

Abstract
Objective
To test the hypothesis that rs573116164 will have disease-modifying effects in patients with superoxide dismutase 1 (SOD1) familial amyotrophic lateral sclerosis (fALS), we characterized rs573116164 within a cohort of 190 patients with fALS and 560 healthy age-matched controls to assess the variant for association with various measures of disease.
Methods
Using a previously described bioinformatics evaluation algorithm, a polymorphic short structural variant associated with SOD1 was identified according to its theoretical effect on gene expression. An 12–18 poly-T repeat (rs573116164) within the 3′ untranslated region of serine and arginine rich proteins-related carboxy terminal domain associated factor 4 (SCAF4), a gene that is adjacent to SOD1, was assessed for disease association and influence on survival and age at onset in an fALS cohort using PCR, Sanger sequencing, and capillary separation techniques for allele detection.
Results
In a North American cohort of predominantly SOD1 fALS patients (n =190) and age-matched healthy controls (n = 560), we showed that carriage of an 18T SCAF4 allele was associated with disease within this cohort (odds ratio [OR] 6.6; 95% confidence interval [CI] 3.9–11.2; p = 4.0e-11), but also within non-SOD1 cases (n = 27; OR 5.3; 95% CI 1.9–14.5; p = 0.0014). This finding suggests genetically SOD1-independent effects of SCAF4 on fALS susceptibility. Furthermore, carriage of an 18T allele was associated with a 26-month reduction in survival time (95% CI 6.6–40.8; p = 0.014), but did not affect age at onset of disease.
Conclusions
The findings in this fALS cohort suggest that rs573116164 could have SOD1-independent and broader relevance in ALS, warranting further investigation in other fALS and sporadic ALS cohorts, as well as studies of functional effects of the 18T variant on gene expression.
Short structural variants (SSVs) are emerging as critical genetic determinants in complex neurodegenerative diseases.1–6 SSVs contribute to pathophysiologic mechanisms responsible for complex genetic diseases by modifying gene expression and messenger RNA (mRNA) regulation.7–11 Given their potential significance, investigating these uncharacterized genetic markers may uncover some of the missing heritability in amyotrophic lateral sclerosis (ALS) and reveal an explanation for the vast diversity within ALS populations.12,13
Studies of common superoxide dismutase 1 (SOD1)-ALS–associated variants such as aspartic acid 91 alanine (D91A) and alanine 5 valine (A5V) (previously known as A4V) suggest a disease-modifying factor close to the SOD1 locus, which contributes broadly to ALS risk.14,15 In Scandinavian populations, the D91A variant is inherited in either a recessive or dominant pattern16; consequently, it has been suggested that a tightly linked disease-modifying factor lying close to SOD1 exists, acting to reduce the penetrance of the mutant D91A allele in recessive families.14 Subsequent high-throughput sequencing has failed to uncover a disease-modifying risk factor within or around SOD117; nevertheless, these results are not conclusive as high-throughput sequencing methods lack precision and accuracy in resolving SSVs.12,13,18 To this end, the existence of disease-associated SSVs within and around SOD1 as potential sources of missing heritability in ALS remains unresolved.
The largest transcript for the serine and arginine rich proteins-related carboxy terminal domain associated factor 4 (SCAF4) gene is situated 55 base pairs away from SOD1 and is known to encode a member of the serine/arginine-rich splicing factor family, which functions as an antiterminator during transcription.19 In view of the close proximity of SCAF4 to SOD1, and its essential role in transcription, SSVs within SCAF4 are potential disease-modifying candidates for ALS. We therefore investigated the association of a polymorphic SSV located within the 3′ untranslated region (UTR) of SCAF4 (rs573116164) in a predominately SOD1 variant–positive familial ALS (fALS) cohort.
