

Abstract
Here, cation exchange (CE) reactions are exploited to radiolabel ZnSe, ZnS, and CuFeS2 metal chalcogenide nanocrystals (NCs) with 64Cu. The CE protocol requires one simple step, to mix the water‐soluble NCs with a 64Cu solution, in the presence of vitamin C used to reduce Cu(II) to Cu(I). Given the quantitative cation replacement on the NCs, a high radiochemical yield, up to 99%, is reached. Also, provided that there is no free 64Cu, no purification step is needed, making the protocol easily translatable to the clinic. A unique aspect of the approach is the achievement of an unprecedentedly high specific activity: by exploiting a volumetric CE, the strategy enables to concentrate a large dose of 64Cu (18.5 MBq) in a small NC dose (0.18 µg), reaching a specific activity of 103 TBq g−1. Finally, the characteristic dielectric resonance peak, still present for the radiolabeled 64Cu:CuFeS2 NCs after the partial‐CE reaction, enables the generation of heat under clinical laser exposure (1 W cm−2). The synergic toxicity of photo‐ablation and 64Cu ionization is here proven on glioblastoma and epidermoid carcinoma tumor cells, while no intrinsic cytotoxicity is seen from the NC dose employed for these dual experiments.
Keywords: cation exchange reactions, dual therapy, internal radiotherapy, metal chalcogenides nanocrystals, photo‐thermal therapy
The first examples of cation exchange reactions used for quantitative radiolabelling with 64Cu aqueous stable metal chalcogenides of ZnSe, ZnS and CuFeS2 nanocrystals are reported here, setting protocols readily transferable to the clinic. The exploitation of radiolabeled nanocrystals for dual therapy combining internal radiotherapy and phototherapy is also demonstrated.
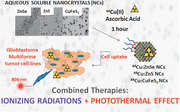
1. Introduction
Non‐stoichiometric copper chalcogenide nanocrystals NCs are mainly known for their localized surface plasmonic resonance band, which peaks in the near infrared region (NIR).[ 1 ] Thanks to this band, the energy of a NIR source can be transduced into macroscopic heat, making these materials promising heat mediators for photo‐thermal therapy.[ 2 ] The effectiveness of the photo‐thermal behavior of both copper sulfide[ 3 ] and copper selenide[ 4 ] NCs has already been demonstrated in vitro and in vivo. A large variety of copper chalcogenide NCs can be prepared either by a direct synthesis approach (i.e., hot decomposition of copper and sulfur or selenium precursors) or by an indirect approach exploiting cation exchange (CE) reactions,[ 5 ] that is, by replacing pristine cations with copper ions on preformed NCs,[ 6 ] making it possible to access a large variety of NC compositions that cannot be prepared by direct synthesis.[ 7 ]
In clinic, radioisotopes are extensively used for both imaging and therapeutic applications. Depending on the imaging or therapeutic purpose, the radionuclide has to fulfill certain requirements in terms of the type of ionizing particles that are released and their decay time.[ 8 ] 64Cu decays though β− particles (0.573 MeV, 39%) and electron capture (44%) emission, which are useful for radiotherapy applications[ 9 ] but it can also produce a β+ decay (0.655 MeV, 17%), therefore it is also suitable for positron emission tomography (PET) imaging.[ 10 ] Furthermore, 64Cu is currently produced in many cyclotron facilities on a routine basis,[ 11 ] and it is much more widely available than 67Cu, making 64Cu an ideal candidate for preclinical research and for the development of radio‐therapeutic agents.[ 12 ]
Merging the properties of plasmonic photo‐thermal NCs and radionuclides in one single nano‐object can offer new combined approaches to cancer therapy.[ 13 ] To date, 64Cu radiolabeled NCs were mostly used as imaging tools for PET.[ 14 ] In some studies, theranostic applications have been pursued: the positron emission behavior of low dopant 64Cu was exploited for PET imaging, while the intrinsic material's properties were used for therapeutic purposes (i.e., for photo‐thermal performances).[14b, 15] Only one work has explored the synergistic effect of radiation and NIR (near infrared light) photo‐thermal therapy of 64Cu‐radiolabeled NCs.[ 16 ] Zhou et al.[ 16 , 17 ] prepared 64Cu:CuS PEG‐coated NCs by aqueous co‐precipitation of copper (both “hot” 64Cu and “cold” Cu) and sulfur‐containing salts, and administered intra‐tumorally these radiolabeled NCs using a xenograft BT474 breast tumor mice model. They showed that the combination of both treatments was necessary to completely eradicate the tumor mass, as each individual therapy, either radiotherapy or heat therapy, was not sufficient. Moreover, this work suggests that 64Cu associated with NCs can greatly affect the retention time of radioisotopes, enabling a longer persistence of the 64Cu radionuclide at the tumor site than when using the bare 64CuCl2 solution.
Accumulating a high radionuclide dose at the tumor site, by increasing the specific activity of 64Cu:NC, is the key to maximize the radio‐therapeutic efficacy. The specific or molar activity is defined as the amount of radioactivity per unit mass or mole of material. In other words, it represents the amount of radionuclides associated to a carrier (e.g., a nanoparticle, antibody, chelating ligand etc.). The use of radiolabeled NCs with a high specific activity would enable to administer a lower NC dose, thus minimizing cytotoxic side effects, without compromising the radionuclide dose that must be delivered for a more effective therapy.
