

Abstract
Objective
To report safety, pharmacokinetics, exon 53 skipping, and dystrophin expression in golodirsen-treated patients with Duchenne muscular dystrophy (DMD) amenable to exon 53 skipping.
Methods
Part 1 was a randomized, double-blind, placebo-controlled, 12-week dose titration of once-weekly golodirsen; part 2 is an ongoing, open-label evaluation. Safety and pharmacokinetics were primary and secondary objectives of part 1. Primary biological outcome measures of part 2 were blinded exon skipping and dystrophin protein production on muscle biopsies (baseline, week 48) evaluated, respectively, using reverse transcription PCR and Western blot and immunohistochemistry.
Results
Twelve patients were randomized to receive golodirsen (n = 8) or placebo (n = 4) in part 1. All from part 1 plus 13 additional patients received 30 mg/kg golodirsen in part 2. Safety findings were consistent with those previously observed in pediatric patients with DMD. Most of the study drug was excreted within 4 hours following administration. A significant increase in exon 53 skipping was associated with ∼16-fold increase over baseline in dystrophin protein expression at week 48, with a mean percent normal dystrophin protein standard of 1.019% (range, 0.09%–4.30%). Sarcolemmal localization of dystrophin was demonstrated by significantly increased dystrophin-positive fibers (week 48, p < 0.001) and a positive correlation (Spearman r = 0.663; p < 0.001) with dystrophin protein change from baseline, measured by Western blot and immunohistochemistry.
Conclusion
Golodirsen was well-tolerated; muscle biopsies from golodirsen-treated patients showed increased exon 53 skipping, dystrophin production, and correct dystrophin sarcolemmal localization.
Clinicaltrials.gov identifier
Classification of evidence
This study provides Class I evidence that golodirsen is safe and Class IV evidence that it induces exon skipping and novel dystrophin as confirmed by 3 different assays.
Duchenne muscular dystrophy (DMD) is a degenerative neuromuscular disease caused by mutations in the DMD gene1 that affects ∼1/3,500–5,000 male births worldwide.2–4 Mutations causing DMD disrupt the dystrophin messenger RNA (mRNA) reading frame and prevent production of functional dystrophin.5–7 Dystrophin, a crucial component of a protein complex, links muscle cytoskeleton to the extracellular matrix, and ameliorates muscle membrane damage during eccentric contraction.5,8–10 The effect of dystrophin loss is progressive muscle wasting and ultimately premature death.1,10,11
Through selection of sequences complementary to the desired target, phosphorodiamidate morpholino oligomers (PMOs) are designed to bind to pre-mRNA, alter the splicing process, and skip the targeted exon from the mature mRNA sequence. Such exon skipping agents enable rational sequence-guided targeting of exons to be excluded from mature mRNA, restore the reading frame of dystrophin transcripts, and allow production of internally shortened, yet functional, proteins in patients with eligible out-of-frame deletions.12–15 Eteplirsen is the first PMO approved in the United States for DMD in patients with confirmed DMD mutations amenable to exon 51 skipping.16–18
Golodirsen (formerly SRP-4053) is a new PMO that hybridizes to exon 53 of dystrophin pre-mRNA, and restores the mRNA reading frame in patients with confirmed DMD mutations amenable to exon 53 skipping,15 which includes 7.7% of individuals with DMD.19 The aim of golodirsen treatment is to facilitate production of an internally shortened dystrophin protein and eventually slow disease progression. This report presents safety, pharmacokinetics, and biological activity data of golodirsen in patients with DMD and confirmed genetic mutations eligible for exon 53 skipping.
Methods
Study design
This multicenter phase 1/2 clinical trial was designed with involvement of Sarepta Therapeutics, Inc. (Cambridge, MA), advocacy groups from each participating country, and academic members of the SKIP-NMD project, to include 2 parts (figure 1A). Part 1 was a randomized, double-blind, placebo-controlled, dose titration study. Its primary objective was to assess safety, tolerability, and pharmacokinetics of 4 escalating dose levels of golodirsen or placebo over 12 weeks of treatment. Part 2 is a long-term, 168-week, open-label evaluation designed to assess biological efficacy (primary biological outcome measure [at week 48] and clinical efficacy [at week 144] and safety of once-weekly IV) infusions of golodirsen 30 mg/kg in patients with a deletion mutation amenable to exon 53 skipping. An untreated group of patients with a deletion mutation not amenable to exon 53 skipping was recruited to part 2 of the study to explore the value of exploratory clinical biomarkers.
Figure 1. Golodirsen study details.

(A) Study design. (B) Study flow diagram (part 1). 6MWT = 6-minute walk test; DSMB = data safety monitoring board; PDPF = percentage dystrophin-positive fibers.
Standard protocol approvals, registrations, and patient consents
The study is registered at clinicaltrials.gov (NCT02310906) and is being conducted in accordance with the principles of the Declaration of Helsinki and the International Council for Harmonisation Good Clinical Practice guidelines. Ethical approval was obtained in each of the participating centers (London and Newcastle, United Kingdom; Paris, France; and Rome, Italy) and parents or legal guardians of all patients provided written informed consent before study participation and genetic testing; all patients provided written assent.
Study patients
Patients eligible for part 1 enrollment were boys aged 6–15 years (inclusive) with a diagnosis of DMD and out-of-frame deletions amenable to exon 53 skipping, as confirmed by genomic multiplex ligation-dependent probe amplification or DMD gene sequencing. Additional inclusion criteria included a mean 6-minute walk test (6MWT) distance ≥250 meters at screening and baseline visits and either a North Star Ambulatory Assessment total score >17 or a Rise (Gowers) time of <7 seconds. Additional requirements were stable cardiac function (left ventricular ejection fraction ≥50% based on screening echocardiogram and QTc <450 ms based on screening electrocardiogram), percent predicted forced vital capacity of at least 50% and no requirement for nocturnal ventilation, and taking a stable dose regimen of oral corticosteroids for at least 24 weeks prior to week 1.
Exclusion criteria were the use of any treatment other than corticosteroids that may affect muscle strength or function, planned major surgery, change in contracture treatment within 3 months, or other clinically significant illness that would interfere with participation in the study.
All patients who completed part 1 transitioned to part 2. Additional patients enrolled in part 2 included treatment-naive boys aged 6–15 years who met the same criteria for part I, as well as patients with deletion mutations not amenable to exon 53 skipping who served as the untreated group for the exploratory efficacy endpoints (part 2).
