

Abstract
Purpose:
The response to acute and long-term arginine starvation results in a conditional adaptive metabolic reprogramming that can be harnessed for therapeutic opportunities in ASS1-negative tumors. Here, we investigate the underlying biology of priming ASS1− tumors with arginine deiminase (ADI-PEG20) before treatment with gemcitabine (GEM) and docetaxel (DTX) in sarcoma, pancreatic cancer, and melanoma cell lines.
Experimental Design:
ASS1− tumor cell lines were treated to create LTAT (long-term ADI treated) cell lines (ASS1+) and used for drug combination studies. Protein expression of ASS1, dCK, RRM2, E2F1, c-MYC, and hENT1 was measured. c-MYC activity was determined, live-cell immunofluorescent studies for hENT1, uptake assays of FITC-cytosine probe, and rescue studies with a c-MYC inhibitor were all determined in the presence or absence of the ADI-PEG20:GEM:DTX.
Results:
In examining modulations within the pyrimidine pathway, we identified that the addition of DTX to cells treated with ADI-PEG20 resulted in translocation of stabilized c-Myc to the nucleus. This resulted in an increase of hENT1 cell-surface expression and rendered the cells susceptible to GEM. In vivo studies demonstrate that the combination of ADI-PEG20:GEM: DTX was optimal for tumor growth inhibition, providing the preclinical mechanism and justification for the ongoing clinical trial of ADI-PEG20, GEM, and DTX in sarcoma.
Conclusions:
The priming of tumors with ADI-PEG20 and DTX results in the stabilization of c-MYC potentiating the effect of GEM treatment via an increase in hENT1 expression. This finding is applicable to ASS1-deficient cancers that are currently treated with GEM.
Introduction
Sarcomas are a rare heterogeneous class of solid tumors of mesenchymal origin that arise from either soft tissue or bone. These tumors represent between 1% and 2% of all adult cancers and 15% of all pediatric cancers (1, 2). In the metastatic setting, most high-grade soft-tissue tumors are initially treated palliatively with an anthracycline-based chemotherapy. Second- and third-line therapies include agents such as combination gemcitabine (GEM) and docetaxel (DTX; ref. 3), pazopanib (4), eribulin (5), trabectedin, or dacarbazine (6). Given the molecular heterogeneity of this group of tumors (7), identifying agents with therapeutic efficacy across this wide variety of sarcoma subtypes has been challenging.
The most common metabolic change that occurs in sarcomas is the lack of expression of arginine succinate synthetase 1 (ASS1), the rate-limiting enzyme in the urea cycle, necessary for arginine production. This occurs in approximately 90% of all high-grade sarcomas. Several other cancers have been shown to be largely ASS1-negative, including melanoma, lung cancer, renal cell carcinoma, prostate cancer, and hepatocellular carcinoma (8-11). Cancers that lack the expression of ASS1 are highly sensitive to arginine deprivation via arginine deiminase (ADI-PEG20) and other arginine-depleting enzymes (12-16). ADI-PEG20 converts extracellular arginine into citrulline, starving ASS1-deficient tumors of extracellular arginine, and resulting in tumor growth inhibition (11, 17, 18). Although initially tumors respond to ADI-PEG20, ASS1-deficient tumors eventually become resistant to this treatment by reexpressing ASS1 (12, 14, 19-21). ADI-PEG20 has not been shown to have any single-agent activity, and clinical trials of ADI-PEG20 alone have been negative to date (22).
GEM is recommended as a single agent for first-line therapy in pancreatic cancer, non–small lung cancer, breast cancer, bladder cancer, and ovarian cancer; however, all of these cancers eventually develop resistance (23-26). In 2007, Maki and colleagues published SARC02, a randomized phase II clinical trial comparing the responses in patients with advanced sarcoma to treatment with GEM or a combination of GEM and DTX. Patients who received GEM and DTX had an increase in progression-free and overall survival compared with GEM alone, thus establishing the regimen of GEM/DTX as a standard second-line therapy for most high-grade soft-tissue sarcomas (STS; refs. 3, 27). Although results with GEM alone or in combination with DTX continue to be inconsistent between trials, it remains a standard second-line therapy for metastatic STS (28-34).
GEM is a hydrophilic molecule that cannot cross the plasma membrane by passive diffusion, so cellular uptake of GEM requires the presence of a specialized plasma membrane nucleoside transporter (35). The main transporter of GEM is human equilibrative transporter 1 (hENT1). After intracellular entry, GEM is phosphorylated by deoxycytidine kinase (dCK) to its active state, and can be further phosphorylated to its diphosphate and triphosphate forms. The major mechanism of GEM cytotoxicity is chain termination as a result of GEM triphosphate incorporation into DNA (36, 37). In addition, GEM diphosphate inhibits ribonucleotide reductase (RRM1 and RRM2), causing a decrease in the cellular pool of deoxycytidine triphosphate that can compete with GEM triphosphate for incorporation into DNA (ref. 38; schematic diagram of pathway in Fig. 1A).
Figure 1.

ASS1 expression determines the efficacy of GEM and DTX therapies. A, Schematic diagram of the GEM pathway: arrows represent stimulation and T-bars represent inhibition. hENT1, human equilibrative transporter 1. dCK, deoxycytidine kinase; NMPK, nucleoside monophosphate kinase; NDPK, nucleoside diphosphate kinase; RRM2, ribonucleotide reductase; CDP, Cytidine diphosphate; dCDP, deoxycytidine diphosphate; dCTP, deoxycytidine triphosphate. B-D, Protein expression of main biomarkers in the GEM pathway were compared across SK-LMS-1 WT (−ADI) WT(+ADI) and LTAT cell lines. Protein extracts were analyzed by WES automated blotting system. Band density differences were expressed as relative densitometry normalized to the total protein in each capillary. E-G, Nuclear (Nuc) and cytoplasmic (Cyto) protein expression of main biomarkers in the GEM pathway were compared across SK-LMS-1 WT (−ADI) and LTAT cell lines. Protein extracts were analyzed by WES automated blotting system. Band density differences were expressed as relative densitometry normalized to the total protein in each capillary. Data are from 3 independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Preclinical modeling in pancreatic and bladder cancers has shown that ADI-PEG20 increases the effectiveness of GEM, in vitro and in vivo, by blocking the increase in expression of RRM2, a known resistance marker of GEM (39, 40). The effects of ADI-PEG20 in combination with DTX have only been studied in prostate cancer, demonstrating reduced tumor growth in vivo (41). Given the efficacy of ADI-PEG20 in combination with these two agents separately, these data suggest that a direct analysis of the triple combination of ADI-PEG20, DTX, and GEM could offer a new therapeutic option in sarcoma.
