblast viability of various microcages with AgNP loading and the control (50 × 10−6 m AgNP suspensions). c) S. aureus killing kinetics of LC3 microcages loaded with AgNPs. Cell density of d) osteoblast cells and e) hMSCs on control substrate and substrates coated with microcages incorporated with SiNPs within 12 h. f) Proliferation of hMSC and g) percentage of live cells on various substrates at 1, 3, and 5 days. h) CLSM images of hMSC adhesion at 12 h and 3 days on samples in (e)–(g). Scale bars in (h) are 200 µm. i) Mineralized extracellular matrix production and j) evaluation of osteogenic potential of hMSCs cultured on various nanocoatings. Scale bars in (i) are 50 µm. The mineralized extracellular matrix was stained with Alizarin Red S at day 14. Glyceraldehyde‐3‐phosphate‐dehydrogenase (GAPDH) was carried out as an endogenous control. & p < 0.01 compared to LC microcages. % p < 0.05 compared to LC2 microcages. * p < 0.01 compared to the control. ^ p < 0.05 compared to LC3 microcages. @ p < 0.01 compared to 0.5 h. # p < 0.01 compared to the controls.
The capability of pinecone‐inspired microcages can be further demonstrated by their promising bone regeneration function after loading with bioactive SiNPs (Figure 4d–h). Compared to the control (uncoated implants), substantially more hMSCs or osteoblast cells were attached on the implants coated with SiNP‐loaded microcages (Figure 4d,e; Figure S15, Supporting Information) due to their hierarchical structures, hydrophilic surfaces, and bioactive SiNP stimulation. At 6 h, the hMSC density on controls, LC, LC2, and LC3 microcage incorporated films loaded with SiNPs, was 900, 2680, 3750, and 3880 cell cm−2, respectively. After 12 h, LC3 microcages even resulted in a cell adhesion density of 5050 cell cm−2. In striking contrast to control implants, the cell proliferation was significantly enhanced by almost 200% (Figure 4f), probably due to the delivery of SiNPs. A rapid and sustained cell growth was observed from 1 to 5 days, with a cell density of 11 050 cell cm−2 at day 5 on LC3 microcage incorporated films. These results were supported by CLSM images of hMSCs after different culture times (Figure 4h). More hMSCs attached on the surfaces of microcage incorporated films compared to that of the controls, and much better cell spread was observed on LC2 and LC3 microcage‐based surfaces. Moreover, the viabilities of both hMSC and osteoblast using SiNP‐loaded microcages were significantly higher compared to the control, i.e., 50 × 10−6 m AgNP suspension (Figure 4g; Figure S14, Supporting Information). The percentage of live hMSCs on microcage coated films was in the range of 80–85% at 1, 3, and 5 days compared to the controls (70–75%). This result suggested that the encapsulation of bioactive SiNPs using pinecone‐inspired microcages could significantly reduce the cytotoxicity of conventional implants.
We further evaluated the osteogenic differentiation of hMSCs cultured on the SiNP‐loaded microcage coated films, control substrates, and SiNP suspensions. After culturing for 14 days, the mineralization production of hMSCs was analyzed using Alizarin Red S staining to mark inorganic calcium, a characteristic common to bone structures (Figure 4i). In contrast to the implants (no obvious nodule formation), obviously more nodules and mineralized matrix were formed owing to the presence of SiNPs. Similar bioactive results of SiNPs, both released from LC3 microcages and added as suspension (20 µg mL−1), were observed, underlying the effective delivery of SiNPs using pinecone‐inspired microcages and its resultant enhanced osteogenic differentiation. The osteogenic potential of hMSCs was also evaluated according to the expression levels of secreted phosphoprotein 1 (SPP1) and Runt‐related transcription factor 2 (RUNX2) genes, as indicated in Figure 4j. With the addition of SiNPs, a significant increase in expression levels of SPP1 and RUNX2 was noticed, for example, 200% increase of SPP1 and 140% increase of RUNX2 of hMSCs cultured using LC3 microcages, compared to those using the controls. This unique delivery of bioactive SiNPs based on pinecone‐inspired microcages may be processed to construct devices and regenerative tissues, such as injectable tissue repair matrixes, implants, and on‐demand release of therapeutic agents toward bone‐related tissue engineering.