Randomization and masking
In part 1, patients were randomized (2:1) to receive golodirsen or placebo prior to dosing in week 1 using an interactive voice response system. All patients, parents/caregivers, investigators, and study staff not involved with preparation of study medication were blinded to treatment assignment. Qualified pharmacists who were authorized to verify treatment and dose were unblinded to treatment assignment and not allowed to interact with study participants. In part 2, all eligible patients received open-label treatment with golodirsen.
Tissue sections allocated for reverse transcription PCR (RT-PCR), Western blot analysis, and immunofluorescence tissue staining were assigned blinding codes generated and provided by PharPoint Research, Inc. (Durham, NC). Investigators and laboratory personnel remained blinded to patient identification, time point of biopsy, and treatment status throughout all analyses.
Study treatment
In part 1, randomized patients received once-weekly IV infusions of golodirsen or placebo. After at least 2 weeks of treatment at the initial 4 mg/kg dose, patients randomized to golodirsen received doses of 10, 20, or 30 mg/kg for 2 weeks each prior to escalation to the next dose. Prior to initiation of golodirsen dosing in part 2, an independent data safety monitoring board was required to review safety data from the dose titration phase. During this safety review, all patients who completed part 1 continued to receive blinded weekly IV infusions of golodirsen 30 mg/kg or placebo per their original randomization. Following completion of the safety review, all patients from part 1, and treatment-naive patients newly enrolled in part 2, initiated open-label treatment with weekly IV infusions of golodirsen 30 mg/kg. Doses of golodirsen and placebo were calculated based on the most recently recorded patient weight, and patients were closely monitored for at least 1 hour following completion of each infusion. Patients enrolled in the untreated control group did not receive any study medication but continued their current standard of care.
Study assessments
Primary and secondary safety and clinical efficacy endpoints of parts 1 and 2 are summarized in figure 1A. The current report describes safety, tolerability, and pharmacokinetic endpoints evaluated in part 1 and key, predetermined biological endpoints evaluated through week 48 of part 2 following the collection of muscle biopsies. Part 1 safety endpoints were evaluated in all randomized patients (level of evidence I). Pharmacokinetic endpoints were evaluated in active-treated patients at 4, 10, 20, or 30 mg/kg on visits 1, 3, 5, and 7, respectively. The primary biological endpoint was the blinded change from baseline in dystrophin protein levels at week 48 as measured by Western blot. Secondary biological endpoints were evaluation of exon 53 skipping, as measured by RT-PCR, and dystrophin sarcolemmal localization assessed using immunohistochemistry (level of evidence IV). The clinical efficacy endpoint, which will be completed at week 144, will be the focus of a separate publication when this timepoint is reached.
Safety assessments
Patients were assessed for safety and tolerability throughout the study. Adverse events (AEs) were considered treatment-emergent if they occurred between initiation of the first dose of golodirsen to 28 days after last dose. Clinical laboratory measures, vital signs, physical examinations, 12-lead EKG, and echocardiogram/echocardiography were routinely monitored. Safety data from part 1 were blinded at the time of writing of this manuscript.
Pharmacokinetic methodology
Validated methods for analysis of concentration of golodirsen in patient plasma were employed featuring both a high- and low-range assay (1–100 μg/mL and 10–1,000 ng/mL, respectively). A detailed description of the pharmacokinetic methodology used is available from Dryad (https://doi.org/10.5061/dryad.g003051).
Muscle biopsy methodology
Muscle biopsy specimens were collected from one biceps brachii muscle at baseline and from the contralateral muscle at week 48 of part 2 using an optimized, standardized surgical procedure developed to avoid technical issues previously experienced during other studies in the field.20 For each biopsy surgery, 2 pieces of muscle (samples A and B) were excised, allocated, and analyzed separately. Patients in the untreated group did not have muscle biopsies and were not included in dystrophin assessments. Muscle biopsy tissue was mounted with minimal OCT, cryosectioned, and blind-labeled to patient ID and treatment status and allocated for analysis by Western blot, RT-PCR, or immunohistochemistry.
Western blot analysis
The Western blot assay was performed under Good Clinical Laboratory Practice standards. Western blots were executed according to validated methodology adapted from Charleston et al.21 Dystrophin levels of treatment-blinded samples were calculated from a 5-point standard curve ranging from 0.25% to 4%. Reported dystrophin levels were the average value of both biological replicates and 2 technical gel replicates for each sample result.
Exon skipping analysis
Cryosectioned tissue sections from muscle biopsy samples were treatment-blinded and samples homogenized in TRIzol reagent (Thermo Fisher Scientific, Waltham, MA) to isolate total RNA. RNA quality was evaluated using a DNA 5K/RNA/Charge Variant Assay LabChip and LabChip GXII Touch HT (PerkinElmer, Inc., Waltham, MA). A detailed description of the methodology used for exon skipping analysis, including patient genotypes, is shown in table e-1 and percentage of exon 53 skipping in figure e-1 (https://doi.org/10.5061/dryad.g003051).
Immunofluorescence staining and image capture
To evaluate dystrophin localization, muscle biopsy tissue cryosections of treatment-blinded samples were dual-stained (performed at University of Iowa Hospitals and Clinics by Dr. Steven A. Moore) with antidystrophin antibody at 1:25 (MANDYS106, clone 2C6, mouse monoclonal antibody obtained from Glenn Morris, Wolfson Centre for Inherited Neuromuscular Disease, Oswestry, UK, and now commercially available from Millipore [MABT827, clone 2C6]) and anti-laminin α2 antibody at 1:400 (ab11576, clone 4H8, lot GR95776-14, rat monoclonal antibody; Abcam, Cambridge, MA) followed by incubation with the detection cocktail that included Alexa Fluor 594 goat anti-mouse IgG2a (A21135, lot 1366503; Thermo Fisher Scientific) and Alexa Fluor 488 rabbit anti-rat IgG (H + L) (A21210, lot 53122A; Thermo Fisher Scientific).20
Tissue sections were imaged using a validated whole-slide scanner method at 20× magnification in fluorescein (Alexa Fluor 488; laminin α2 signal) and tetramethylrhodamine (Alexa Fluor 594; MANDYS106 signal) detection channels using a 3DHISTECH Panoramic MIDI fluorescent scanner (PANMIDI; Perkin Elmer) at a fixed exposure time (imaging performed at Flagship Biosciences, Inc., Westminster, CO). Contrast-inverted, MANDYS106-stained images in the Alexa Fluor 594 channel were processed to correct for background staining before analysis was conducted. An outer region of analysis (ROA) was drawn at least 1 mm outside the tissue while an inner exclusion ROA was drawn around the tissue. A grid of 0.5 mm2 was overlaid on the tissue and 1 box in each quadrant was identified for analysis of background signal (figure e-2, https://doi.org/10.5061/dryad.g003051). A detailed description of the methodology utilized for the correction of the background and the image capture, including figure e-3 showing selection of high-power fields and figure e-4 showing an example of a background-corrected inverted Alexa Fluor image, is available from Dryad (https://doi.org/10.5061/dryad.g003051).