In this study, the mechanism and efficacy of ADI-PEG20, DTX, and GEM in sarcoma, pancreatic cancer, and melanoma cells were examined in vitro and in vivo in order to develop a novel therapeutic strategy for treating cancers deficient in ASS1. Our results show tumors lacking ASS1 develop resistance to arginine deprivation due to forced reexpression of ASS1 (14). The expression of ASS1 results in the stabilization of c-Myc (19). The effects of c-Myc stabilization are enhanced by the addition of DTX, which aids in the translocation of stabilized c-Myc to the nucleus (42, 43). Nuclear c-Myc increases transcription and translation of the nucleotide/gemcitabine transporter gene, SLC29A1, expressing hENT1, the main transporter for GEM uptake. This suggests that treatment of tumors with ADI-PEG20 and DTX potentiates the anticancer activities of GEM by increasing GEM uptake. Thus, ADI-PEG20 in combination with GEM and DTX is shown to be more effective than the combination of GEM and DTX alone. Further, this treatment is translatable to other ASS1-deficient cancers, with parallel findings in sarcoma, pancreatic cancer, and melanoma cell lines. These data provide the preclinical mechanism and justification for the clinical trial of the combination of ADI-PEG20, GEM, and DTX in STS (NCT03449901).
Materials and Methods
Cell culture conditions
Cell lines (SK-LMS-1, SK-UT-1, HTB-93, HT-1080, SK-MEL-2, AS-Pc-1, MiaPaCa-2, MNNG, RDES, and RD HPAC) were all purchased from ATCC. SYO-1 and FUJI were kindly provided by Dr. Akira Kawai (National Cancer Centre Hospital, Tokyo, Japan) and Dr. Kazuo Nagashima (Hokkaido University School of Medicine, Sapporo, Japan), LUPI was a gift from Dr. John Pfeiffer (Washington University in St. Louis). RH28 were a gift from Lee J. Helman (USC Children's Hospital, Los Angeles, California). All cell lines were cultured at 37°C in 5% CO2, in Dulbecco Modified Eagle Medium (DMEM, Life Technologies) supplemented with 10% FBS (Gibco, Thermo Fisher). For experiments, Modified Eagle Medium (MEM; Life Technologies), supplemented with 10% FBS, was used. All LTAT cell lines were cultured in DMEM or MEM with 1 μg/mL ADI-PEG20 (Polaris) until ASS1 expression was detected, and were then continuously cultured in ADI-PEG20–supplemented media. All cell lines other than LUPI, RD, SYO-1, FUJI, RDES, and RH28 are from ATCC and cultured less than 6 months. The SYO-1 and FUJI cell lines were authenticated by confirming the expression of the pathognomic SYT-SSX fusion gene by RT-PCR, LUPI, and RD were authenticated by confirming the expression of the pathognomic EWS-FLI fusion gene by RT-PCR. All cell lines were determined to be Mycoplasma free using the LookOut Mycoplasma PCR Detection kit (Sigma-Aldrich).
Immuno assays
For analysis of dCK (sc-393099, Santa Cruz), RRM2 (OAAN01167, Aviva Systems Biology), hENT1 (sc-4849, Santa Cruz), c-Myc (ab11917, abcam), and ASS1 (Polaris), cells were seeded at 3 × 105 in a 6-well plate 1 day prior to collection. On day of collection, media were removed and cells were washed 3 × with 1 × PBS. Cells were collected and lysed per reagent protocol of 1 × Cell Lysis Buffer (9803, Cell Signaling Technology) or fractionated using NE-PER Nuclear and Cytoplasmic Extraction Reagents (78833, Thermo Scientific). Lysates were run on a ProteinSimple Wes Capillary electrophoresis instrument using instrument default settings and manufacturer's standard protocol. For plasma membrane isolation, manufacturer's protocol was followed (ab65400, abcam).
Drug treatment and cell death assays
For IC50 assays and combination index (CI) measurements, transduced cells with NucLight Red (4476, NucLight Lentivirus, Sartorius) were plated at 10,000 cells/well in a 96-well plate. Cells were treated with 1 μmol/L of ADI-PEG20 (Polaris), increasing doses of DTX (S1148, Selleckchem), and increasing doses of GEM (S1714, Selleckchem), either alone or in combination, in phenol red-free MEM (11966-025, Thermo Fisher). Cell death was determined using an IncuCyte FLR imaging system (Sartorius). For cell death assays, cells were plated in medium containing 50 nmol/L YOYO-1 (Thermo Fisher Y3601). Cells were treated as described, imaged every hour over a period of 3 days, and analyzed using IncuCyte image analysis software (Sartorius). For quantification of death, YOYO-1 florescence was normalized to Nuclear Red counts for each respective well. For quantification of proliferation assays, only Nuclear Red counts were used. Once IC50 values were established, cell death assays were performed as previously stated, except cells were treated with 1 μmol/L of ADI-PEG20, 3.32 μmol/L DTX, and 5.42 μmol/L GEM alone or in combination. All inhibition studies used 100 μmol/L of a c-Myc inhibitor, 10058-F4 (F3680, Sigma-Aldrich) 1 hour prior to treatment and maintained within culture medium throughout the experiment.
Clonogenic assays
Nuclight Red cell lines were seeded at 200 cells/6 well (triplicates) in 3 mL DMEM. Cells were treated with combination treatment (DTX:GEM:ADI) for 24 hours after treatment media were replaced with fresh media, and cells were incubated for 2 weeks in a humidified incubator with 95% air/5% CO2 at 37° C. Thereafter, cells were analyzed using IncuCyte image analysis software (Sartorius). For quantification of colonies, Nuclear Red counts were calculated for each respective well.
Live-cell immunocytochemistry
Nuclight Red cell lines were treated as previously described and incubated with FabFluor-488 antibody labeling reagents per manufacturer's instructions (4743, Sartorius). hENT1 (sc-4849, Santa Cruz). Surface expression was quantified using an IncuCyte FLR imaging system (Sartorius), imaged every hour over a period of 3 days, and analyzed using IncuCyte image analysis software (Sartorius). Representative pictures were taken from image analysis software.
Probe uptake
Nuclight Red cell lines were treated as previously described and incubated with 5-Propargylamino-cytidine-5-carboxyfluorescein per manufacturer's protocol (Jena Bioscience). Fluorescence was measured with a Microplate Fluorimeter FLX-800 (Bio-Tek Instruments, Inc.) at an excitation wavelength of 492 nm at various time points. Representative pictures were taken from image analysis software.