Methods
Standard protocol approvals, registrations, and patient consents
This study was approved by the ethical standards of the relevant institutional review board, the Human Research Ethics Committee of the University of Western Australia (RA/4/20/5308). Participants were enrolled after informed consent was obtained. Clinical and sample collection data were obtained according to the US Health Insurance Portability and Accountability Act standards of confidentiality and disclosure and approved by the Northwestern University institutional review board (approval number: STU0012722/CR5_STU00012722). All patients were diagnosed by board-certified neurologists and met the revised El Escorial World Federation of Neurology criteria for diagnosis with ALS.20
Identification of SCAF4 SSV
A previously described SSV bioinformatics evaluation algorithm was used to identify a variant around the SOD1 gene region.21 Briefly, the bioinformatics software evaluated and prioritized SSVs likely to modulate expression regulated within SOD1. The rs573116164 variant was selected because of its proximity to SOD1, its high potential impact score (PIS), and poly-T nature, based on our previous experience with a poly-T SSV in translocase of outer mitochondrial membrane 40 homolog (TOMM40).6 The PIS was calculated according to 24 different properties, including variability (number, size, and simple sequence repeat slippage), synergy of consecutive variants, trait association, nearby regulatory elements, signal for transcription factor binding sites, conservation among mammals and primates, local drop in conservation value, and intron size.
Participants and DNA extraction
A total of 190 patients with fALS and 560 age-matched, neurologically normal community and spousal controls with no family history of ALS and dementia were collected from the Northwestern University School of Medicine (Chicago, IL) ALS Registry and biobank. Demographic details of the study participants are summarized in table 1, and further details of previously identified disease-associated variants in this patient cohort are provided in table e-1 (http://links.lww.com/NXG/A274). ALS cases were screened for variant status in the laboratory of Prof. Siddique, for various ALS genes in previous studies.22 As c9orf72 GGGCC repeats are reported in patients without a family history, controls were also tested for c9orf72 repeats. All controls had less than 20 repeats and were not tested for variants in other ALS genes. This data set contained 41 ALS families encompassing a total of 158 siblings and first-degree relatives, in addition to 32 unrelated ALS cases. The data set also includes 1 control family encompassing 33 related individuals and first-degree relatives, in addition to 527 unrelated controls. DNA was extracted and purified from transformed lymphocytes according to standard protocols (Qiagen, Valencia, CA).23
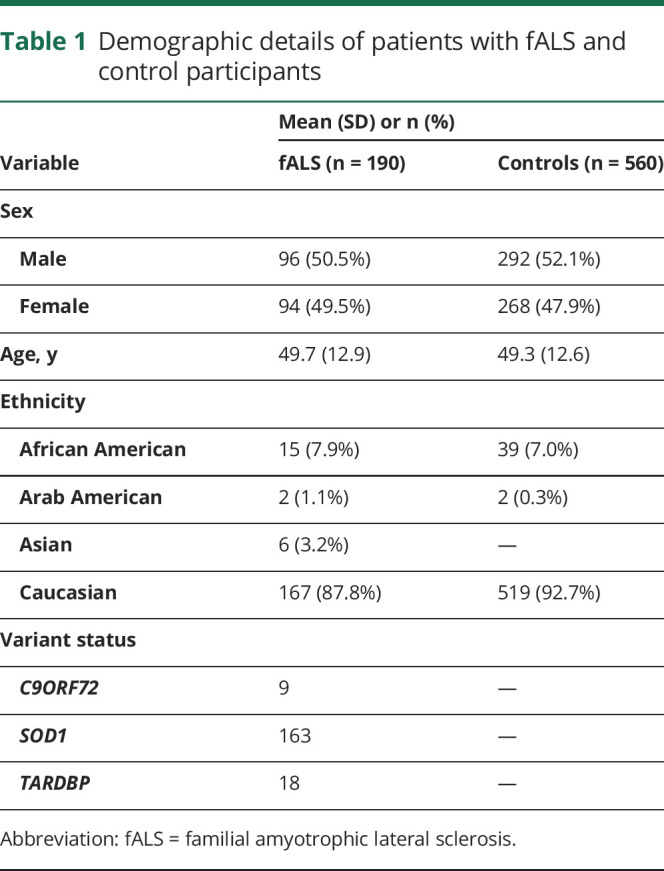
Table 1.
Demographic details of patients with fALS and control participants
PCR amplification, capillary separation, and sequencing
End point PCR reactions were prepared to a final volume of 20 µL, containing; 0.1 µL of MyFi DNA Polymerase (Meridian Bioscience Inc., Cincinnati, OH) and 10–50 ng human genomic DNA. Thermocycling was performed with the following conditions: 95°C for 6 minutes, followed by 30 cycles of 95°C for 40 seconds, 58°C for 30 seconds, and 72°C for 1 minute. Primers used were FWD: 5′FAM-GTTTTGAGGAGCTTTCACCGTT-3′ and RVS: 5′-GCAATTTGATTGCTTCCCTTC-3′ (Integrated DNA Technologies Inc., Coralville, IA). Capillary separation, fragment analysis, and subsequent Sanger sequencing were conducted at the Australian Genome Research Facility (figure e-1, http://links.lww.com/NXG/A274).
