

Abstract
There is compelling evidence that oncogenic MET and PIK3CA signaling pathways contribute to breast cancer. However, the activity of pharmacological targeting of either pathway is modest. Mechanisms of resistance to these monotherapies have not been clarified. Currently commonly used mouse models are inadequate for studying the HGF-MET axis because mouse HGF does not bind human MET. We established human HGF-MET paired mouse models. In this study, we evaluated the cooperative effects of MET and PIK3CA in an environment with involvement of human HGF in vivo. Oncogenic MET/PIK3CA synergistically induced aggressive behavior and resistance to each targeted therapy in an HGF-paracrine environment. Combined targeting of MET and PI3K abrogates resistance. Associated cell signaling changes were explored by functional proteomics. Consistently, combined targeting MET and PI3K inhibited activation of associated oncogenic pathways. We also evaluated the response of tumor cells to HGF-stimulation using breast cancer patient-derived xenografts (PDXs). HGF-stimulation induced significant phosphorylation of MET for all PDX lines detected to varying degrees. However, the levels of phosphorylated MET are not correlated with its expression, suggesting that MET expression level cannot be used as a sole criterion to recruit patients to clinical trials for MET-targeted therapy. All together, our data suggests that combined targeting of MET and PI3K could be a potential clinical strategy for breast cancer patients, where phosphorylated MET and PIK3CA mutation status would be biomarkers for selecting patients who are most likely to derive benefit from these co-targeted therapy.
Keywords: HGF, PIK3CA, RTK, breast cancer, drug resistance
Introduction
PIK3CA mutations are present in 30%-40% of hormone receptor-positive breast cancers representing about 15%-20% of HER2-positive breast cancers (1–3). Hyperactivation of PI3K promotes escape from hormone dependence in estrogen receptor–positive human breast cancer (4,5). Pharmacological targeting of PI3K has mostly been tested in combination with hormonal therapy and showed modest benefit with about a 2-month improvement in disease-free survival (6,7). However, progression in most patients underscores the need to improve upon single pathway blockade. PIK3CA and c-MET (MET) are frequently co-aberrant in different types of cancer, including breast cancer which is associated with worse survival compared with either aberration alone, although it is not known if this represents a biclonal phenomenon (8, 9). Upregulation of MET was also observed in recurrent tumors in a PIK3CA-H1047R inducible mouse model, suggesting that MET is involved in resistance to PI3K-targeted therapy (10).
Upregulation of the tyrosine kinase oncogene MET has been reported in different types of cancer (11–13, http://resources.vai.org/Met/Index.aspx). A meta-analysis of 6010 cases and a review of 8281 cases of breast cancer patients exhibited a correlation of overexpression of MET in breast cancer with higher histologic grade and significantly poorer clinical outcomes (14,15). However, targeting MET has not shown the anticipated efficacy, with short-lived responses in about 10% of patients using the VEGFR2/MET inhibitor, cabozantinib (16,17). The underlying mechanism is not well known. We found that a high level of phospho-MET is associated with higher risk of recurrence in breast cancer patients (18). We also identified MET-T1010I, a germline functional single-nucleotide polymorphism (SNP) in 2% of breast cancer patients with metastatic disease. The frequency of MET-T1010I in patients with metastatic breast cancer was twice that in the general population (19). Compared to overexpression of wild-type (WT) MET, MET-T1010I significantly induces cells invasion both in vitro and in vivo.
Hepatocyte growth factor (HGF)/scatter factor, the only known ligand for MET (20, 21), is produced and secreted mainly by stromal cells and acts primarily on cells of epithelial origin, although some cancers derived from epithelial cells, such as breast cancer, express both HGF and MET (22–26). Increased HGF levels are observed in the serum of patients with breast cancer, which and is associated with progression of the disease (27–29). However, currently used mouse models are inappropriate for studying the HGF-MET axis because mouse HGF does not bind human MET. We recently developed an HGF-MET paired breast cancer mouse model, in which human HGF is expressed as a transgene in mice with severe combined immunodeficiency background (hHGF Tg/SCID) (30) while variants of human MET genes are expressed in orthotopic xenografts. Using the model, we demonstrated that MET-T1010I or MET overexpression of WT MET induces tumor growth and progression in an hHGF-overexpressed environment (19).
In this study, we generated and used orthotopic xenograft models expressing variants of human MET and PIK3CA in the hHGF Tg/SCID mice to evaluate the cooperative oncogenic effect of MET and PIK3CA and their response to the targeted therapies in breast cancer with HGF-overexpression.
Materials and Methods
Generation of recombinant lentiviruses expressing WT or mutant PIK3CA genes
The lentiviral constructs expressing PIK3CA variants (pLenti6/V5-DEST/PIK3CA-WT, pLenti6/V5-DEST PIK3CA-E545K, pLenti6/V5-DEST PIK3CA-H1047R, and the control, pLenti6/V5-DEST/Lac Z) were gifts from Dr. G. Wu (Karmanos Cancer Institute, Detroit, MI; ref. 31) and were used to make lentiviruses and establish stable cell lines expressing various types of PIK3CA with the ViraPower Lentiviral Expression System (Invitrogen, Carlsbad, CA) following the manufacturer’s instructions. Lentivirus-containing supernatants were collected after 48 hours, filtered and used to infect MCF-10A cells. Selection began 48 hours after infection with 10μg/ml blasticidin (Invitrogen) for two weeks. The stable cell lines expressing variants of PIK3CA were also used to establish cell lines with co-expressing PIK3CA and MET genes.
Generation of lentiviruses expressing WT MET or MET-T1010I
We designed, and GeneArt synthesized, constructs expressing Flag epitope-tagged full-length human MET and MET-T1010I that locate the juxtamembrane (19, Supplementary Fig. S1A). The gene cassette (Supplementary Fig. S1B) was transferred into a lentiviral vector, pLVX-tdTomato-N1 (Clontech, Mountain View, CA). The lentiviruses expressing WT MET or MET-T1010I were used for transduction of HCC1954 cells or MCF-10A–derived cells expressing variants of PIK3CA genes (19). To ensure that the majority of the cells were only infected with one aimed gene-carrying virus, we optimized conditions to achieve ~20-30% transduction efficiency in the titering assay, and then performed the experiment with 3-4 times more cells than infecting viral particles. Selection began 48 hours after infection with 1 μg/ml puromycin for 2 weeks. Expression of exogenous MET fusion protein was detected with Td Tomamo fluorescence (Supplementary Fig. S1C) and Western blot analysis (Supplementary Fig. S1D)
Cell culture
The cell lines used in the studies were from MDACC Characterized Cell Line Core at the University of Texas MD Anderson Cancer Center. All cell lines were authenticated by fingerprinting using short tandem repeat testing and were verified to be free of mycoplasma contamination regularly by the Characterized Cell Line Core.
MCF-10A and HCC1954 were grown at 37°C in humidified 5% CO2. The MCF-10A cells were maintained in Dulbecco Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12; Thermo Fisher Scientific, Waltham, MA) supplemented with 5% horse serum (Invitrogen, Carlsbad, CA), 20 ng/ml epidermal growth factor (EGF) (PeproTech, Rocky Hill, NJ), 10 μg/ml insulin (Sigma-Aldrich, St. Louis, MO), 100 ng/ml cholera toxin (Sigma-Aldrich), 0.5 μg/ml hydrocortisone (Sigma-Aldrich), 100 units/ml penicillin, and 100 μg/ml streptomycin. The HCC1954 cells were maintained in Roswell Park Memorial Institute medium supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 μg/ml streptomycin. Cells were frozen at early passages and used for less than 4 weeks in continuous culture.
Drugs
Onartuzumab (METMAb), a monovalent, humanized, anti-MET monoclonal antibody (Genentech, South San Francisco, CA; ref. 32), was used following the manufacturer’s instructions. For in vivo studies, onartuzumab (10 mg/kg, intraperitoneal injection) was administered twice weekly. MET tyrosine kinase inhibitor (TKI) tepotinib [EMD1214063; ref. 33; EMD Serono, Rockland, MA), PI3Kα/δ TKI pictilisib [GDC-0941; ref. 34; Genentech) and class 1 PI3K/mTOR TKI apitolisib [GDC-0980; ref. 34; Genentech) were dissolved in dimethyl sulfoxide (DMSO) (Sigma-Aldrich, St. Louis, MO) and stored at −20°C. The stocked solution was further diluted to an appropriate final concentration upon use. For in vitro studies, DMSO in the final solution was 0.1% (v/v). For in vivo use, pictilisib at 100 mg/kg (in 100 μl of 5% DMSO) was given daily by oral gavage. All drugs were provided by the companies. Human recombinant HGF was purchased from R&D Systems (Minneapolis, MN).
Clonogenic assay
In total 1000 cells were seeded in 60-mm dishes in growth medium for 11 days. For inhibitory assays, after attaching on the dish, cells were treated for 2 days with drugs in variable combinations as indicated. Then, the drugs were washed away, and cells were allowed to grow in growth medium for 11 days. The cells were rinsed with PBS, followed by staining with 0.25% crystal violet/20% ethanol. Quantitative analysis of the total number and size of clones was performed with AlphaView SA software (Cell Biosciences). Dose-response curves and synergistic index were analyzed using CalcuSyn software (Biosoft, Ferguson, MO), following the manufacturer’s guide. Combination Index (CI) < 1, =1, and >1 indicate synergism, additive, and antagonism, respectively.
Cell growth assay
The MCF-10A–derived cells were seeded in triplicate at the density of 2 × 104 per well in 12-well plates in low-serum medium (2.5% horse serum) lacking EGF, insulin, and hydrocortisone with a supplement of HGF (40 ng/ml) for 3 days. Cells were trypsinized and counted each day with an automated cell counter and cell analyzer, Cellometer Vision (Nexcelom, Lawrence, MA).
