

Abstract
Sortase-mediated ligation is a powerful method for generating site-specifically modified proteins. However, this process is limited by the inherent reversibility of the ligation reaction. To address this, here we report the continued development and optimization of an experimentally facile strategy for blocking reaction reversibility. This approach, which we have termed metal-assisted sortase-mediated ligation (MA-SML), relies on the use of a solution additive (Ni2+) and a C-terminal tag (LPXTGGHH5) that is widely used for converting protein targets into sortase substrates. In a series of model systems utilizing a 1:1 molar ratio of sortase substrate and glycine amine nucleophile, we find that MA-SML consistently improves the extent of ligation. This enables the modification of proteins with fluorophores, PEG, and a bioorthogonal cyclooctyne moiety without the need to use precious reagents in excess. Overall, these results demonstrate the potential of MA-SML as a general strategy for improving reaction efficiency in a broad range of sortase-based protein engineering applications.
Graphical Abstract
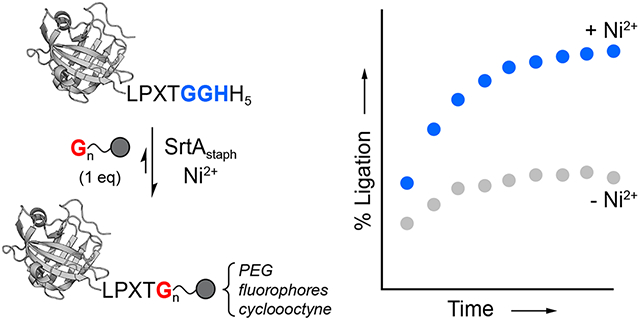
Introduction
Site-specific protein modification is a critical step in the preparation of unique protein derivatives for a range of therapeutic and basic research applications. Among the numerous approaches available for modifying proteins, sortase-mediated ligation (SML) has continued to see widespread use.1–3 This strategy requires only modest changes to the protein of interest in order to render it compatible with SML, typically an LPXTG motif at the protein C-terminus or one or more glycine residues at the protein N-terminus. With these features in place, the target protein can be ligated to a broad range of useful modifications. The applications of the SML strategy are numerous, with recent examples including the detection of cell-cell interactions,4 biomaterials fabrication,5 and the synthesis of ubiquitylated/SUMOylated proteins.6 The choice of SML for these applications is due to its many advantageous features, which include excellent site selectivity, mild reaction conditions, and ease of use. Indeed, the barrier for implementing an SML approach is relatively low; robust protocols have been reported,7–13 sortase enzymes can be obtained from commercial sources or online gene repositories, and SML-compatible peptides decorated with a range of useful modifications can be readily synthesized or simply purchased.
While easy access to reagents and straightforward experimental guidelines have simplified the SML process, the default protocols for SML continue to rely on using one of the ligation partners (either a protein or peptide building block) in excess in order to achieve high levels of the desired modified protein products.7–10,12,13 This need for excess reagents remains a frequently cited criticism of SML, and is due to the inherent reversibility of the ligation reaction.3,14–16 In response, multiple strategies have been described for limiting SML reversibility.17–28 These approaches rely on careful removal or deactivation of certain SML products and by-products, thus blocking their ability to participate in the reverse reaction. While a number of these techniques have been effective in reducing the need for excess reagents, they typically require deviations from standard SML procedures that make them experimentally more cumbersome. These additional requirements include the need to fabricate flow-based devices for performing the SML reaction,23 the need to synthesize more complex peptide reagents containing esters in place of specific amide bonds,18,24 or the need to re-engineer target proteins such that they contain certain secondary structure elements or fused polypeptide domains.17,19,20
With these considerations in mind, here we sought to develop a simple and practical method for blocking the reversibility of SML reactions by expanding on our prior work using metal-coordinating peptides.28 As illustrated in Scheme 1, this strategy relies on using an LPXTGGH sequence in place of the standard LPXTG sortase substrate motif. Upon cleavage by sortase A from S. aureus (SrtAstaph) an embedded GGH peptide is released that can be sequestered by Ni2+ ions in solution. High affinity binding by Ni2+ deactivates the GGH fragment, thus preventing the reverse SML reaction and favoring the formation of the desired ligation products. This approach, which we here term metal-assisted sortase-mediated ligation (MA-SML), was previously shown to improve the extent of ligation for a small panel of model SML reactions when the LPXTG substrate and glycine amine nucleophile were utilized in a 1:1 molar ratio.28 However, the compatibility of MA-SML with a variety of useful protein modifications and protein targets was not established. To address this, the present work describes a series of studies focused on the continued development and optimization of MA-SML. This includes thorough peptide model studies to establish the structural features of ligation substrates and nucleophiles that are required for MA-SML. We further provide multiple examples demonstrating that MA-SML is superior to standard SML for appending fluorophores, PEG, or cyclooctyne to full size protein targets. Finally, we also find that MA-SML is compatible with a common C-terminal fusion tag consisting of the LPXTG motif adjacent to a hexahistidine (His6) affinity handle. Therefore, this approach may be readily accessible for many users of sortase-based methods without the need to redesign any of the protein or peptide reagents currently on hand.
Scheme 1.
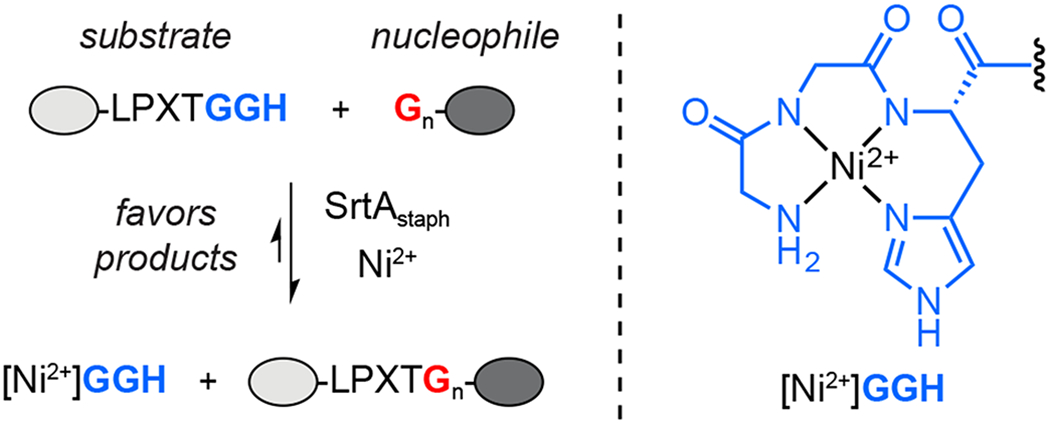
Metal-assisted sortase-mediated ligation (MA-SML).
Results and Discussion
Peptide Model Studies.
The MA-SML strategy is enabled by using an extended LPXTGGH substrate motif. In order to determine whether additional structural features were required in this substrate or the corresponding glycine amine nucleophile, we began with a series of model reactions using peptide ligation partners. As a basis for comparison, we first synthesized a peptide substrate (1) containing the masked GGH unit and a dinitrophenyl (Dnp) chromophore to assist with reaction monitoring by RP-HPLC (Figure 1A). As anticipated, a standard sortase-mediated ligation reaction of 1 with 1 equivalent of a simple diglycine nucleophile (GG-OH) exhibited modest formation of the desired ligation product (2), which peaked at 38% conversion. Repeating this reaction with 2 equivalents of NiSO4 resulted in a significant increase in product formation, with ligation product 2 representing 77% of the final reaction mixture after 6 h at room temperature (Figure 1A). In both reactions, the remaining material consisted of unreacted substrate 1, along with low levels of a hydrolysis by-product (3) that remained at <5% even after a 14 h incubation period. As shown in Figure 1B, the advantages of the MA-SML strategy were clearly evident when the reactions were monitored over time, with reactions in the presence of the Ni2+ additive consistently showing higher ligation efficiency at all reaction time points.
Figure 1.
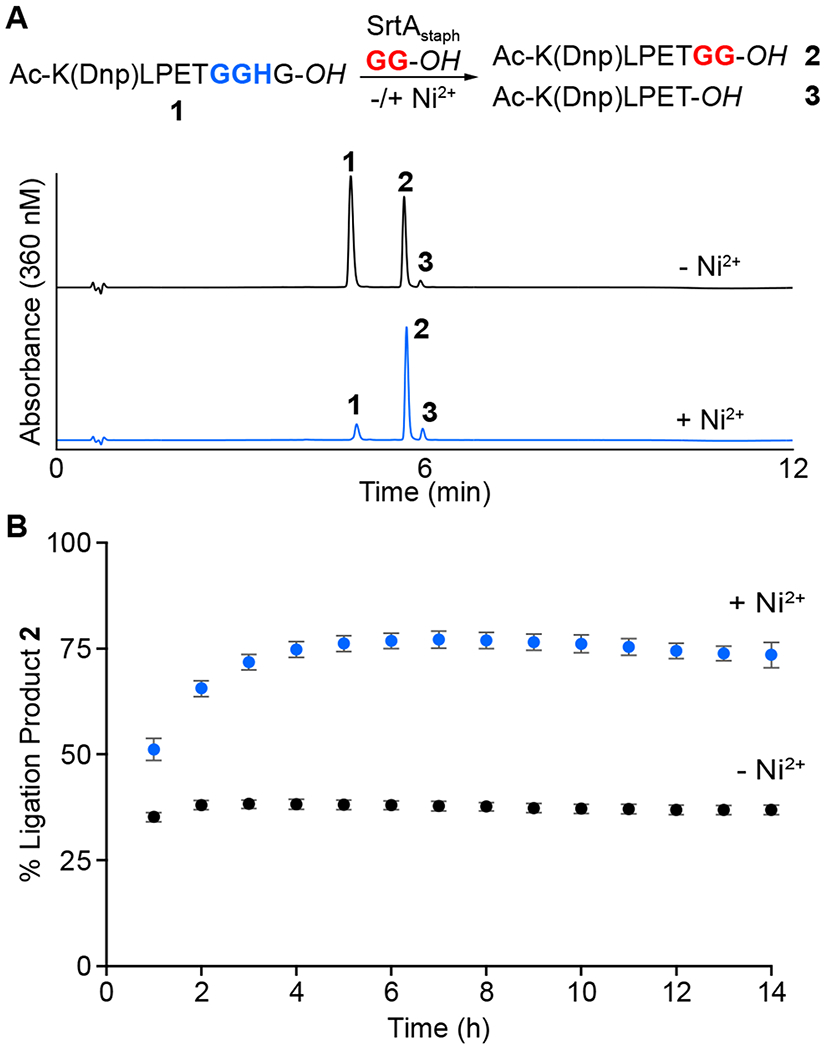
(A) RP-HPLC analysis comparing MA-SML (+Ni2+) versus standard sortase-mediated ligation (−Ni2+) in a ligation reaction utilizing a 1:1 stoichiometry of a GGH-containing peptide substrate (1) and a diglycine nucleophile (GG-OH). Chromatograms represent the 6 h reaction timepoint. All peptide starting materials and products contained native carboxylic acid C-termini (−OH). (B) Time course of reactions in panel A demonstrating increased formation of ligation product 2 in the presence of Ni2+ as estimated by RP-HPLC. Data points represent three independent experiments (mean ± standard deviation).