NCs coupled with proper chelating agents that can bind 64Cu at their surface[ 18 ] are expected to be less nephrotoxic[ 19 ] than antibodies or proteins bearing a radionuclide.[ 20 ] These NCs, however, due to the limited number of chelating units per NC surface, have specific activity values around 0.7 TBq g−1.[ 21 ] Moreover, the detachment of the radiometal from the NC surface should be taken into account when the radiolabeled NCs are exposed to the biological fluid.[ 22 ] The insertion of 64Cu ions inside the NCs has been proposed as an alternative method to produce stable radiolabeled NCs.[ 23 ] Radionuclides can be inserted into the NCs either during the NC synthesis[15a,24] or in a post‐synthesis step, following CE or intercalation reactions.[14a,25] The direct incorporation of 64Cu into a NC's core during the synthesis has provided NC with a higher specific activity (2 TBq g−1) than chelating methods.[ 16 ] However, no in situ synthesis has led to a quantitative insertion of 64Cu but only radiotracer doses for PET imaging were reported. CE or intercalation reactions are feasible for a quantitative replacement of cations, thus significantly increasing the specific activity of the radiolabeled NCs. Indeed, exchanging only 10% of the cations in a 6 nm spherical shaped zinc selenide (ZnSe) NC with a cubic sphalerite structure, using 37 MBq of a 64Cu solution (50 GBq µmol−1) would produce a material with a specific activity of around 43 TBq g−1, assuming that a quantitative cation exchange (QCE) process occurs.
Here, we demonstrate that performing a quantitative CE reaction is feasible using a 64CuCl2 solution of high specific activity, on water‐dispersible chalcogenide‐based NCs of different materials, including ZnS, CuFeS2, and ZnSe. In this work, we show that, by using a constant amount of 64Cu on a µg dose of NCs, the specific activity can be varied in a wide range (from 2 to 100 TBq g−1), especially designed for a radiotherapeutic use. The use of a proper surface coating was crucial to develop radiolabeling protocols that provide NCs with an optimal colloidal and radiolabeling stability upon a 64Cu exchange. Among the various ligands that were available, we selected a multi‐anchoring cysteamine‐containing polyethylene glycol (PEG) ligand, as it does not interfere with the CE reaction and, at the same time, it provides the NCs with high colloidal stability in aqueous buffer or serum solutions. Finally, we want to highlight the simplicity of the radiolabeling procedure herein reported, which involves a single step consisting in the mixing of the NCs with the 64CuCl2 solution, in buffer at 37 °C for 1 h, with no further need of purification steps. Such simplicity makes is easily implementable in any clinical hospital where 64Cu is available.
2. Results and Discussion
For our radiolabeling protocol, we selected Cd‐free chalcogenides NCs having different compositions to minimize toxicity effects of the NCs, including spherical Zn‐based chalcogenides (ZnS and ZnSe, with a diameter of 10 and 6 nm, respectively),[ 26 ] and tetrahedral CuFeS2 chalcopyrite NCs with an edge length of around 13 nm.[26b] These NCs were synthetized following non‐hydrolytic hot decomposition protocols reported in the literature, with minor modifications (Figures S1 and S2, Supporting Information).[ 26 ] The as‐synthetized NCs, coated with hydrophobic ligands (e.g., oleylamine and/or oleic acid), were transferred into water following a ligand exchange procedure.[ 27 ] We synthetized (by adapting an already reported procedure) and characterized a multi‐anchoring ligand made of poly‐isobutylene maleic anhydride (PIMA), equipped with cysteamine (cys) units, which act as anchors for the NC surface and having amine‐polyethylene glycol‐methoxy‐terminated (PEG) molecules as water soluble units (Figure 1a), (Figure S3, Supporting Information, for more details).[ 28 ] Water‐transferred NCs deposited from an aqueous solution on a transmission electron microscopy (TEM) grid formed a monolayer of well‐separated NCs, indicating homogenous NC samples, with no presence of aggregates (Figure 1c,f,i). Dynamic light scattering (DLS) measurements indicated average hydrodynamic sizes for each of the three NC samples in between 15 and 25 nm, with low polydispersity (PDI) index (Figure 1d,g,l). Both data confirm the absence of aggregates in the water transferred NCs. The multi‐dentate cys‐PIMA‐PEG provided also a stable polymer shell to the NCs, minimizing the yield loss during the NCs water transfer to less than 20% (only a small fraction of NC was lost during the washing steps on an amicon cartridge) in accordance with other studies in which multidentate ligands were reported.[ 28 , 29 ]
Figure 1.
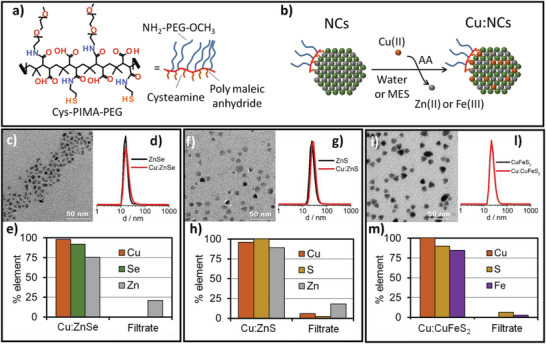
a) Cys‐PIMA‐PEG polymer ligand structure used for the water transfer of all NCs involved in this study. b) Schematic sketch of the CE reaction on the water‐dispersible NCs. c,f,i) TEM images of water‐dispersible NCs used for the CE reaction. d,g,l) DLS hydrodynamic sizes (in number weighted) before (black line) and after (red line) the partial (13%) Cu exchange reaction showed no change in size, indicating the absence of aggregation after CE. e,h,m) Elemental analysis to follow the partial CE reaction (13%): Cu, Se, S, Zn or Fe element quantification, after CE reactions, reported as a percentage of the initial elements on the NC fraction (Cu:ZnSe, Cu:ZnS, and Cu:CuFeS2) and on the washing solutions (filtrate) after NCs separation for e) ZnSe, h) ZnS and m) CuFeS2 NCs.