Percent dystrophin-positive fibers
Assessment of percentage dystrophin-positive fibers (PDPFs) was executed according to methodology adapted from Charleston et al.21 by 3 trained pathologists who each scored all blinded high-power fields (HPFs). Training ensured that the pathologists approached the evaluation and scoring of muscle fibers by the same criteria for identification of dystrophin-positive muscle fibers and muscle fiber total counts. The scoring of randomly duplicated images was used to assess intrapathologist precision. Pathologists determined positive muscle fibers based on visual examination of the background-corrected inverted Alexa Fluor 594 digital images. Total fiber counts were defined by the structural appearance of the fiber cross-section. The total fiber counting was done while the pathologists viewed the Alexa Fluor 488–only HPFs. Scoring of the dystrophin-positive fibers on the background-corrected Alexa Fluor 594 HPFs was completed before scoring of total muscle fibers using the Alexa Fluor 488 HPFs.
Statistical analyses
For each patient sample at the 2 biopsy time points (baseline vs part 2 [week 48]), replicate runs were performed to determine dystrophin level (% normal) by Western blot, % exon skipping by RT-PCR, and manual scoring of PDPFs. The average of replicate values from available samples was used in the analyses.
Change from baseline and fold change from baseline for each patient was calculated as follows:
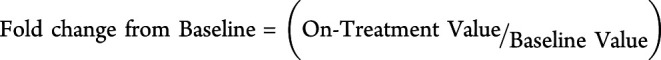 |
Dystrophin level (% normal) determined by Western blot, % exon skipping by RT-PCR, and manual scoring of PDPFs was averaged across all patients for baseline and for part 2, week 48. In addition, change from baseline to part 2, week 48, and fold change from baseline to part 2, week 48, was averaged across all patients.
Changes from baseline in dystrophin level (% normal) were assessed using a 1-sample permutation t test. A sign test was employed to determine if exon skipping at part 2, week 48, was greater than the mean ratio at baseline. Statistical significance was set at α < 0.05 for all analyses. Correlations between exon 53 skipping and dystrophin level (% normal) and between PDPF and dystrophin level (% normal) were performed using the Spearman method. All statistical analyses were performed using SAS version 9.4 (Cary, NC) except the permutation t test using R package.
Data availability
Study results and individual de-identified patient data will not be available in a publicly accessible repository to protect the interests of the patients and investigators, in accordance with the policies of Sarepta Therapeutics, Inc., and in line with the General Data Protection Regulation.
Results
Baseline characteristics and safety
Demographics and disease characteristics of all 25 study patients who received golodirsen described in this report are summarized in table 1. In part 1, 13 patients were screened and 12 patients were randomized to receive once-weekly IV infusions of golodirsen 4 mg/kg followed by dose escalation to golodirsen 10, 20, and 30 mg/kg (n = 8) or placebo (n = 4; figure 1B). All patients reported at least 1 treatment-emergent AE (TEAE), none of which was serious. No patients discontinued due to a TEAE. Moderate TEAEs were reported in 2 patients: Staphylococcus aureus infection of a Port-A-Cath and pyrexia. TEAEs were assessed as related to study drug by the investigator in 8 patients. In general, the TEAEs reported in part 1 of this study were consistent with what would be expected in a pediatric DMD population.
Table 1.
Baseline demographics and clinical characteristics of study patients

Pharmacokinetics
Pharmacokinetics were evaluated in 8 active-treated patients at dose levels 4, 10, 20, and 30 mg/kg on visits 1, 3, 5, and 7, respectively (table e-2, https://doi.org/10.5061/dryad.g003051). Following each dose level, concentrations at 24 hours postdose were at or near the lower limit of quantitation (10 ng/mL). A dose-proportional increase in maximum concentration (Cmax) was observed with an average of 8,501 ng/mL at the lowest dose of 4 mg/kg and 56,550 ng/mL at the highest dose of 30 mg/kg. When evaluated across all dose levels, plasma clearance (CLp) and volume of distribution of study drug at steady-state (Vss) averaged 365 ± 133 mL/h/kg and 684 ± 252 mL/kg, respectively, and half-life averaged 3.09 ± 1.30 hours. The short half-life also indicates that with weekly dosing no accumulation in plasma is expected. Exposure increased in a proportional manner with dose increment (table e-3, https://doi.org/10.5061/dryad.g003051).
Most of the recovered study drug was excreted in the first 4 hours (table e-4, https://doi.org/10.5061/dryad.g003051). The percent of dosed golodirsen recovered in urine over 24 hours was between 54.5% and 60.4%, and renal clearance (CLR) accounted for 54.5%, 54.7%, 60.6%, and 59.0% of total systemic clearance (CL) at visits 1, 3, 5, and 7 (4, 10, 20, and 30 mg/kg), respectively (table e-5, https://doi.org/10.5061/dryad.g003051).
There was a trend of increasing CLp and Vss with body weight when CLp and Vss were evaluated without body weight normalization. There was a trend of decreasing CLp and Vss (normalized to body weight) with age. Half-life showed little dependence on body weight or age.
Dystrophin production
With once-weekly golodirsen treatment, mean percent of normal dystrophin protein as measured by Western blot was 1.019% (SD 1.033%), with a range across patients of 0.09%–4.30% (figure 2, A and B), representing a significant increase over baseline (p < 0.001; mean baseline dystrophin level estimate of 0.095% [SD 0.068%], range 0.02–0.31). Utilizing the methodology in the statistical analyses section, we observed an approximately 16-fold increase in dystrophin. Baseline biopsy dystrophin levels as measured by Western blot were consistently very low (figure 2B), with only one patient's baseline sample above the lowest standard curve point of 0.25% (figure 2A). Assessing drug effect as an increase over baseline dystrophin levels22 is challenging due to the lack of a quantitative protein assay to accurately measure the low levels of dystrophin that are detectable by immunohistochemistry in most patients with DMD. In certain samples that fell below 0.25%, the Western blot method utilized in this analysis was able to detect a signal at the appropriate molecular weight for dystrophin. This enabled estimation of extrapolated mean value of 0.095% for baseline samples.