Statistical analysis
To compare binary outcomes across the 2 groups (patient/control), while accommodating correlations between outcomes on related family members, a weighted quasi-binomial (logistic) regression was used with weights inversely proportional to the numbers of members in the family. Joint associations of allele combinations with groups (patient/control) were based on similarly weighted case-control quasi-binomial regression. p Values were corrected using Bonferroni correction and were considered statistically significant when <0.05. Within the affected cases, associations of explanatory variables with the continuous variables onset age and survival duration were assessed using linear mixed (repeated measures) models to accommodate familial correlations. Analyses were performed in R version 3.4.3. To analyze linkage disequilibrium (LD) between the 18T allele and the A5V SOD1 variant, genotype data were directly imported into PLINK v1.07.24 Pairwise LD was calculated using the genotype correlation coefficient (r2).
Data availability
Study data for the primary analyses presented in this report are available on reasonable request from the corresponding author.
Results
Evaluation of rs573116164
A 12–18T repeat SSV (rs573116164) was identified within the 3′UTR of SCAF4 and was evaluated on the National Center for Biotechnology Information (NCBI) database to determine whether it had been reported previously and whether a disease association was established (figure 1). Although the region had been extensively reported as polymorphic in NCBI GRCH38.p12 (NC_000021.9), there was uncertainty as to any clinical significance. In silico analysis confirmed that rs573116164 lies close (2 kb downstream) to SOD1, in the 3′UTR region of SCAF4, and that binding sites for POLR2A, the largest subunit of RNA polymerase II, flank both sides of rs573116164, with the 3′ and 5′ POLR2A sites 145 and 350 bp away, respectively.25 Analysis of CLIP and mRNA data on UCSC supported these findings and reports multiple RNA-binding protein binding sites across the rs573116164 region (figure e-2, http://links.lww.com/NXG/A274).25 Because of the combination of the SSV genomic location, associated regional features, and its poly-T nature, investigation into the regional implication between SCAF4 and SOD1 was warranted.6
Figure 1. Schematic representation of rs573116164 (red) within the SCAF4 gene and associated regional features.

Rs573116164 is common to both the long and short 3′UTR SCAF4 isoforms and is located in between 2 POLR2A sites (purple) approximately 2 kbp away from the SOD1 gene on the reverse DNA strand. There are 12 isoforms of SCAF4, some of which do not contain the variant in the 3′UTR (figure e-2, http://links.lww.com/NXG/A274). ALS-associated variants within the SOD1 locus that are present in the cohort analyzed are also pictured (blue). This figure was generated from the National Center for Biotechnology Information genomic sequence NC000021.9 chromosome 21 GRCh38.p13 (GCF_000001405.39). ALS = amyotrophic lateral sclerosis; SCAF4 = SR-related CTD associated factor 4; SOD1 = superoxide dismutase 1.
Carriage of 18T and the 17T/18T genotype correlates with disease in both SOD1 and non-SOD1 cases
Carriage of the 18T allele was associated with fALS disease expression (odds ratio [OR] 6.6; 95% confidence interval [CI] 3.9–11.2; p = 4.0e-11) (table 2, figure e-3, http://links.lww.com/NXG/A274). Genotypically, only joint carriage of the 17T allele was associated with disease expression (OR 13.0; 95% CI 6.9–24.6; p = 6.8e-14) (table 3, figure e-3, http://links.lww.com/NXG/A274). Carriage of the 18T allele was investigated for patients without the SOD1-A5V variant (n = 126) and was also found to be associated with disease expression when controlling for ethnicity (OR 1.972; 95% CI 1.0–3.9; p = 0.044). In addition, carriage of 18T was investigated for patients without any SOD1 variants (n = 27) and was also found to be associated with disease expression (OR 5.3; 95% CI 1.9–14.5; p = 0.0014). Of interest, the only 2 patients homozygous for the 18T allele also carried an A5V SOD1 variant. Imbalances in ethnicities among patients/control groups were controlled for and did not change the results. Given the low numbers for some allele pairs, only those listed in table 3 were considered for analysis. The lower proportions of 17T/17T in the unadjusted analysis results from the excess 17T/18T cases among the patients with fALS, which is amended once the 17T/18T cases are omitted or adjusted for. After adjusting for the 17T/18T cases, the only other association with fALS disease expression was carriage of the 13T/18T genotype (OR 7.1; 95% CI 1.8–29.1; p = 0.0062), although the numbers of patients and controls with 13T/18T are small (n = 4 and n = 4, respectively) and significance is lost with adjustment for ethnicity and multiple comparisons (table 3). This analysis points to carriage of the 18T allele as a key determinant of fALS disease expression.