Using peptide substrate 1, we next assessed whether the structure of the glycine nucleophile impacted the efficacy of MA-SML (Table 1, Entries 1-6). Simple mono-, di-, and triglycine nucleophiles were evaluated, including derivatives with either a C-terminal carboxylic acid (−OH) or derivatives terminating with a primary amide (−NH2). Reactions were monitored by RP-HPLC, and the identity of all relevant reaction components was confirmed by LC-ESI-MS (Table S1). For all di- and triglycine nucleophiles, MA-SML provided superior results, exhibiting a 1.6-2.0 fold enhancement in extent of ligation as compared to controls lacking the Ni2+ additive. Results with monoglycine derivatives (Table 1, Entries 1-2) varied, with glycinamide (G-NH2) giving results similar to the larger nucleophiles, whereas free glycine (G-OH) gave overall poor performance in the presence or absence of Ni2+. This incompatibility of G-OH with sortase-mediated ligations has been described, as has the fact that substrates that terminate with a native carboxylic acid at the glycine of the LPXTG motif generally show poor reactivity.29,30 The low reactivity of these minimalist LPXTG motifs has been attributed to the presence of a charged carboxylate at the C-terminal glycine.30 We suspect that a similar phenomenon impairs the reactivity of G-OH, as switching to a derivative with a charge neutral primary amide (G-NH2) restores ligation efficiency. We also note that the poor of reactivity of G-OH is likely an insignificant limitation of MA-SML, as nearly all glycine amine nucleophiles utilized for sortase-based protein modification would be extended beyond a single free glycine residue.
Table 1.
Effect of Substrate and Nucleophile on MA-SML.a
| Entry | Substrate (~GGXX) | Nucleophile (GnX) | % Ligation (+Ni2+) | % Ligation (−Ni2+) |
|---|---|---|---|---|
| 1 | ~GGHG-OH | G-OH b | 5.9 ± 0.1 | 1.8 ± 0.1 |
| 2 | ~GGHG-OH | G-NH2 | 68 ± 0.4 | 43 ± 1.8 |
| 3 | ~GGHG-OH | GG-OH | 77 ± 1.8 | 38 ± 1.0 |
| 4 | ~GGHG-OH | GG-NH2 | 74 ± 5.8 | 42 ± 0.7 |
| 5 | ~GGHG-OH | GGG-OH | 69 ± 0.5 | 39 ± 0.1 |
| 6 | ~GGHG-OH | GGG-NH2 | 64 ± 1.6 | 41 ± 0.7 |
| 7 | ~GGH-OH | GG-NH2 | 65 ± 0.8 | 44 ± 0.6 |
| 8 | ~GGH-NH2 | GG-NH2 | 81 ± 1.2 | 45 ± 2.6 |
| 9 | ~GGH-NH2 c | GG-NH2 | 65 ± 4.4 | 44 ± 0.5 |
| 10 | ~GGG-NH2 c | GG-NH2 | 46 ± 0.9 | 46 ± 0.2 |
| 11 | ~GGS-NH2 c | GG-NH2 | 45 ± 0.1 | 45 ± 0.3 |
| 12 | ~GGD-NH2 c | GGG-NH2 | 44 ± 1.0 | 43 ± 1.1 |
Unless otherwise indicated, reaction conditions were 100 μM substrate, 100 μM nucleophile, 10 μM SrtAstaph, 0/200 μM NiSO4, sortase reaction buffer (pH 7.5), glycerol (0.2% v/v), 6 h at room temperature. Substrates and nucleophiles contained either native carboxylic acids (−OH) or primary amides (−NH2) at their C-termini. Estimates of reaction progress were obtained by comparing RP-HPLC peak areas for the unreacted substrate peptide, ligation product, and hydrolysis by-product. All data points represent three independent experiments (mean ± standard deviation).
Data for the G-OH nucleophile reported for the 17 h timepoint.
Conditions: 50 μM substrate, 50 μM nucleophile, 10 μM SrtAstaph, 0/200 μM NiSO4, sortase reaction buffer (pH 7.5), glycerol (0.2% v/v), 3 h at room temperature.
Having established that a range of nucleophiles were well tolerated by MA-SML, we turned our attention to the structure of the peptide substrate. First, two truncated variants of substrate 1 were generated that lacked the C-terminal glycine residue and terminated in either a carboxylic acid or amide (Table 1, Entries 7-8). Both peptides exhibited an increase in ligation efficiency in the presence of Ni2+, albeit with a slight reduction in the effect for the substrate terminating in a free carboxylate (Entry 7). We also prepared a panel of substrates in which the histidine of the MA-SML motif was replaced with glycine, or residues with potentially coordinating side chains such as serine or aspartic acid (Table 1, Entries 9-12). As anticipated, these alternate substrates showed no difference in the extent of ligation when the Ni2+ additive was included, confirming that the LPXTGGH motif was uniquely suited for our MA-SML strategy. We note in this case that there is literature precedent that the replacement of histidine with cysteine could also lead to the release of a viable ligand for Ni2+ following sortase-mediated substrate cleavage.31 However, to avoid complications from undesired disulfide bond formation we elected to exclude cysteine from the current study.
Taken together, the peptide model studies provided important design principles for the successful implementation of MA-SML. First, the LPXTGGH motif was found to be strictly required, and removal of the critical histidine eliminated any response to the included Ni2+ ions. As further evidence of the unique properties of LPXTGGH paired with Ni2+, we also found that switching to other divalent or trivalent metal ions failed to produce the same level of ligation enhancement as obtained with Ni2+ salts such as NiSO4 or NiCl2 (Figure S3). Finally, with the exception of free glycine (G-OH), the MA-SML strategy was effective for all other glycine amine nucleophiles tested. This suggested that MA-SML retained the same broad nucleophile tolerance as standard sortase-mediated ligation, and would likely be compatible with appending diverse modifications to larger protein targets.
A Multifunctional C-terminal Tag for MA-SML.
Building from our peptide studies, we next explored the modification of proteins. Having previously shown that MA-SML was effective for fluorescent labeling of a single protein target,28 the goal of the present work was to assess the generality of this strategy in the context of other proteins and a broader array of modifications. Additionally, we sought to highlight the ease with which MA-SML could be implemented using materials and reagents that were frequently used in the sortase literature. To that end, we found several studies in which the same C-terminal extension (LPXTGGHHHHHH, hereafter abbreviated as LPXTGGHH5) was utilized to convert a protein of interest (POI) into a viable sortase substrate (a non-exhaustive list of examples is provided in references 32–37). In this tag, the protein is often fused via a flexible spacer to an LPXTGG motif, followed directly by a hexahistidine (His6) purification handle (Figure 2). The extension of the canonical LPXTG motif by one additional glycine in this case serves to improve reaction rates.22 With respect to MA-SML, we recognized that this frequently used sequence also contained the requisite GGH unit required for Ni2+ binding. Thus, it suggested that a significant number of reported sortase substrates were already compatible with MA-SML, and that the simple addition of a Ni2+ salt could be used to improve sortase-mediated modification of these types of protein targets (Figure 2). Notably, recent studies have provided evidence that this type of multifunctional tag can be used for MA-SML.38–41 In these reports, however, reactions lacking the Ni2+ additive were not described and therefore the advantage of MA-SML versus more standard sortase-mediated ligation protocols was not clearly established.
Figure 2.
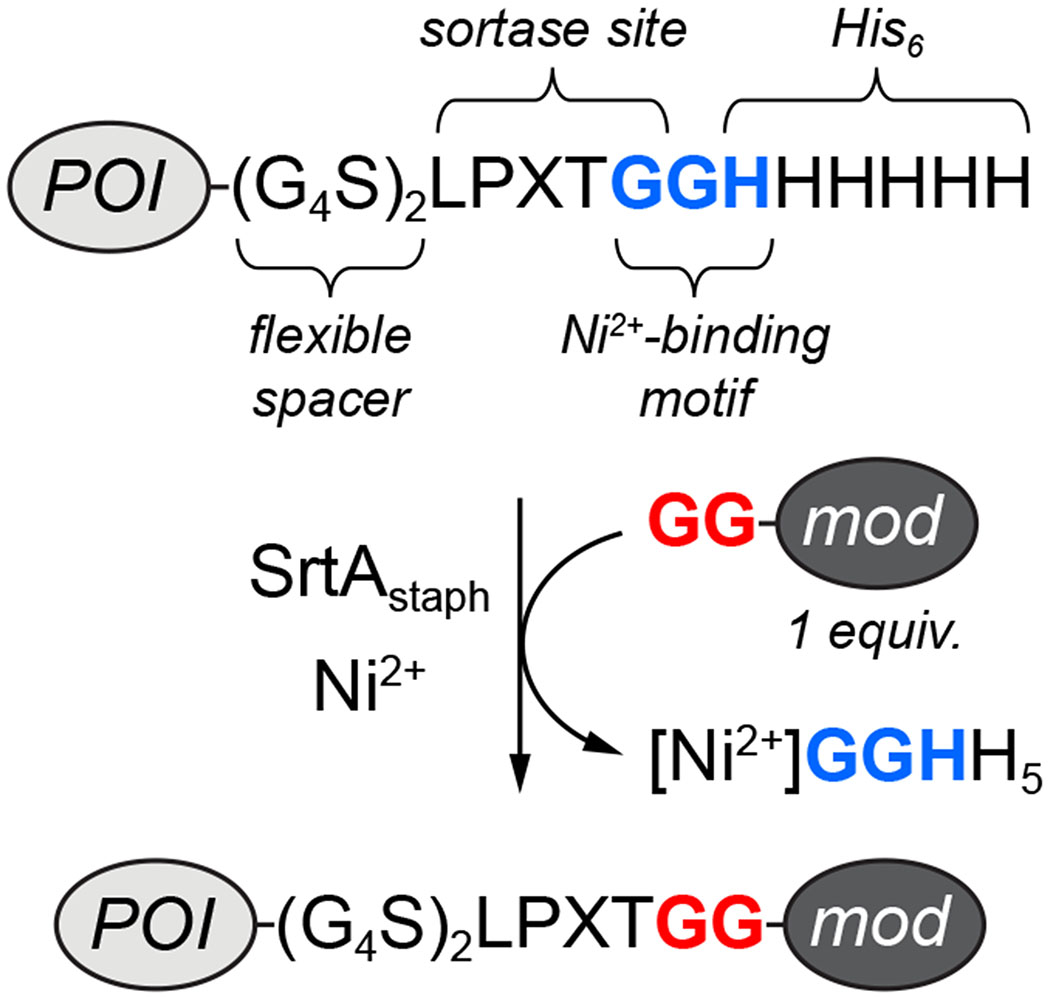
A commonly used tag to render proteins compatible with sortase-mediated ligation contains the embedded GGH motif required for MA-SML (POI = protein of interest, mod = modification of interest).