3. Conclusion
In summary, the work described here demonstrates a novel strategy for creating pinecone‐inspired smart microcages from anisotropic CNFs to deliver nanoparticle drugs using LbL assembly, for preventing implant‐associated infection and promoting bone regeneration. For the first time, the chemically functionalized CNFs with controllable size and surface physical/chemical properties are synthesized and assembled into open network shells with haystack‐like morphology, large‐through pores, and tunable pore structures. pH‐triggered, postpreparation loading, and on‐demand release are achieved for nanoparticles using such microcages, resulting in high and controllable loading of nanoparticles with diameters ranging from 10 to 100 nm (for instance, 58.8 µg cm−2 of 10 nm AgNPs and 29.7 µg cm−2 of 100 nm AgNPs) and sustained release profiles (up to 2 weeks or longer). This effective across‐shell transport of antimicrobial AgNPs and bioactive SiNPs also enables S. aureus killing efficacy of 87% while maintaining cell viability of >84%, and more than twofold cell adhesion and proliferation capability. We envision that such advanced delivery carriers based on pinecone‐inspired microcages will open up numerous opportunities for a range of applications in musculoskeletal injuries, infections, cancers, and tissue engineering.
4. Experimental Section
Materials
Poly‐l‐lysine hydrobromide (PL, M w 150–300 kDa), cellulose microcrystalline (20 µm), sulfuric acid (H2SO4, 95–98%), sodium chloride (NaCl), and methanol (≥99.0%) were purchased from Sigma‐Aldrich (St. Louis, MO). Silver nanoparticles (AgNPs) of diameter of 10, 40, and 100 nm and capped with polyvinylpyrrolidone (PVP) were obtained from nanoComposix, Inc. (San Diego, CA), and designated as 10‐AgNPs, 40‐AgNPs, and 100‐AgNPs, respectively. Calcium carbonate (CaCO3) particles were supplied by PlasmaChem Gmbh (Rudower Chaussee, Berlin, Germany). The SiNPs (Laponite XLG, 25–30 nm in diameter and 1 nm thick) were obtained from Southern Clay Products Inc. (Louisville, KY). Dulbecco's modified eagle medium/nutrient mixture F‐12 and penicillin–streptomycin were bought from Life Technologies (Grand Island, NY). Fetal bovine serum and Dulbecco's phosphate buffered saline (PBS, pH = 7.0) were obtained from Corning (Manassas, VA). Quartz slides and implant discs were bought from Electron Microscopy Sciences (Hatfield, PA), and cleaned and cut into 10 × 25 mm2. N‐type 〈100〉 silicon wafers were provided by Montco Silicon Technologies Inc. (Spring City, PA) and cut into 10 × 25 mm2 rectangles. Disodium ethylenediaminetetraacetic acid (EDTA) and dialysis tubing (12 000–14 000 MWCO) were purchased from Thermo Fisher Scientific (Fair Lawn, NJ).
Synthesis of CNFs
The preparation of CNFs from cellulose fibers was carried out using typical acid hydrolysis. Briefly, 1.0 g cellulose powder was dissolved in 3 mL H2SO4 (64% w/w) and homogenized using KA‐Homogenizer‐24 (20 000 rpm, 2 min). Then, the hydrolyzing CNF suspensions were halted by 5 °C water, and repeatedly washed, centrifuged (10 000 rpm, 4 °C, 10 min), and followed by dialysis against deionized water until neutrality. Sonication was performed on the CNF aqueous media using a Crest P1100H‐45 Ultrasonic Cleaner for 30 min prior to further use.
Fabrication of PL/CNF Microcages
CaCO3 particles were dispersed in 1 mg mL−1 PL solution (50 × 10−3 m NaCl, pH 7.4) for 20 min to prepare a prime shell layer to facilitate the following assembly of CNFs during LbL assembly process. Then, PL coated particles were immersed in 1 mg mL−1 CNF solution for 20 min, followed by two rinsing processes with 50% and 100% methanol by centrifugation at 3000 rpm for 1 min, and dried in a vacuum oven (85 °C, 3 Torr, 15 min). This process was repeated to fabricate the multilayer films, which were designated as PL/CNFm (m, the coating cycles of CNF layer). Moreover, after the 5 cycle‐coating of CNFs (i.e., PL/CNF5), a PL layer was assembled to stabilize the subsequent LbL assembly process, followed by the next round of assembly of CNF multilayers. After that, the shelled CaCO3 particles were incubated in 0.1 m EDTA solution for 1.5 h, followed by centrifugation and removal of supernatants to dissolve the CaCO3 templates. This typical procedure is shown in Figure 1a. The final microcages were referred to as (PL/CNFm)n, where PL represents PL layers, CNF indicates CNF layers, and m and n are the numbers of CNF layers and (PL/CNFm) multilayers. PL/CNF5, (PL/CNF5)2, (PL/CNF5)3, and (PL/CNF5)4 were further simplified as LC, LC2, LC3, and LC4, respectively. In addition, the incorporation of microcages on model implants was achieved by first dipping the precleaned substrates (such as quartz slides, silica wafers, and implant discs) into PL solution to obtain a prime layer, followed by immersing them into suspensions of PL/CNF shelled particles (particle density of 8 × 106 mL−1), and then removing the template and drying it in air.