Table 2.
Differential carriage of rs573116164 alleles in the control and fALS groups

Table 3.
Differential carriage of rs573116164 genotypes in the control and fALS groups

A5V and the 18T allele are in LD
As the majority of A5V variant–positive patients carried at least 1 18T allele, the cohort was assessed to determine whether the A5V variant and 18T allele were in LD. The 2 alleles were in LD (D′ = 0.909); however, because of differences in allele frequencies, the 18T allele was not a strong predictor of the presence of A5V (r2 = 0.375), with the 18T allele occurring independently of the A5V variant.
Carriage of the 18T allele is a key determinant of survival but not age at onset
To determine whether rs573116164 is associated with ALS patient outcomes, survival (measured as duration from first symptom onset) and age at onset data were analyzed. Although no statistically significant relationship was noted with age at onset and rs573116164 carriage, a significant correlation was observed with survival times (figure 2). When accounting for age at onset, patients who carried the 18T allele reported an estimated median decrease in the survival duration of 26.3 months (95% CI 6.6–40.8; p = 0.014) when compared against patients without an 18T allele. This data points to carriage of the 18T allele as a key determinant of shorter survival among those with ALS. This effect is due to the high SOD1 variant–positive group, mostly comprised of A5V.
Figure 2. Survival of patients with fALS with and without an 18T allele on the log2 scale.
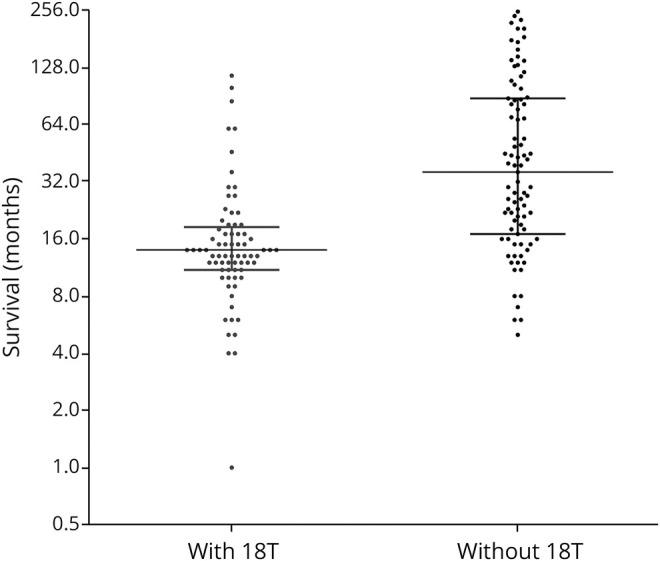
After adjusting for age at onset and familial correlations, patients with an 18T reported an estimated shorter median survival of 26.3 months (95% confidence interval 6.6–40.8; p = 0.014). fALS = familial amyotrophic lateral sclerosis.
Discussion
Our study investigated the role of a poly-T SSV in the 3′UTR region of the SCAF4 gene in a large cohort of predominantly SOD1 fALS patients, based on the premise that a risk-modifying factor located close to SOD1 may contribute to variable penetrance and phenotypic variability of the disease.14,15 The rs573116164 SSV was selected using a bioinformatics algorithm, which predicted a high probability that it would have an influence on disease expression and alter gene expression; in addition to this, we were particularly interested in poly-T SSVs due to our previous experience with a poly-T SSV in TOMM40.6,21 Our findings confirmed an association between carriage of the 18T allele of rs573116164 in SOD1 fALS. However, the association with the 18T allele was not confined to SOD1 fALS cases and was also present in the smaller group of non–SOD1 variant–positive cases, discounting a founder haplotype hypothesis. Finally, we observed that carriage of 18T negatively affects disease survival, although it does not affect age at onset of disease. Overall, this suggests more widespread genomic effects and that rs573116164 may be relevant to other forms of fALS and to the larger sporadic ALS population.