Fluorescent Labeling.
As an initial test application for MA-SML, we first explored the site-specific attachment of different fluorophores. A designed ankyrin repeat protein (DARPin) with known specificity for human epidermal growth factor receptor 2 (Her2) was selected as a substrate for these studies.42 The sequence of this protein was extended with the C-terminal tag described above to generate a target for MA-SML (DARP-LPETGGHH5, Figure 3). This protein was paired with three diglycine nucleophiles tethered via a lysine residue to either fluorescein (GGK-6FAM), cyanine 3 (GGK-Cy3), or 7-(diethylamino)coumarin (GGK-DEAC). All nucleophiles were synthesized via a combination of solid-phase and solution methods, and purified to homogeneity by RP-HPLC prior to use (Figure S4–S6).
Figure 3.
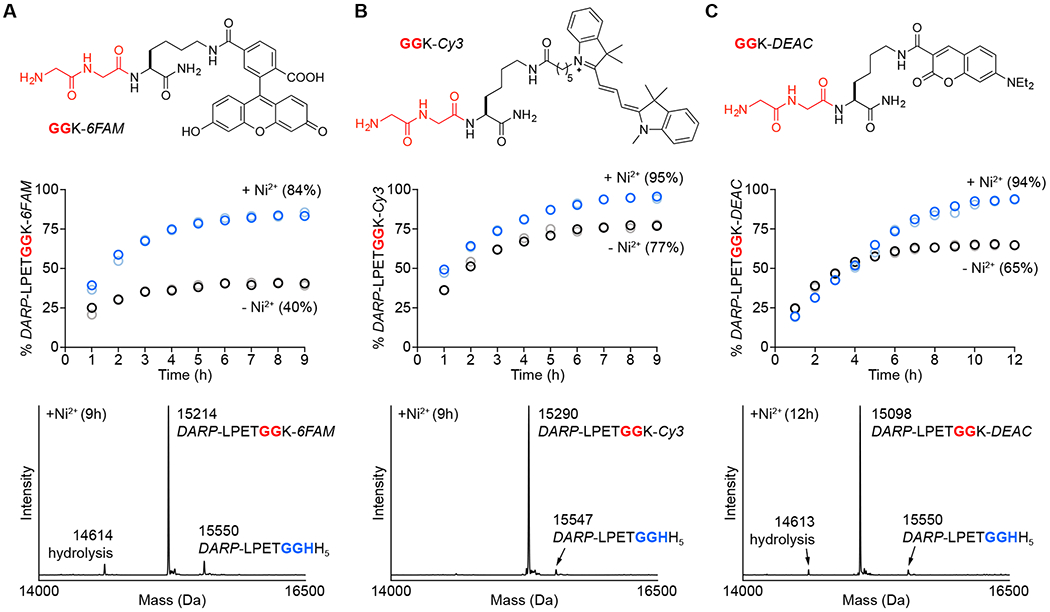
Comparison of MA-SML versus standard sortase-mediated ligation for fluorescent labeling of a DARPin substrate (DARP-LPETGGHH5). Diglycine nucleophiles (top) modified with (A) fluorescein (GGK-6FAM), (B) cyanine 3 (GGK-Cy3), or (C) 7-(diethylamino)coumarin (GGK-DEAC) were reacted with DARP-LPETGGHH5 in a 1:1 molar ratio using 20 mol% SrtAstaph in the presence or absence of 4 equivalents of NiSO4. The addition of Ni2+ significantly improves product formation (middle) as determined by LC-ESI-MS (blue/light blue circles represent two independent reactions containing Ni2+, black/grey circles represent two reactions in the absence of Ni2+; values in parentheses represent average % product formation for the two independent trials at the final timepoint). Deconvolved mass spectra (bottom) of the 9 or 12 h timepoints for reactions with Ni2+ confirm formation of the desired conjugates, with minimal contamination from unreacted DARP-LPETGGHH5 and competing substrate hydrolysis (calculated MWs: DARP-LPETGGHH5 = 15550 Da, hydrolysis = 14613 Da, DARP-LPETGGK-6FAM = 15213 Da, DARP-LPETGGK-Cy3 = 15294 Da, DARP-LPETGGK-DEAC = 15098 Da).
A standard reaction mixture consisting of 50 μM DARP-LPETGGHH5 and 1 molar equivalent of diglycine nucleophile was employed to evaluate the efficacy of MA-SML versus a control lacking the Ni2+ additive. Beginning with the GGK-6FAM nucleophile (Figure 3A), reactions in the absence of Ni2+ reproducibly exhibited a maximum of ~40% conversion to the desired ligation product as estimated by LC-ESI-MS. In this case, peak reaction conversion was reached after roughly 5 h at room temperature, and remained unchanged for an additional 3-4 hours. By simply adding Ni2+ to the reaction, we were able to boost the reaction conversion to 84% (Figure 3A, middle). Importantly, ESI-MS spectra of crude MA-SML reactions confirmed formation of the desired ligation product (DARP-LPETGGK-6FAM), with low levels of unreacted DARP-LPETGGHH5 substrate (10%) and the expected hydrolysis by-product (7%) (Figure 3A, bottom). Similar reactivity trends were observed when using Cy3 (GGK-Cy3) and coumarin (GGK-DEAC) modified nucleophiles. Specifically, MA-SML reactions including Ni2+ showed higher levels of ligation product formation as compared to controls lacking the metal additive (Figure 3B–C, middle). Crude reaction mixtures were exceptionally clean, with 94-95% of the DARPin content corresponding to the fluorescent ligation products, along with trace levels of unreacted substrate and hydrolysis (Figure 3B–C, bottom).
Interestingly, whereas MA-SML reactions gave consistently high levels of product formation (84-95%), controls run without Ni2+ showed a much wider variation in the extent of ligation (40-77%). This suggested that the fluorophore itself plays a role in controlling the equilibrium of sortase-mediated ligations. To our knowledge, the influence of residues or modifications beyond the N-terminal glycines has not been explored extensively in the literature, though some effects arising from residues distant from the nucleophilic N-terminal amine have been reported.22 We suspect that these phenomena are typically masked when one of the sortase ligation partners is used in excess, which is often the case for most sortase-based applications. When reagents are used in equimolar ratios as described here, reactivity differences between structurally distinct glycine amine nucleophiles become more apparent. That said, a notable advantage of our MA-SML procedure is the ability to coax uniformly higher ligation yields out of both inherently less successful (GGK-6FAM) and more successful (GGK-Cy3, GGK-DEAC) nucleophiles.
As an additional demonstration of the enhancement in fluorophore labeling provided by MA-SML, we conducted a series of standard sortase-mediated ligations in which excess glycine nucleophile was used to drive the reaction to completion. To that end, the ligation of DARP-LPETGGHH5 and GGK-DEAC was performed in the absence of Ni2+ using up to 20 molar equivalents of the nucleophile. Reactions were monitored by LC-ESI-MS over the course of 12 hours, and results were compared to our initial trials employing a 1:1 molar ratio of the ligation partners (Table 2). As expected, increasing the loading of GGK-DEAC in reactions without Ni2+ led to continuous improvements in the amount of DARP-LPETGGK-DEAC (4) formed. However, at least 5 equivalents of GGK-DEAC were required to approach the reaction efficiency of the corresponding MA-SML reaction (+Ni2+) utilizing only 1 equivalent of nucleophile. The levels of undesired substrate hydrolysis (5) remained low (≤ 3%) in all cases, albeit it was slightly elevated in the MA-SML reaction. Overall, these results provide clear evidence that MA-SML can substantially reduce the need for excess reagents in sortase-mediated labeling applications without compromising the formation of the desired protein conjugate.
Table 2.
Comparison of MA-SML versus Higher Nucleophile Loading in the Absence of Ni2+.a
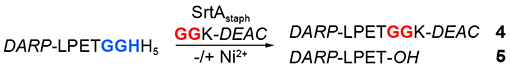 | |||
|---|---|---|---|
| Equivalents GGK-DEAC | Ni2+ | % Ligation (4) | % Hydrolysis (5) |
| 1 | + | 94 | 3 |
| 1 | − | 65 | <1 |
| 2 | − | 78 | <1 |
| 3 | − | 84 | <1 |
| 5 | − | 88 | <1 |
| 20 | − | 91 | ndb |
Conditions: 50 μM DARP-LPETGGHH5, 1-20 equivalents GGK-DEAC nucleophile, 10 μM SrtAstaph, 0/200 μM NiSO4, sortase reaction buffer (pH 7.5), glycerol (0.2% v/v), residual DMSO (0.2-3% v/v) from GGK-DEAC stock solution, 12 h at room temperature. Reactions were analyzed by LC-ESI-MS and % ligation (4) and % hydrolysis (5) were estimated from peak areas derived from the corresponding deconvoluted mass spectra.
nd = hydrolysis by-product not detected.