Cell growth inhibition assays
HCC1954-derived cells were seeded in triplicate at a density of 2000 cells per well in 96-well plates in complete growth medium and were allowed to attach for 24 hours. The medium was changed to low-serum medium (5% FBS) for overnight culture, followed by the addition of serial dilutions of pictilisib or apitolisib. Growth inhibition was determined using the CellTiter-Blue viability assay according to the manufacturer’s protocol (Promega, Madison, WI). Each experiment was repeated three times. Cell growth was calculated as [(T−T0)/(C−T0)] × 100, with T0 as the readout on day 0, T as the readout of the treated on day 3, and C as the readout of the control on day 3. Growth inhibition of 50% (GI50) was analyzed using Prism 7.
Morphogenesis assay
In total 4 × 103 cells were resuspended in modified growth medium (2.5% horse serum and 5 ng/ml EGF) containing 2% growth factor reduced Matrigel (BD Biosciences, Franklin Lakes, NJ), supplemented with HGF (40 ng/ml) and seeded onto Matrigel matrix in eight-well chamber slides (BD Bioscience), with or without variable treatment as indicated. Medium with drugs was replaced every 3 days. Photographs of representative fields were taken as indicated. Acini were photographed and counted in 10 randomly chosen fields and expressed as means of triplicates, representative of three independent experiments.
In vitro invasion assay
In vitro cell invasion was analyzed with 24-well Biocoat Matrigel invasion chambers with 8-μm polycarbonated filters (Becton Dickinson, Franklin Lakes, NJ). Briefly, MCF-10A–derived cells were starved for 20 hours in serum-free DMEM/F12 with all growth factors withdrawn. A total of 1 × 105 cells in 0.6 ml of serum-free medium were inoculated into the upper chamber, and 0.75 ml of serum-free medium containing HGF (40 ng/ml) and fibronectin (5 μg/ml) was added to the lower chamber. For invasion-inhibition assays, drugs or vehicle was added to both the upper and lower chambers. The cells were allowed to pass through the Matrigel at 37°C, 5% CO2, for 22 hours. Non-invasive cells on the upper surface of the filter were removed by wiping with a cotton swab. The cells that penetrated through the pores of the Matrigel to the underside of the filter were stained with 0.25% crystal violet in 20% methanol for 30 minutes. Invasive cells were photographed and counted in 10 random fields. We tested the HCC1954-derived cells using the same method but used Roswell Park Memorial Institute medium instead of the DMEM F12.
Establishment of HGF-MET paired mouse model and targeted therapies in vivo
The hHGF Tg/SCID mice (30) were bred in a room specifically for SCID mice. Female mice at 6 weeks of age were used for experiments. All animal studies were carried out under Animal Care and Use Form-approved protocols. HCC1954-derived cells expressing endogenous PIK3CA-H1047R and exogenous WT MET or MET-T1010I genes with exponential growth were collected. After being washed with phosphate-buffered saline (PBS), the cells were resuspended in PBS and injected into the mammary fat pads of the mice (1 × 107 cells in 150 μl) for tumor formation assays, or 8 × 106 cells for drug tests, per mouse). For drug tests, 3 days after implantation, the mice were randomized to treatment with vehicle, pictilisib, onartuzumab, or their combination. Tumor sizes were measured with calipers twice weekly. Tumor volume was calculated with the formula V = lw2/2. Differences in tumor volume between groups were analyzed using two-way analysis of variance (ANOVA). At the end of the experiment, mice were sacrificed with CO2. The tumors were harvested and then were cut and flash-frozen in liquid nitrogen for signal testing or fixed in 10% neutral-buffered formalin for paraffin embedding. Xenograft tumors and all the organs of each mouse were subjected to double-blind histopathologic analysis by a veterinary pathologist.
Establishment of stable cell lines using patient-derived xenograft
We selected seven lines from a panel of 23 PDXs from different subtypes of breast cancer patients (35) to represent a spectrum of MET expression levels. To detect response to HGF, we established stable cells for each PDX line by maintaining their growth in Dulbecco Modified Eagle Medium/Nutrient Mixture F-12 (DMEM/F12; Thermo Scientific, Waltham, MA) supplemented with 10% fetal bovine serum (FBS), 20 ng/ml epidermal growth factor (EGF; PeproTech, Rocky Hill, NJ), 10 μg/ml insulin (Sigma-Aldrich), 100 ng/ml cholera toxin (Sigma-Aldrich), 0.5 μg/ml hydrocortisone (Sigma-Aldrich), 100 units/ml penicillin, and 100 μg/ml streptomycin.
Immunoprecipitation and Western blot analyses
Cells were washed twice with cold phosphate-buffered saline and lysed in ice-cold lysis buffer (36). After the cellular protein concentration was tested, the cell lysates with equal amount of protein were immunoprecipitated with anti-V5 (Invitrogen). Immunocomplexes were collected on Protein A/G plus-conjugated agarose beads (Santa Cruz Biotechnology). The immunocomplexes or cell lysates were separated by SDS-PAGE, and followed by Western blot to detect the specific expression (36). Quantitative analysis of the bands was performed with AlphaView SA software.
Reverse phase protein array (RPPA)
MCF-10A–derived cells expressing variants of PIK3CA and/or MET genes were treated with or without PI3K inhibitors and/or MET antibody or MET inhibitor, followed by stimulation with 10% FBS or HGF (40 ng/ml) for 30 minutes. The cell lysis was prepared and analyzed by RPPA as described previously (36–38). The antibodies used for RPPA and Western blot are listed in Supplementary Table S1.
Statistical analyses
The data from clonogenic assays, invasion assays, and cell growth and inhibition assays were analyzed with one-way ANOVA. Tumor growth curves were analyzed with two-way ANOVA. The RPPA data were analyzed as previously described (36–38) and followed by further analysis with one-way ANOVA to compare different groups. All other statistical tests were performed using Prism (GraphPad Software, La Jolla, CA).
Results
Expression of oncogenic MET genes along with endogenous mutation of PIK3CA induces aggressive behavior in breast cancer cells
To study the effect of exogenous oncogenic MET genes on survival ability of tumor cells with endogenous PIK3CA-H1047R, the most common PIK3CA mutation in breast cancer, we performed clonogenic assays using HCC1954 cells transfected with WT MET or MET-T1010I (Supplementary Fig. S1) as shown in Fig. 1A. Compared with control cells transfected with empty vectors, both WT MET and MET-T1010I showed a significant increase in colony formation in growth medium supplemented with HGF (40 ng/ml) (P < 0.001 each, Fig. 1B). We next tested cell invasive activity in vitro. Cell invasion induced by HGF and fibronectin was significantly and dramatically increased by MET-T1010I expression and modestly but still significantly increased by WT MET expression (P < 0.001 and P < 0.01, respectively) (Fig. 1C, D).
Figure 1. Characterization of breast cancer cells with endogenous PIK3CA-H1047R and exogenous MET.
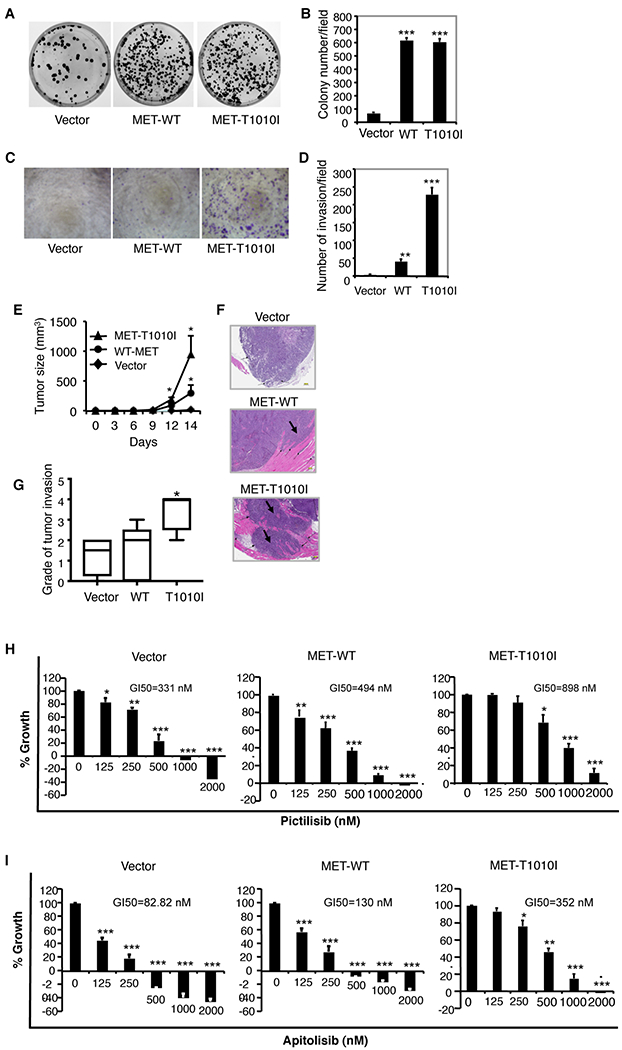
HCC1954 cells, with endogenous PIK3CA-H1047R, were transfected by WT MET or MET-T1010I genes as indicated. A. The clonogenic assay was performed as described in Material and Methods. The HCC1954-derived cells were seeded in triplicate at a density of 1000 cells in 60-mm dishes in Roswell Park Memorial Institute medium supplemented with 5% FBS and 40 ng/ml HGF. The dishes were scanned on day 10. B. Quantitative analysis of the total number of clones was performed with AlphaVIEW SA software. Data are mean ± standard deviation of triplicates, representative of two independent experiments (***, P < 0.001 vs. control cells, ANOVA). C. An invasion assay was performed as described in Materials and Methods. The HCC1954-derived cells that penetrated Matrigel to the underside of the filter were photographed and counted in 10 random fields. D. Data are mean ± standard deviation of triplicates, representative of two independent experiments (**, P < 0.01, ***, P < 0.001 vs. control cells, ANOVA). E. A total of 1 × 107 HCC1954-derived cells were injected into the mammary fat pads of hHGF Tg/SCID female mice. Each group consisted of 5 mice. Tumor volume was calculated with the formula V = lw2/2. Differences in tumor volume between groups were analyzed using ANOVA (*, P < 0.05, vs. control group). F. Hematoxylin and eosin staining for histologic images from a representative tumor from MET-transfected groups show tumor invasion (black arrows) to adjacent skeletal muscle (pink). G. Data show grade of tumor invasion (*, P < 0.05 vs. control group, ANOVA). H, I. The HCC1954-derived cells were seeded in 96-well plates (2000 cells per well) in complete growth medium and were incubated at 37°C for 24 hours. The medium was changed to low-serum medium (2% FBS). Cells were incubated overnight, followed by the addition of serial dilutions of pictilisib (H) or apitolisib (I) in variable combinations for 72 hours. Growth inhibition was determined (details in Materials and Methods). The data are mean ± standard deviation of triplicates, representative of two independent experiments (*, P < 0.05; **, P < 0.01; ***, P < 0.001 vs. control).