Figure 2. Treatment with golodirsen demonstrated an increase in de novo dystrophin protein.

(A) Western blot examples performed with baseline (labeled as BL) and on treatment (Tx) paired samples from 4 patients. Numbers above Tx gel lanes indicate the percent normal control dystrophin, calculated from the standard curve on each blot (lanes denoted with 4%, 2%, 1%, 0.5%, 0.25%). In both panels, lane 0 represents baseline untreated DMD control tissue with no normal control lysate. Arrows and values above gel indicate the percent normal dystrophin measured for the indicated lane. (B) Western blot data shown as averaged dystrophin percent of normal for each individual patient at baseline (open circles) and at part 2, week 48, (solid squares), with dystrophin levels on y-axis presented as linear values. *Sample reading was above the upper limit of quantification.
Alternative imputation methods were used in a sensitivity analysis of the increase in dystrophin levels from baseline to part 2, week 48. In addition to using extrapolated estimates, the more conservative analysis method only used values within the standard curve range (0.25%–4%). Data analysis using more conservative imputations all resulted in a significant increase (p < 0.001) from baseline in dystrophin protein at part 2, week 48 (table e-6, https://doi.org/10.5061/dryad.g003051).
We investigated whether differences in the duration of drug exposure (table e-7, https://doi.org/10.5061/dryad.g003051) could account for differences in dystrophin expression by comparing results for patients who participated in both parts 1 and 2 vs only part 2 of the clinical study. While the 2 patients who received the highest number of golodirsen doses also had higher levels of dystrophin as measured by Western blot, overall, no correlation was observed between the number of doses or the duration of exposure and dystrophin expression.
Alteration of dystrophin mRNA splicing in patients treated with golodirsen can be measured using semiquantitative endpoint RT-PCR as the amount of dystrophin mRNA that omits exon 53 relative to the amount of mRNA that includes exon 53. The mean percentage of exon skipping for all patients increased from 2.590% (SD, 4.0864%; range, 0.00%–14.69%) at baseline to 18.953% (SD, 13.2245%; range, 2.62%–48.03%) at part 2, week 48, representing a mean per-patient 28.897-fold increase (SD, 39.6763; range, 2.59–150.36) in exon 53 skipping (figure 3A). Sanger DNA sequence analysis of PCR-amplified products of the exon-skipped bands across the new exon junction formed by skipping exon 53 confirmed restoration of reading frame in the dystrophin transcript in 100% of assessed patients (data not shown).
Figure 3. Exon 53 skipping and dystrophin.
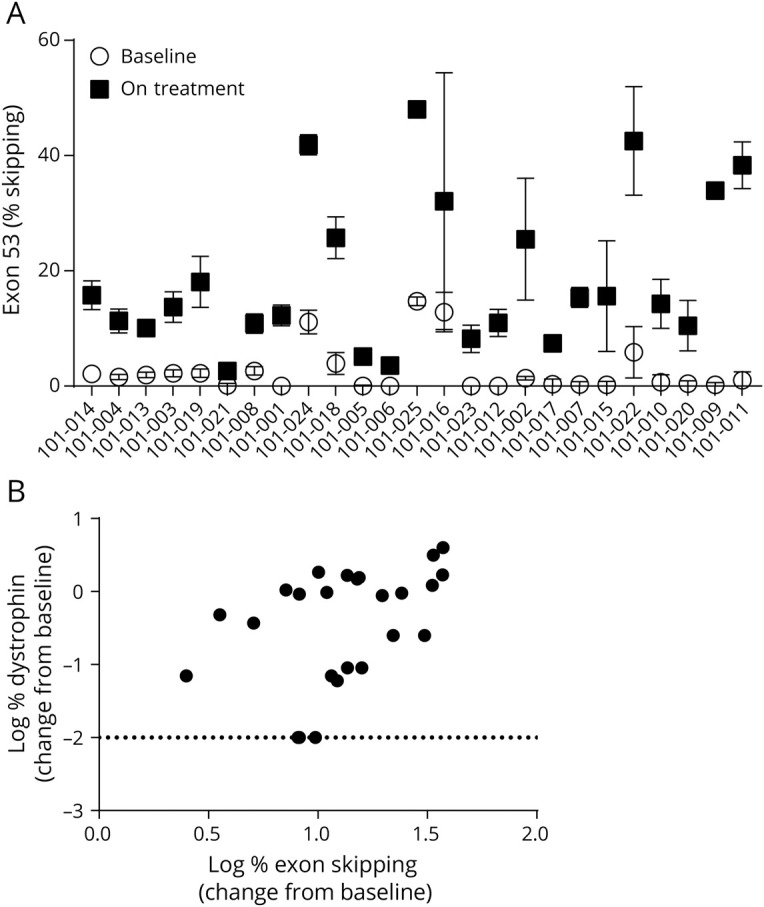
(A) Treatment with golodirsen demonstrates an increase in skipping of exon 53 in dystrophin mRNA. Percent exon 53 skipping for individual patients at baseline (open circles) and on treatment (filled squares). Data represent mean ± SD values for 4–8 replicates. (B) Positive correlation between percent exon 53 skipping and production of dystrophin protein. Percent exon 53 skipping change from baseline was plotted against the change in dystrophin protein from baseline, as measured by Western blot (Spearman r correlation coefficient: 0.500; p = 0.011).
Consistent with golodirsen’s mechanism of action, a significant, positive correlation between exon skipping and de novo dystrophin protein expression was observed with a Spearman r correlation coefficient of 0.500 (p = 0.011; figure 3B). In addition, dystrophin expression was localized to the sarcolemma in patients treated with golodirsen. Whole-slide scan images at baseline and part 2, week 48, from 2 patients (with average and high dystrophin levels as measured by Western blot) are shown as examples in figure 4, A and B. An apparent increase in staining intensity of dystrophin above baseline levels is evident and is clearly localized to the sarcolemma.
Figure 4. Dystrophin expression, baseline and treated part 2, week 48, biopsy samples from the same patient.