In vivo studies
Mouse xenograft experimental protocols were approved by the Institutional Animal Care and Use Committee (IACUC) at Washington University in St. Louis. Animals were maintained and evaluated under pathogen-free conditions following IACUC guidelines. Athymic nude mice (4- to 6-week-old females) were obtained from Jackson Laboratories. In all, 1.0 × 106 cells in media and 50% Matrigel (BD Biosciences) were injected subcutaneously into the right flank of each mouse. Tumors were measured in length and width with calipers several times per week and volumes were calculated using the formula (length × width2)/2. Tumors were allowed to reach an approximate volume of 200 mm3 before being randomized into treatment groups. Mice were treated with ADI-PEG20 (Polaris) intramuscularly at 320 international units per meter cubed (IU/m3) twice weekly, GEM (Lilly) by intraperitoneal injections at 125 milligram/kilogram (mg/kg) twice weekly, and DTX (Sandoz) at 10 mg/kg weekly, or combinations of the drugs based on treatment group. Mice were sacrificed according to the IACUC-approved protocol upon reaching 2 cm diameter in one dimension or 2 days after last treatment. Tumor material was harvested for both protein analyses by snap freezing in liquid nitrogen for Western blots and histologic studies by formalin fixation.
Statistical analysis
Data were analyzed using GraphPad Prism 7 Software or Calcusyn (Biosoft). In vitro protein expression, cell death, probe uptake, CI, activity assay, and live-cell immunocytochemistry data are expressed as mean ± standard deviation and Student t test for the individual group comparison. Statistical analysis for in vivo study data was performed by analysis of variance for multiple-group comparison and two-way ANOVA for the individual group comparison with data expressed as standard error of the mean. *, P < 0.05; **, P < 0.01; ***, P < 0.001 were considered to represent statistically significant group differences.
Results
Effect of acute and chronic arginine starvation on the GEM pathway in ASS1− cell lines
Based on previous findings, alterations in the pyrimidine pathway have been seen in response to arginine starvation (12, 44, 45). Using our established model of arginine starvation in sarcoma of acute and long-term ADI-PEG20 treated (LTAT), the alterations in the GEM pathway were examined (Fig. 1A; refs. 12-14). Based on the importance of the biomarkers involved in GEM metabolism, the expression of dCK, RRM2, and hENT1 was assessed within the SK-LMS-1 cell line in three treatment conditions (WT, WT(ADI), and LTAT). dCK expression increased significantly in acute and long-term treatment of ADI-PEG20 (WT(ADI) and LTAT; Fig. 1B), while RRM2 expression decreased significantly in both acute and long-term treatment of ADI-PEG20 (Fig. 1C). Finally, ADI-PEG20 alone did not change the overall expression of the GEM transporter hENT1 (Fig. 1D).
Cytoplasmic and nuclear fractions were also analyzed in WT and LTAT cell lines to determine if subcellular localization of these biomarkers was altered. From acute to long-term treatment of ADI-PEG20, dCK localization was increased in the cytoplasmic fraction (Fig. 1E), while RRM2 decreased within the nuclear fraction (Fig. 1F). hENT1 overall expression did not change and was mainly in the cytoplasmic fraction (Fig. 1G). These results indicate that upon ADI-PEG20 treatment, SK-LMS-1 cells increase dCK and decrease RRM2, rendering the cells sensitive to GEM treatment, but lack the upregulation of the hENT1 transporter that would allow GEM into the cell, suggesting intrinsic transporter-associated resistance.
Combination of ADI-PEG20, GEM, and DTX is increasingly synergistic with prolonged arginine starvation
WT and LTAT cell lines were subjected to combination studies with DTX and increasing doses of GEM to establish if treatment with ADI-PEG20 sensitized the cells to GEM/DTX treatment. The half-maximal growth inhibition (GI50) for DTX alone (Fig. 2A) and half-maximal inhibitory concentration (IC50) for GEM alone (Fig. 2B) were determined in SK-LMS-1 cells. Both WT and short-term WT + ADI were resistant to GEM, whereas the LTAT demonstrated some cell death sensitivity to GEM. The addition of a fixed dose of DTX to ADI-PEG20 treatment increased the sensitivity of SK-LMS-1 to GEM treatment (Fig. 2C), with the effect being stronger with longer exposure to ADI-PEG20 (LTAT; Fig. 2D).
Figure 2.

Addition of ADI-PEG20 decreases the IC50 of GEM and DTX treatment and increases synergistic effects. A, GI50 values of incremental doses of DTX in SK-LMS-1 without ADI (WT), acute ADI treatment (WT + ADI), or chronic ADI treatment (>3 days; LTAT) at 24 hours. B, IC50 values incremental doses of GEM treated in SK-LMS-1 without ADI (WT), acute ADI treatment (WT + ADI), or chronic ADI treatment (>3 days; LTAT) at 24 hours. C, IC50 values incremental doses of GEM in SK-LMS-1 in the presence of 3.3 μmol/L DTX without ADI (WT), acute ADI treatment (WT + ADI), or chronic ADI treatment (>3 days; LTAT) at 24 hours. D, Comparison of cell death from the GEM (DTX ± ADI) IC50 curves. Data are from 3 independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001. E, CI plots for the interaction between the combination of GEM and DTX administered on SK-LMS-1 WT, WT (+ADI) and LTAT for 24 hours. CI > 1.3, antagonism; CI 1.1–1.3, moderate antagonism; CI 0.9–1.1, additive effect; CI 0.8–0.9, slight synergism; CI 0.6–0.8, moderate synergism; CI 0.4–0.6, synergism; CI 0.2–0.4, strong synergism.
The synergistic combined effects of GEM and DTX in SK-LMS-1 with and without ADI-PEG20 were evaluated by isobolographic analysis. The CI values for WT ranged from 0.32 to 563, for WT (+ADI) ranged from 0.201 to 5.339, and for LTAT ranged from 0.127 to 1.583. Only CI values that were 3 or below were plotted. Out of 50 values, this resulted in only 8 CI values for WT, 38 CI values for WT(+ADI), and 36 values for LTAT out of 40. Of the included CI values, 6 of the 8 for WT were located below the line of additivity, while 20 of the 38 included CI values in WT(+ADI) and 28 of the 36 CI values in LTAT were below the additive line (Fig. 2E). This shows that synergy of ADI-PEG20 combined with GEM and DTX increases with ADI-PEG20 treatment from acute to long term, and suggests strong synergy associated with longer term ADI-PEG20 treatment in SK-LMS-1 cells.