We next explored the effect of oncogenic MET and PIK3CA in vivo. The HCC1954-derived cells with or without expression of aberrant MET along with endogenous PIK3CA-H1047R were tested. Tumor size was largest in the MET-T1010I group, smallest in the vector control group, and in between in the WT MET group (P < 0.05) (Fig. 1E). Once tumors formed in the MET-T1010I group, they grew rapidly. All tumors from the MET-T1010I group, and three of the five tumors from MET-WT group showed marked invasion of adjacent tissues (Fig. 1F). Tumor invasion grade was highest in the MET-T1010I group (P < 0.05), lowest in the vector group, and in between in the WT MET group (Fig. 1G).
MET aberrations confer cell resistance to PI3K-targeted therapy
Our in vitro and in vivo studies showed that dysregulation of HGF-MET signaling induces aggressive biologic behavior in breast cancer cells with endogenous oncogenic PI3KCA-H1047R. These findings led us to investigate whether the MET genotype affects response to PI3K inhibitors. We tested proliferation of the HCC1954-derived cells treated with pictilisib, a pan class I PI3K inhibitor, using cell growth inhibition assays. The control cells transfected with empty vector exhibited the highest sensitivity to pictilisib (concentration for 50% growth inhibition [GI50] = 331 nM), while the cells transfected by WT MET (GI50 = 494 nM) or MET-T1010 (GI50 = 898 nM) showed greater resistance to pictilisib (Fig. 1H). Cells transfected with MET were also resistant to apitolisib, a dual PI3K/mTOR inhibitor (Fig. 1I).
Combined targeting of MET and PI3K inhibits growth and invasion of tumor cells and xenografts with exogenous aberration of MET and endogenous PIK3CA-H1047R
To test whether co-targeting of MET and PI3K would increase therapeutic efficacy on cell survival, we performed colony formation assay. The HCC1954-derived cells expressing exogenous MET-T1010I and endogenous PIK3CA-H1047R were treated with variable doses of pictilisib and the MET TKI tepotinib. The dose-effect curve (Fig. 2A) and the combination index (Table 1) from the synergy analyses exhibited synergistic interactions. The combination of tepotinib and pictilisib significantly increased the inhibitory effect than each of them alone (P < 0.0001 vs. tepotinib and P < 0.05, < 0.01 vs. pictilisib, respectively, Fig. 2 B, C).
Figure 2. Effect of targeting PI3K and/or MET in HCC1954-derived cells in vitro and in vivo.
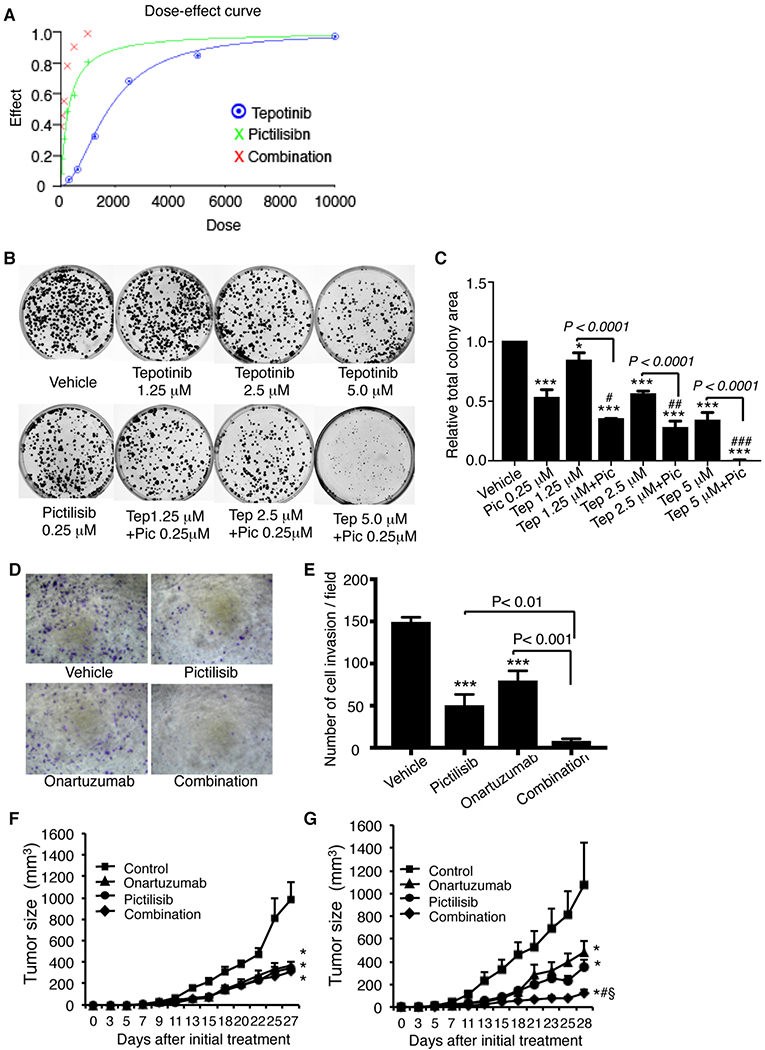
The HCC1954-derived cells were treated as indicated with PI3K inhibitor pictilisib and/or either MET TKI tepotinib or MET antibody onartuzumab and then tested for cell survival and invasion (as described in Figure 1 and Material and Methods). The combined effect of tepotinib and pictilisib with variable doses on colony formation was analyzed with CalcuSyn Dose Effect Analyzer (detail see Materials and Methods). A. Dose effect. B. Colony formation assay with tepotinib (variable doses) combined with pictilisib (fixed dose) C. Quantitative analysis of the area of total clones was performed with AlphaVIEW SA software. Data are mean ± standard deviation of triplicates, representative of two independent experiments (*, P < 0.05; ***, P < 0.0001 vs. vehicle; #, P < 0.05, ##, P < 0.01, ###, P < 0.001 vs. pictilisib, ANOVA). D. Invasion assay (pictilisib 0.25 μM, onartuzumab 1.5 μM). E. Data are mean ± standard deviation of triplicates, representative of two independent experiments (***, P < 0.001 vs. control cells, ANOVA). In vivo studies, HCC1954-derived cells with expression of MET-WT/PIK3CA-H1047R (F) or MET-T1010I/PIK3CA-H1047R (G) were implanted into mammary fat pad of hHGF Tg/SCID female. Three days later, the mice were randomized to treatment with vehicle, onartuzumab (10 mg/kg, intraperitoneal injection, twice weekly), pictilisib (100 mg/kg in 100 μl of 5% DMSO, daily by oral gavage), or their combination. Tumor sizes were measured with calipers twice weekly. Tumor volume was calculated with the formula V = lw2/2. Data were analyzed with two-way ANOVA (*, P < 0.0001 vs. vehicle; §, P < 0.0001 vs. onartuzumab alone; #, P < 0.01 vs. pictilisib alone)
Table 1.
Combination index (CI)
| Tepotinib (μM) | Pictilisib (μM) | CI |
|---|---|---|
| 0.625 | 0.25 | 0.804 |
| 1.25 | 0.25 | 0.63 |
| 1.25 | 0.5 | 0.431 |
| 1.25 | 1.0 | 0.101 |
| 2.5 | 0.25 | 0.635 |
| 5.0 | 0.25 | 0.179 |
| 10.0 | 0.25 | 0.048 |
Cell invasion assays showed that targeting PI3K with pictilisib or targeting MET with its antibody onartuzumab inhibited cell invasion (P < 0.001 each); however, the combination of pictilisib and onartuzumab significantly increased the inhibitory effect (P < 0.01 vs. pictilisib and P < 0.001 vs. onartuzumab, Fig. 2D and E).
To assess effects of targeting PI3K and MET in a human HGF-containing microenvironment, we used hHGF Tg/SCID mice. The mean of serum human HGF concentration of the mice was 1.148 ng/ml (Supplementary Fig. 2), consistent with previous study (30). The levels of HGF in the mice are similar with that in breast cancer patients (27–29). We established xenografts in the mice by orthotopic transplantation of the HCC1954-derived cells expressing PIK3CA-H104R/WT MET or PIK3CA-H104R/MET-T1010I. Three days after implantation, mice were randomly assigned to treatment with vehicle, pictilisib, onartuzumab, or their combination. Pictilisib or onartuzumab alone significantly inhibited growth of xenografts with MET-WT/PIK3CA-H1047R (P < 0.0001 each), and the combination of pictilisib and onartuzumab did not further increase the inhibitory effect (Fig. 2F). However, in mice with xenografts expressing MET-T1010I/PIK3CA-H1047R, pictilisib or onartuzumab alone again inhibited growth of the tumors (P < 0.0001 each); however, the combination of pictilisib and onartuzumab further induced tumor regression (P < 0.0001 vs. onartuzumab alone; P < 0.01 vs. pictilisib alone; Fig. 2G).