(A) Example 1: Low magnification image of whole tissue section stained for laminin α2 (green, all fibers stained) and dystrophin (red). A total of 2,843 muscle fibers were present in the baseline whole-slide image (A.a) and 1,077 fibers in the part 2, week 48, image (A.b). Three highlighted regions from the whole tissue sections (green) are magnified below each image to illustrate the corresponding dystrophin staining in these regions (red). Image intensity was not enhanced and represents original captured images. Dystrophin protein at part 2, week 48, was 4.3% of normal, as measured by Western blot for this patient. (B) Example 2: Low magnification image of whole tissue section stained for laminin α2 (green, all fibers stained) and dystrophin (red). A total of 4,945 muscle fibers were present in the baseline whole-slide image (B.a) and 2,167 fibers in the part 2, week 48, image (B.b). Three highlighted regions from the whole tissue sections (green) are magnified below each image to highlight the corresponding dystrophin staining in these regions (red). Image intensity was not enhanced and represents original captured images. Dystrophin protein at part 2, week 48, was 1.91% of normal, as measured by Western blot for this patient.
PDPF scoring indicated that weekly treatment of patients with golodirsen at week 48 resulted in a significant increase in positive dystrophin fibers (p < 0.001). The mean baseline level for scoring PDPF was 1.430% (SD, 2.042; range, 0.06%–9.75%), whereas mean scoring PDPF at week 48 was 10.471% (SD, 10.102; range, 0.87%–32.59%), a mean per-patient 13.461-fold increase (SD, 11.9171; range, 1.88–49.67). The PDPF data for each individual patient at baseline and part 2, week 48, are shown in figure 5A. The intraclass correlation coefficient (ICC) for interpathologist consistency estimates the correlation of scores on the same section scored by different pathologists. The interpathologist ICC for the positive dystrophin fibers was 0.8134 (95% CI, 0.8080–0.8192).
Figure 5. Manual percentage dystrophin-positive fiber (PDPF) analysis.

(A) Baseline values of PDPF for each patient are shown as open circles and part 2, week 48, intensity is shown as solid squares. This graphical representation provides a ready way to visualize relative changes in each patient. Data represent the mean of 3–4 replicates with SD bars. (B) Western blot protein change from baseline is plotted on the x-axis and percent dystrophin-positive fiber change from baseline as assessed by manual scoring is plotted on the y-axis. Spearman analysis shows a significant correlation (p < 0.001; r = 0.663). Each point represents a single patient.
A positive correlation and linear relationship between dystrophin protein as measured by Western blot and dystrophin localization to the membrane (PDPF) was observed with a Spearman correlation coefficient of 0.663 (p < 0.001), as shown in figure 5B.
Discussion
In this first-in-human, multicenter trial, we evaluated dystrophin production (primary biological outcome measure), exon 53 skipping, and dystrophin localization at week 48 in muscle biopsies from patients who received weekly IV treatments with golodirsen. The week 48 time point was prespecified as the primary biological assessment to allow sufficient time for dystrophin to be produced based upon previous experience with eteplirsen, an antisense PMO targeting exon 51 of the DMD gene.17,23 Our findings demonstrated the robust pharmacologic activity of golodirsen using 3 independent, complementary methods. All 25 patients had an increase in skipping of exon 53, demonstrating clear evidence of target engagement by golodirsen. The primary biological endpoint of the study was achieved, as statistically significant increases of approximately 16-fold over baseline were observed in de novo dystrophin protein expression as measured by Western blot at week 48, with a mean of 1.019% of normal and a range across patients of 0.09%–4.30%. The de novo dystrophin protein correctly localized to the sarcolemma with a significant increase in mean change in PDPF from baseline to 10.471%, for a mean increase of 13.461-fold. The consistency of the dystrophin response measured by the complementary assays and the positive correlation between exon skipping and dystrophin protein as measured by Western blot and dystrophin localization to the sarcolemma as measured by manual PDPF support the robustness of the pharmacologic activity of golodirsen.
While the response of the golodirsen-treated patient population was unequivocal, the level of skipping and protein restoration varied across individual patients. This finding could be due to factors such as biological variation across muscle tissue in the therapeutic response of particular biopsies and sectioned muscle areas, as previously noted in preclinical models.24,25 Nevertheless the levels of dystrophin detected using Western blot exceed the levels previously obtained using eteplirsen, where, at week 180, levels of dystrophin on blot ranged from 0 to 2.47 (mean, 0.93; median [calculated], 0.96).21
While the level of dystrophin restoration necessary to alter the course of disease for patients with DMD is not known, the clinical and preclinical literature clearly indicates that the presence of very low or trace levels of dystrophin is associated with improved clinical outcome. In preclinical studies, very low levels of dystrophin (between 1% and 4%) demonstrated clear benefit in improving outcome, not only in the classical mdx mouse model of DMD but also in the double dystrophin/utrophin mdx mouse model, with clear survival benefit.26 In humans, several groups have demonstrated that spontaneous exon skipping in patients with DMD with certain mutation types leads to a milder course of disease, with divergence in the age at loss of ambulation of several years compared to the typical DMD population.27,28 In particular, dystrophin expression in muscle fibers is detectable by the highly sensitive immunohistochemical methods in patients with DMD with genetic mutations amenable to exon 44 skipping, presumably due to spontaneous exon 44 skipping.27–30 Importantly, the low level of dystrophin detected by immunohistochemistry in these patients is typically below the level of detection using Western blot methodology used in our study.24
We also recently confirmed a significant difference in the 6MWT outcome when comparing patients with mutations amenable to exon 53 skipping to those amenable to exon 44 skipping. This latter group of patients (in whom higher levels of dystrophin protein are typically found27) had better results both at baseline and at follow-up.31
The data from ongoing eteplirsen studies are in keeping with these predictions, as eteplirsen treatment was associated with delayed loss of ambulation18 and halving of the annual decline in respiratory function.32 A pivotal aspect of the conditional approval of eteplirsen was the demonstration of posttreatment dystrophin restoration.
Nevertheless, higher levels of dystrophin are necessary to correct other aspects of dystrophin deficiency: preclinical studies suggest that levels of ∼15% are necessary to protect the muscle against eccentric contraction-induced injury, while higher levels, in the range of ∼40%, also normalize muscle force production.33 These data therefore indicate the hierarchical implication of different levels of dystrophin in muscle, from improving muscle pathology, to higher levels that also protect muscle from further damage, to even higher levels associated with normalization of force generation.