DTX treatment in ADI-PEG20 cell line drives c-Myc translocation and hENT1 expression
To determine the mechanism by which GEM-induced cell death occurs in the presence of ADI-PEG20 and DTX, we hypothesized that the effect of this combination relied on c-Myc stabilization, as c-Myc has been shown to be stabilized in cell lines that are treated with ADI-PEG20 (19). c-Myc expression did increase significantly in acute or long-term ADI-PEG20–treated cells when compared with WT SK-LMS-1 (Fig. 3A). To further explore the role of c-Myc within this pathway, cytoplasmic and nuclear fractions of WT and LTAT were compared in response to drug combinations. ADI-PEG20 long-term treatment stabilized c-Myc within the nucleus (Fig. 3B; NT-WT vs. NT-LTAT). DTX has also been reported to play a role in c-Myc translocation (42, 43). To determine if DTX was impacting c-Myc expression or localization, SK-LMS-1 LTAT (ADI-PEG20) cell line was treated with DTX or DTX with GEM [combo (ADI-PEG20/GEM/DTX)]. In both DTX and combo treatments, nuclear c-Myc expression increased significantly (Fig. 3B). To determine if this increase could be prevented, SK-LMS-1 cells were pretreated with a c-Myc/Max heterodimerization inhibitor, 10058-F4, 1 hour prior to combo treatment. This inhibition did prevent the expression and translocation of c-Myc to the nucleus (Fig. 3B). Furthermore, c-Myc activity was assessed on SK-MLS-1 LTAT cells with the combo treatment. In the combo treatment, c-Myc activity significantly increased within the nucleus (Fig. 3C) and decreased when cells were pretreated with a c-Myc inhibitor. To assess the importance of c-Myc over other Myc proteins, such as N-Myc, protein expression of N-Myc in the presence of treatment was evaluated. As the expression of N-Myc decreased during combination treatment, there is no direct link to the expression of hENT1 by N-Myc (Supplementary Fig. S2). To evaluate if the increase in GEM sensitivity is due to c-Myc stabilization (by ADI-PEG20) and translocation (by DTX) specifically, hENT1 surface expression was analyzed in the SK-LMS-1 LTAT cell line. DTX and combo treatment did significantly increase hENT1 surface expression compared with NT (Fig. 3C and D). When cells were pretreated with the c-Myc inhibitor, hENT1 surface expression decreased, shown quantitatively and with representative pictures (Fig. 3C-E). Taken together, these data show that c-Myc expression and localization is coordinated by ADI-PEG20 and DTX treatment. This increase in activity of c-Myc due to translocation to the nucleus increases hENT1 expression at the surface of the cell membrane.
Figure 3.

DTX translocates c-Myc to the nucleus, driving hENT1 expression. A, Protein expression of c-Myc expression in SK-LMS-1WT (NT), WT (+ADI), and LTAT cell lines. Protein extracts were analyzed by WES automated blotting system. Band density differences were expressed as relative densitometry normalized to the total protein in each capillary. B, Nuclear (Nuc) and cytoplasmic (Cyto) protein expression of c-MYC were compared across SK-LMS-1WT (NT) and LTAT cell lines, LTAT cells treated with DTX alone (45 μmol/L for 6 hours), ADI-PEG-20, DTX, and GEM (combo) together (1, 45, and 3.25 μmol/L, respectively) or combo pretreated with 100 μmol/L c-Myc inhibitor for 1 hour [combo (+c-MYC inhibitor)]. Protein extracts were analyzed by WES automated blotting system. Band density differences were expressed as relative densitometry normalized to the total protein in each capillary. C, SK-LMS-1 LTAT cells were treated with combo or combo (+ c-Myc inhibitor) for 6 hours. c-Myc activity was determined in the cytoplasmic (Cyto) and nuclear (Nuc) fractions by the ELISA-based TransAM-c-Myc binding activity assay. D, SK-LMS-1 LTAT cells were treated with DTX, DTX (+c-Myc inhibitor), combo or combo (+c-Myc inhibitor), and plasma membrane fractionation was performed to demonstrate hENT1 surface expression. E and F, SK-LMS-1 LTAT cells were treated with DTX, DTX (+c-Myc inhibitor), combo or combo (+c-Myc inhibitor) and real-time increase in expression of hENT1 was quantified by live-cell immunofluorescence. Data are from 3 independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
hENT1 surface expression increases uptake of a cytidine-5-carboxyfluorescein probe
To directly relate the increased efficacy of GEM in the combo treatment to hENT1 surface expression, WT and LTAT cell lines were either treated with combo, or pretreated with a c-Myc inhibitor before the combo treatment. The cells were incubated with a cytidine-5-carboxyfluorescein probe (chemical structure in Supplementary Fig. S1). This probe allows for the measurement of cytidine uptake, and can be directly correlated to increased hENT1 expression and transporter activity. Uptake of the probe increased with combo treatment in both WT and LTAT cell lines, and was blunted when cells were pretreated with a c-Myc inhibitor, shown quantitatively and with representative pictures (Fig. 4A and B). These data support the hypothesis that when the combo treatment is given, c-Myc facilitates an increase in hENT1 surface expression, resulting in an increase in cell death due to increased GEM uptake. Additionally, cell death was measured in SK-LMS-1 WT and LTAT cell lines, shown quantitatively and with representative pictures (Fig. 4C and D). Although the WT cell line is significantly affected by the combo treatment, long-term treatment of ADI-PEG20 increased cell death by 98%. Cell death was protected against when cells were pretreated with the c-Myc inhibitor (Fig. 4C and D), confirming that the mechanism of cell death was reliant on c-Myc activity. To further understand if the combination treatment was specific to a population of the tumor cells or the population as a whole, clonogenic assays were performed. The combination treatment showed to devastate the population as a whole after 7 days of treatment. In WT as well as LTAT, the colonies formed post-treatment were significantly null after a 24-hour treatment (Fig. 4E and 4).
Figure 4.