Expression of oncogenic MET and PIK3CA transforms mammary epithelial cells
To examine whether concurrent aberration of MET and PIK3CA transform mammary epithelial cells, we established MCF-10A cells stably expressing WT PIK3CA, PIK3CA-E545K, or PIK3CA-H1047R as well as MCF-10A cells expressing WT MET or MET-T1010I. We then further transfected each PIK3CA-variant line to express WT MET or MET-T1010I. Expression of PIK3CA and MET was confirmed (Supplementary Fig. S3A,B). Without stimulation, mutant PIK3CA-transformed cells exhibited constitutive activation of PI3K and STAT3 pathways, while co-expressing MET-T1010I further increased this activation, as indicated by increased phosphorylation of AKT and STAT3 (Supplementary Fig. S3B). Without stimulation, all the cells exhibited low baseline of phosphorylation of MAPK. However, after stimulation with HGF, MET-transformed cells showed significantly increased the level of phospho-MAPK, with MET-T1010I stronger than WT-MET. Co-transfection with PIK3CA genens did not further increase the level (Supplementary Fig. S3C).
Oncogenic MET and PIK3CA cooperate in promoting cell proliferation and abnormal morphogenesis of mammary acini
MCF-10A non-tumorigenic mammary epithelial cells can grow only in an optimal growth medium supplemented with 5% horse serum, EGF, insulin, and hydrocortisone. The EGF requirement can be overcome by exogenous expression of mutant PIK3CA-E545K and PIK3CA-H1047R (31, 39), a molecular criterion of oncogenes (39). To explore whether aberrations of MET cooperate with mutant PIK3CA on cell proliferation, we cultured the cells transformed with PIK3CA-E545K (Fig. 3 A) or / PIK3CA-H1047R (Fig. 3B) alone or co-transformed with WT MET or MET-T1010I in a suboptimal environment with decreased horse serum medium (2.5%), devoid of all growth factors (EGF, insulin, and hydrocortisone), and provided with a supplement of HGF (40 ng/ml). In this poor culture environment, cells with expression of PIK3CA-E545K alone (E545K/vector) or PIK3CA-H1047R alone (H1047R/vector) exhibited only modest proliferation. In contrast, concurrent expression of MET-T1010I or WT MET with PIK3CA-E545K (Fig. 3A) or with PIK3CA-H1047R (Fig. 3B) significantly increased cell proliferation (P < 0.001 vs. mutant PIK3CA expression alone each).
Figure 3. Characterization of breast epithelial cells transfected with oncogenic PIK3CA with MET.
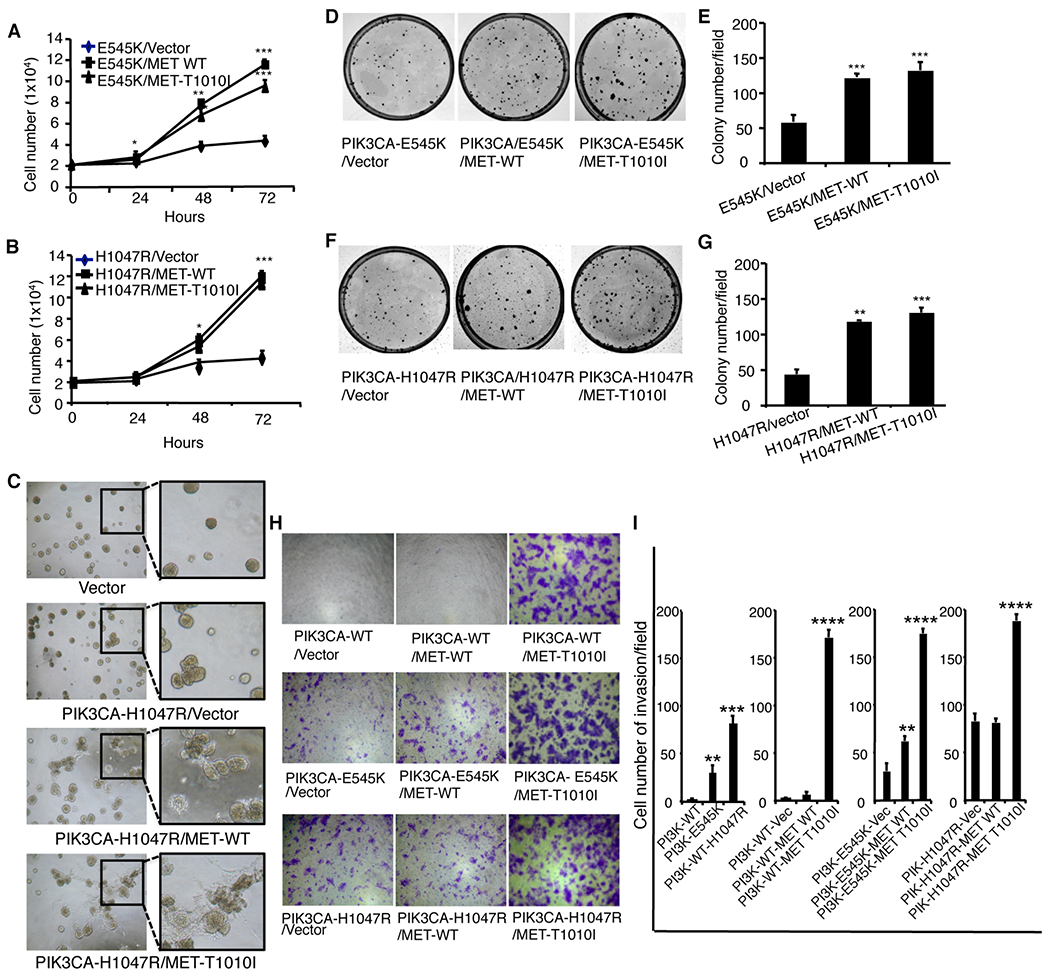
MCF-10A–derived cells co-transfected with various MET genotypes and PIK3CA-E545K (A) or PIK3CA-H1047R (B) were seeded in 12-well plates in triplicate with a density of 2 × 104 per well in 2.5% horse serum with withdrawn EGF, insulin, and hydrocortisone, supplemented with HGF (40 ng/ml) for 3 days. Cells were counted each day. Data are mean ± standard deviation of triplicates, representative of two independent experiments (*, P < 0.05; **, P < 0.01; ***, P < 0.001 vs. control cells, ANOVA). C. Effects of MET aberrations on mammary acinar morphogenesis were tested as described in material and Methods. MCF-10A cells transfected with PIK3CA-H1047R and various MET genes were resuspended in modified growth medium (2.5% horse serum and 5 ng/ml EGF) containing 2% Matrigel supplemented with HGF (40 ng/ml). Representative field images of acini were taken on day 8; original magnification, ×40. The clonogenic assay was analyzed as described in Materials and Methods. The MCF-10A–derived cells (D, F) were seeded in triplicate at a density of 1000 cells in 60-mm dishes in DMEM/F12 medium supplemented with 2.5% horse serum and 40 ng/ml HGF. The dishes were scanned on day 10. Quantitative analysis of the total number of clones was performed with AlphaVIEW SA software (E, G). Data are mean ± standard deviation of triplicates, representative of two independent experiments (**, P < 0.01; ***, P < 0.001, vs. cells transfected with mutant PIK3CA alone, ANOVA). Cell invasion in vitro was analyzed as indicated in Materials and Methods. The MCF-10A–derived cells were induced to invade with HGF (40 ng/ml) and fibronectin (5 μg/ml). H. Cells invading through Matrigel were photographed at × 100 magnification. I. Data are mean ± standard deviation of triplicates, representative of two independent experiments (***, P < 0.001; ****, p < 0.0001 vs. vector, ANOVA).
We next evaluated the effect of concurrent aberration of MET and mutant PIK3CA genes on mammary acinar morphogenesis using the MCF-10A–derived cells. Expression of mutant PIK3CA increased cell proliferation in three-dimensional culture and mildly changed the structure of acini. However, overexpression of WT MET or expression of MET-T1010I induced significant abnormal structures with obvious cell scattering. The effect of MET-T1010I was stronger than overexpression of WT MET (Fig. 3C).
Oncogenic MET and PIK3CA act coordinately in inducing cell survival and invasion
We next examined whether expression of MET-T1010I or overexpression of WT MET with mutant PIK3CA coordinately affects cell survival using clonogenic assays. Cells were seeded at low density in a suboptimal growth environment (Materials and Methods). Co-expression of different MET genotypes and PIK3CA-E545K in MCF-10A cells significantly increased colony formation (P < 0.001; Fig. 3D, E). These findings were recapitulated with PIK3CA-H1047R (Fig. 3F, G).
To determine whether expression of MET-T1010I or overexpression of WT MET and expression of mutant PIK3CA coordinately affect cell invasion, we performed invasion assays by seeding MCF-10A–derived cell lines expressing these variants (as indicated) in invasion chambers (Fig. 3H, I). Since WT PIK3CA does not induce cell invasion, we used cells expressing WT PIK3CA as controls. Compared with WT PIK3CA, PIK3CA-E545K or PIK3CA-H1047R enhanced cell invasion ability (P < 0.01 and P < 0.001, respectively). Co-expression of WT MET with PIK3CA-E545K, but not with PIK3CA-H1047R, mildly increased invasion ability (P < 0.01 and P > 0.05, respectively). However, co-expression of MET-T1010I with the PIK3CA variants markedly increased invasion (P < 0.0001 each).