Drug Loading and In Vitro Study
To load AgNPs, the microcages (coated on 1 × 1 cm2 quartz slides) were immersed into 0.2 mg mL−1 AgNP suspension of pH 2 (PBS buffered). SiNP loading was conducted by incubating the microcages (coated on 1 × 1 cm2 quartz slides) in 0.5 mg mL−1 SiNP suspension (pH 2). After incubating for a predetermined time period, the samples were rinsed in PBS to remove the loosely attached nanoparticles. A clinical isolate of S. aureus (SA 1004) reported in previous work[ 11 , 51 , 52 ] was studied and cultured in tryptic soy broth (TSB) to achieve log‐phase growth. The control AgNP suspensions and AgNP‐loaded microcage coated implants were added to S. aureus inoculum (1.0 × 105 colony forming units per milliliter or CFU mL−1) and incubated for 24 h at 37 °C. Human osteoblast cells and hMSCs were obtained from American Type Cell Culture (ATCC; Manassas, VA) and cultured in Dulbecco's modified Eagle's medium/nutrient mixture F‐12 and Alpha Modified Eagle's Medium (α‐MEM), respectively, supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin in 24‐well culture plates, and incubated at 37 °C in 5% CO2 atmosphere. The drug‐loaded microcage coated and uncoated implants were placed into the 24‐well culture plates to investigate the cell adhesion and proliferation, and culture medium was replaced every other day.
Characterization
The morphology of CaCO3 particles, CNFs, and microcages was characterized using S‐4700 SEM (Hitachi Ltd., Japan), NanoScope AFM (tapping mode, PicoSPM II, Tempe, AZ), and JEM‐2100 TEM (JEOL Ltd., Japan). The thickness of CNF multilayer films was examined using ellipsometry (M‐2000, JA Woollam Co., Lincoln, NE), and their WCA (3 µL) was measured using SL200B contact angle goniometer (Kino Industry Co., Ltd., MA). The size distribution and zeta‐potential of AgNPs, SiNPs, and CNFs were collected with Malvern NanoSight NS300 instrument and Malvern Zetasizer Nano Z (Malvern Instruments). The microindentation property of the multilayer films was examined using FSF001.1 single‐nanofiber mechanical tester (precision of 0.0001 cN, Suzhou Intel‐Rising Technology Co., Ltd., China). The observation of assembly of fluorescence labeled polypeptides was performed using confocal laser scanning microscopy (CLSM, LSM 510, Zeiss, Thornwood, NY). The BET surface area and porous structures of the microcages were measured based on N2 adsorption–desorption isotherms using an automatic adsorption system (ASAP 2020, Micromeritics Co.). The loading and release profiles of AgNPs were obtained by measuring the absorption spectra at 410 nm using a hybrid multimode microplate reader (Synergy H4, Winooski, VT), as described previously.[ 11 ] In brief, the drug‐loaded samples were sonicated in PBS and the amounts of encapsulated nanoparticles were quantitatively determined. The drug loading efficiency was calculated: drug‐loading efficiency = the amount of nanoparticles encapsulated in the microcages/the total amount of microcages × 100%. The S. aureus killing efficacy was assessed by dilution of samples from 24‐well culture plates and then agar plate counting, and cell adhesion, proliferation, and viability were evaluated after detaching from the substrates and counted using a hemocytometer under a light microscope, as previously reported.[ 11 , 35 ] TaqMan real‐time polymerase chain reaction (PCR) was carried out using iCycler iQ Multi Color Real‐Time PCR Detection system (Perkin‐Elmer, Wellesley, MA). The relative quantification of the targeted genes, such as SPP1 and RUNX2, was performed using the 2−ΔΔCT method. The data presented were mean ± standard deviation for triplicate experiments. Statistical significance was determined using Student's t‐test, one‐way ANOVA followed by Tukey's honestly significant difference means comparison (p < 0.05).
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
This work was supported by the Office of the Assistant Secretary of Defense for Health Affairs, through the Peer Reviewed Medical Research Program, Discovery Awards under Award No. W81XWH1810203. The authors also acknowledge financial support from AO Foundation, Osteosynthesis & Trauma Care Foundation, the West Virginia National Aeronautics and Space Administration Experimental Program to Stimulate Competitive Research (WV NASA EPSCoR), WVU PSCoR, and WVCTSI. In addition, the authors acknowledge the use of the WVU Shared Research Facilities that are supported by NIH grants 2U54GM104942‐02, 5P20RR016477, U57GM104942, P30GM103488, P20GM109098, and P20GM103434. Opinions, interpretations, conclusions, and recommendations are those of the authors and are not necessarily endorsed by the funding agencies. The authors acknowledge Suzanne Danley for proofreading.