A first set of CE reactions were carried out employing “cold” copper, which is a non‐radioactive copper salt (CuCl2). CuCl2 0.021 m in HCl (0.01 m) was chosen because it mimics the clinically available 64CuCl2 solution. These CE reactions were based on an in situ reduction of Cu(II) in the presence of a mild reducing agent, namely ascorbic acid (AA, Figure 1b for the reaction scheme) to Cu(I) in a MES buffer. Differently from Cu(II), Cu(I) is known to exchange with Zn ions in ZnSe and ZnS NCs.[ 30 ] In a typical reaction, 10 µL of 2–15 µm NCs (corresponding to an anion concentration of sulfur or selenium in between 0.02 to 0.05 m) were diluted with 100 µL of a MES buffer (0.1 m, pH 5.5) or water and mixed with the proper amount of AA (0.1 m). The amount of CuCl2 was adjusted in order to vary the final Cu/S or Cu/Se ratio so that a partial (13%, Figure 1e,h,m) or a total (100%, Figure S5, Supporting Information) nominal Cu exchanged with the NCs cations was achieved.
Cu, Zn, Fe, S, and Se elemental analyses (by means of inductively‐coupled plasma optical emission spectrometry, ICP‐OES) were performed on the pristine NCs, on the NCs fraction after CE reaction (the exchanged NC samples are indicated as Cu:ZnSe, Cu:ZnS, and Cu:CuFeS2) and on the filtrate after a cleaning step (the reaction mixtures were subjected to three cycles of concentration/dilution on amicon centrifuge filter) to carefully separate the cation exchanged NCs (Cu:ZnSe, Cu:ZnS, and Cu:CuFeS2) from the eventually unbound Cu ions and from the cations freed in solution (Zn and Fe). After CE reaction, the copper amount was found entirely in the NC fractions and no Cu was present in the washing solutions, indicating a complete incorporation of Cu into the NCs (Figure 1, panels e–h for Cu:ZnSe, Cu:ZnS, and panel m for Cu:CuFeS2 for an exchange at 13%). The replaced ions, Zn2+ in case of ZnSe and ZnS and the Fe3+ for CuFeS2, as expected, were found instead in the washing solutions (named Filtrate in Figure 1e,h,m). The Cu:NCs did not show any variation in colloidal stability (Figure 1d,g,l). The same trend was also observed when increasing the initial amount of Cu to have a nominal exchange of almost 100% (Figure S5, Supporting Information). It is worth mentioning that, for the complete CE reaction, the amount of Cu per NC can be ideally exchanged to the stoichiometric ratio Cu/Se or Cu/S of 2:1. However, the Cu2Se and Cu2NCs would be easily oxidized to the copper deficient chalcogenides Cu2− xSe or Cu2− xS,[ 31 ] and there would be a concomitant release of Cu ions in the solution (data not shown). Therefore, for a QCE, we chose to perform a copper CE reaction at a final Cu/Se or Cu/S ratio of 1.8:1 (Figure S5, Supporting Information), which should produce copper exchanged NCs that are stable under air‐equilibrated media.[31a] Even after a quasi‐quantitative exchange, the samples remained stable in solution, with no sign of aggregation, as evidenced by TEM and DLS characterization (Figure S5, Supporting Information). Further confirmation of the quantitative CE reaction came from the X‐ray diffraction patterns of the samples before and after the exchange reaction. Indeed, when working at a Cu/Se or Cu/S ratio of 1.8, a quantitative transformation from ZnSe to Cu2− xSe, from ZnS to Cu2− xS and from CuFeS2 to Cu2− xS NCs occurred, as shown by the characteristic diffractometer patterns of the exchanged Cu:NCs (Figure S5, Supporting Information).
Finally, further proofs of NC transformation are found in the optical spectral changes: Cu2− xSe and Cu2− xS NCs, obtained upon CE exchange, exhibit an NIR localized surface plasmon resonance band (LSPR) that was not present in the spectra of the initial ZnSe and ZnS NCs (Figure S6, Supporting Information). On the contrary, for CuFeS2 NCs, the dielectric resonance at 496 nm attributed to the intermediate band formed by the presence of Fe,[ 32 ] disappears when an almost quantitative exchange occurs (Figure S6, Supporting Information).
These data, all together, suggest that under the experimental conditions set by us, the copper CE reactions on the NCs occurred quantitatively, irrespective of the materials composition and the initial percentage of Cu to be exchanged (13% or almost 100%).
To verify the chemical stability of the Cu‐exchanged NCs, after the CE reactions (total exchange Cu/Se or Cu/S fixed at 1.8, corresponding to about 90% of the fully stoichiometric Cu2S), the Cu2− xSe and Cu2− xS NCs were kept in pure water or in human serum at 37 °C for well‐defined time points. Elemental analysis revealed that, in water, a minor copper leakage from the NCs started to occur only after 24 h (around 5%; Figure S7, Supporting Information). Copper release, however, was more pronounced in the human serum, as indeed 68% and 85% of the total copper on the Cu2− xSe and Cu2− xS NCs respectively was released after 24 h (Figure S8, Supporting Information). Conversely, the copper leakage was lower when applying the same stability tests to partially exchanged NCs (Cu/Se or Cu/S 13%, Figure 2e).
Figure 2.