Combined targeting of MET and PI3K reverses the aggressive behavior of cells transformed with MET and PIK3CA variants
To explore whether combined targeting of MET and PI3K could inhibit the cell growth or scatter induced by aberrations of MET and PIK3CA, we seeded MCF-10A–derived cells expressing MET-WT/PIK3CA-H1047R or MET-T1010I/PIK3CA-H1047R in modified growth medium (2.5% horse serum and 5 ng/ml EGF) containing 2% growth factor-reduced Matrigel supplemented with HGF (40 ng/ml). The cells were treated with PI3K inhibitor pictilisib and/or either MET antibody onartuzumab or MET inhibitor tepotinib. Targeting PI3K or MET alone inhibited growth of the acini. However, targeting both PI3K and MET further increased the inhibitory effects (P < 0.0001 vs. pictilisib and vs. onartuzumab) in cells expressing PIK3CA-H1047R/MET-T1010I (Fig. 4A, B), and cells expressing PIK3CA-H1047R/MET-WT exhibited a similar pattern (Fig. 4C, D). These findings were repeated in cells expressing PIK3CA-E545K/MET-T1010I (Supplementary Fig. S4A). The enhanced inhibition due to combined targeting was further confirmed using another PI3K inhibitor, apitolisib (Supplementary Fig. S4B).
Figure 4. Effects of combined targeting of PI3K and MET on cell growth in Matrigel or invasion in MCF-10A derived cells.
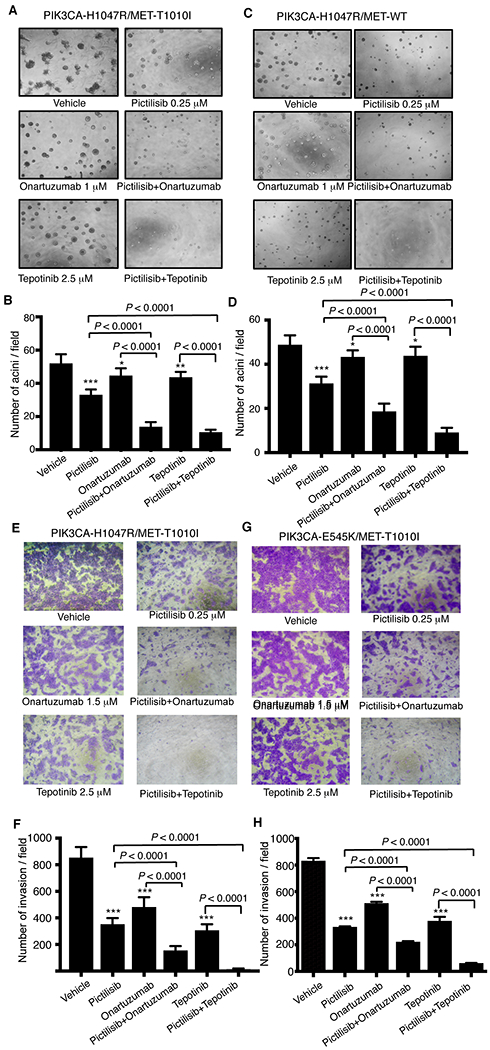
A total of 4 × 103 MCF-10A–derived cells expressing PIK3CA-H1047R/MET-T1010I (A) or PIK3CA-H1047R/MET-WT genes (C) were resuspended in modified growth medium (2.5% horse serum and 5 ng/ml EGF) supplemented with HGF (40 ng/ml) and 2% growth factor reduced Matrigel and treated with various drugs as indicated. The medium was exchanged every 3 days. Photographs (×40) of representative fields were taken on day 7. The figures shown are mean ± standard deviation of triplicates, representative of two independent experiments (*, P < 0.01; **, P < 0.001; ***, P < 0.0001 vs. vehicle, ANOVA) (B, D). The MCF-10A–derived cells expressing PIK3CA-H1047R/MET-T1010I (E) or PIK3CA-E545K/MET-T1010I (G) were starved for 20 hours in serum-free DMEM-F12 lacking EGF, insulin, and hydrocortisone. A total of 1 × 105 cells were inoculated into the upper chamber. pictilisib, onartuzumab, or tepotinib alone or in combinations as indicated were added into both the upper and lower chambers. HGF and fibronectin were added in the lower chamber as the inducer. Invasive cells were photographed and counted in 10 random fields. The figures shown are mean ± standard deviation of triplicates, representative of two independent experiments (***, P < 0.0001 vs. vehicle, ANOVA) (F, H).
We next tested whether targeting MET and PI3K would also inhibit cell invasion, again using MCF-10A–derived cells expressing PIK3CA-H1047R/MET-T1010I (Fig. 4E, F) or PIK3CA-E545K/MET-T1010I (Fig. 4G, H). The invasion assays showed a similar pattern to that seen in cell growth assays. Pictilisib (0.25 μM), MET antibody onartuzumab (1.5 μM), or MET inhibitor tepotinib (2.5 μM) alone inhibited invasion at various levels. However, the combination of pictilisib with onartuzumab or tepotinib markedly inhibited invasion (P < 0.0001 vs. pictilisib and vs. onartuzumab/tepotinib). These findings were recapitulated using apitolisib (Supplementary Fig. S4C, D).
Cooperative oncogenic effect of MET and PIK3CA on cellular signaling pathways in an hHGF-enriched microenvironment
Our in vitro and in vivo studies showed that oncogenic MET and PIK3CA act synergistically to induce aggressive cell behavior in an hHGF-enriched microenvironment. To elucidate the oncogenic signaling that leads to this aggressive behavior, we assessed functional proteomics using RPPA. The MET-T1010I/PIK3CA-H1047R-expressing MCF-10A–derived cells received stimulation with HGF (40 ng/ml) or 10% FBS after overnight serum starvation. Unsupervised hierarchical clustering revealed that conditions without stimulation (control) formed a cluster at the left of the dendrogram. The cluster at the right comprised two sub-clusters representing stimulation with HGF and with (Fig. 5A). As expected, both HGF and FBS activated a wide range of oncogenic pathways, including PI3K-AKT and RAS-MEK-MAPK pathways, to varying degrees. However, compared with FBS, HGF more markedly activated the Ras-Raf-MAPK pathway, as indicated by increased levels of p-MAPK (P < 0.001) and p-MEK1 (P < 0.001), increased phosphorylation of Y-box binding protein 1 (YB1, YBX1) and 4EBP-1 (EIF4EBP1) (P < 0.01 and P < 0.05, respectively), and increased expression of B-cell lymphoma 2–related protein A1 (Bcl2A1; P < 0.05) (Fig. 5B).
Figure 5. Cooperative oncogenic effect of MET and PIK3CA on cellular signaling pathways in an environment with or without HGF.
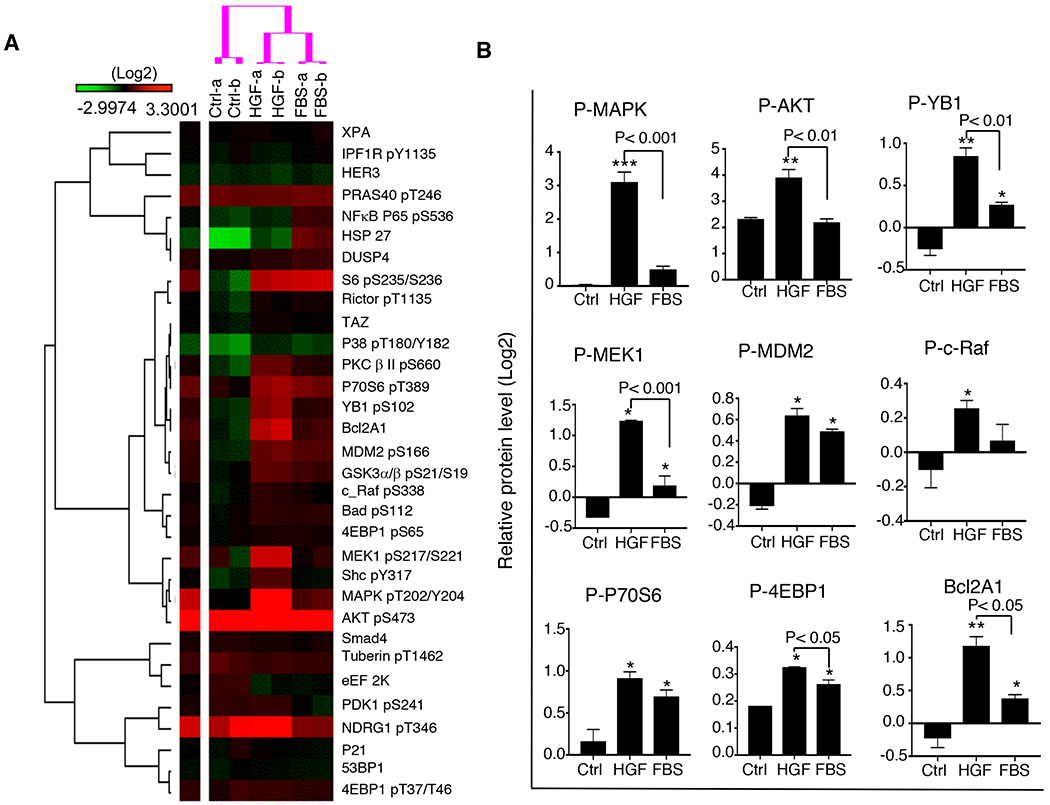
PIK3CA-H1047R/MET-T1010I-expressing MCF-10A–derived cells were starved overnight followed by stimulation with HGF (40 ng/ml) or 10% FBS for 30 minutes. Cell lysates were subject to RPPA as described in Materials and Methods. A. Data are presented in a matrix format: each row represents an antibody target and each column a sample. In each sample, the ratio of the abundance of the molecule to its median abundance across all samples is represented by the color of the corresponding cell in the matrix (see the scale for expression levels). B. The RPPA data were further analyzed (as described in Materials and Methods) (mean ± standard deviation; P value based on the log2 data).