Increase in hENT1 expression leads to an increase in GEM uptake and death, which is rescued by c-Myc inhibition. A and B, Functional assays using 5-proparglyamino-cytidine-5-carboxyfluorescein probe. Probe uptake was measured at 492 nmol/L and normalized to cell count within the well (NucLight Red). SK-LMS-1 WT and LTAT cells treated with combo or combo (+c-Myc inhibitor) for 22 hours. Pictures were taken on the IncuCyte system at 12 hours of uptake. C and D, Cell death activity was measured using YOYO-1 nucleic acid dye on the IncuCyte system. Green counts/well were measured and normalized to total nuclear count within the well (NucLight Red). Representative pictures directly correlating to the assay were captured. E, SK-LMS1 WT and LTAT cells with and without combo treatment for 24 hours, after which fresh media were replaced without drug. Analysis done at day 7. Data are from 3 independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Efficacy of GEM and DTX and ADI-PEG20 in multiple cancers
To determine if the sensitivity to GEM and DTX could be correlated to ASS1 negativity as a biomarker, and therefore have efficacy across a wider range of cancer systems, we explored if the combination of ADI-PEG20/GEM/DTX would be effective in a broad range of cancer subtypes where GEM and DTX were used. We also tested pancreatic cancer, where GEM and nab-paclitaxel are used, as well as chemotherapy-resistant melanoma. For this panel, eight ASS1-low to -null expressing cell lines were utilized: a nontraditional synovial sarcoma without the SYT:SSX translocation (HTB-93), fibrosarcoma (HT-1080), uterine spindle cell sarcoma (SK-UT-1), leiomyosarcoma (SK-LMS-1), melanoma (SK-MEL-2), and several pancreatic cell lines (As-PC-1, MiaPaCa-2, and HPAC). To broaden our panel of sarcomas, several complex and translocation-driven sarcomas were also assessed; MNNG (osteosarcoma), FUJI (Synovial Sarcoma), LUPI and RDES (Ewing), and RD and RD28 (rhabdomyosarcoma). These cell lines were treated with ADI-PEG20 until ASS1 was reexpressed or an increase in levels of ASS1 expression was observed (Fig. 5A; Supplementary Fig. S3). To evaluate if increased expression of ASS1 in these cell lines would improve GEM uptake via increased hENT1 surface expression, WT and LTAT cell lines were treated with ADI-PEG20, GEM, and DTX (combo). All cell lines were then incubated with the cytidine-5-carboxyfluorescein probe, and all demonstrated increased uptake as a result of the combo treatment (Fig. 5B). To test if this probe uptake was linked to an increase in hENT1 surface expression, live-cell immunocytochemistry was performed (Fig. 5C). hENT1 surface expression was shown to increase across cell lines from WT to LTAT treated with combo compared with NT. Furthermore, when c-Myc is inhibited, both probe uptake and hENT1 surface expression are blunted (Fig. 5C). Next, cell death analysis with the combination treatment demonstrated a robust increase in cell death across all cancer types, with both acute and long-term treatment of ADI-PEG20 compared with NT (Fig. 5D; Supplementary Fig. S4), these findings are independent of TP53 status of the cell lines (Supplementary Table S1). These results indicate that the treatment of ADI-PEG20, GEM, and DTX within multiple cancer types increases hENT1 surface expression, which renders the cells susceptible to this combination treatment. Furthermore, when c-Myc is inhibited, hENT1 expression is blunted and cell death is protected from this treatment.
Figure 5.
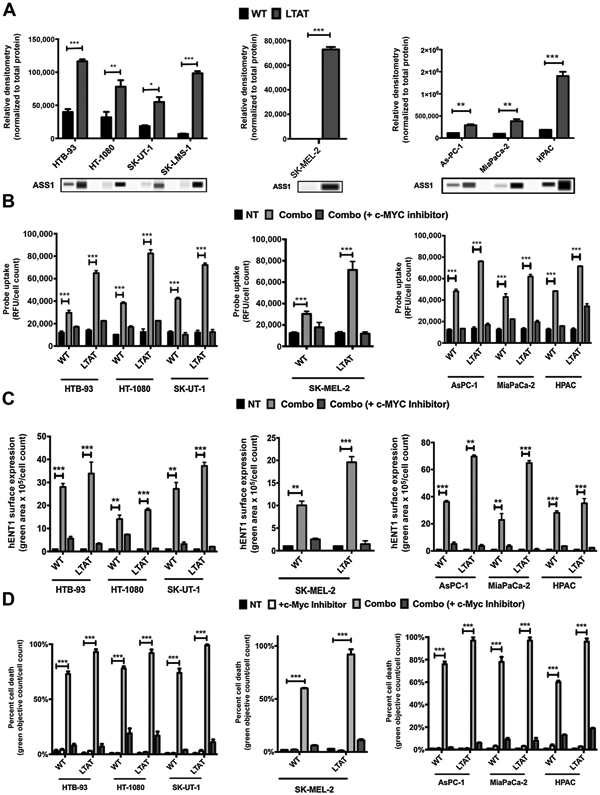
Efficacy of ADI-PEG20, DTX, and GEM combination across cancers. A, Protein expression of ASS1 in sarcoma cell lines and the corresponding LTAT cell lines [HTB-93 (WT and LTAT), HT-1080 (WT and LTAT), SK-UT-1 (WT and LTAT), and SK-LMS-1 (WT and LTAT)], a melanoma cell line [SK-MEL-2 (WT and LTAT)], and pancreatic cell lines [As-PC-1 (WT and LTAT), MiaPaCa-2 (WT and LTAT), and HPAC (WT and LTAT)]. Protein extracts were analyzed by a WES automated blotting system. Band density differences were expressed as relative densitometry normalized to the total protein in each capillary. B, Functional assays using 5-proparglyamino-cytidine-5-carboxyfluorescein probe. Probe uptake was measured at 492 nmol/L and normalized to cell count within the well (NucLight Red). Sarcoma, melanoma, and pancreatic WT and LTAT cells were treated with combo or combo (+c-Myc inhibitor) for various time points. C, Sarcoma, melanoma, and pancreatic cells were treated with combo or combo (+c-Myc inhibitor) and real-time increase in expression of hENT1 was quantified by live-cell immunocytochemistry on the IncuCyte system. D, Cell death activity was measured using YOYO-1 nucleic acid dye on the IncuCyte system. Green counts/well were measured and normalized to total nuclear count within the well (NucLight Red). Sarcoma, melanoma, and pancreatic cell lines were treated with c-Myc inhibitor, combo or combo (+c-Myc inhibitor). Data are from 3 independent experiments. *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Effect of ADI-PEG20, GEM, and DTX on tumor growth in vivo
The evaluation of ADI-PEG20, GEM, and DTX as a combination therapy effects on local tumor growth revealed that the combination of all three agents given together yielded the maximal growth inhibition in vivo (Fig. 6A). The SK-LMS-1 cell line was chosen for xenografting as it is the best example of a sarcoma cell line exhibiting no ASS1 expression that has a predictable xenograft growth rate (14). In examining the effects of arginine depletion therapy on the individual treatment components, ADI-PEG20 alone had a significant but short-term (21-day) growth inhibitory effect compared with control (Fig. 6A and B). Next, the effects of arginine starvation with DTX treatment were examined. Similar to ADI-PEG20 alone, there is a short-term but significant effect on tumor growth for the combination of ADI-PEG20 and DTX; DTX alone had little effect on tumor growth (Fig. 6C). Unlike in vitro results, which suggested that the SK-LMS-1 cell line is GEM resistant (Fig. 2B), in vivo GEM treatment demonstrates a prolonged and significant effect on tumor growth compared with the other single agents (Fig. 6D). Furthermore, the addition of ADI-PEG20 to GEM treatment was not any better than single-agent GEM, suggesting that DTX is needed for effective treatment response (Fig. 6D). Finally, the effect of ADI-PEG20 combined with GEM and DTX demonstrated an improvement over GEM and DTX alone (Fig. 6E). These in vivo studies support the addition of ADI-PEG20 to the standard line of treatment, GEM + DTX. For full statistical analysis, please see Supplementary Table S2.