Variants within SOD1 have been identified in ∼20% of fALS cases, providing evidence for a key role in ALS pathogenesis. It is widely accepted that the pathogenesis of SOD1 variants is related to a toxic gain of function of the SOD1 protein.26 However, studies into the mechanism behind this toxic gain of function are limited.26–29 We hypothesized that SOD1 variants where no toxic gain of function has been ascribed may be tagging undiscovered disease-causing variants. Studies of common SOD1 variants such as D91A and A5V have previously suggested the existence of a disease-modifying factor close to the SOD1 locus that may explain the marked variability in the disease phenotype with both variants.14,15 In addition, the vast phenotypic variability observed in ALS could be explained by variable penetrance of common SOD1 variants.14,15 To explore these hypotheses, we investigated the SOD1 loci for SSVs that may have disease-modifying effects and contribute to variability in disease phenotype.
In silico SSV analysis of the SOD1 loci included investigation of the SCAF4 gene, which encodes the SCAF4 protein. Recent evidence has elucidated the role of SCAF4 in the prevention of early mRNA termination during transcription, acting with SCAF8 to suppress the use of alternative early polyadenylation sites.19 In the current study, a poly-T SSV (rs573116164) within the 3′UTR region of SCAF4 was found to be associated with ALS risk. Although rs573116164 occurs after the SCAF4 termination codon, variants within the 3′UTR region can alter transcript regulation, decreasing mRNA stability and potentially leading to low levels of SCAF4 protein.30 Loss of SCAF4 function results in read-through of canonical polyadenylation sites during transcription, leading to cleavage at a downstream alternative site.19 In the event where there is loss of both SCAF4 and SCAF8, early transcription termination is likely to occur.19 In addition, rs573116164 is flanked on both sides by binding sites for POLR2A, and as such, varying the length of rs573116164 may affect the availability of the region to POLR2A binding. Consequently, rs573116164 may subtly affect the transcription of nearby genes, including SOD1.31 Given the importance of SCAF4, further investigation is required to determine whether rs573116164 has functional effects on the expression of SCAF4 and SOD1.
In our fALS cohort, rs573116164 was found to be significantly associated with survival time. Specifically, carriage of the 18T allele was associated with a reduction in survival time from symptom onset when compared to patients carrying shorter poly-T alleles. Of interest, patients positive for the SOD1-A5V variant, which is typically associated with a severe ALS phenotype, almost always carried at least 1 18T allele.32 Furthermore, only patients with an A5V variant were homozygous for the 18T allele. This suggests that there is some common link between SOD1 variants such as A5V and rs573116164, yet our data demonstrate that non–SOD1 variant–positive patients also carry the 18T allele. As such, the relationship between A5V and rs573116164 warrants further investigation.
There is growing evidence surrounding the importance of noncoding variants in complex genetic traits, such as ALS.33,34 These noncoding variants within 3′UTR regions are highly conserved between mammals and primates, can affect mRNA expression and regulation, and have been shown to cause disease by a single base pair change.30,35,36 In our findings, rs573116164, proximal to SOD1, has a strong association with fALS disease expression and survival duration and as such presents a tantalizing new variant to consider for further studies. It remains to be determined whether these findings are confirmed in other fALS cohorts and importantly in sporadic ALS. A significant association with sporadic ALS would provide additional evidence that rs573116164 is mechanistically involved at a broader level in ALS pathogenesis. Further studies are also required to investigate the effects of the rs573116164 variant on expression of SCAF4 and SOD1 and to determine the mechanism by which rs573116164 has an effect on disease.
Our findings provide another example of the importance of identifying structural variants in ALS-related genes that have an effect on disease expression and/or have disease-modifying effects.34 The recognition of such variants has implications for uncovering some of the missing heritability of ALS, and importantly for the identification of novel therapeutic targets, as well as a more reliable basis for stratification of patients in clinical trials of new therapies.
Acknowledgments
The authors acknowledge funding support provided by the Giumelli Foundation. They also acknowledge Prof. Sue Fletcher and Prof. Steve Wilton for their ongoing support. Finally, the authors gratefully recognize the leadership role of Prof. Allen D. Roses in the design of the rs573116164 study. The concept of rs573116164 as a genetic marker to identify individuals at risk for ALS was conceived and put into effect by Prof. Roses. His dedication to alleviating the burden of ALS, and other neurodegenerative conditions, culminated in the design and execution of this and similar studies. Unfortunately, Prof. Roses died before completion of this study.