Combined targeting of MET and PIK3CA abrogates cancer-associated signaling induced by HGF compared with that induced by FBS
We next examined molecular response to targeting MET and/or PI3K in an environment with or without HGF involvement by RPPA. MCF-10A–derived cells with expression of PIK3CA-H1047R/MET-T1010I were treated with PI3K inhibitor pictilisib and/or MET antibody onartuzumab with or without stimulation with HGF (40 ng/ml) or 10% FBS (Fig. 6A). In contrast to targeting MET with onartuzumab, targeting PI3K with pictilisib showed a more pronounced effect on signaling induced by FBS, due to a considerable reduction in phosphorylation of AKT, MDM2, Bcl2A1, PRAS40, P70S6, S6, Bad, NDRG1, GSK3α/β, and 4EBP1. However, onartuzumab had a stronger inhibitory effect on HGF-induced signaling than pictilisib did, as indicated by decreased phosphorylation levels of MAPK, MEK1, YB1, PKCβII, MDM2, Bcl2A1, Bad, and S6. Combined targeting of MET and PI3K increased the inhibitory effect on several HGF-induced oncogenic pathways, as indicated by further decreases in phosphorylation levels of MAPK, PKC β II, MDM2, P70S6, S6, GSK3α/β, and 4EBP1, compared with that in cells treated with single agents. However, the targeting of MET with onartuzumab did not increase the inhibitory effect on FBS-induced oncogenic signaling compared with that of pictilisib alone. The findings were repeated using the MET inhibitor tepotinib instead of onartuzumab (Fig. 6B) and using apitolisib instead of pictilisib (Supplementary Fig. S5A, B). A dendrogram based on unsupervised hierarchical clustering shows the effect of targeting MET and/or PI3K on signaling induced by HGF (Fig. 6C). As expected, targeting PI3K with pictilisib significantly inhibited phosphorylation of AKT. Compared with targeting PI3K, targeting MET with onartuzumab significantly inhibited phosphorylation of MAPK, MEK1, S6, YB1, and Bcl2A1. However, combined targeting of MET and PI3K further increased the inhibitory effects compared with targeting of either alone as indicated by dramatic decrease in phosphorylation of the downstream molecules S6 and 4EBP1. Using tepotinib instead of onartuzumab yielded the same pattern (Fig. 6D). MCF-10A derived cells with expression of MET-Y1253D/PI3KCA-H1047R exhibited similar response to pictilisib and /or tepotinib (Supplementary Fig. S6).
Figure 6. Cellular signaling response to targeting of MET and/or PIK3CA in an environment with or without HGF.
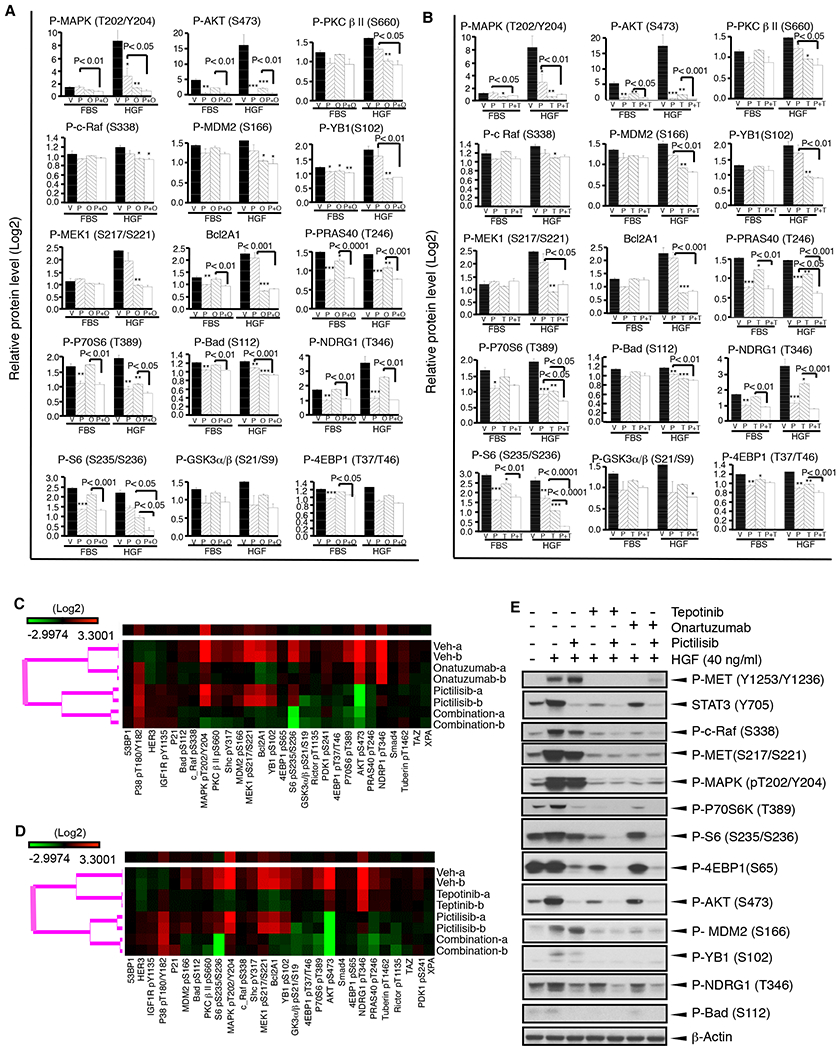
PIK3CA-H1047R/MET-T1010I-expressing MCF-10A–derived cells were starved overnight followed by treatment with or without drugs for 6 hours and then were stimulated with HGF (40 ng/ml) or 10% FBS for 30 minutes. Cell lysates were subject to RPPA as described in Materials and Methods (mean ± standard deviation; P value based on the log2 data). A. Cells were treated with pictilisib (P) or onartuzumab (O) alone or their combination (P+O) before stimulation with HGF or FBS. B. Cells were treated with pictilisib (P) or tepotinib (T) alone or their combination (P+T) before stimulation with HGF or FBS. C. Cells were treated with pictilisib and/or onartuzumab followed by stimulation with HGF. Data are presented in a matrix format: each row represents a sample and each column an antibody target. There are four sub-clusters as indicated from top to bottom: vehicle, onartuzumab, pictilisib, and their combination. Red represents high and green represents low activation. D. Using tepotinib instead of onartuzumab. E. A portion of the RPPA data was validated by Western blot.
We further assessed MET/PI3K–associated downstream molecules by Western blot (Fig. 6E). Consistent with the data from RPPA, stimulation with HGF markedly increased phosphorylation of MET and activated its downstream pathways RAS-MEK-MAPK, PI3K-AKT, and STAT3. As expected, PI3K inhibition with pictilisib blocked the PI3K-AKT and STAT3 pathways, but not the MAPK pathway. Targeting MET with onartuzumab or tepotinib completely inhibited phosphorylation of MET and significantly inhibited phosphorylation levels of c-Raf, MET, MAPK, P70S6K, S6, MDAM2, and YB1. Targeting MET also inhibited phosphorylation of STAT3, AKT, 4EBP1, and Bad. The combined targeting of both MET and PI3K further increased the inhibitory effect.
High expression of MET is not equivalent of activation of MET, while HGF is one of key factors that determine activation of MET in breast cancer
Our RPPA data revealed that HGF not only regulated activation of MET signaling but also affected the response of cells to MET/PI3K-targeted therapies. To further identify factors affect activation of MET signaling in breast cancer, we chose 8 PDXs (WHIM2, WHIM3a, WHIM3b, WHIM13, WHIM21, WHIM22, WHIM27, and WHIM31) to represent different levels of MET expression. After establishing PDX-derived stable cell lines using the PDXs, we treated the cells with or without HGF for 20 minutes. MET and phospho-MET were guantified with Western blotting (Fig. 7A). As expected, the lines showed varying expression levels of MET; in order from highest to lowest expression were WHIM22, WHIM31, WHIM2, WHIM21, WHIM3a, WHIM3b, and WHIM13, while MET expression was hard to detect in WHIM27. Stimulation with HGF slightly decreased the level of MET in each line, possibly owing to ligand-dependent internalization and degradation. As expected, stimulation with HGF induced significant phosphorylation of MET for each line (P < 0.0001, respectively). The responses to HGF among the lines exhibited massive difference. However, intriguingly, there was no correlation between the levels of phospho-MET and its expression except in the MET-negative line, as indicated the lines in order from highest to lowest phosphorylation: WHIM3a, WHIM3b, WHIM31, WHIM13, WHIM22, WHIM21, WHIM2, and WHIM27 (Fig. 7B).
Figure 7. Characterization of HGF/MET axis in patients-derived xenografts.