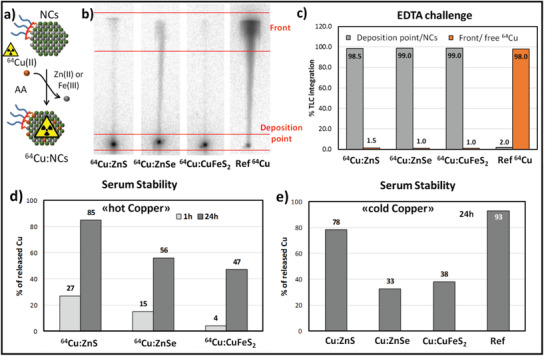
a) Sketch of the 64Cu radiolabeling via CE reaction on the water‐dispersible NCs. b) Radio‐TLC of the crude of 13% CE reactions (after 1h of incubation at 50 °C) for 64Cu:ZnS, 64Cu:ZnSe, and 64Cu:CuFeS2 (EDTA 0.1M as a mobile phase); free 64CuCl2 is also spotted as a reference control. c) Integration of peaks from radio‐TLC chromatogram of “NCs previously exposed for 20 min to an EDTA 0.1 m solution; most of the Cu is still associated to the NCs fractions, indicating stable radiolabeled NCs products. d) Results from radio‐TLC integration (EDTA 0.1 m mobile phase) of 64Cu released upon incubation of 64Cu:ZnS, 64Cu:ZnSe and 64Cu:CuFeS2 in human serum at 37 °C for 1 and 24 h. e) Copper released upon 24 h serum incubation of Cu:NCs; amicon wash was performed using EDTA 0.1 m solution. The free copper incubation experiment is also reported (Ref).
For the first set of CE reactions with the 64Cu ions, the so called “hot” copper, we have therefore considered a partial CE reaction with a 64Cu/Se or 64Cu/S ratio corresponding to 13%. For each radiolabeling experiment, we used 37 MBq of a 64CuCl2 solution in 10 mm HCl (specific activity 50 GBq µmol−1). Prior to the addition of the radiocopper solution, the pH of the 64Cu solution had been adjusted to pH 5–6 with a NaOH solution. Due to the low amount of total copper in this radiolabeled solution (7.4 × 10−10 moles), the amount of NCs was reduced with respect the one used in the cold experiments, while keeping the same ratio of copper/sulfur (or copper/selenium) to 13%. In a typical reaction, 20–10 µL of the NC solution (0.3–0.6 mm in sulfur or selenium respectively, 0.9–0.5 µg of NCs) was diluted in 100 µL of an MES buffer (0.05 m at pH 5.5), then it was mixed with 2 µL of AA (0.1 m) and finally a 64Cu solution was added to the reaction mixture. In this case, the CE reaction temperature was set at 50 °C and the reaction was run for 1 h. The radiolabeling yield was assessed via radio‐TLC, using glass microfiber chromatography paper impregnated with silica gel (iTLC‐SG) as stationary phase and 0.1 m EDTA as mobile phase (Figure 2b). The radio‐TLC run revealed that, in the presence of NCs, the radioactivity was mostly located at the origin, while in case of free 64Cu the spot migrated with the solvent front. The integration of the TLC peaks at the deposition point and at the front of the solvent enabled us to estimate the radiochemical yield (RCY) that is associated with the NCs. For all types of NCs, quantitative radiolabeling was found (RCY > 99% ∼just after the reaction; Figure 2b; Figure S9, Supporting Information).
The stability of the radiolabeled NCs was tested by incubating the reaction mixture (after CE reaction) in EDTA 0.1 m for 20 min (Figure S9, Supporting Information). The integration of peaks from radio‐TLC chromatogram of radio‐TLC showed that less than 2% of 64Cu was released, meaning that non‐specific Cu absorption by the polymer at the surface of NCs could be excluded (the adsorbed 64Cu ions would be easily washed out by EDTA) and a high RCY can be achieved with this CE protocol (Figure 2c).
To verify the colloidal stability of 64Cu:NCs during the CE reaction and after the following cleaning steps (which could consequently cause a loss of the radiolabeled NCs), all plastic parts involved in the radiolabeling (cartridge, vials, pipette tips, etc.) were measured with a gamma counter (Figures S4 and S10a, Supporting Information). Most of the activity was recorded in the NC fraction (53–57%), but some radioactivity (5.9–16%) was also found on the amicon centrifuge cartridge, indicating a minimal loss of radiolabeled NC. Another significant amount of activity (≈30%) was collected in the empty plastic vials and pipette tips, indicating a partial adhesion of the NCs to the plastic materials. Overall, the sum of the recovered activity was close to 97% of the initial one. We used a similar test not only for NCs with multidentate ligands (Figure 1) but also for NCs coated with a mono‐dentate PEG‐thiol polymer (SH‐PEG2000‐OCH3) and exposed to the same CE reaction protocol. In the case of NCs coated with SH‐PEG2000‐OCH3, 60% of the radiolabeled material adhered to the amicon cartridge, and just 3% of the radioactivity was recovered with the NC solution (Figure S4, Supporting Information). This higher aggregation and instability of mono‐dentate PEG coated NCs is likely caused by the ligand depletion from NCs surface upon the CE reaction, as shown in our previous paper.[ 25 ] Thus the multi‐anchoring polymer with cysteamine was preferred to a monodentate polymer for all the following studies, due to the higher colloidal stability during CE reactions, especially when using 64Cu‐labeling.
Purified 64Cu:NCs were also tested in human serum to evaluate 64Cu leakage. The NCs were diluted in pure serum (200 µL) and incubated for 1 or 24 h at 37 °C. The amount of 64Cu that was released was determined via radio‐TLC in EDTA 0.1 m (Figure S11, Supporting Information). 64Cu:CuFeS2 is quite stable for 1 h in the presence of human serum. On the other hand, a significant 64Cu release is observed for 64Cu:ZnS and 64Cu:ZnSe after just 1 h, but especially after 24 h. It is worth noting that the 64Cu leakage is comparable to the leakage that was measured by ICP when carrying out cold copper reactions (Figure 2d,e). Data from both tests suggest a lower copper release for 64Cu:CuFeS2 NCs in comparison to 64Cu:ZnSe and 64Cu:ZnS NCs (Figure 2e).