PEGylation.
Having successfully conjugated a range of fluorophores, we next evaluated MA-SML in the context of ligations involving polyethylene glycol (PEG). PEGylation remains an important strategy for modulating the pharmacokinetic and immunogenic properties of therapeutically important proteins,43,44 and efforts to directly ligate PEG-modified glycine nucleophiles using sortase have been reported.45–47 However, prior studies utilizing sortase have either relied on a large excess of the PEG nucleophile, or shown modest conversion to the desired PEGylated protein conjugate when the ligation partners were used in equimolar amounts.45–47 Thus, we hypothesized that MA-SML would provide a superior method for PEGylation by eliminating the need for using precious PEG reagents in excess while maintaining high ligation efficiency.
To test this approach, a diglycine nucleophile was synthesized tethered to a branched, monodisperse PEG moiety (GGKY-PEG, Figure 4A). A single tyrosine chromophore was included in this structure to assist with RP-HPLC purification of GGKY-PEG (Figure S8) and to allow estimation of its concentration in solution via UV-Vis. The PEG-modified nucleophile was then combined in a 1:1 ratio with our DARP-LPETGGHH5 substrate in an MA-SML reaction analogous to those described above. After a 12 h incubation at room temperature, LC-ESI-MS revealed excellent conversion (90%) to the desired PEG conjugate, with only trace levels (~4%) of competing hydrolysis (Figure 4B). In contrast, control ligations lacking Ni2+ showed only 45% conversion to the PEG-modified product, despite otherwise identical reaction conditions (Figure S12). Interestingly, all deconvoluted ESI-MS spectra of the PEG conjugate (DARP-LPETGGKY-PEG) showed a distribution of minor peaks that were consistent with ammonium adducts. A similar pattern of ammonium adducts was observed for the free GGKY-PEG peptide (Figure S8). This phenomenon has been observed previously for other PEG oligomers, and has been attributed to trace ammonium contamination in the components of the LC-ESI-MS mobile phase.48
Figure 4.
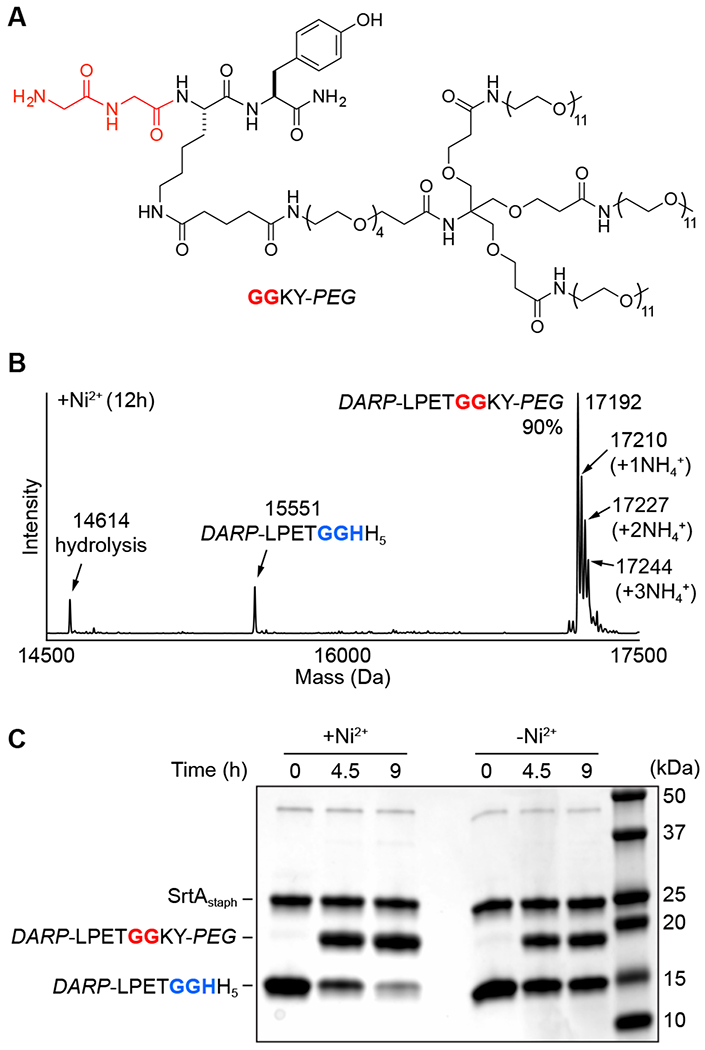
(A) Structure of PEG-modified diglycine nucleophile (GGKY-PEG) for attachment of a branched PEG unit via MA-SML. (B) Representative deconvoluted mass spectrum of 12 h timepoint from MA-SML reaction utilizing 20 mol% SrtAstaph, 4 equivalents of NiSO4, and a 1:1 stoichiometry of DARP-LPETGGHH5 and GGKY-PEG. The extent of product formation (90%) was estimated from peak areas derived from the deconvoluted mass spectrum. Peak areas for the observed ammonium adducts were included as part of the total product formed (calculated MWs: DARP-LPETGGHH5 = 15550 Da, hydrolysis = 14613 Da, DARP-LPETGGKY-PEG = 17191 Da). (C) SDS-PAGE gel demonstrating higher PEGylation in the presence of Ni2+ for reactions using a 1:1 ratio of DARP-LPETGGHH5 to GGKY-PEG.
Given the large increase in protein molecular weight afforded by modification with PEG, we were able to further verify the increase in ligation efficiency afforded by MA-SML via SDS-PAGE. As shown in Figure 4C, reactions containing the Ni2+ additive showed near complete consumption of the DARP-LPETGGHH5 substrate and the appearance of a higher molecular band consistent with PEG attachment. As expected, reaction progress was substantially reduced when Ni2+ omitted, and significant levels of unreacted DARP-LPETGGHH5 persisted in the reaction mixture after a 9 h incubation.
One-Pot MA-SML / Strain-Promoted Azide-Alkyne Cycloaddition (SPAAC).
As a final application of MA-SML, we next evaluated the attachment of a bioorthogonal cyclooctyne moiety.49–51 Biomolecules decorated with functional groups such as azides, cyclooctynes, trans-cyclooctenes, tetrazines and others are valuable reagents capable of subsequent derivatization with a range of functional labels via bioorthogonal ligation reactions.52–54 The initial modification of the biomolecular target with a bioorthogonal reaction handle is a critical step in this process, and is ideally conducted in a site-specific and efficient fashion. To demonstrate the suitability of MA-SML for this purpose, we began by synthesizing a diglycine nucleophile containing a dibenzocyclooctyne group (GGK-DBCO, Figure 5A). The ligation of GGK-DBCO to DARP-LPETGGHH5 was then assessed in the presence and absence of the Ni2+ additive. As anticipated, the MA-SML approach (+Ni2+) produced high levels (85%) of the desired conjugate (6) when only 1 equivalent of GGK-DBCO was utilized (Figure 5A,B). Removal of Ni2+ led to a more than two-fold reduction in the extent of ligation (Figure S13). Importantly, competing substrate hydrolysis in the MA-SML reaction remained low, reaching levels of only 4% after 9 h (Figure 5B) and 10% after 20 h (Figure S13).
Figure 5.
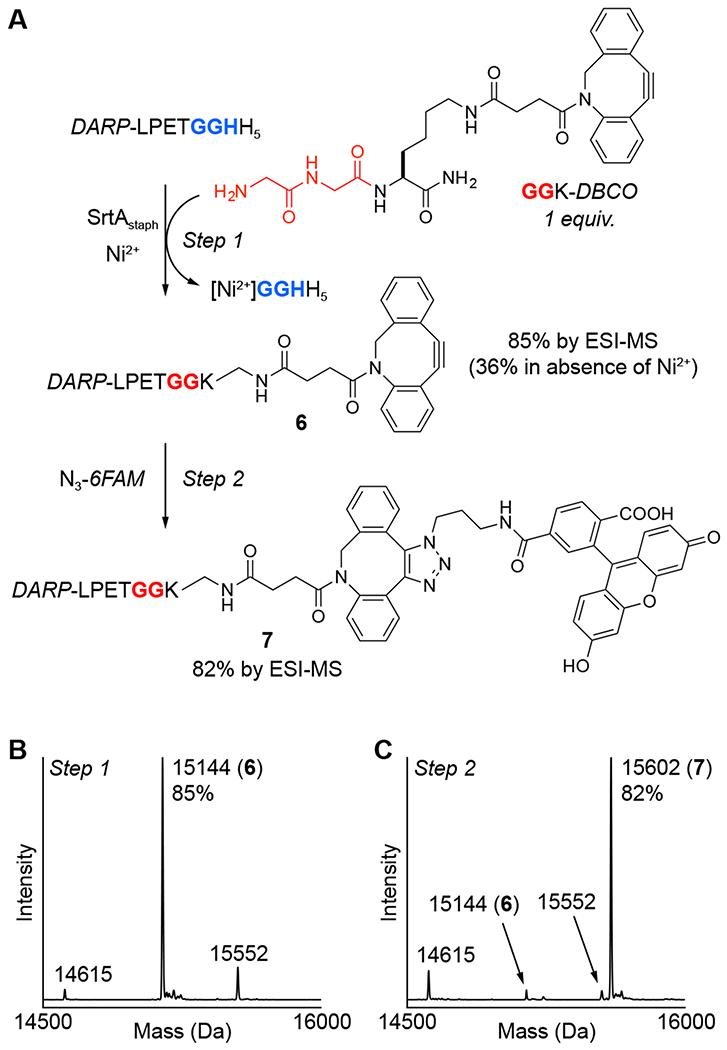
One-pot strategy for sequential MA-SML and strain-promoted azide-alkyne cycloaddition (SPAAC). (A) An initial MA-SML reaction using 20 mol% SrtAstaph, 4 equivalents of NiSO4, and a 1:1 ratio of DARP-LPETGGHH5 and GGK-DBCO generates the desired DBCO conjugate (6) with more than a two-fold increase in reaction conversion compared to controls lacking Ni2+. Direct addition of two equivalents of fluorescent azide (N3-6FAM) to the crude MA-SML reaction mixture produces the final SPAAC product (7) as 82% of the total crude protein mixture. Deconvoluted mass spectra of the crude reaction mixtures from the (B) MA-SML (spectrum shown is for the 9 h reaction time point) and (C) SPAAC steps confirm formation of the desired protein conjugates (calculated MWs: DARP-LPETGGHH5 = 15550 Da, hydrolysis = 14613 Da, DARP-LPETGGK-DBCO (6) = 15142, SPAAC product (7) = 15600 Da).