Zhang S., Zhou S., Liu H., Xing M., Ding B., Li B., Pinecone‐Inspired Nanoarchitectured Smart Microcages Enable Nano/Microparticle Drug Delivery. Adv. Funct. Mater. 2020, 30, 2002434 10.1002/adfm.202002434
References
- 1. Mao A. S., Shin J. W., Utech S., Wang H., Uzun O., Li W., Cooper M., Hu Y., Zhang L., Weitz D. A., Nat. Mater. 2017, 16, 236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Amstad E., Science 2018, 359, 743. [DOI] [PubMed] [Google Scholar]
- 3. Mura S., Nicolas J., Couvreur P., Nat. Mater. 2013, 12, 991. [DOI] [PubMed] [Google Scholar]
- 4. Li J., Mooney D. J., Nat. Rev. Mater. 2016, 1, 16071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blanco E., Shen H., Ferrari M., Nat. Biotechnol. 2015, 33, 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sun C., Lee J. S., Zhang M., Adv. Drug Delivery Rev. 2008, 60, 1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang Y., Kohane D. S., Nat. Rev. Mater. 2017, 2, 17020. [Google Scholar]
- 8. Giri S., Trewyn B. G., Stellmaker M. P., Lin V. S. Y., Angew. Chem., Int. Ed. 2005, 44, 5038. [DOI] [PubMed] [Google Scholar]
- 9. Arciola C. R., Campoccia D., Montanaro L., Nat. Rev. Microbiol. 2018, 16, 397. [DOI] [PubMed] [Google Scholar]
- 10. Henkel J., Woodruff M. A., Epari D. R., Steck R., Glatt V., Dickinson I. C., Choong P. F. M., Schuetz M. A., Hutmacher D. W., Bone Res. 2013, 1, 216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zhang S., Xing M. M. Q., Li B., ACS Appl. Mater. Interfaces 2018, 10, 44267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jiang B., Defusco E., Li B., Biomacromolecules 2010, 11, 3630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ye C., Malak S. T., Hu K., Wu W., Tsukruk V. V., ACS Nano 2015, 9, 10887. [DOI] [PubMed] [Google Scholar]
- 14. Postma A., Yan Y., Wang Y., Zelikin A. N., Tjipto E., Caruso F., Chem. Mater. 2009, 21, 3042. [Google Scholar]
- 15. De Koker S., De Cock L. J., Rivera‐Gil P., Parak W. J., Velty R. A., Vervaet C., Remon J. P., Grooten J., De Geest B. G., Adv. Drug Delivery Rev. 2011, 63, 748. [DOI] [PubMed] [Google Scholar]
- 16. Richardson J. J., Maina J. W., Ejima H., Hu M., Guo J., Choy M. Y., Gunawan S. T., Lybaert L., Hagemeyer C. E., De Geest B. G., Adv. Sci. 2015, 2, 1400007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Jiang C., Tsukruk V. V., Adv. Mater. 2006, 18, 829. [Google Scholar]
- 18. Cui W., Li J., Decher G., Adv. Mater. 2016, 28, 1302. [DOI] [PubMed] [Google Scholar]
- 19. Ye C., Shchepelina O., Calabrese R., Drachuk I., Kaplan D. L., Tsukruk V. V., Biomacromolecules 2011, 12, 4319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu W., Choi I., Plamper F. A., Synatschke C. V., Muller A. H., Tsukruk V. V., ACS Nano 2013, 7, 598. [DOI] [PubMed] [Google Scholar]
- 21. Lin X., Wu Z., Wu Y., Xuan M., He Q., Adv. Mater. 2016, 28, 1060. [DOI] [PubMed] [Google Scholar]
- 22. Paulraj T., Riazanova A. V., Yao K., Andersson R. L., Mullertz A., Svagan A. J., Biomacromolecules 2017, 18, 1401. [DOI] [PubMed] [Google Scholar]
- 23. Gu L., Mooney D. J., Nat. Rev. Cancer 2016, 16, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Qi R., Wang Y., Bruno P. M., Xiao H., Yu Y., Li T., Lauffer S., Wei W., Chen Q., Kang X., Nat. Commun. 2017, 8, 2166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Gaharwar A. K., Mihaila S. M., Swami A., Patel A., Sant S., Reis R. L., Marques A. P., Gomes M. E., Khademhosseini A., Adv. Mater. 2013, 25, 3329. [DOI] [PubMed] [Google Scholar]