These CE reactions were first performed at 50 °C. However, such temperature can be harmful for radiolabeling of NCs bearing temperature‐sensitive biomolecules (e.g., an antibodies, peptides, etc.).[ 25 ] We have therefore repeated the CE reaction at body temperature 37 °C and used experimental conditions similar to those of the first set of reactions performed at 50 °C (Figure 1). As expected, at 37 °C, for all NC compositions, lower RCYs were found as compared to the reaction performed at 50 °C (RCYs between 30% and 50% with respect to almost 100%) (Figure 3 ; Figure S11, Supporting Information).
Figure 3.
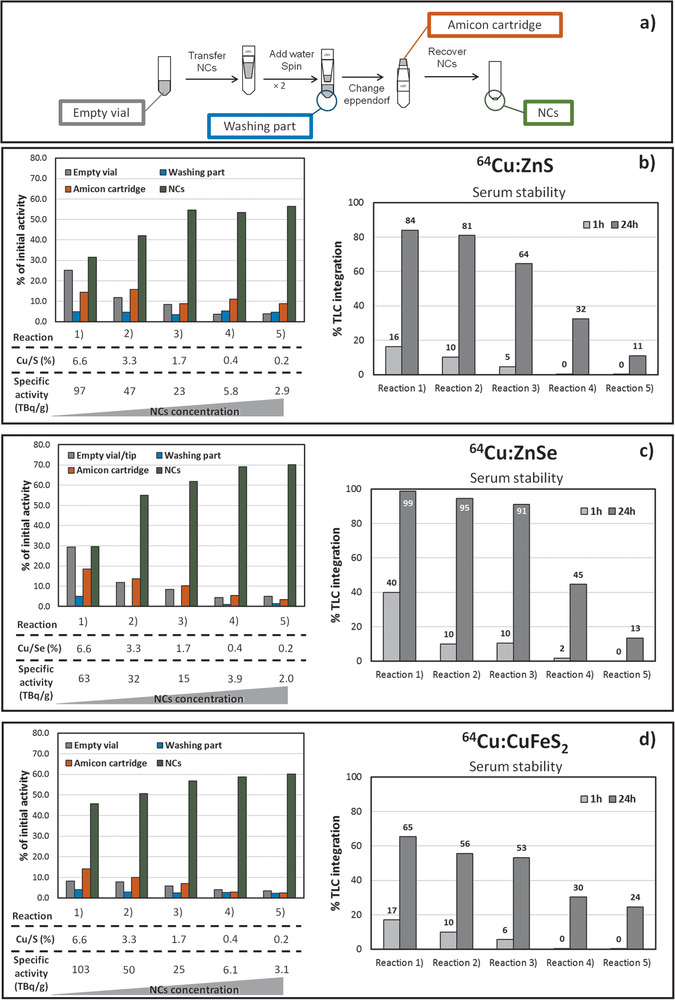
a) Scheme of amicon purification upon CE reaction. Set of CE reactions performed at 37 °C on different NCs compositions. To tune the specific activity, by keeping the 64Cu activity (18.5 MBq) the amount of NCs was varied in the range from 0.2 to 9.4 µg. Radio‐distribution test: Monitoring the radioactivity on the reaction components after CE reactions as a percentage of the initial 64Cu activity: NCs (green); washing solutions (blue); amicon filters (orange); and the vials (grey). Stability of 64Cu:NCs in serum. 64Cu leakage is given as percentage of the total radioactivity in a single TLC strip. Radio‐stability test: Radio‐TLC integration after incubation for 1h or 24h for b) 64Cu:ZnS, c) 64Cu:ZnSe, and d) 64Cu:CuFeS2 NCs fraction after CE reaction.
In this case, however, the lower RCY was not related to the loss of stability of the NCs, as indeed only negligible fractions of radioactivity were found either in the reaction vials or in the amicon cartridge after purification, but it is clearly related to the lower temperature used for the CE reaction. However, even at 37 °C, by increasing the amount of NCs, the RCY improved, and the NCs aggregated less on the amicon cartridge and vial (Figure 3). Keeping constant the overall feeding 64Cu activity, by varying the amount of NCs from 0.2 to 6–9 µg, we could tune the specific activity of NCs from 60–100 to 2–3 TBq g−1. A specific activity of 60–100 TBq g−1 NC is a record value, never reported so far (Table S2, Supporting Information). However, the radiolabeling stability of the NCs in the human serum was strongly compromised and a substantial leakage of 64Cu was measured at 24 h (Figure 3). On the other hand, for local tumor treatment using intra‐tumoral injection, NCs at higher specific activities (60–100 TBq g−1) may be used, since the 64Cu:NCs are trapped in the tumor.
Instead, when investigating the 64Cu:NCs with a specific activity as low as 2–3 TBq g−1, there was an increase of stability in human serum for all the three material compositions (Figure 3). The 64Cu percentage that was released into the serum media ranged from 11% to 24% after 24 h incubation. These leakage values are in line with those of other radiolabeling approaches. It is worth highlighting that, usually, the specific activity of radiolabeled NCs is not considered. But this value can be easily calculated based on the total activity associated to the NC fraction and knowing the corresponding amount of the nanomaterial used in the radiolabeling experiment (Table S2, Supporting Information). We would like to stress that, when using our CE protocols, even for values as low as 2–3 TBq g−1, the specific activity is still higher than that of other 64Cu‐labeled NCs prepared using a surface‐bound chelating ligands[ 33 ] and some chelator‐free radiolabeled NCs (Table S2, Supporting Information).[15a, 24c]
As previously mentioned, for ZnSe and ZnS NCs complete CE on those materials leads to the formation of copper‐deficient chalcogenides, with the appearance of an LSPR band in the near infrared. Even in case of partial exchange, upon 13% of Cu exchange, the absorption spectra are modified (Figure 4 ): The initial ZnSe and ZnS NCs, that are not photo‐thermally active, upon slightly exchanged with Cu, showed a thermal response upon 808 nm laser irradiation. Instead, chalcopyrite CuFeS2 NCs that are already photo‐thermally active prior to the Cu exchange,[26b] upon replacement of 13% of Cu showed a slight shift in the absorption spectrum, but the partially exchanged Cu:CuFeS2 NCs are still thermally active under laser irradiation (Figure 4c). These spectral features also guarantee that the Cu exchanged NCs are all thermally active under an IR laser irradiation, as recorded by the temperature changes under an IR camera (Figure 4). The temperature that could be reached, after the Cu exchange by the same NC dose, was higher for Cu:CuFeS2 NCs than for Cu:ZnS and Cu:ZnSe NCs (76 °C for Cu:CuFeS2 versus 33 °C for Cu:ZnSe and 26 °C for Cu:ZnS). For this reason, we have selected the CuFeS2 NCs, and on this sample the exchange was performed with 64Cu ions (ratio 64Cu/NCs at 0.2%, see Supporting Information for more details).