Glossary
- A5V
alanine 5 valine
- ALS
amyotrophic lateral sclerosis
- CI
confidence interval
- D91A
aspartic acid 91 alanine
- fALS
familial amyotrophic lateral sclerosis
- LD
linkage disequilibrium
- mRNA
messenger RNA
- NCBI
National Center for Biotechnology Information
- OR
odds ratio
- PIS
potential impact score
- SCAF4
serine and arginine rich proteins-related carboxy terminal domain associated factor 4
- SOD1
superoxide dismutase 1
- SSV
short structural variant
- TOMM40
translocase of outer mitochondrial membrane 40 homolog
Appendix. Authors
Study funding
The study was funded in part by the Perron Institute for Neurological and Translational Science, NIH grants to TS (NS050641, NS046535), the Les Turner ALS Foundation/Herbert C. Wenske Foundation Professorship, and the Les Turner ALS Research and Patient Care Center. The funders have no role in the design of the study and collection, analysis, decision to publish, interpretation of data, or preparation of the manuscript.
Disclosure
Miss. Pytte, Dr. Flynn, Dr. Anderton, Prof. Mastaglia, Miss. Theunissen, Prof. James, Dr. Pfaff, Prof. Koks, Dr. Saunders, Prof. Bedlack, Dr. Burns, Dr. Lutz, and Mrs. N. Siddique report no disclosures. Prof. T. Siddique participated in advisory meetings to Biohaven, Novartis, and Stealth Pharmaceuticals. Dr. Roses is deceased; disclosures are not included for this author. Prof. Akkari reports no disclosures. Go to Neurology.org/NG for full disclosures.
References
- 1.Lutz MW, Crenshaw DG, Saunders AM, Roses AD. Genetic variation at a single locus and age of onset for Alzheimer's disease. Alzheimers Dement 2010;6:125–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.De Jesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron 2011;72:245–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gijselinck I, Van Langenhove T, van der Zee J, et al. A C9orf72 promoter repeat expansion in a Flanders-Belgian cohort with disorders of the frontotemporal lobar degeneration-amyotrophic lateral sclerosis spectrum: a gene identification study. Lancet Neurol 2012;11:54–65. [DOI] [PubMed] [Google Scholar]
- 4.Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21-linked ALS-FTD. Neuron 2011;72:257–268 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sproviero W, Shatunov A, Stahl D, et al. ATXN2 trinucleotide repeat length correlates with risk of ALS. Neurobiol Aging 2017;51:178.e1–178.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Roses AD, Lutz MW, Amrine-Madsen H, et al. A TOMM40 variable-length polymorphism predicts the age of late-onset Alzheimer's disease. Pharmacogenomics J 2010;10:375–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lupiáñez DG, Kraft K, Heinrich V, et al. Disruptions of topological chromatin domains cause pathogenic rewiring of gene-enhancer interactions. Cell 2015;161:1012–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ricard G, Molina J, Chrast J, et al. Phenotypic consequences of copy number variation: insights from Smith-Magenis and Potocki-Lupski Syndrome mouse models. PLoS Biol 2010;8:e1000543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gheldof N, Witwicki RM, Migliavacca E, et al. Structural variation-associated expression changes are paralleled by chromatin architecture modifications. PLoS One 2013;8:e79973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carvalho CMB, Lupski JR. Mechanisms underlying structural variant formation in genomic disorders. Nat Rev Genet 2016;17:224–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Feuk L, Carson AR, Scherer SW. Structural variation in the human genome. Nat Rev Genet 2006;7:85–97. [DOI] [PubMed] [Google Scholar]
- 12.Mejzini R, Flynn LL, Pitout IL, et al. ALS genetics, mechanisms, and therapeutics: where are we now? Front Neurosci 2019;13:1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Theunissen F, Flynn LL, Anderton RS, et al. Structural variants may be a source of missing heritability in sALS. Front Neurosci 2020;14:47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Al-Chalabi A, Andersen PM, Chioza B, et al. Recessive amyotrophic lateral sclerosis families with the D90A SOD1 mutation share a common founder: evidence for a linked protective factor. Hum Mol Genet 1998;7:2045–2050. [DOI] [PubMed] [Google Scholar]