Figure 6.

Xenograft studies of single and combination treatments. A, Tumor volume of SK-LMS-1 WT xenografts under various conditions: untreated (n = 5), ADI-PEG20 (n = 10), DTX (n = 5), ADI + DTX (N = 8), GEM (n = 5), GEM + ADI-PEG20 (n = 7), GEM + DTX (n = 4), GEM + DTX + ADI-PEG20 (n = 5). B, Tumor volumes for ADI-PEG20 vs. control from days 0–42. C, Tumor volumes of control, DTX, and DTX + ADI-PEG20 from days 0–42. D, Early tumor volumes from days 0–84, comparing GEM and GEM + ADI-PEG20. E, Comparison of GEM + DTX vs. GEM + DTX-ADI-PEG20 to day 84.
Discussion
ASS1 deficiency is a recurrent feature across many types of cancers and is associated with decreased overall survival and an earlier appearance of metastases (8-11, 15, 16). Clinically, the ASS1 expression has been identified as a metabolic vulnerability that can be exploited with arginine-depleting enzymes such as ADI-PEG20. One of the problems that arise with arginine starvation strategies is that ASS1-deficient cells will force reexpression of ASS1 when exposed to an arginine-depleted environment, as a mechanism of resistance. This expression of ASS1 results in the rewiring of the innate metabolism of the tumor. Understanding this conditional metabolic reprogramming allows for the development of metabolic therapies based on arginine starvation from ADI-PEG20 in cancers deficient in ASS1 expression.
Previous work has identified changes in the GEM pathway that occur as a result of arginine starvation induced by ADI-PEG20 (12, 44, 45). This study confirmed that the degradation of arginine from ASS1-deficient, arginine auxotrophic cancers stabilizes c-Myc (19), and that the addition of DTX facilitates c-Myc translocation to the nucleus, which results in increased hENT1 surface expression. This is an unexpected finding of DTX, in that a mitotic inhibitor induced and translocated c-Myc in the condition of arginine starvation. Overall, this allows for the augmentation of GEM efficacy in ASS1− tumors. Furthermore, these data show that even when ASS1 reexpresses in long-term treatment with ADI-PEG20 (LTAT), the combination treatment remains significantly effective. The proposed model is summarized in Fig. 7A and B.
Figure 7.

Schematic diagram of the proposed model. A, Known model: arrows represent stimulation and T-bars represent inhibition. hENT1, human equilibrative transporter 1; dCK, deoxycytidine kinase; NMPK, nucleoside monophosphate kinase; NDPK, nucleoside diphosphate kinase; RRM2, ribonucleotide reductase; CDP, cytidine diphosphate; dCDP, deoxycytidine diphosphate; dCTP, deoxycytidine triphosphate. B, Proposed model in ASS1− tumors: ADI-PEG20 converts extracellular arginine to citruline. Arginine starvation stabilizes c-Myc. DTX induces translocation of c-Myc to nucleus. c-Myc stimulates hENT1 expression. hENT1 expression increases at cell surface. Increase in hENT1 allows for an increase in GEM uptake, leading to cell death.
Prior work from our laboratory demonstrated that ADI-PEG20 was able to induce a conditional synthetic lethality in ASS1-deficient cells placed in a prolonged state of arginine starvation. This results in a downregulating Warburgian biology and creates a dependency on oxidative phosphorylation as cells reprogram themselves for biomass production (12). The arginine-depleted environment induced expression of ASS1, and the conditional biology persisted for the duration of arginine starvation. It is this adaptation that may be most useful clinically. ADI-PEG20 used as a single agent in ASS1-deficient cancers quickly loses efficacy due to metabolic adaptation and developed resistance via ASS1 reexpression. Within our studies, we have demonstrated that this adaptation can be therapeutically exploited, and that it holds promises for future drug development. There are now two clinical trials using mitotic inhibitors that should allow for increased uptake of GEM based on our findings, which include NCT03449901, a phase II trial of ADI-PEG 20 in combination with GEM and DTX for the treatment of STS, and NCT02101580, a phase IB trial with ADI-PEG20 plus nab-paclitaxel and GEM in subjects with pancreatic cancer.
Finally, these findings support the suggested paradigm in Kremer and colleagues that there are adaptations in metabolic pathways unique to the cancer and not the host, that can be exploited to develop multidrug therapies based on arginine starvation (12). As ASS1 deficiency is primarily found in cancer cells of patients, rather than normal tissue, our laboratory continues to explore biomarker-driven metabolic therapies that allow for an effective multiagent treatment in this ASS1-deficient population. Continued understanding of the biological consequences and adaptations of arginine starvation will allow for the development of less toxic therapies.
Supplementary Material
Translational Relevance.
The cellular response to arginine starvation in ASS1-deficient tumors allows for a conditional adaptive metabolic reprogramming that upregulates hENT1 surface expression to overcome intrinsic gemcitabine transporter-related resistance across multiple tumors. This work provides the preclinical justification for the ongoing clinical trial utilizing ADI-PEG20, gemcitabine, and docetaxel in sarcoma.
Acknowledgments
This project was supported by grants from CJ's Journey (B.C. Prudner, R. Rathore, A.M. Robinson, S.F. Chang, B.A. Van Tine), The Sarcoma Foundation of America (B.C. Prudner, B.A. Van Tine), The Sarcoma Alliance For Research and Collaboration (SARC); (B.A. Van Tine) and Siteman Cancer Center Investment Program (funded by the Foundation for Barnes-Jewish Hospital Cancer Frontier Fund and Barnard Trust) (B.C. Prudner, R. Rathore, A.M. Robinson, S. F. Chang, B.A. Van Tine).