Figure 4.
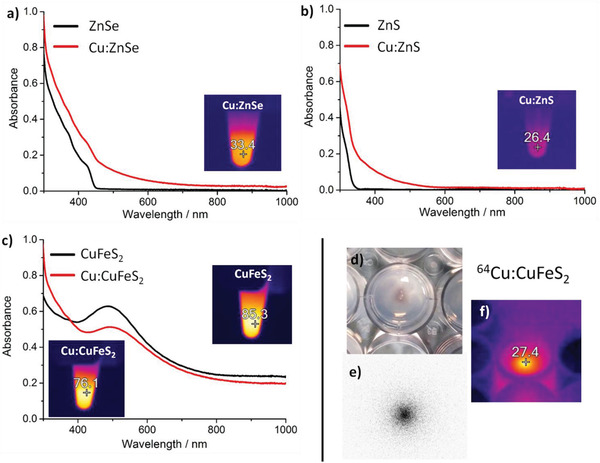
a) Absorption spectra of ZnSe and Cu:ZnSe NCs,b) ZnS and Cu:ZnS NCs and c) CuFeS2 and Cu:CuFeS2, before (black line) and after (red line) undergoing a 13% Cu exchange; the inset panel is a IR image of the Cu:NCs after 3 min of continuous exposure to an 808 nm laser at 2 W cm−2. A digital picture (panel d) and the radioluminography (panel e) of a tiny drop (35 µL) of 64Cu:CuFeS2 NCs (exchanged at 0.2%) loaded on agarose gel (0.5% in PBS 1X). Also, the IR image taken after 20 min of exposure to an 800 nm laser at 2 W cm−2 is shown (f).
Under an NIR‐laser irradiation (808 nm, 2 W cm−1), on a concentrated spot of the 64Cu/NCs sample loaded on agarose gel phantom, a light purple spot appeared under the IR camera, indicating the temperature rise from room temperature (20 °C) to 27 °C (Figure 4d). At the same time, the radioluminography image shows that radioactivity was localized in the deposition point (Figure 4e). Given the temperature reached under laser irradiation even when only a tiny fraction of 64Cu ions (0.2%) is exchanged, these data highlight the possible use of the exchanged 64Cu:CuFeS2 chalcopyrite NCs as radio‐ and photo‐thermal therapeutic tools, in which the two therapeutic modalities can be combined by using a single nanoplatform.
To provide a proof of concept for such dual therapeutic effects, an in vitro cellular study was performed on two cancer cell lines, the human glioblastoma cell line (U87) and the human epidermoid carcinoma (A431). For these tests, the cell pellets (two million cells) mimicking a tiny tumor mass were incubated with radiolabeled 64Cu:CuFeS2 NCs or with 64CuCl2 solution with or without the exposure to an IR laser irradiation of clinical use (808 nm and 1.7 W cm−2 for 13 min) for three short cycles (Figure 5a).
Figure 5.
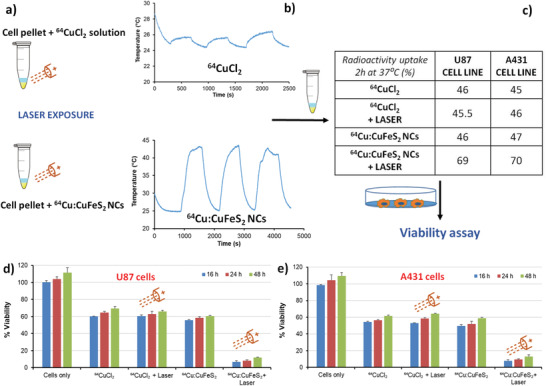
a) Schematic figure depicting the 808 nm laser irradiation on cell pellet treated with either 64CuCl2 or 64Cu:CuFeS2 NC solution. b) Temperature profiles upon laser irradiation for a cell pellet treated with a 64CuCl2 (top) or with a 64Cu:CuFeS2 NC solution (bottom). c) Radio‐labeling test for cell uptake on two different cell lines for 64CuCl2 or 64Cu:CuFeS2 NC with and without the effect of laser irradiation. Cell viability upon the dual action of radiotherapy and laser treatment on d) U87 cells and e) A431 cells.
During the laser exposure, the temperature of the pellet treated with 64Cu:CuFeS2 NCs increased remarkably when exposed to the 808 nm laser. In contrast, the cell samples exposed to 64CuCl2 solution showed only a few degrees (3 °C) temperature variation due to laser light effect (Figure 5b). Interestingly, at 2 h post‐irradiation at 37 °C, the 64Cu cellular uptake measured in terms of radioactivity associated to the cell pellet for the 64Cu:CuFeS2 NCs in the presence of the laser increased up to 70% with respect to the total 64Cu dose initially administered (Figure 5c). This activity uptake percentage was certainly higher than that of the pellet sample treated with 64Cu:CuFeS2 NCs which was not exposed to IR irradiation (in this case only 45% of the radioactive dose was associated to the cell pellet when no laser was used, Figure 5c). Moreover, in this latter case, the radioactive percentage of uptake was very similar to that of the 64CuCl2 solutions, with or without laser exposure (≈45–47%, Figure 5c data in the table). Reasonably, the hyperthermia effect induced by the photo‐thermal treatment led to enhanced cell membrane permeability with consecutively enhanced uptake of NCs and, in turn, 64Cu radioisotopes uptake associated to the NCs.