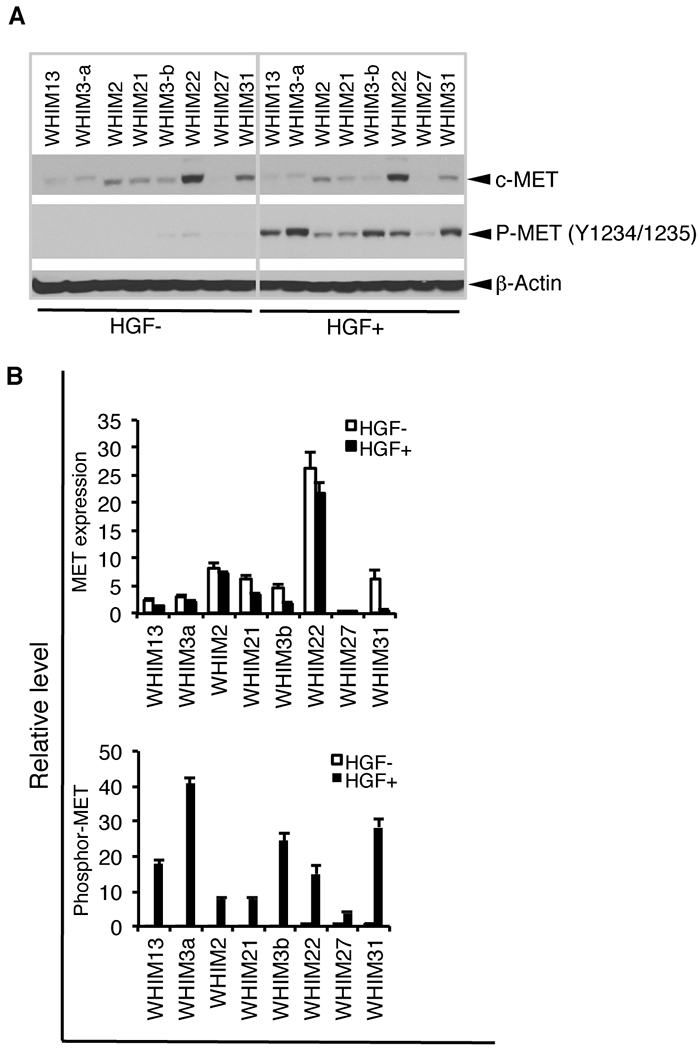
A. MET protein expression and activation by stimulation with HGF in PDXs. The cell lines from the PDX received starvation for over night by withdrawing serum and all growth factors, including insulin, EGF, and hydrocortisone, followed by stimulation with or without HGF (40 ng/ml) for 20 minutes. Cell lysis was collected and assessed expression and phosphorylation of MET with Western blotting. B. Quantitative densitometric analysis of the density was performed with AlphaView SA software.
Discussion
In this study, we demonstrate that aberrant MET and PIK3CA mutations coordinately induce aggressive cell behavior in an HGF-enriched microenvironment by broadly activating oncogenic pathways. Our preclinical findings could explain the clinical reports that concurrent dysregulation of oncogenic MET and PIK3CA is associated with worse recurrence-free survival in patients with breast cancer, while PIK3CA mutation status alone is not (3,7). We also provided direct evidence that combined targeting of both MET and PI3K abrogates resistance to individual targeted therapies.
By systematically analyzing oncogenic signaling, we demonstrated that concurrent expression of oncogenic MET and PIK3CA synergistically activates multiple oncogenic signals, including RAS-MEK-MAPK, STAT3, and PI3K-AKT pathways in an HGF-enriched environment, and further markedly increases activation of Y-box binding protein 1 (YB1) that induces progression and metastasis of different types of cancer through angiogenesis, resistance of cell death, tumor-promoting inflammation, and evasion of immune destruction (40–42). Targeting PI3K significantly inhibits the PI3K-AKT pathway, while targeting MET dramatically inhibits the MAPK pathway and also inhibits the PI3K-AKT pathway. However, the combination of targeting PI3K and MET dramatically further inhibited their downstream molecules as indicated by phosphorylation of p70S6K, S6, 4EBP-1, suggesting these substrates represent a convergence for combinatorial blockade of the MET and PI3K pathways is an HGF- enriched microenvironment.
Despite that targeting MET has yielded exciting results in preclinical studies, clinical trials of MET inhibitors have been. Immunohistochemical MET positivity has been used as a criterion to recruit patients in clinical trials for MET or HGF targeted therapy (43, 44). In this study, we assessed the response to HGF-stimulation in multiple PDX lines with variable degrees of MET expression. MET expression did not correlate with MET signaling activation. Thus, MET expression level alone may not be an idea qualifier for MET-targeting in a clinical trial.
Elevation of HGF was found in serum/plasma of patients with different types of cancer (20–24) and cancer tissue microenvironment (45–47). Higher serum HGF level is associated with therapy resistance and poorer prognosis in multiple types of cancer (22, 48–50). As the only known ligand of MET, HGF is necessary for MET activation, especially for WT MET, so tumoral/microenvironment assessment may be important for preclinical and clinical studies. Even this may not be sufficient because our data exhibited that the response of different tumor cells to HGF is extremely variable. Thus, activated phospho-MET is a potential biomarker for optimizing therapies for targeting MET signaling and improving treatment efficacy in individual breast cancer patients.
In summary, using two- and three-dimensional in vitro models and an HGF-MET paired mouse model, we demonstrate that oncogenic MET and PIK3CA cooperatively contribute to breast cancer in an HGF-dominant environment. Combined targeting of MET and PI3K is a rational strategy that merits clinical testing for breast cancer with concurrent aberrations of the HGF-MET axis and PI3K pathway. In multiple PDX lines, MET expression did not predict MET pathway activation. HGF in microenvironment is basic necessary for MET activation, but variable responses to HGF are cross tumor models. Thus, the levels of MET phosphorylation and its downstream signaling are potential predictive or qualifying biomarkers for therapeutic purposes.
Supplementary Material
Acknowledgments
We thank Dr. Guojun Wu (Karmanos Cancer Institute, Department of Pathology, Wayne State University) for providing PIK3CA plasmids; Genentech (South San Francisco, CA) provided onartuzumab, pictilisib and apitolisib as well as communication; EMD Serono (Rockland, MA) provided tepotinib; Dr. Prahlad Ram (Department of Systems Biology, The University of Texas MD Anderson Cancer Center) for valuable discussion.
Financial support: This work was supported by an Institutional Research Grant of The University of Texas MD Anderson Cancer Center to S. Liu (IRG-116731); The Commonwealth Foundation for Cancer Research (to A.M. Gonzalez-Angulo) and philanthropic support from Mr. A. Ray Weeks, Jr. (to D. Tripathy). MD Anderson Cancer Center is supported in part by the National Institutes of Health through Cancer Center Support Grant P30CA016672.
Footnotes
Conflicts of interest: The authors declare no potential conflicts of interest other than GBM being on the advisory board of Symphogen and AstraZenca and receiving research support from GSK and AstraZeneca.
References
- 1.Saal LH, Holm K, Maurer M, Memeo L, Su T, Wang X, et al. PIK3CA mutations correlate with hormone receptors, node metastasis, and ERBB2, and are mutually exclusive with PTEN loss in human breast carcinoma. Cancer Res 2005;65:2554–9. [DOI] [PubMed] [Google Scholar]
- 2.Stemke-Hale K, Gonzalez-Angulo AM, Lluch A, Neve RM, Kuo WL, Davies M, et al. An integrative genomic and proteomic analysis of PIK3CA, PTEN, and AKT mutations in breast cancer. Cancer Res 2008;68:6084–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zardavas D, Te Marvelde L, Milne RL, Fumagalli D, Fountzilas G, Kotoula V, et al. Tumor PIK3CA Genotype and Prognosis in Early-Stage Breast Cancer: A Pooled Analysis of Individual Patient Data. J Clin Oncol 2018;36:981–90. [DOI] [PubMed] [Google Scholar]
- 4.Guerrero-Zotano A, Mayer IA, Arteaga CL. PI3K/AKT/mTOR: role in breast cancer progression, drug resistance, and treatment. Cancer Metastasis Rev 2016;35:515–24. [DOI] [PubMed] [Google Scholar]
- 5.Miller TW, Hennessy BT, González-Angulo AM, Fox EM, Mills GB, Chen H, et al. Hyperactivation of phosphatidylinositol-3 kinase promotes escape from hormone dependence in estrogen receptor-positive human breast cancer. J Clin Invest 2010;120:2406–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Di Leo A, Johnston S, Lee KS, Ciruelos E, Lønning PE, Janni W, et al. Buparlisib plus fulvestrant in postmenopausal women with hormone-receptor-positive, HER2-negative, advanced breast cancer progressing on or after mTOR inhibition (BELLE-3): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2018;19:87–100. [DOI] [PubMed] [Google Scholar]
- 7.Baselga J, Im SA, Iwata H, Cortes J, De Laurentiis M, Jiang Z, et al. Buparlisib plus fulvestrant versus placebo plus fulvestrant in postmenopausal, hormone receptor-positive, HER2-negative, advanced breast cancer (BELLE-2): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2017;18:904–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zenali M, deKay J, Liu Z, Hamilton S, Zuo Z, Lu X, et al. Retrospective Review of MET Gene Mutations. Oncoscience 2015;2:533–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gonzalez-Angulo AM, Chen H, Karuturi MS, Chavez-MacGregor M, Tsavachidis S, Meric-Bernstam F, et al. Frequency of mesenchymal-epithelial transition factor gene (MET) and the catalytic subunit of phosphoinositide-3-kinase (PIK3CA) copy number elevation and correlation with outcome in patients with early stage breast cancer. Cancer 2013;119:7–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liu P, Cheng H, Santiago S, Raeder M, Zhang F, Isabella A, et al. Oncogenic PIK3CA-driven mammary tumors frequently recur via PI3K pathway-dependent and PI3K pathway-independent mechanisms. Nat Med 2011; 7:1116–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dean M, Park M, Le Beau MM, Robins TS, Diaz MO, Rowley JD, et al. The human met oncogene is related to the tyrosine kinase oncogenes. Nature 1985;318:385–8. [DOI] [PubMed] [Google Scholar]
- 12.Koochekpour S, Jeffers M, Rulong S, Taylor G, Klineberg E, Hudson EA, et al. Met and hepatocyte growth factor/scatter factor expression in human gliomas. Cancer Res 1997;57:5391–8. [PubMed] [Google Scholar]
- 13.Tsao MS, Liu N, Chen JR, Pappas J, Ho J, To C, et al. Differential expression of Met/hepatocytegrowth factor receptor in subtypes of non-small cell lung cancers. Lung Cancer 1998;20:1–16. [DOI] [PubMed] [Google Scholar]