Footnotes
Disclosure of Potential Conflicts of Interest
B.A. Van Tine reports receiving commercial research grants from Merck, Pfizer, and Tracon; reports receiving speakers bureau honoraria from Lilly, Janssen, and Caris; is a consultant/advisory board member for Epizyme, Lilly, CytRx, Janssen, Immune Design, Daiichi, Plexxicon, and Adaptimmune; and reports receiving other remuneration from Lilly. No potential conflicts of interest were disclosed by the other authors.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2018. CA Cancer J Clin 2018;68:7–30. [DOI] [PubMed] [Google Scholar]
- 2.von Mehren M, Randall RL, Benjamin RS, Boles S, Bui MM, Ganjoo KN, et al. Soft tissue sarcoma, version 2.2018, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2018;16:536–63. [DOI] [PubMed] [Google Scholar]
- 3.Maki RG, Wathen JK, Patel SR, Priebat DA, Okuno SH, Samuels B, et al. Randomized phase II study of gemcitabine and docetaxel compared with gemcitabine alone in patients with metastatic soft tissue sarcomas: results of sarcoma alliance for research through collaboration study 002 [corrected]. J Clin Oncol 2007;25:2755–63. [DOI] [PubMed] [Google Scholar]
- 4.van der Graaf WTA, Blay J-Y, Chawla SP, Kim D-W, Bui-Nguyen B, Casali PG, et al. Pazopanib for metastatic soft-tissue sarcoma (PALETTE): a randomised, double-blind, placebo-controlled phase 3 trial. Lancet 2012;379:1879–86. [DOI] [PubMed] [Google Scholar]
- 5.Schoffski P, Chawla S, Maki RG, Italiano A, Gelderblom H, Choy E, et al. Eribulin versus dacarbazine in previously treated patients with advanced liposarcoma or leiomyosarcoma: a randomised, open-label, multicentre, phase 3 trial. Lancet 2016;387:1629–37. [DOI] [PubMed] [Google Scholar]
- 6.Demetri GD, von Mehren M, Jones RL, Hensley ML, Schuetze SM, Staddon A, et al. Efficacy and safety of trabectedin or dacarbazine for metastatic liposarcoma or leiomyosarcoma after failure of conventional chemotherapy: results of a phase III randomized multicenter clinical trial. J Clin Oncol 2016;34:786–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Abeshouse A, Adebamowo C, Adebamowo SN, Akbani R, Akeredolu T, Ally A, et al. Comprehensive and integrated genomic characterization of adult soft tissue sarcomas. Cell 2017;171:950–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ensor CM, Holtsberg FW, Bomalaski JS, Clark MA. Pegylated arginine deiminase (ADI-SS PEG20,000 mw) inhibits human melanomas and hepatocellular carcinomas in vitro and in vivo. Cancer Res 2002;62: 5443–50. [PubMed] [Google Scholar]
- 9.Kelly MP, Jungbluth AA, Wu BW, Bomalaski J, Old LJ, Ritter G. Arginine deiminase PEG20 inhibits growth of small cell lung cancers lacking expression of argininosuccinate synthetase. Br J Cancer 2012;106:324–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoon CY, Shim YJ, Kim EH, Lee JH, Won NH, Kim JH, et al. Renal cell carcinoma does not express argininosuccinate synthetase and is highly sensitive to arginine deprivation via arginine deiminase. Int J Cancer 2007; 120:897–905. [DOI] [PubMed] [Google Scholar]
- 11.Bowles TL, Kim R, Galante J, Parsons CM, Virudachalam S, Kung HJ, et al. Pancreatic cancer cell lines deficient in argininosuccinate synthetase are sensitive to arginine deprivation by arginine deiminase. Int J Cancer 2008; 123:1950–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kremer JC, Prudner BC, Lange SES, Bean GR, Schultze MB, Brashears CB, et al. Arginine deprivation inhibits the Warburg effect and upregulates glutamine anaplerosis and serine biosynthesis in ASS1-deficient cancers. Cell Rep 2017;18:991–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Prudner BC, Sun F, Kremer JC, Xu J, Huang C, Sai KKS, et al. Amino acid uptake measured by [(18)F]AFETP increases in response to arginine starvation in ASS1-deficient sarcomas. Theranostics 2018;8:2107–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bean GR, Kremer JC, Prudner BC, Schenone AD, Yao JC, Schultze MB, et al. A metabolic synthetic lethal strategy with arginine deprivation and chloroquine leads to cell death in ASS1-deficient sarcomas. Cell Death Dis 2016;7:e2406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kobayashi E, Masuda M, Nakayama R, Ichikawa H, Satow R, Shitashige M, et al. Reduced argininosuccinate synthetase is a predictive biomarker for the development of pulmonary metastasis in patients with osteosarcoma. Mol Cancer Ther 2010;9:535–44. [DOI] [PubMed] [Google Scholar]
- 16.Huang HY, Wu WR, Wang YH, Wang JW, Fang FM, Tsai JW, et al. ASS1 as a novel tumor suppressor gene in myxofibrosarcomas: aberrant loss via epigenetic DNA methylation confers aggressive phenotypes, negative prognostic impact, and therapeutic relevance. Clin Cancer Res 2013;19: 2861–72. [DOI] [PubMed] [Google Scholar]
- 17.Kim JE, Kim SY, Lee KW, Lee HJ. Arginine deiminase originating from Lactococcus lactis ssp. lactis American Type Culture Collection (ATCC) 7962 induces G1-phase cell-cycle arrest and apoptosis in SNU-1 stomach adenocarcinoma cells. Br J Nutr 2009;102:1469–76. [DOI] [PubMed] [Google Scholar]
- 18.Feun L, You M, Wu CJ, Kuo MT, Wangpaichitr M, Spector S, et al. Arginine deprivation as a targeted therapy for cancer. Curr Pharm Des 2008;14: 1049–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsai WB, Aiba I, Long Y, Lin HK, Feun L, Savaraj N, et al. Activation of Ras/PI3K/ERK pathway induces c-Myc stabilization to upregulate argininosuccinate synthetase, leading to arginine deiminase resistance in melanoma cells. Cancer Res 2012;72:2622–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Long Y, Tsai WB, Wangpaichitr M, Tsukamoto T, Savaraj N, Feun LG, et al. Arginine deiminase resistance in melanoma cells is associated with metabolic reprogramming, glucose dependence, and glutamine addiction. Mol Cancer Ther 2013;12:2581–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yeh TH, Chen YR, Chen SY, Shen WC, Ann DK, Zaro JL, et al. Selective intracellular delivery of recombinant arginine deiminase (ADI) using pH-sensitive cell penetrating peptides to overcome ADI resistance in hypoxic breast cancer cells. Mol Pharm 2016;13:262–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Abou-Alfa GK, Qin S, Ryoo BY, Lu SN, Yen CJ, Feng YH, et al. Phase III randomized study of second line ADI-PEG 20 plus best supportive care versus placebo plus best supportive care in patients with advanced hepatocellular carcinoma. Ann Oncol 2018;29:1402–8. [DOI] [PubMed] [Google Scholar]