To evaluate the toxicity of the radioactive NCs, a viability test (presto blue) was carried out. For these trials, the same amount of radioactivity (37 MBq) was administered to the cells either in the form of 64CuCl2 solutions or as 64Cu:CuFeS2 NCs. After irradiation and 2 h incubation at 37 °C (Figure 5a), the cells were re‐cultured and the viability was measured at well‐defined time points post‐incubation and compared to cells exposed to radioactive NCs without NIR irradiation. After 16 h, while 64CuCl2 alone could kill 40% (U87) and 45% (A431) of the cells, the addition of laser irradiation to the 64CuCl2 solutions did not cause any additional cytotoxic effects to the cell populations (viability was of 40% for U87 and 47% for A431 and very similar to the 64CuCl2 solutions with no laser exposure). Radiolabeled 64Cu:CuFeS2 NCs alone could eliminate 45% (U87) and 51% (A431) of the cells, a percentage slightly higher than that of free 64CuCl2 solutions. In case of additional laser irradiation, the combination of radiotoxicity and photo‐thermal heat effects mediated by 64Cu:CuFeS2 could significantly accelerate the cell killing (percentage of viability as low as less than 10% indicates remarkable toxicity effects). The difference in cytotoxicity appears already after 16 h of incubation (93% of cell elimination for U87 and 92% for A431) and it manages to reduce the cell densities to less than 10% viability up to 48 h (Figures 5d and 4e). These data indicate that damages by laser and radioactivity were so critical that the cells were not able to recover even after the half‐life of the 64Cu radioisotopes were significantly reduced (the half life of 64Cu is 12.7 h and of the average dose associated to the cells the radioactivity at 48 h should be in the range of 1.1–1.85 MBq).
We also wondered about the intrinsic toxicity of the NCs at a dose range close to the amount of NCs needed for the radiolabeling experiments on the cells. To evaluate this toxicity, pristine ZnS, ZnSe, and CuFeS2 NCs were administered to the two tumor cells (U87 and A431 cells also used for the dual modal therapeutic experiments and the presto blue proliferation assays was assessed (Figure S12, Supporting Information). The NCs were added to the cell culture media, and incubated at 37 °C for 24 h. On both cell lines, for all the three materials' compositions, the toxicity was not significant as the viability was more than 80% (Figure S12, Supporting Information). These data suggest that the doses employed for the radio experiments on cells using the three different compositions of NCs are safe.
3. Conclusions
We have here set simple post‐synthesis CE protocols to prepare 64Cu‐radiolabeled NCs, on Cd‐free chalcogenide NCs made of ZnSe, ZnS, and CuFeS2 NCs. This method relies on the exchange of the pristine cations of the NCs with 64Cu ions when employing physiologically stable chalcogenide NCs. The CE reactions were performed by simply mixing the NCs in MES buffer (pH 5.5) in the presence of ascorbic acid (used as a mild reducing agent for 64CuCl2). Radiolabeling can occur at 50 °C or at 37 °C and in a short time (60 min). The radiolabeled NCs had a high radiochemical yield (RCY, >99%) and high radiochemical purity (close to 100%). The high RCY enables to use these NCs without any post‐radiolabeling purifications steps. This is advantageous, as it reduces the time lag between 64Cu production and the use of the 64Cu‐NCs, particularly suitable for 64Cu having a half‐life of about 13 h. Moreover, by fixing the amount of 64Cu and by tuning the NCs dose we could tune the specific activity from 2 to 103 TBq g−1, reaching the highest and unprecedented specific activity when using a very tiny dose of NC material (µg amounts). Finally, the radiolabeling protocol being the last step of the preparation pipeline and being performed on physiologically stable NCs, this post‐synthesis radio‐protocol could be easily implemented in hospitals.
Among the three NC compositions studied, 64Cu:CuFeS2 chalcopyrite NCs maintained their photo‐thermal properties even after the partial radiolabeling reaction (13% exchange) while producing remarkable ionizing effects. This may pave the way for the production of a double‐therapeutic tools in which the intrinsic photo‐thermal properties of the material and the radiation from the radionuclide are combined in a single nano‐object as here shown in vitro on two cancer cell lines. At the same time, negligible intrinsic material toxicity has been recorded at the µg dose of NCs used for the dual therapeutic effects.
With regard to the stability of 64Cu:NCs in serum, it is worth highlighting that tuning the 64Cu/NC ratio could be a useful parameter to be adjusted based on the aimed application of the NCs. At lower specific activity it is possible to have serum‐stable radiolabeled 64Cu:NCs useful for imaging applications (PET). Instead, at higher specific activity serum stability is compromised. However these fully exchanged 64Cu:NCs may be more suitable for local tumor treatment upon intratumoral injection as in this treatment no direct exposure to serum is expected while reaching a high specific activity and photo‐thermal hyperthermia with a very tiny µg dose of NCs is achievable.
Conflict of Interest
The authors declare no conflict of interest.
Supporting information
Supporting Information
Acknowledgements
T.A. and N.S. contributed equally to this work. This work was partially funded by the European Research Council (starting grant ICARO, Contract No. 678109) and partially by the Programme for research and Innovation Horizon 2020 (2014–2020) under the Marie Skłodowska‐Curie Grant Agreement COMPASS No. 691185. The HZDR is thanked for supporting the studies. The authors thank Karin Landrock for excellent technical assistance.