Encouraged by the efficiency of the MA-SML reaction between DARP-LPETGGHH5 and GGK-DBCO, we next evaluated the ability of conjugate 6 to participate in a strain-promoted azide-alkyne cycloaddition (SPAAC) with a suitable azide.49–51 Ultimately, a one-pot sequence was developed in which the crude reaction mixture from the MA-SML reaction was simply treated with azide-modified fluorescein (N3-6FAM) to produce the final fluorescent protein conjugate (7, Figure 5A). Characterization of the final crude reaction mixture by LC-ESI-MS indicated that fluorescent derivative 7 comprised 82% of the total DARPin content, suggesting good purity without any additional purification (Figure 5C). Moreover, the high level of conversion for the SPAAC step (>95% based solely on peaks for 6 and 7 in the deconvoluted mass spectrum) indicated that the azide-alkyne cycloaddition was not impaired by the SrtAstaph and NiSO4 additive remaining in solution.
Additional Protein Substrates.
Three additional proteins were investigated as targets for MA-SML. This included a Fynomer with binding specificity for human chymase,55 the FH8 protein from Fasciola hepatica,56 and an Affibody engineered for high affinity binding to Her2.57 All proteins were extended with the C-terminal motif depicted in Figure 2 to generate the sortase-compatible substrates Fyn-LPETGGHH5, FH8-LPETGGHH5, and Aff-LPETGGHH5. For consistency, each target was modified using the same coumarin-labeled nucleophile (GGK-DEAC) that had shown success with the DARP-LPETGGHH5 substrate described above (Figure 3C).
Using LC-ESI-MS to estimate reaction progress, we compared sortase-mediated ligation efficiency in the presence or absence of the Ni2+ additive (Figure 6). In all cases, reactions maintained an identical stoichiometry of 1:1 protein substrate to GGK-DEAC. While all three targets showed evidence of successful labeling, the advantage conferred by the MA-SML (+Ni2+) reaction conditions proved to be varied. Specifically, the Fyn-LPETGGHH5 substrate showed excellent labeling using MA-SML, reaching >90% conversion within approximately 10-12 h (Figure 6A). Removal of Ni2+ reduced the extent of ligation for Fyn-LPETGGHH5 by more than two-fold within the same time period, indicating a significant benefit derived from the MA-SML approach. The FH8-LPETGGHH5 substrate also exhibited good ligation efficiency (74%) under MA-SML conditions, however this represented a relatively minor improvement over controls lacking the metal additive, which plateaued at approximately 60% conversion after 6 h (Figure 6B). Interestingly, ligations involving FH8-LPETGGHH5 and GGK-DEAC consistently showed the formation of an unexpected by-product at 10130 Da. This represented a mass loss of 57 Da from the desired FH8-LPETGGK-DEAC conjugate, and we speculate that this corresponds to a derivative lacking a single glycine residue. Estimates of reaction progress for the FH8-LPETGGHH5 / GGK-DEAC system did account for the formation of this undesired species, and its origin was not investigated further. Finally, the least successful ligation results were obtained using the Aff-LPETGGHH5 substrate. MA-SML was not found to provide a significant advantage, and the extent of ligation was <30% irrespective of the presence of the Ni2+ additive (Figure 6C).
Figure 6.
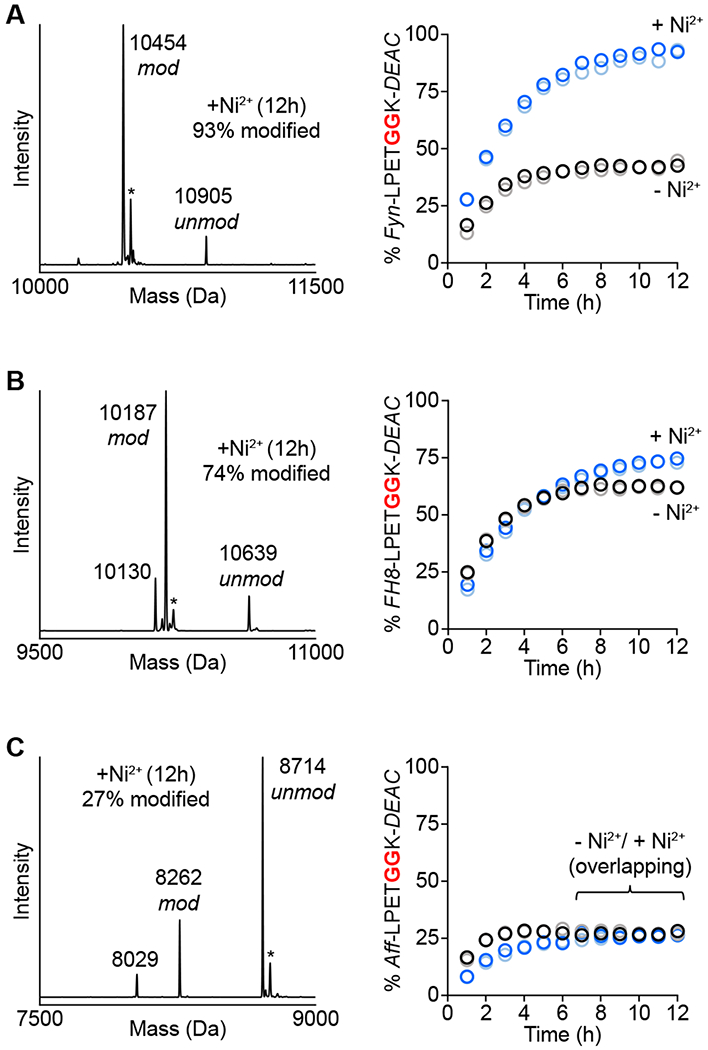
MA-SML versus standard sortase-mediated ligation for modification of additional protein targets. One equivalent of coumarin nucleophile (GGK-DEAC) was reacted with (A) Fynomer (Fyn-LPETGGHH5), (B) FH8 (FH8-LPETGGHH5), or (C) Affibody (Aff-LPETGGHH5) using 20 mol% SrtAstaph in the presence or absence of Ni2+. The addition of Ni2+ improves reaction conversion (right) for Fynomer and FH8 substrates as estimated by LC-ESI-MS (blue/light blue circles represent two independent reactions containing Ni2+, black/grey circles represent two reactions in the absence of Ni2+). Representative deconvoluted mass spectra (left) of MA-SML reactions at 12 h confirm formation of the desired DEAC conjugates (calculated MWs: modified/unmodified Fyn-LPETGGHH5 = 10454/10907 Da, modified/unmodified FH8-LPETGGHH5 = 10187/10640 Da, modified/unmodified Aff-LPETGGHH5 = 8261/8714 Da). Additional notes: * = MeCN adducts from LC-ESI-MS mobile phase; peak at 10130 Da in B is consistent with the FH8-LPETGGK-DEAC conjugate lacking a glycine residue (−57 Da); peak at 8029 Da in C corresponds to a truncated Affibody substrate that co-purified with full length Aff-LPETGGHH5.
Considered alongside the DARP-LPETGGHH5 ligations described above, our results with the Fynomer, FH8, and Affibody substrates clearly indicate that MA-SML efficacy can vary by target. In contrast, for those substrates that exhibited the best performance (DARP-LPETGGHH5 and Fyn-LPETGGHH5), MA-SML exhibited broad compatibility with a range of structurally diverse glycine amine nucleophiles. This was evident in all reactions involving DARP-LPETGGHH5, where the extent of ligation under MA-SML conditions was uniformly high for fluorescent, PEGylated, and DBCO-modified nucleophiles. This also appears to be the case for Fyn-LPETGGHH5, where in addition to the results provided in Figure 6A for reactions with GGK-DEAC, preliminary studies using GGK-DBCO and Fyn-LPETGGHH5 showed a similar boost in ligation efficiency when the MA-SML strategy was employed (data not shown).
With respect to less successful substrates, in particular Aff-LPETGGHH5, it is known that structural elements of the protein target and solvent accessibility of the LPXTG motif can impact the success of sortase-mediated ligations.17,58,59 The use of an extended (G4S)2 linker between the LPXTG sequence and the body of the protein target in this work was intended to mitigate these effects, however this appears to be ineffective in the case of Affibody. While not explored here, we envision that further optimization of the linker structure could improve sortase-mediated ligations involving this target. Additionally, we note that our results are not the first to show difficulty in using Affibody as a sortase substrate. Previous work using a highly similar anti-Her2 Affibody also reported an uncharacteristically low reaction efficiency even when one of the sortase ligation partners was used in excess.60 Interestingly, there is evidence that MA-SML may still provide a viable approach for ligations involving the Affibody target, as later reports described improved reaction yields via a reaction using an excess of the Affibody substrate, a catalytically enhanced mutant of SrtAstaph, and the same Ni2+ coordination strategy described here.38,41 In these examples, direct comparisons to reactions lacking Ni2+ were not reported and it is not clear to what extent MA-SML was responsible for the observed gains in reaction efficiency.
Conclusion
In summary, this work describes multiple studies focused on the continued optimization and development of MA-SML for use in sortase-based protein engineering. First, using peptide models we have confirmed that this strategy relies on the use of Ni2+ in combination with a modified sortase substrate motif containing an embedded GGH sequence. Provided these conditions are met, MA-SML is able to accommodate a range of simple mono-, di-, and triglycine nucleophiles. In addition to peptide studies, we have further demonstrated that MA-SML can be applied to the modification of full size proteins possessing a C-terminal tag (LPXTGGHH5) that is prevalent in the sortase literature. Notably, out of eight unique pairings of protein substrate and functionalized glycine amine nucleophile, seven examples were found in which MA-SML provided superior ligation results when the substrate and nucleophile were used in equimolar amounts. In these cases, the extent of ligation fell within the range of 74-95%, and enabled the site-specific attachment of modifications such as PEG, fluorophores, and a bioorthogonal cyclooctyne moiety. Overall, these results highlight the utility of MA-SML, and we envision that this technique will provide a valuable and easily implemented strategy for users of sortase-based methods.