The validated methodology that we developed in concordance with current regulatory guidance is the same that was used in previous studies of eteplirsen, therefore allowing a more direct comparison between our current results and the previously published eteplirsen studies.17,34 Applying the same methodology used in these studies, we recently demonstrated that a patient with a nonsense mutation in exon 42 had low levels of spontaneous exon skipping that resulted in an in-frame deletion removing the nonsense mutation from the transcript (skipping levels 6%–9.8%). On Western blot, this patient, who at his age appears to follow a milder disease course, had dystrophin levels of 3.2%, well within the range of levels found in several of the patients in this study.30 Future studies using this validated methodology and applied to a range of patients with DMD or Becker muscular dystrophy will be helpful to refine the correlation between the amount of dystrophin produced and clinical course of dystrophinopathies, including the roles of genetic modifiers such as LTPB4 and CD40.34,35
Following our previous experience with eteplirsen and recent guidance put forth by the US Food and Drug Administration and the European Medicines Agency, 48 weeks is not a sufficient duration of time to allow the trajectories of treated patients to diverge from those of untreated patients.22,36 A longer duration of treatment is needed to characterize the clinical benefits of exon skipping leading to low levels of dystrophin expression.18,23 The number of patients enrolled in the current study was relatively small but comparable to most other DMD phase I/II studies and was adequate to meet the primary endpoint of biochemical efficacy.
This study is ongoing and efficacy, additional safety, and functional outcomes will be described in a separate publication when the week 144 data cut has been performed.
Golodirsen is the second PMO shown to increase dystrophin expression and sarcolemmal localization through the initiation of exon skipping, further validating the potential of the PMO antisense oligonucleotide platform in DMD. The consistency of results across these endpoints supports the conclusion that golodirsen will increase levels of functional dystrophin for patients with DMD with genetic mutations amenable to exon 53 skipping therapy. The levels produced are expected to slow the rate of decline of this devastating disease, but this will need to be confirmed in longer and larger studies. Progress in advancing the rigor of methods to measure dystrophin expression should facilitate evaluation of therapies based on dystrophin restoration and will ultimately assist in clarifying the relationship between dystrophin production and clinical outcome measures.
Acknowledgment
The authors thank the MRC Centre for Neuromuscular Diseases for its support through the Neuromuscular Disease BioBank; Muscular Dystrophy UK for its support to the Neuromuscular Centre at UCL; Dr. Silvia Torelli for the supervision of the muscle biopsy storage and processing techniques in London; Steven A. Moore and the Wellstone Muscular Dystrophy Cooperative Research Center (Iowa City, IA) for assistance with analysis of muscle biopsies; and the additional investigators who participated in this study as part of the SKIP NMD Study Group. Editorial assistance (which included editing and formatting the manuscript, updating the references, contacting the various coauthors to solicit their contribution, updating the manuscript following the comments of the reviewers, and submitting the manuscript) was provided by Aji Nair (Sarepta Therapeutics, Inc.) and Peloton Advantage, Parsippany, NJ, and Health & Wellness Partners, LLC, Upper Saddle River, NJ, which were funded by Sarepta Therapeutics, Inc. Joana Domingos is deceased.
Glossary
- 6MWT
6-minute walk test
- AE
adverse event
- CLp
plasma clearance
- Cmax
maximum concentration
- DMD
Duchenne muscular dystrophy
- HPF
high-power field
- ICC
intraclass correlation coefficient
- mRNA
messenger RNA
- PDPF
percentage dystrophin-positive fibers
- PMO
phosphorodiamidate morpholino oligomer
- ROA
region of analysis
- RT-PCR
reverse transcription PCR
- TEAE
treatment-emergent adverse event
- Vss
volume of distribution of study drug at steady-state
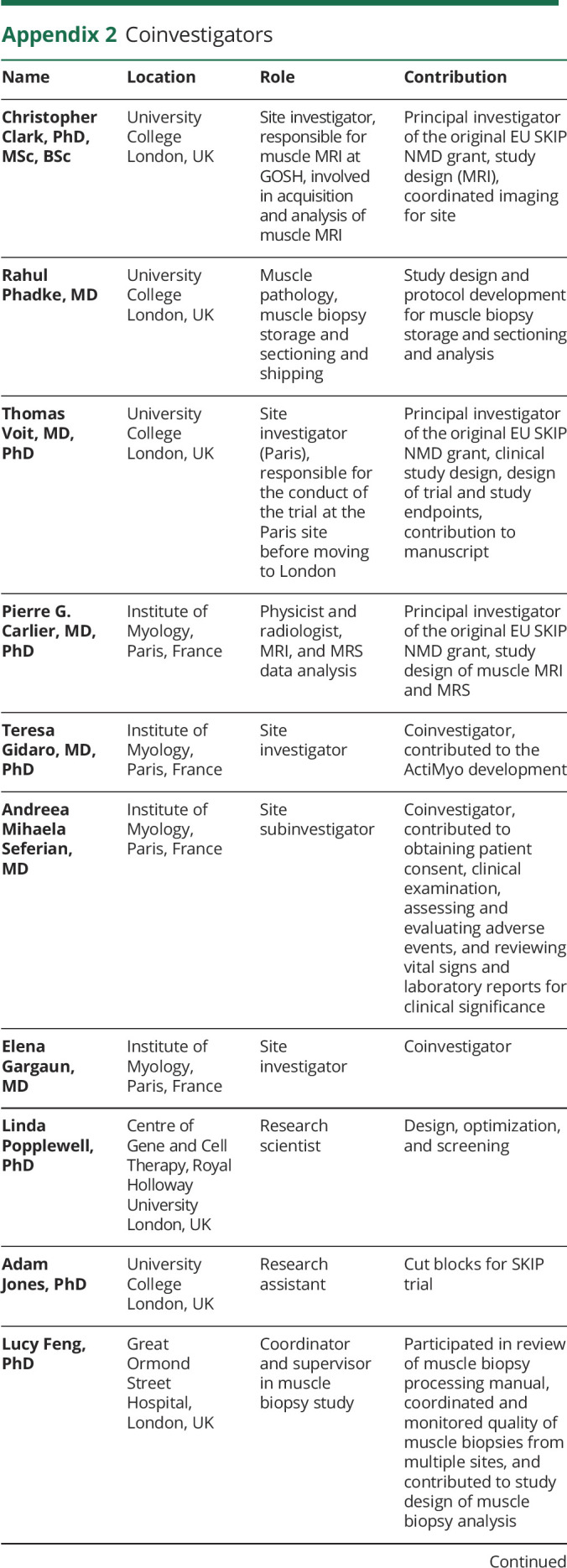
Appendix 1. Authors
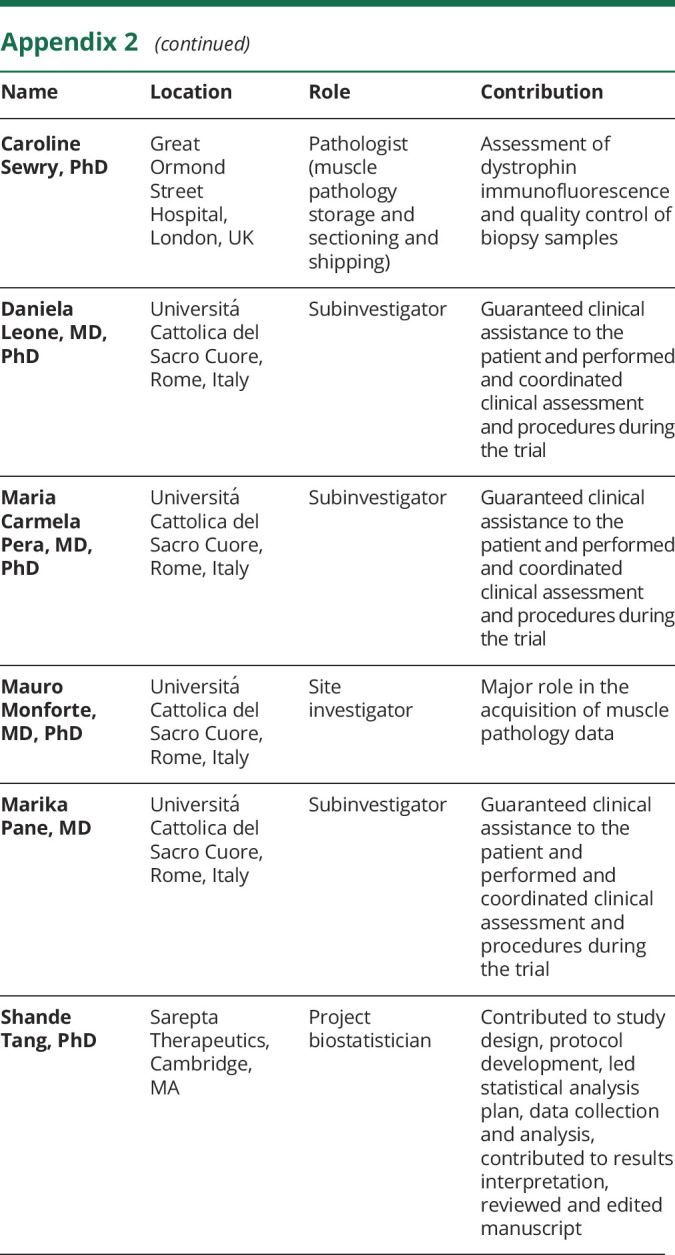
Appendix 2. Coinvestigators
Footnotes
Class of Evidence: NPub.org/coe
Study funding
This study was sponsored by Sarepta Therapeutics, Inc., and the European Union Framework Project 7 (EU FP7) SKIP-NMD grant (no. HEALTH-F4-2012-30537).
Disclosure