- 23.Burris HA 3rd, Moore MJ, Andersen J, Green MR, Rothenberg ML, Modiano MR, et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: a randomized trial. J Clin Oncol 1997;15:2403–13. [DOI] [PubMed] [Google Scholar]
- 24.Sandler AB, Nemunaitis J, Denham C, von Pawel J, Cormier Y, Gatzemeier U, et al. Phase III trial of gemcitabine plus cisplatin versus cisplatin alone in patients with locally advanced or metastatic non-small-cell lung cancer. J Clin Oncol 2000;18:122–30. [DOI] [PubMed] [Google Scholar]
- 25.Nagourney RA, Flam M, Link J, Hager S, Blitzer J, Lyons W, et al. Carboplatin plus gemcitabine repeating doublet therapy in recurrent breast cancer. Clin Breast Cancer 2008;8:432–5. [DOI] [PubMed] [Google Scholar]
- 26.Rueff J, Rodrigues AS. Cancer drug resistance: a brief overview from a genetic viewpoint. Methods Mol Biol 2016;1395:1–18. [DOI] [PubMed] [Google Scholar]
- 27.von Mehren M, Randall RL, Benjamin RS, Boles S, Bui MM, Conrad EU 3rd, et al. Soft tissue sarcoma, version 2.2016, NCCN Clinical Practice Guidelines in Oncology. J Natl Compr Canc Netw 2016;14:758–86. [DOI] [PubMed] [Google Scholar]
- 28.Ducoulombier A, Cousin S, Kotecki N, Penel N. Gemcitabine-based chemotherapy in sarcomas: a systematic review of published trials. Crit Rev Oncol Hematol 2016;98:73–80. [DOI] [PubMed] [Google Scholar]
- 29.Pautier P, Floquet A, Penel N, Piperno-Neumann S, Isambert N, Rey A, et al. Randomized multicenter and stratified phase II study of gemcitabine alone versus gemcitabine and docetaxel in patients with metastatic or relapsed leiomyosarcomas: a Federation Nationale des Centres de Lutte Contre le Cancer (FNCLCC) French Sarcoma Group Study (TAXOGEM study). Oncologist 2012;17:1213–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Takano T, Niikura H, Ito K, Nagase S, Utsunomiya H, Otsuki T, et al. Feasibility study of gemcitabine plus docetaxel in advanced or recurrent uterine leiomyosarcoma and undifferentiated endometrial sarcoma in Japan. Int J Clin Oncol 2014;19:897–905. [DOI] [PubMed] [Google Scholar]
- 31.Hensley ML, Miller A, O'Malley DM, Mannel RS, Behbakht K, Bakkum-Gamez JN, et al. Randomized phase III trial of gemcitabine plus docetaxel plus bevacizumab or placebo as first-line treatment for metastatic uterine leiomyosarcoma: an NRG Oncology/Gynecologic Oncology Group study. J Clin Oncol 2015;33:1180–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.van Hoesel QG, Verweij J, Catimel G, Clavel M, Kerbrat P, van Oosterom AT, et al. Phase II study with docetaxel (Taxotere) in advanced soft tissue sarcomas of the adult. EORTC Soft Tissue and Bone Sarcoma Group. Ann Oncol 1994;5:539–42. [DOI] [PubMed] [Google Scholar]
- 33.Spath-Schwalbe E, Genvresse I, Koschuth A, Dietzmann A, Grunewald R, Possinger K. Phase II trial of gemcitabine in patients with pretreated advanced soft tissue sarcomas. Anticancer Drugs 2000;11:325–9. [DOI] [PubMed] [Google Scholar]
- 34.Lee EM, Rha SY, Lee J, Park KH, Ahn JH. Phase II study of weekly docetaxel and fixed dose rate gemcitabine in patients with previously treated advanced soft tissue and bone sarcoma. Cancer Chemother Pharmacol 2012;69:635–42. [DOI] [PubMed] [Google Scholar]
- 35.Mackey JR, Mani RS, Selner M, Mowles D, Young JD, Belt JA, et al. Functional nucleoside transporters are required for gemcitabine influx and manifestation of toxicity in cancer cell lines. Cancer Res 1998;58: 4349–57. [PubMed] [Google Scholar]
- 36.Huang P, Plunkett W. Induction of apoptosis by gemcitabine. Semin Oncol 1995;22(4 Suppl 11):19–25. [PubMed] [Google Scholar]
- 37.Ruiz van Haperen VW,Veerman G,Vermorken JB, Peters GJ. 2′,2′-Difluoro-deoxycytidine (gemcitabine) incorporation into RNA and DNA of tumour cell lines. Biochem Pharmacol 1993;46:762–6. [DOI] [PubMed] [Google Scholar]
- 38.Plunkett W, Huang P, Searcy CE, Gandhi V. Gemcitabine: preclinical pharmacology and mechanisms of action. Semin Oncol 1996;23(5 Suppl 10):3–15. [PubMed] [Google Scholar]
- 39.Phillips MM, Sheaff MT, Szlosarek PW. Targeting arginine-dependent cancers with arginine-degrading enzymes: opportunities and challenges. Cancer Res Treat 2013;45:251–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Locke M, Ghazaly E, Freitas MO, Mitsinga M, Lattanzio L, Lo Nigro C, et al. Inhibition of the polyamine synthesis pathway is synthetically lethal with loss of argininosuccinate synthase 1. Cell Rep 2016;16: 1604–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim RH, Coates JM, Bowles TL, McNerney GP, Sutcliffe J, Jung JU, et al. Arginine deiminase as a novel therapy for prostate cancer induces autophagy and caspase-independent apoptosis. Cancer Res 2009;69: 700–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Geng H, Rademacher BL, Pittsenbarger J, Huang CY, Harvey CT, Lafortune MC, et al. ID1 enhances docetaxel cytotoxicity in prostate cancer cells through inhibition of p21. Cancer Res 2010;70:3239–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hatano K, Yamaguchi S, Nimura K, Murakami K, Nagahara A, Fujita K, et al. Residual prostate cancer cells after docetaxel therapy increase the tumorigenic potential via constitutive signaling of CXCR4, ERK1/2 and c-Myc. Mol Cancer Res 2013;11:1088–100. [DOI] [PubMed] [Google Scholar]
- 44.Allen MD, Luong P, Hudson C, Leyton J, Delage B, Ghazaly E, et al. Prognostic and therapeutic impact of argininosuccinate synthetase 1 control in bladder cancer as monitored longitudinally by PET imaging. Cancer Res 2014;74:896–907. [DOI] [PubMed] [Google Scholar]
- 45.Daylami R, Muilenburg DJ, Virudachalam S, Bold RJ. Pegylated arginine deiminase synergistically increases the cytotoxicity of gemcitabine in human pancreatic cancer. J Exp Clin Cancer Res 2014;33:102. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.