- 15.Saeed M, Yang Y, Deng H, et al. Age and founder effect of SOD1 A4V mutation causing ALS. Neurology 2009;72:1634–1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Andersen P, Nilsson P, Keränen ML, et al. Phenotypic heterogeneity in motor neuron disease patients with CuZn-superoxide dismutase mutations in Scandinavia. Brain 1997;120:1723–1737. [DOI] [PubMed] [Google Scholar]
- 17.Broom WJ, Johnson DV, Garber M, et al. DNA sequence analysis of the conserved region around the SOD1 gene locus in recessively inherited ALS. Neurosci Lett 2009;463:64–69. [DOI] [PubMed] [Google Scholar]
- 18.Chaisson MJP, Sanders AD, Zhao X, et al. Multi-platform discovery of haplotype-resolved structural variation in human genomes. Nat Commun 2019;10:1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gregersen LH, Mitter R, Ugalde AP, et al. SCAF4 and SCAF8, mRNA anti-terminator proteins. Cell 2019;177:1797–1813.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brooks BR, Miller RG, Swash M, et al. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord 2000;1:293–299. [DOI] [PubMed] [Google Scholar]
- 21.Saul R, Lutz MW, Burns DK, et al. The SSV evaluation system: a tool to prioritize short structural variants for studies of possible regulatory and causal variants. Hum Mutat 2016;37:877–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Deng HX, Tainer JA, Mitsumoto H, et al. Two novel SOD1 mutations in patients with familial amyotrophic lateral sclerosis. Hum Mol Genet 1995;4:1113–1116. [DOI] [PubMed] [Google Scholar]
- 23.Fecto F, Yan J, Vemula SP, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol 2011;68:1440–1446. [DOI] [PubMed] [Google Scholar]
- 24.Purcell S, Neale B, Todd-Brown K, et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet 2007;81:559–575. 10.1086/519795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kent W, Sugnet C, Furey T, et al. The human genome browser at UCSC. Genome Res 2002;12:996–1006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bunton-Stasyshyn R, Saccon R, Fratta P, Fisher E. SOD1 function and its implications for amyotrophic lateral sclerosis pathology. Neurosci 2015;21:519–529. [DOI] [PubMed] [Google Scholar]
- 27.Tsang CK, Liu Y, Thomas J, et al. Superoxide dismutase 1 acts as a nuclear transcription factor to regulate oxidative stress resistance. Nat Commun 2014;5:3446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Volkening K, Leystra-Lantz C, Yang W, et al. Tar DNA binding protein of 43 kDa (TDP-43), 14-3-3 proteins and copper/zinc superoxide dismutase (SOD1) interact to modulate NFL mRNA stability. Implications for altered RNA processing in amyotrophic lateral sclerosis (ALS). Brain Res 2009;1305:168–182. [DOI] [PubMed] [Google Scholar]
- 29.Kiskinis E, Sandoe J, Williams L, et al. Pathways disrupted in human ALS Motor neurons identified through genetic correction of mutant SOD1. Stem Cell 2014;14:781–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chatterjee S, Pal JK. Role of 5′- and 3′-untranslated regions of mRNAs in human diseases. Biol Cell 2009;101:251–262. [DOI] [PubMed] [Google Scholar]
- 31.Albert FW, Kruglyak L. The role of regulatory variation in complex traits and disease. Nat Rev Genet 2015;16:197–212. [DOI] [PubMed] [Google Scholar]
- 32.Valentine JS, Doucette PA, Zittin Potter S. Copper-zinc superoxide dismutase and amyotrophic lateral sclerosis. Annu Rev Biochem 2005;74:563–93. [DOI] [PubMed] [Google Scholar]
- 33.Li Y, van de Geijn B, Raj A, et al. RNA splicing is a primary link between genetic variation and disease. Science 2016;352:600–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Pytte J, Anderton RS, Flynn LL, et al. Association of a structural variant within the SQSTM1 gene with amyotrophic lateral sclerosis. Neurol Genet 2020;6;e404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Suraweera N, Iacopetta B, Duval A, et al. Conservation of mononucleotide repeats within 3′ and 5′ untranslated regions and their instability in MSI-H colorectal cancer. Oncogene 2001;20:7472–7477. [DOI] [PubMed] [Google Scholar]
- 36.Ruggiero T, Olivero M, Follenzi A, et al. Deletion in a (T)8 microsatellite abrogates expression regulation by 3′-UTR. Nucleic Acids Res 2003;31:6561–6569. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Study data for the primary analyses presented in this report are available on reasonable request from the corresponding author.