D.E. Frank is an employee of Sarepta Therapeutics, Inc., and may own stock/stock options in the company. F.J. Schnell is an employee of Sarepta Therapeutics, Inc., and may own stock/stock options in the company. C. Akana was an employee of Sarepta at the time of the study. S.H. El-Husayni is an employee of Sarepta Therapeutics, Inc., and may own stock/stock options in the company. C.A. Desjardins was an employee of Sarepta at the time of the study. J. Morgan is supported by the NIHR Great Ormond Street Hospital Biomedical Research Centre; the views expressed in this paper are hers and not necessarily those of the NHS, the NIHR, or the Department of Health. J. Charleston is an employee of Sarepta Therapeutics, Inc., and may own stock/stock options in the company. V. Sardone received funding through EU FP 7 No. HEALTH-F4-2012-30537 and from Sarepta Therapeutics, Inc. J. Domingos has no conflicts to disclose. G. Dickson works for an institution that received funding from SKIP-NMD and unrelated research funding from Sarepta Therapeutics, Inc.; and is an inventor on a related Royal Holloway, University of London patent. V. Straub has served as an investigator for Sarepta Therapeutics, Inc., and received honoraria and serves/served on advisory boards for Sarepta. M. Guglieri has no conflicts to disclose. E. Mercuri has served as a remunerated consultant for Sarepta Therapeutics, Inc. L. Servais participated in a scientific advisory board funded by Sarepta Therapeutics, Inc. F. Muntoni has served as a remunerated consultant for Sarepta Therapeutics, Inc. and is supported by the NIHR Great Ormond Street Hospital Biomedical Research Centre; the views expressed in this paper are his and not necessarily those of the NHS, the NIHR, or the Department of Health. D.E. Frank and F. Muntoni had full access to all of the data in the study and had final responsibility for the decision to submit the manuscript for publication. Go to Neurology.org/N for full disclosures.
References
- 1.Bushby K, Finkel R, Birnkrant DJ, et al. Diagnosis and management of Duchenne muscular dystrophy, part 1: diagnosis, and pharmacological and psychosocial management. Lancet Neurol 2010;9:77–93. [DOI] [PubMed] [Google Scholar]
- 2.Emery AE. Population frequencies of inherited neuromuscular diseases: a world survey. Neuromuscul Disord 1991;1:19–29. [DOI] [PubMed] [Google Scholar]
- 3.Mendell JR, Shilling C, Leslie ND, et al. Evidence-based path to newborn screening for Duchenne muscular dystrophy. Ann Neurol 2012;71:304–313. [DOI] [PubMed] [Google Scholar]
- 4.Mah JK, Korngut L, Dykeman J, Day L, Pringsheim T, Jette N. A systematic review and meta-analysis on the epidemiology of Duchenne and Becker muscular dystrophy. Neuromuscul Disord 2014;24:482–491. [DOI] [PubMed] [Google Scholar]
- 5.Kobayashi YM, Campbell KP. Skeletal muscle dystrophin-glycoprotein complex and muscular dystrophy. In: Hill JA, Olson EN, eds. Muscle: Fundamental Biology and Mechanisms of Disease, vol. 2. New York: Academic Press; 2012:935–942. [Google Scholar]
- 6.Emery AE. The muscular dystrophies. Lancet 2002;359:687–695. [DOI] [PubMed] [Google Scholar]
- 7.Monaco AP, Bertelson CJ, Liechti-Gallati S, Moser H, Kunkel LM. An explanation for the phenotypic differences between patients bearing partial deletions of the DMD locus. Genomics 1988;2:90–95. [DOI] [PubMed] [Google Scholar]
- 8.Allen DG, Whitehead NP, Froehner SC. Absence of dystrophin disrupts skeletal muscle signaling: roles of Ca2+, reactive oxygen species, and nitric oxide in the development of muscular dystrophy. Physiol Rev 2016;96:253–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rando TA. The dystrophin-glycoprotein complex, cellular signaling, and the regulation of cell survival in the muscular dystrophies. Muscle Nerve 2001;24:1575–1594. [DOI] [PubMed] [Google Scholar]
- 10.Mah JK. Current and emerging treatment strategies for Duchenne muscular dystrophy. Neuropsychiatr Dis Treat 2016;12:1795–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Falzarano MS, Scotton C, Passarelli C, Ferlini A. Duchenne muscular dystrophy: from diagnosis to therapy. Molecules 2015;20:18168–18184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wilton SD, Fall AM, Harding PL, McClorey G, Coleman C, Fletcher S. Antisense oligonucleotide-induced exon skipping across the human dystrophin gene transcript. Mol Ther 2007;15:1288–1296. [DOI] [PubMed] [Google Scholar]
- 13.Wilton SD, Fletcher S. RNA splicing manipulation: strategies to modify gene expression for a variety of therapeutic outcomes. Curr Gene Ther 2011;11:259–275. [DOI] [PubMed] [Google Scholar]
- 14.Wilton SD, Lloyd F, Carville K, et al. Specific removal of the nonsense mutation from the mdx dystrophin mRNA using antisense oligonucleotides. Neuromuscul Disord 1999;9:330–338. [DOI] [PubMed] [Google Scholar]
- 15.Popplewell LJ, Trollet C, Dickson G, Graham IR. Design of phosphorodiamidate morpholino oligomers (PMOs) for the induction of exon skipping of the human DMD gene. Mol Ther 2009;17:554–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Exondys 51 [package insert]. Cambridge: Sarepta Therapeutics, Inc.; 2016. [Google Scholar]