- 14.Yan S, Jiao X, Zou H, Li K. Prognostic significance of c-Met in breast cancer: a meta-analysis of 6010 cases. Diagn Pathol 2015;10:62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhao X, Qu J, Hui Y, Zhang H, Sun Y, Liu X, et al. Clinicopathological and prognostic significance of c-Met overexpression in breast cancer. Oncotarget 2017;8:56758–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tolaney SM, Nechushtan H, Ron IG, Schoffski P, Awada A, Yasenchak CA, et al. Cabozantinib for metastatic breast carcinoma: results of a phase II placebo-controlled randomized discontinuation study. Breast Cancer Res Treat 2016;160:305–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tolaney SM, Ziehr DR, Guo H, Ng MR, Barry WT, Higgins MJ, et al. Phase II and Biomarker Study of Cabozantinib in Metastatic Triple-Negative Breast Cancer Patients. Oncologist 2017;22:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Raghav KP, Wang W, Liu S, Chavez-MacGregor M, Meng X, Hortobagyi GN, et al. cMET and phospho-cMET protein levels in breast cancers and survival outcomes. Clin Cancer Res 2012;18, 2269–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu S, Meric-Bernstam F, Parinyanitikul N, Wang B, Eterovic AK, Zheng X, et al. Functional consequence of the MET-T1010I polymorphism in breast cancer. Oncotarget 2015;6:2604–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bottaro DP, Rubin JS, Faletto DL, Chan AM, Kmiecik TE, Vande Woude GF, et al. Aaronson SA. Identification of the hepatocyte growth factor receptor as the c-met proto-oncogene product. Science 1991;251:802–4. [DOI] [PubMed] [Google Scholar]
- 21.Naldini L, Vigna E, Narsimhan RP, Gaudino G, Zarnegar R, Michalopoulos GK, et al. Hepatocyte growth factor (HGF) stimulates the tyrosine kinase activity of the receptor encoded by the proto-oncogene c-MET. Oncogene 1991;6:501–4. [PubMed] [Google Scholar]
- 22.Matsumoto K, Umitsu M, De Silva DM, Roy A, Bottaro DP. Hepatocyte growth factor/MET in cancer progression and biomarker discovery. Cancer Sci 2017;108:296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rahimi N, Saulnier R, Nakamura T, Park M, Elliott B. Role of hepatocyte growth factor in breast cancer: a novel mitogenic factor secreted by adipocytes. DNA Cell Biol 1994;13:1189–97. [DOI] [PubMed] [Google Scholar]
- 24.Tuck AB, Park M, Sterns EE, Boag A, Elliott BE: Coexpression of hepatocyte growth factor and receptor (Met) in human breast carcinoma. Am J Pathol 1996;148:225–32. [PMC free article] [PubMed] [Google Scholar]
- 25.Li G, Schaider H, Satyamoorthy K, Hanakawa Y, Hashimoto K, Herlyn M. Downregulation of E-cadherin and Desmoglein 1 by autocrine hepatocyte growth factor during melanoma development. Oncogene 2001;20:8125–35. [DOI] [PubMed] [Google Scholar]
- 26.Ferracini R, Di Renzo MF, Scotlandi K, Baldini N, Olivero M, Lollini P, et al. The Met/HGF receptor is over-expressed in human osteosarcomas and is activated by either a paracrine or an autocrine circuit. Oncogene 1995;10:739–49. [PubMed] [Google Scholar]
- 27.Ahmed HH, Metwally FM, Mahdy ES, Shosha WG, Ramadan SS. Clinical value of serum hepatocyte growth factor, B-cell lymphoma-2 and nitric oxide in primary breast cancer patients. Eur Rev Med Pharmacol Sci 2012;16:958–65. [PubMed] [Google Scholar]
- 28.El-Attar HA, Sheta MI. Hepatocyte growth factor profile with breast cancer. Indian J Pathol Microbiol 2011; 54:509–13. [DOI] [PubMed] [Google Scholar]
- 29.Taniguchi T, Toi M, Inada K, Imazawa T, Yamamoto Y, Tominaga T. Serum concentrations of hepatocyte growth factor in breast cancer patients. Clin Cancer Res 1995;1:1031–4 [PubMed] [Google Scholar]
- 30.Zhang YW, Su Y, Lanning N, Gustafson M, Shinomiya N, Zhao P, Cao B, et al. Enhanced growth of human met-expressing xenografts in a new strain of immunocompromised mice transgenic for human hepatocyte growth factor/scatter factor. Oncogene 2005;24:101–6. [DOI] [PubMed] [Google Scholar]
- 31.Zhang H, Liu G, Dziubinski M, Yang Z, Ethier SP, Wu G. Comprehensive analysis of oncogenic effects of PIK3CA mutations in human mammary epithelial cells. Breast Cancer Res Treat 2008;112:217–27. [DOI] [PubMed] [Google Scholar]
- 32.Merchant M, Ma X, Maun HR, Zheng Z, Peng J, Romero M, et al. Monovalent antibody design and mechanism of action of onartuzumab, a MET antagonist with anti-tumor activity as a therapeutic agent. Proc Natl Acad Sci U S A 2013;110:E2987–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bladt F, Faden B, Friese-Hamim M, Knuehl C, Wilm C, Fittschen C, et al. EMD_1214063_and_EMD_1204831 constitute a new class of potent and highly selective c-Met inhibitors. Clin Cancer Res. 2013;19:2941–51. [DOI] [PubMed] [Google Scholar]
- 34.Sutherlin DP, Bao L, Berry M, Castanedo G, Chuckowree I, Dotson J, et al. Discovery of a potent, selective, and orally available class I phosphatidylinositol 3-kinase (PI3K)/mammalian target of rapamycin (mTOR) kinase inhibitor (GDC-0980) for the treatment of cancer. J Med Chem. 2011;54:7579–87. [DOI] [PubMed] [Google Scholar]
- 35.Li S, Shen D, Shao J, Crowder R, Liu W, Prat A, et al. Endocrine-therapy-resistant ESR1 variants revealed by genomic characterization of breast-cancer-derived xenografts. Cell Rep 2013;4:1116–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu S, Umezu-Goto M, Murph M, Lu Y, Liu W, Zhang F, et al. Expression of autotaxin and lysophosphatidic acid receptors increases mammary tumorigenesis, invasion, and metastases. Cancer Cell 2009;15:539–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012;490:61–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tibes R, Qiu Y, Lu Y, Hennessy B, Andreeff M, Mills GB, et al. Reverse phase protein array: validation of a novel proteomic technology and utility for analysis of primary leukemia specimens and hematopoietic stem cells. Mol Cancer Ther 2006;5:2512–21. [DOI] [PubMed] [Google Scholar]
- 39.Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, Pearline RV, et al. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res 2005;65:10992–1000. [DOI] [PubMed] [Google Scholar]
- 40.Mylona E, Melissaris S, Giannopoulou I, Theohari I, Papadimitriou C, Keramopoulos A, et al. Y-box-binding protein 1 (YB1) in breast carcinomas: relation to aggressive tumor phenotype and identification of patients at high risk for relapse. Eur J Surg Oncol 2014;40:289–96. [DOI] [PubMed] [Google Scholar]
- 41.Bledzka K, Schiemann B, Schiemann WP, Fox P, Plow EF, Sossey-Alaoui K. The WAVE3-YB1 interaction regulates cancer stem cells activity in breast cancer. Oncotarget 2017;8:104072–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lasham A, Print CG, Woolley AG, Dunn SE, Braithwaite AW. YB-1: oncoprotein, prognostic marker and therapeutic target? Biochem J 2013;449:11–23. [DOI] [PubMed] [Google Scholar]
- 43.Shah MA, Bang YJ, Lordick F, Alsina M, Chen M, Hack SP, et al. Effect of Fluorouracil, Leucovorin, and Oxaliplatin With or Without Onartuzumab in HER2-Negative, MET-Positive Gastroesophageal Adenocarcinoma: The METGastric Randomized ClinicalTrial. JAMA Oncol. 2017;3:620–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Catenacci DVT, Tebbutt NC, Davidenko I, Murad AM, Al-Batran SE, Ilson DH, Rilotumumab plus epirubicin, cisplatin, and capecitabine as first-line therapy in advanced MET-positive gastric or gastro-oesophageal junction cancer (RILOMET-1): a randomised, double-blind, placebo-controlled, phase 3 trial. Lancet Oncol 2017;18:1467–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamashita J, Ogawa M, Yamashita S, Nomura K, Kuramoto M, Saishoji T, et al. Immunoreactive hepatocyte growth factor is a strong and independent predictor of recurrence and survival in human breast cancer. Cancer Res 1994;54:1630–3. [PubMed] [Google Scholar]
- 46.Yao Y, Jin L, Fuchs A, Joseph A, Hastings HM, Goldberg ID, et al. Scatter factor protein levels in human breast cancers: clinicopathological and biological correlations. Am J Pathol 1996;149:1707–17. [PMC free article] [PubMed] [Google Scholar]
- 47.Parr C, Watkins G, Mansel RE, Jiang WG. The hepatocyte growth factor regulatory factors in human breast cancer. Clin Cancer Res 2004;10:202–11. [DOI] [PubMed] [Google Scholar]
- 48.Nagy J, Curry GW, Hillan KJ, McKay IC, Mallon E, Purushotham AD, et al. Hepatocyte growth factor/scatter factor expression and c-met in primary breast cancer. Surg Oncol 1996;5:15–21. [DOI] [PubMed] [Google Scholar]
- 49.Edakuni G, Sasatomi E, Satoh T, Tokunaga O, Miyazaki K. Expression of the hepatocyte growth factor/c-Met pathway is increased at the cancer front in breast carcinoma. Pathol Int 2001;51:172–78. [DOI] [PubMed] [Google Scholar]
- 50.Kang JY, Dolled-Filhart M, Ocal IT, Singh B, Lin CY, Dickson RB, et al. Tissue microarray analysis of hepatocyte growth factor/Met pathway components reveals a role for Met, matriptase, and hepatocyte growth factor activator inhibitor 1 in the progression of node-negative breast ancer. Cancer Res 2003;63: 1101–5. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.