- 26. Sheridan C., Nat. Biotechnol. 2012, 30, 471. [DOI] [PubMed] [Google Scholar]
- 27. Bobo D., Robinson K. J., Islam J., Thurecht K. J., Corrie S. R., Pharm. Res. 2016, 33, 2373. [DOI] [PubMed] [Google Scholar]
- 28. Lin W., Chem. Rev. 2015, 115, 10407. [DOI] [PubMed] [Google Scholar]
- 29. Shao Z., Vollrath F., Nature 2002, 418, 741. [DOI] [PubMed] [Google Scholar]
- 30. Steven E., Saleh W. R., Lebedev V., Acquah S. F., Laukhin V., Alamo R. G., Brooks J. S., Nat. Commun. 2013, 4, 2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhang S., Liu H., Tang N., Ge J., Yu J., Ding B., Nat. Commun. 2019, 10, 1458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nishiyama Y., Sugiyama J., Chanzy H., Langan P., J. Am. Chem. Soc. 2003, 125, 14300. [DOI] [PubMed] [Google Scholar]
- 33. Larrañaga A., Lomora M., Sarasua J. R., Palivan C. G., Pandit A., Prog. Mater. Sci. 2017, 90, 325. [Google Scholar]
- 34. Yang L., Gong Z., Lin Y., Chinthapenta V., Li Q., Webster T. J., Sheldon B. W., Adv. Funct. Mater. 2017, 27, 1702689. [Google Scholar]
- 35. Likibi F., Jiang B., Li B., J. Mater. Res. 2008, 23, 3222. [Google Scholar]
- 36. An X., Wen Y., Cheng D., Zhu X., Ni Y., Cellulose 2016, 23, 2409. [Google Scholar]
- 37. Richardson J. J., Björnmalm M., Caruso F., Science 2015, 348, aaa2491. [DOI] [PubMed] [Google Scholar]
- 38. Lee Y. L., Lin T. X., Hsu F. M., Jan J. S., Nanoscale 2016, 8, 2367. [DOI] [PubMed] [Google Scholar]
- 39. Laugel N., Betscha C., Winterhalter M., Voegel J.‐C., Schaaf P., Ball V., J. Phys. Chem. B 2006, 110, 19443. [DOI] [PubMed] [Google Scholar]
- 40. Habibi Y., Lucia L. A., Rojas O. J., Chem. Rev. 2010, 110, 3479. [DOI] [PubMed] [Google Scholar]
- 41. Martines E., Seunarine K., Morgan H., Gadegaard N., Wilkinson C. D., Riehle M. O., Nano Lett. 2005, 5, 2097. [DOI] [PubMed] [Google Scholar]
- 42. Zhao L., Zhao J., Liu Y., Guo Y., Zhang L., Chen Z., Zhang H., Zhang Z., Small 2016, 12, 3327. [DOI] [PubMed] [Google Scholar]
- 43. Medronho B., Lindman B., Adv. Colloid Interface Sci. 2015, 222, 502. [DOI] [PubMed] [Google Scholar]
- 44. Liu J., Zhao Y., Guo Q., Wang Z., Wang H., Yang Y., Huang Y., Biomaterials 2012, 33, 6155. [DOI] [PubMed] [Google Scholar]
- 45. Franci G., Falanga A., Galdiero S., Palomba L., Rai M., Morelli G., Galdiero M., Molecules 2015, 20, 8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Schreurs W., Rosenberg H., J. Bacteriol. 1982, 152, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bragg P., Rainnie D., Can. J. Microbiol. 1974, 20, 883. [DOI] [PubMed] [Google Scholar]
- 48. Mohanta V., Madras G., Patil S., ACS Appl. Mater. Interfaces 2014, 6, 20093. [DOI] [PubMed] [Google Scholar]
- 49. Mihaila S. M., Gaharwar A. K., Reis R. L., Khademhosseini A., Marques A. P., Gomes M. E., Biomaterials 2014, 35, 9087. [DOI] [PubMed] [Google Scholar]
- 50. Dawson J. I., Kanczler J. M., Yang X. B., Attard G. S., Oreffo R. O., Adv. Mater. 2011, 23, 3304. [DOI] [PubMed] [Google Scholar]
- 51. Noore J., Noore A., Li B., Antimicrob. Agents Chemother. 2013, 57, 1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kang J., Dietz M. J., Hughes K., Xing M., Li B., J. Antimicrob. Chemother. 2019, 74, 1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information