EXPERIMENTAL PROCEDURES
Instrumentation.
Reversed-phase HPLC purifications and analyses were performed on a Dionex Ultimate 3000 HPLC system. Specific columns, mobile phases, and gradient details are described in the appropriate sections below or in Supporting Information. For LC-ESI-MS analyses, the Dionex Ultimate 3000 HPLC was interfaced with an Advion CMS expressionL mass spectrometer. LC-ESI-MS data were analyzed using Advion Data Express software, and protein charge ladders were deconvoluted using a maximum-entropy algorithm provided by Analyst 1.4.2 software.
Desalting of protein solutions and immobilized metal affinity chromatography (IMAC) utilizing an imidazole gradient were carried out on an NGC Quest™ 10 Plus FPLC system (Bio-Rad) equipped with a 10 mL Bio-Scale™ Mini Bio-Gel® P-6 Desalting Cartridge or a 5 mL Bio-Scale™ Mini Nuvia™ IMAC Ni-Charged column. Additional details on mobile phase conditions, flow rates, and gradients are provided in the appropriate sections below.
UV-Vis measurements for estimating the concentration of protein and peptide solutions were obtained using either a Nanodrop ND-1000 spectrophotometer (ThermoFisher) or a Biotek® Epoch™ Microplate Spectrophotometer.
Imaging of SDS-PAGE gels stained with Coomassie Brilliant Blue R-250 was performed using a Gel Doc™ EZ imager (Bio-Rad).
Water used in all experimental procedures was purified using a Milli-Q Advantage A10 system (Millipore).
Expression and Purification of SrtAstaph.
The pET-28a(+) expression vector encoding Δ59-sortase A from S. aureus (SrtAstaph) with an N-terminal His6 tag was obtained from Hidde Ploegh (Addgene plasmid #51138).8 Expression and purification of SrtAstaph was performed as previously reported.28 Prior to use, SrtAstaph was dialyzed against 50 mM Tris (pH 7.5) and 150 mM NaCl at 4 °C. Glycerol was then added to a final concentration of 10% (v/v), and aliquots were stored at −80 °C. Protein concentration was estimated by absorbance at 280 nm using a calculated extinction coefficient of 14440 M−1cm−1 (Expasy ProtParam). The identity and purity of SrtAstaph was confirmed by SDS-PAGE and LC-ESI-MS (Figure S10, Figure S11).
Expression and Purification of LPETGGHH5 Protein Substrates.
Expression vectors for DARP-LPETGGHH5, Fyn-LPETGGHH5, Aff-LPETGGHH5, and FH8-LPETGGHH5 were obtained by commercial gene synthesis from ATUM. All constructs were synthesized in the pD441-SR plasmid backbone, and contained the protein domain of interest fused at its C-terminus to a GGGGSGGGGSLPETGGHHHHHH sequence.
Expression and purification of DARP-LPETGGHH5, Fyn-LPETGGHH5, and Aff-LPETGGHH5 were achieved using an identical protocol. Expression vectors were first transformed (heat shock) into E. coli BL21(DE3) cells, and cells were then plated on LB agar plates containing kanamycin (50 μg/mL). After overnight incubation at 37 °C, single colonies were selected and used to inoculate 50 mL of sterile LB containing kanamycin (50 μg/mL). These starter cultures were incubated at 37 °C for approximately 18 h, and then added to 1 L of sterile LB containing kanamycin (50 μg/mL). Cells were grown at 37 °C in a shaking incubator to an optical density of ~0.8-0.9 at 600 nm. Protein expression was then induced with IPTG (1 mM final concentration) for 3 h at 37 °C. The cells were harvested by centrifugation, and the pellets were frozen at −80 °C. Cell pellets were then thawed and resuspended on ice using 30 mL of lysis buffer A (20 mM Tris (pH 7.5), 150 mM NaCl, and 0.5 mM EDTA). Lysozyme was then added to a final concentration of 1 mg/mL, and the suspensions were gently shaken for 1 h at room temperature. The cells were further lysed by sonication and then centrifuged. The clarified supernatants were applied to a nickel-nitrilotriacetic acid (Ni-NTA) column containing 5 mL of HisPur™ resin (ThermoFisher) that had been equilibrated with wash buffer A (20 mM Tris (pH 7.5), 150 mM NaCl, and 20 mM imidazole). The column was then flushed with 10 column volumes of wash buffer A, and the protein eluted with 10 mL of elution buffer A (20 mM Tris (pH 7.5), 150 mM NaCl, and 300 mM imidazole). Eluted protein was then desalted on a Bio-Scale™ Mini Bio-Gel® P-6 Desalting Cartridge to remove imidazole. Desalting was performed following the manufacturer’s directions using a mobile phase consisting of 20 mM Tris (pH 7.5) and 150 mM NaCl at a flow rate of 2 mL/min. Desalted fractions were then evaluated by LC-ESI-MS to confirm protein identity and to assess purity. In cases where impurities where detected, an additional IMAC purification was performed utilizing an imidazole gradient for elution. Briefly, a 5 mL Bio-Scale™ Mini Nuvia™ IMAC column was first equilibrated with wash buffer A. Protein was then loaded onto the column, followed by gradient elution (20-300 mM imidazole over 50 mL) at a flow rate of 7 mL/min. Pure fractions were pooled and desalted as described above. The final storage buffer for these substrate proteins was 20 mM Tris (pH 7.5) and 150 mM NaCl. Protein concentrations were estimated by absorbance at 280 nm using calculated extinction coefficients (Expasy ProtParam). The identity and purity of all proteins stocks were evaluated by LC-ESI-MS and SDS-PAGE (Figure S10, Figure S11).
FH8-LPETGGHH5 was expressed and purified in a similar fashion. Following transformation of E. coli BL21(DE3) cells with the appropriate expression vector, cells were grown at 37 °C in six 1 L portions of sterile LB containing kanamycin (10 μg/mL). Cells were grown to an optical density of 0.4-0.5 at 600 nm, and then protein expression was induced with IPTG (1.6 mM final concentration) for 5 h at 37 °C. The cells were harvested by centrifugation, and the combined pellets were resuspended in 60 mL of lysis buffer B (50 mM NaH2PO4 (pH 8.0), 300 mM NaCl, and 10 mM imidazole). Lysozyme was then added (50 μg/mL final concentration) and the suspensions were incubated at 4 °C for 30 minutes with gentle agitation. The cells were then further lysed by sonication. Lysates were next treated with DNAse I (0.1 units/mL) and incubated at room temperature for 30 minutes, followed by centrifugation. The clarified supernatant was then mixed with 3 mL of Ni-NTA resin that had been equilibrated with lysis buffer B. This mixture was incubated at 4 °C for 30 minutes with gentle agitation. The lysate/Ni-NTA slurry was then poured into a fritted glass column and the flow through was discarded. The column was washed with 24 mL of wash buffer B (50 mM NaH2PO4 (pH 8.0), 300 mM NaCl, and 20 mM imidazole), and the protein eluted with 9 mL of elution buffer B (50 mM NaH2PO4 (pH 8.0), 300 mM NaCl, and 300 mM imidazole). Eluted protein was then desalted on a Bio-Scale™ Mini Bio-Gel® P-6 Desalting Cartridge to remove imidazole. Desalting was performed following the manufacturer’s directions using a mobile phase consisting of 50 mM Tris (pH 7.5) and 150 mM NaCl at a flow rate of 2 mL/min. Desalted fractions were pooled and the identity and purity of FH8-LPETGGHH5 were confirmed by LC-ESI-MS and SDS-PAGE (Figure S10, Figure S11). Due to the low number of aromatic residues in FH8-LPETGGHH5, the concentration was estimated by absorbance at 205 nm using the method of Anthis et al.61
Model Peptide Substrates and Nucleophiles.
All mono-, di-, and triglycine nucleophiles used in peptide model studies were obtained from commercial sources and used without further purification. The preparation of substrates with the general structure Ac-K(Dnp)-LPETGGXX was done using manual Fmoc solid phase peptide synthesis as described previously.28 All synthetic peptides were purified by RP-HPLC and their identity was confirmed by LC-ESI-MS (Figure S1, Figure S2). Prior to use in sortase-mediated ligation reactions, peptide materials were prepared as concentrated stock solutions in water. For peptides containing the Dnp chromophore, solution concentrations were estimated using the absorbance of the Dnp chromophore at 365 nm (extinction coefficient = 17,300 M−1cm−1).62
Sortase-Mediated Ligations using Model Peptide Substrates and Nucleophiles.
Reactions were generally conducted on 200 μL scale. Stock solutions of peptide substrate, glycine nucleophile, and metal additive were prepared in water and combined with SrtAstaph in appropriate ratios to give the desired final reagent concentrations. Unless indicated otherwise, final reagent concentrations of 100 μM peptide substrate, 100 μM glycine nucleophile, 10 μM SrtAstaph, and 0/200 μM NiSO4 were employed. All reactions also contained 10% (v/v) 10x sortase reaction buffer (500 mM Tris (pH 7.5), 1500 mM NaCl, 100 mM CaCl2), as well as residual glycerol (0.2% v/v) from the SrtAstaph stock solution. Reactions were incubated at room temperature and analyzed by LC-ESI-MS and RP-HPLC (Phenomenex Kinetex® 2.6 μm C18 100 Å column (100 × 2.1 mm), aqueous (95% H2O, 5% MeCN, 0.1% formic acid) / MeCN (0.1% formic acid) mobile phase at 0.4 mL/min, gradient adjusted for each substrate/nucleophile pair to ensure separation of all reaction components). Estimates of reaction progress were obtained by comparing peak areas for the unreacted substrate peptide, ligation product, and hydrolysis by-product from the 360 nm RP-HPLC chromatograms.
Synthesis of Functionalized Diglycine Nucleophiles.