- 17.Cirak S, Arechavala-Gomeza V, Guglieri M, et al. Exon skipping and dystrophin restoration in patients with Duchenne muscular dystrophy after systemic phosphorodiamidate morpholino oligomer treatment: an open-label, phase 2, dose-escalation study. Lancet 2011;378:595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mendell JR, Goemans N, Lowes LP, et al. Longitudinal effect of eteplirsen versus historical control on ambulation in Duchenne muscular dystrophy. Ann Neurol 2016;79:257–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Aartsma-Rus A, Fokkema I, Verschuuren J, et al. Theoretic applicability of antisense-mediated exon skipping for Duchenne muscular dystrophy mutations. Hum Mutat 2009;30:293–299. [DOI] [PubMed] [Google Scholar]
- 20.Frank D, Dworzak J, Lawlor M, et al. Optimization and implementation of best practices for collection and preparation of muscle biopsies for analysis during clinical trials of neuromuscular disease therapeutics [poster]. International Annual Congress of the World Muscle Society; October 3–7, 2017; Saint Malo, France.
- 21.Charleston JS, Schnell FJ, Dworzak J, et al. Eteplirsen treatment for Duchenne muscular dystrophy: exon skipping and dystrophin production. Neurology 2018;90:e2146–e2154. [DOI] [PubMed] [Google Scholar]
- 22.Guidance for Industry. Duchenne muscular dystrophy and related dystrophinopathies: developing drugs for treatment. 2018. Available at: fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM450229.pdf. Accessed February 5, 2019.
- 23.Mendell JR, Rodino-Klapac LR, Sahenk Z, et al. Eteplirsen for the treatment of Duchenne muscular dystrophy. Ann Neurol 2013;74:637–647. [DOI] [PubMed] [Google Scholar]
- 24.Alter J, Lou F, Rabinowitz A, et al. Systemic delivery of morpholino oligonucleotide restores dystrophin expression bodywide and improves dystrophic pathology. Nat Med 2006;12:175–177. [DOI] [PubMed] [Google Scholar]
- 25.Yokota T, Lu QL, Partridge T, et al. Efficacy of systemic morpholino exon-skipping in Duchenne dystrophy dogs. Ann Neurol 2009;65:667–676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Putten M, Hulsker M, Young C, et al. Low dystrophin levels increase survival and improve muscle pathology and function in dystrophin/utrophin double-knockout mice. FASEB J 2013;27:2484–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Anthony K, Arechavala-Gomeza V, Ricotti V, et al. Biochemical characterization of patients with in-frame or out-of-frame DMD deletions pertinent to exon 44 or 45 skipping. JAMA Neurol 2014;71:32–40. [DOI] [PubMed] [Google Scholar]
- 28.Bello L, Morgenroth LP, Gordish-Dressman H, Hoffman EP, McDonald CM, Cirak S. DMD genotypes and loss of ambulation in the CINRG Duchenne natural history study. Neurology 2016;87:401–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moon D, Hu S, Bange J, et al. Genotype-phenotype associations in a large cohort of Duchenne muscular dystrophy patients. Neuromuscul Disord 2017;27(suppl 2):S104–S105. [Google Scholar]
- 30.Waldrop MA, Gumienny F, El Husayni S, Frank DE, Weiss RB, Flanigan KM. Low-level dystrophin expression attenuating the dystrophinopathy phenotype. Neuromuscul Disord 2018;28:116–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Brogna C, Coratti G, Pane M, et al. ; on behalf on the International DMD Group. Long-term natural history data in Duchenne muscular dystrophy ambulant patients with mutations amenable to skip exons 44, 45, 51 and 53. PLoS One 2019;14:e0218683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Khan N, Eliopoulos H, Han L, et al. Eteplirsen treatment attenuates respiratory decline in ambulatory and non-ambulatory patients with Duchenne muscular dystrophy. J Neuromuscul Dis 2019;6:213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Godfrey C, Muses S, McClorey G, et al. How much dystrophin is enough: the physiological consequences of different levels of dystrophin in the mdx mouse. Hum Mol Genet 2015;24:4225–4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bello L, Flanigan KM, Weiss RB, et al. Association study of exon variants in the NF-κB and TGFβ pathways identifies CD40 as a modifier of Duchenne muscular dystrophy. Am J Hum Genet 2016;99:1163–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Flanigan KM, Ceco E, Lamar KM, et al. LTBP4 genotype predicts age of ambulatory loss in Duchenne muscular dystrophy. Ann Neurol 2013;73:481–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.European Medicines Agency. Guideline on the clinical investigation of medicinal products for the treatment of Duchenne and Becker muscular dystrophy. Available at: ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2015/12/WC500199239.pdf. Accessed November 5, 2018.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Study results and individual de-identified patient data will not be available in a publicly accessible repository to protect the interests of the patients and investigators, in accordance with the policies of Sarepta Therapeutics, Inc., and in line with the General Data Protection Regulation.