Detailed experimental procedures and synthetic schemes for the preparation of GGK-6FAM, GGK-Cy3, GGK-DEAC, GGK-DBCO, and GGKY-PEG are provided in Supporting Information. Prior to use in sortase-mediated ligation reactions, all nucleophiles were prepared as stock solutions in DMSO and /or H2O (see Supporting Information).
Sortase-Mediated Ligations using Protein Substrates and Functionalized Diglycine Nucleophiles.
Reactions were performed on 100 μL scale. Stock solutions of protein substrate, diglycine nucleophile, and NiSO4 (stock solution in water) were combined with SrtAstaph in appropriate ratios to give the desired reagent concentrations. Unless indicated otherwise, final reagent concentrations of 50 μM protein substrate, 50 μM diglycine nucleophile, 10 μM SrtAstaph, and 0/200 μM NiSO4 were employed. All reactions also contained 10% (v/v) 10x sortase reaction buffer (500 mM Tris (pH 7.5), 1500 mM NaCl, 100 mM CaCl2), as well as residual glycerol (0.2% v/v) from the SrtAstaph stock solution. Reactions involving GGK-6FAM, GGK-Cy3, GGK-DEAC, and GGK-DBCO also contained residual DMSO (≤ 3% v/v) from the diglycine nucleophile stock solutions (see Supporting Information for details on the preparation of nucleophile stock solutions). All reactions were incubated at room temperature and monitored by LC-ESI-MS (Phenomenex Aeris™ 3.6 μm WIDEPORE C4 200 Å column (100 × 2.1 mm), aqueous (95% H2O, 5% MeCN, 0.1% formic acid) / MeCN (0.1% formic acid) mobile phase at 0.3 mL/min, method: hold 10% MeCN 0.0-0.5 min, linear gradient of 10-90% MeCN 0.5-7.0 min, hold 90% MeCN 7.0-8.0 min, linear gradient of 90-10% MeCN 8.0-8.1 min, re-equilibrate at 10% MeCN 8.1-13.25 min). Reaction progress was estimated from mass spectra by comparing the deconvoluted peak areas of relevant protein species. Unless indicated otherwise, the peaks used for quantitation corresponded to the unreacted protein substrate, ligation product, and hydrolysis by-product. In addition to LC-ESI-MS characterization, the ligation of DARP-LPETGGHH5 and GGKY-PEG was also characterized by SDS-PAGE.
Sequential GGK-DBCO Ligation and Strain-Promoted Azide-Alkyne Cycloaddition (SPAAC).
A ligation reaction consisting of 50 μM DARP-LPETGGHH5, 50 μM GGK-DBCO, 10 μM SrtAstaph, and 200 μM NiSO4 was prepared as described above. The reaction was incubated at room temperature for 20 h and the progress of the reaction was then assessed by LC-ESI-MS (Figure S13). An 85 μL aliquot of the ligation reaction was removed, and then combined with 0.85 μL of a 10 mM DMSO stock solution of N3-6FAM (Lumiprobe). The reaction was incubated for an additional 30 minutes at room temperature and then analyzed by LC-ESI-MS. The extent of the SPAAC reaction was estimated from mass spectra by comparing the deconvoluted peak areas of relevant protein species.
Supplementary Material
ACKNOWLEDGEMENTS
Research reported in this publication was supported by the National Institute of General Medical Sciences of the National Institutes of Health under award number R15GM119048. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
ABBREVIATIONS
- SML
sortase-mediated ligation
- MA-SML
metal-assisted sortase-mediated ligation
- Dnp
2,4-dinitrophenyl
- Ac
acetyl
- RP-HPLC
reversed-phase high performance liquid chromatography
- LC-ESI-MS
liquid chromatography electrospray ionization mass spectrometry
- 6FAM
6-carboxyfluorescein
- Cy3
cyanine 3
- DEAC
7-(diethylamino)coumarin
- PEG
polyethylene glycol
- DBCO
dibenzocyclooctyne
- SPAAC
strain-promoted azide-alkyne cycloaddition
- IMAC
immobilized metal affinity chromatography
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website.
Characterization (RP-HPLC, LC-ESI-MS, SDS-PAGE) of protein and peptide substrates; mass spectrometry characterization of model peptide reactions; effect of metal ion identity on sortase-mediated ligation; synthesis and characterization (RP-HPLC, LC-ESI-MS) of functionalized diglycine nucleophiles; additional data for conjugation of GGKY-PEG and GGK-DBCO.
The authors declare no competing financial interest
REFERENCES
- (1).Zhang Y, Park KY, Suazo KF, and Distefano MD (2018) Recent progress in enzymatic protein labelling techniques and their applications. Chem. Soc. Rev. 47, 9106–9136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Pishesha N, Ingram JR, and Ploegh HL (2018) Sortase A: A model for transpeptidation and its biological applications. Annu. Rev. Cell. Dev. Biol. 34, 163–188. [DOI] [PubMed] [Google Scholar]
- (3).Dai XL, Boker A, and Glebe U (2019) Broadening the scope of sortagging. RSC Adv. 9, 4700–4721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Ge Y, Chen L, Liu S, Zhao J, Zhang H, and Chen PR (2019) Enzyme-mediated intercellular proximity labeling for detecting cell-cell interactions. J. Am. Chem. Soc. 141, 1833–1837. [DOI] [PubMed] [Google Scholar]
- (5).Shadish JA, Benuska GM, and DeForest CA (2019) Bioactive site-specifically modified proteins for 4D patterning of gel biomaterials. Nat. Mater. 18, 1005–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Fottner M, Brunner AD, Bittl V, Horn-Ghetko D, Jussupow A, Kaila VRI, Bremm A, and Lang K (2019) Site-specific ubiquitylation and SUMOylation using genetic-code expansion and sortase. Nat. Chem. Biol. 15, 276–284. [DOI] [PubMed] [Google Scholar]
- (7).Theile CS, Witte MD, Blom AEM, Kundrat L, Ploegh HL, and Guimaraes CP (2013) Site-specific N-terminal labeling of proteins using sortase-mediated reactions. Nat. Protoc. 8, 1800–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Guimaraes CP, Witte MD, Theile CS, Bozkurt G, Kundrat L, Blom AEM, and Ploegh HL (2013) Site-specific C-terminal and internal loop labeling of proteins using sortase-mediated reactions. Nat. Protoc. 8, 1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Antos JM, Ingram J, Fang T, Pishesha N, Truttmann MC, and Ploegh HL (2017) Site-specific protein labeling via sortase-mediated transpeptidation. Curr. Protoc. Prot. Sci. 89, 15.3.1–15.3.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Koussa MA, Sotomayor M, and Wong WP (2014) Protocol for sortase-mediated construction of DNA-protein hybrids and functional nanostructures. Methods 67, 134–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Wang HH, and Tsourkas A (2019) Overcoming the limitations of sortase with proximity-based sortase-mediated ligation (PBSL). Methods Mol. Biol. 2008, 165–177. [DOI] [PubMed] [Google Scholar]
- (12).Hashad RA, Lange JL, Tan NCW, Alt K, and Hagemeyer CE (2019) Engineering antibodies with C-terminal sortase-mediated modification for targeted nanomedicine. Methods Mol. Biol. 2033, 67–80. [DOI] [PubMed] [Google Scholar]
- (13).Gebleux R, Briendl M, Grawunder U, and Beerli RR (2019) Sortase A enzyme-mediated generation of site-specifically conjugated antibody-drug conjugates. Methods Mol. Biol. 2012, 1–13. [DOI] [PubMed] [Google Scholar]
- (14).Schmohl L, and Schwarzer D (2014) Sortase-mediated ligations for the site-specific modification of proteins. Curr. Opin. Chem. Biol. 22, 122–128. [DOI] [PubMed] [Google Scholar]
- (15).Milczek EM (2018) Commercial applications for enzyme-mediated protein conjugation: New developments in enzymatic processes to deliver functionalized proteins on the commercial scale. Chem. Rev. 118, 119–141. [DOI] [PubMed] [Google Scholar]
- (16).Antos JM, Truttmann MC, and Ploegh HL (2016) Recent advances in sortase-catalyzed ligation methodology. Curr. Opin. Struct. Biol. 38, 111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Yamamura Y, Hirakawa H, Yamaguchi S, and Nagamune T (2011) Enhancement of sortase A-mediated protein ligation by inducing a beta-hairpin structure around the ligation site. Chem. Commun. 47, 4742–4744. [DOI] [PubMed] [Google Scholar]
- (18).Williamson DJ, Fascione MA, Webb ME, and Turnbull WB (2012) Efficient N-terminal labeling of proteins by use of sortase. Angew. Chem. Int. Ed. Engl. 51, 9377–9380. [DOI] [PubMed] [Google Scholar]
- (19).Warden-Rothman R, Caturegli I, Popik V, and Tsourkas A (2013) Sortase-tag expressed protein ligation: combining protein purification and site-specific bioconjugation into a single step. Anal. Chem. 85, 11090–11097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Wang HH, Altun B, Nwe K, and Tsourkas A (2017) Proximity-based sortase-mediated ligation. Angew. Chem. Int. Ed. Engl. 56, 5349–5352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Refaei MA, Combs A, Kojetin DJ, Cavanagh J, Caperelli C, Rance M, Sapitro J, and Tsang P (2011) Observing selected domains in multi-domain proteins via sortase-mediated ligation and NMR spectroscopy. J. Biomol. NMR 49, 3–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Pritz S, Wolf Y, Kraetke O, Klose J, Bienert M, and Beyermann M (2007) Synthesis of biologically active peptide nucleic acid-peptide conjugates by sortase-mediated ligation. J. Org. Chem. 72, 3909–3912. [DOI] [PubMed] [Google Scholar]
- (23).Policarpo RL, Kang H, Liao X, Rabideau AE, Simon MD, and Pentelute BL (2014) Flow-based enzymatic ligation by sortase A. Angew. Chem. Int. Ed. Engl. 53, 9203–9208. [DOI] [PubMed] [Google Scholar]
- (24).Liu F, Luo EY, Flora DB, and Mezo AR (2014) Irreversible sortase A-mediated ligation driven by diketopiperazine formation. J. Org. Chem. 79, 487–492. [DOI] [PubMed] [Google Scholar]
- (25).Li YM, Li YT, Pan M, Kong XQ, Huang YC, Hong ZY, and Liu L (2014) Irreversible site-specific hydrazinolysis of proteins by use of sortase. Angew. Chem. Int. Ed. Engl. 53, 2198–2202. [DOI] [PubMed] [Google Scholar]
- (26).Kobashigawa Y, Kumeta H, Ogura K, and Inagaki F (2009) Attachment of an NMR-invisible solubility enhancement tag using a sortase-mediated protein ligation method. J. Biomol. NMR 43, 145–150. [DOI] [PubMed] [Google Scholar]
- (27).Freiburger L, Sonntag M, Hennig J, Li J, Zou P, and Sattler M (2015) Efficient segmental isotope labeling of multi-domain proteins using sortase A. J. Biomol. NMR 63, 1–8. [DOI] [PubMed] [Google Scholar]
- (28).Row DR, Roark TJ, Philip MC, Perkins LL, and Antos JM (2015) Enhancing the efficiency of sortase-mediated ligations through nickel-peptide complex formation. Chem. Commun. 51, 12548–12551. [DOI] [PubMed] [Google Scholar]
- (29).Huang X, Aulabaugh A, Ding W, Kapoor B, Alksne L, Tabei K, and Ellestad G (2003) Kinetic mechanism of Staphylococcus aureus sortase SrtA. Biochemistry 42, 11307–11315. [DOI] [PubMed] [Google Scholar]
- (30).Biswas T, Pawale VS, Choudhury D, and Roy RP (2014) Sorting of LPXTG peptides by archetypal sortase A: role of invariant substrate residues in modulating the enzyme dynamics and conformational signature of a productive substrate. Biochemistry 53, 2515–2524. [DOI] [PubMed] [Google Scholar]
- (31).Sóvágó I, Kállay C, and Várnagy K (2012) Peptides as complexing agents: Factors influencing the structure and thermodynamic stability of peptide complexes. Coord. Chem. Rev. 256, 2225–2233. [Google Scholar]
- (32).Chen L, Cohen J, Song X, Zhao A, Ye Z, Feulner CJ, Doonan P, Somers W, Lin L, and Chen PR (2016) Improved variants of SrtA for site-specific conjugation on antibodies and proteins with high efficiency. Sci. Rep. 6, 31899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Cole LE, Li L, Jetley U, Zhang J, Pacheco K, Ma F, Zhang J, Mundle S, Yan Y, Barone L, et al. (2019) Deciphering the domain specificity of C. difficile toxin neutralizing antibodies. Vaccine 37, 3892–3901. [DOI] [PubMed] [Google Scholar]
- (34).Galimidi RP, Klein JS, Politzer MS, Bai S, Seaman MS, Nussenzweig MC, West AP Jr., and Bjorkman PJ (2015) Intra-spike crosslinking overcomes antibody evasion by HIV-1. Cell 160, 433–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Li Z, Theile CS, Chen GY, Bilate AM, Duarte JN, Avalos AM, Fang T, Barberena R, Sato S, and Ploegh HL (2015) Fluorophore-conjugated Holliday junctions for generating super-bright antibodies and antibody fragments. Angew. Chem. Int. Ed. Engl. 54, 11706–11710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Pierce NW, Lee JE, Liu X, Sweredoski MJ, Graham RLJ, Larimore EA, Rome M, Zheng N, Clurman BE, Hess S, et al. (2013) Cand1 promotes assembly of new SCF complexes through dynamic exchange of F box proteins. Cell 153, 206–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Rashidian M, Keliher E, Dougan M, Juras PK, Cavallari M, Wojtkiewicz GR, Jacobsen J, Edens JG, Tas JM, Victora G, et al. (2015) The use of (18)F-2-fluorodeoxyglucose (FDG) to label antibody fragments for immuno-PET of pancreatic cancer. ACS Cent. Sci. 1, 142–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Altai M, Westerlund K, Velletta J, Mitran B, Honarvar H, and Karlstrom AE (2017) Evaluation of affibody molecule-based PNA-mediated radionuclide pretargeting: Development of an optimized conjugation protocol and (177)Lu labeling. Nucl. Med. Biol. 54, 1–9. [DOI] [PubMed] [Google Scholar]
- (39).Drabek AA, Loparo JJ, and Blacklow SC (2019) A flow-extension tethered particle motion assay for single-molecule proteolysis. Biochemistry 58, 2509–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Stiller C, Aghelpasand H, Frick T, Westerlund K, Ahmadian A, and Karlstrom AE (2019) Fast and efficient Fc-specific photoaffinity labeling to produce antibody-DNA conjugates. Bioconjugate Chem. 30, 2790–2798. [DOI] [PubMed] [Google Scholar]
- (41).Altai M, Vorobyeva A, Tolmachev V, Karlstrom AE, and Westerlund K (2020) Preparation of conjugates for affibody-based PNA-mediated pretargeting. Methods Mol. Biol. 2105, 283–304. [DOI] [PubMed] [Google Scholar]
- (42).Zahnd C, Wyler E, Schwenk JM, Steiner D, Lawrence MC, McKern NM, Pecorari F, Ward CW, Joos TO, and Pluckthun A (2007) A designed ankyrin repeat protein evolved to picomolar affinity to Her2. J. Mol. Biol. 369, 1015–1028. [DOI] [PubMed] [Google Scholar]
- (43).Turecek PL, Bossard MJ, Schoetens F, and Ivens IA (2016) PEGylation of biopharmaceuticals: A review of chemistry and nonclinical safety information of approved drugs. J. Pharm. Sci. 105, 460–475. [DOI] [PubMed] [Google Scholar]
- (44).Alconcel SNS, Baas AS, and Maynard HD (2011) FDA-approved poly(ethylene glycol)-protein conjugate drugs. Polym. Chem. 2, 1442–1448. [Google Scholar]
- (45).Shi H, Shi QY, Oswald AM, Gao Y, Li LJ, and Li YH (2018) Site-specific PEGylation of human growth hormone by mutated sortase A. Chem. Res. Chin. Univ. 34, 428–433. [Google Scholar]
- (46).Popp MW, Dougan SK, Chuang TY, Spooner E, and Ploegh HL (2011) Sortase-catalyzed transformations that improve the properties of cytokines. Proc. Natl. Acad. Sci. U.S.A. 108, 3169–3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Parthasarathy R, Subramanian S, and Boder ET (2007) Sortase A as a novel molecular “stapler” for sequence-specific protein conjugation. Bioconjugate Chem. 18, 469–476. [DOI] [PubMed] [Google Scholar]
- (48).Thurman EM, Ferrer I, Blotevogel J, and Borch T (2014) Analysis of hydraulic fracturing flowback and produced waters using accurate mass: Identification of ethoxylated surfactants. Anal. Chem. 86, 9653–9661. [DOI] [PubMed] [Google Scholar]
- (49).Ning X, Guo J, Wolfert MA, and Boons GJ (2008) Visualizing metabolically labeled glycoconjugates of living cells by copper-free and fast huisgen cycloadditions. Angew. Chem. Int. Ed. Engl. 47, 2253–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Debets MF, van Berkel SS, Schoffelen S, Rutjes FP, van Hest JC, and van Delft FL (2010) Aza-dibenzocyclooctynes for fast and efficient enzyme PEGylation via copper-free (3+2) cycloaddition. Chem. Commun. 46, 97–99. [DOI] [PubMed] [Google Scholar]
- (51).Baskin JM, Prescher JA, Laughlin ST, Agard NJ, Chang PV, Miller IA, Lo A, Codelli JA, and Bertozzi CR (2007) Copper-free click chemistry for dynamic in vivo imaging. Proc. Natl. Acad. Sci. U.S.A. 104, 16793–16797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Sletten EM, and Bertozzi CR (2009) Bioorthogonal chemistry: fishing for selectivity in a sea of functionality. Angew. Chem. Int. Ed. Engl. 48, 6974–6998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Patterson DM, Nazarova LA, and Prescher JA (2014) Finding the right (bioorthogonal) chemistry. ACS Chem. Biol. 9, 592–605. [DOI] [PubMed] [Google Scholar]
- (54).Gong Y, and Pan L (2015) Recent advances in bioorthogonal reactions for site-specific protein labeling and engineering. Tetrahedron Lett. 56, 2123–2132. [Google Scholar]
- (55).Schlatter D, Brack S, Banner DW, Batey S, Benz J, Bertschinger J, Huber W, Joseph C, Rufer A, van der Klooster A, et al. (2012) Generation, characterization and structural data of chymase binding proteins based on the human Fyn kinase SH3 domain. MAbs 4, 497–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Fraga H, Faria TQ, Pinto F, Almeida A, Brito RMM, and Damas AM (2010) FH8-a small EF-hand protein from Fasciola hepatica. FEBS J. 277, 5072–5085. [DOI] [PubMed] [Google Scholar]
- (57).Eigenbrot C, Ultsch M, Dubnovitsky A, Abrahmsen L, and Hard T (2010) Structural basis for high-affinity HER2 receptor binding by an engineered protein. Proc. Natl. Acad. Sci. U.S.A. 107, 15039–15044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Popp MW, Antos JM, Grotenbreg GM, Spooner E, and Ploegh HL (2007) Sortagging: a versatile method for protein labeling. Nat. Chem. Biol. 3, 707–708. [DOI] [PubMed] [Google Scholar]
- (59).Popp MW, Artavanis-Tsakonas K, and Ploegh HL (2009) Substrate filtering by the active site crossover loop in UCHL3 revealed by sortagging and gain-of-function mutations. J. Biol. Chem. 284, 3593–3602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Westerlund K, Honarvar H, Tolmachev V, and Eriksson Karlstrom A (2015) Design, preparation, and characterization of PNA-based hybridization probes for affibody-molecule-mediated pretargeting. Bioconjugate Chem. 26, 1724–1736. [DOI] [PubMed] [Google Scholar]
- (61).Anthis NJ, and Clore GM (2013) Sequence-specific determination of protein and peptide concentrations by absorbance at 205 nm. Protein Sci. 22, 851–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Johanning K, Juliano MA, Juliano L, Lazure C, Lamango NS, Steiner DF, and Lindberg I (1998) Specificity of prohormone convertase 2 on proenkephalin and proenkephalin-related substrates. J. Biol. Chem. 273, 22672–22680. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.