Avellini T., Soni N., Silvestri N., Fiorito S., De F. Donato, De C. Mei, Walther M., Cassani M., Ghosh S., Manna L., Stephan H., Pellegrino T., Cation Exchange Protocols to Radiolabel Aqueous Stabilized ZnS, ZnSe, and CuFeS2 Nanocrystals with 64Cu for Dual Radio‐ and Photo‐Thermal Therapy. Adv. Funct. Mater. 2020, 30, 2002362 10.1002/adfm.202002362
Contributor Information
Tommaso Avellini, Email: tommaso.avellini@gmail.com.
Teresa Pellegrino, Email: teresa.pellegrino@iit.it.
References
- 1. Kriegel I., Jiang C. Y., Rodriguez‐Fernandez J., Schaller R. D., Talapin D. V., da Como E., Feldmann J., J. Am. Chem. Soc. 2012, 134, 1583. [DOI] [PubMed] [Google Scholar]
- 2.a) Coughlan C., Ibanez M., Dobrozhan O., Singh A., Cabot A., Ryan K. M., Chem. Rev. 2017, 117, 5865; [DOI] [PubMed] [Google Scholar]; b) Yan C. L., Tian Q. W., Yang S. P., RSC Adv. 2017, 7, 37887. [Google Scholar]
- 3.a) Tian Q. W., Jiang F. R., Zou R. J., Liu Q., Chen Z. G., Zhu M. F., Yang S. P., Wang J. L., Wang J. H., Hu J. Q., ACS Nano 2011, 5, 9761; [DOI] [PubMed] [Google Scholar]; b) Wang S. H., Riedinger A., Li H. B., Fu C. H., Liu H. Y., Li L. L., Liu T. L., Tan L. F., Barthel M. J., Pugliese G., De Donato F., D'Abbusco M. S., Meng X. W., Manna L., Meng H., Pellegrino T., ACS Nano 2015, 9, 1788. [DOI] [PubMed] [Google Scholar]
- 4.a) Hessel C. M., Pattani V. P., Rasch M., Panthani M. G., Koo B., Tunnell J. W., Korgel B. A., Nano Lett. 2011, 11, 2560; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang S. H., Sun C. X., Zeng J. F., Sun Q., Wang G. L., Wang Y., Wu Y., Dou S. X., Gao M. Y., Li Z., Adv. Mater. 2016, 28, 8927. [DOI] [PubMed] [Google Scholar]
- 5. De Trizio L., Manna L., Chem. Rev. 2016, 116, 10852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Lubeck C. R., Han T. Y. J., Gash A. E., Satcher J. H., Doyle F. M., Adv. Mater. 2006, 18, 781; [Google Scholar]; b) Tan C. S., Hsiao C. H., Wang S. C., Liu P. H., Lu M. Y., Huang M. H., Ouyang H., Chen L. J., ACS Nano 2014, 8, 9422; [DOI] [PubMed] [Google Scholar]; c) Wang Y. X., Zhukovskyi M., Tongying P., Tian Y., Kuno M., J. Phys. Chem. Lett. 2014, 5, 3608; [DOI] [PubMed] [Google Scholar]; d) Manzi A., Simon T., Sonnleitner C., Doblinger M., Wyrwich R., Stern O., Stolarczyk J. K., Feldmann J., J. Am. Chem. Soc. 2015, 137, 14007; [DOI] [PubMed] [Google Scholar]; e) Shao H. B., Wang C. L., Xu S. H., Wang Z. Y., Yin H. H., Cui Y. P., J. Fluoresc. 2015, 25, 305. [DOI] [PubMed] [Google Scholar]
- 7.a) Li H. B., Zanella M., Genovese A., Povia M., Falqui A., Giannini C., Manna L., Nano Lett. 2011, 11, 4964; [DOI] [PubMed] [Google Scholar]; b) Li H. B., Brescia R., Krahne R., Bertoni G., Alcocer M. J. P., D'Andrea C., Scotognella F., Tassone F., Zanella M., De Giorgi M., Manna L., ACS Nano 2012, 6, 1637. [DOI] [PubMed] [Google Scholar]
- 8.a) Qaim S. M., Radiochim. Acta 2012, 100, 635; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Cutler C. S., Hennkens H. M., Sisay N., Huclier‐Markai S., Jurisson S. S., Chem. Rev. 2013, 113, 858; [DOI] [PubMed] [Google Scholar]; c) Boros E., Packard A. B., Chem. Rev. 2019, 119, 870; [DOI] [PubMed] [Google Scholar]; d) Kostelnik T. I., Orvig C., Chem. Rev. 2019, 119, 902. [DOI] [PubMed] [Google Scholar]
- 9.a) Yoshii Y., Furukawa T., Kiyono Y., Watanabe R., Mori T., Yoshii H., Asai T., Okazawa H., Welch M. J., Fujibayashi Y., Nucl. Med. Biol. 2011, 38, 151; [DOI] [PubMed] [Google Scholar]; b) Thieme S., Walther M., Pietzsch H.‐J., Henniger J., Preusche S., Mäding P., Steinbach J., Appl. Radiat. Isot. 2012, 70, 602; [DOI] [PubMed] [Google Scholar]; c) Guo Y., Parry J. J., Laforest R., Rogers B. E., Anderson C. J., J. Nucl. Med. 2013, 54, 1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wadas T. J., Wong E. H., Weisman G. R., Anderson C. J., Chem. Rev. 2010, 110, 2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) McCarthy D. W., Shefer R. E., Klinkowstein R. E., Bass L. A., Margeneau W. H., Cutler C. S., Anderson C. J., Welch M. J., Nucl. Med. Biol. 1997, 24, 35; [DOI] [PubMed] [Google Scholar]; b) Shokeen M., Anderson C. J., Acc. Chem. Res. 2009, 42, 832; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Matarrese M., Bedeschi P., Scardaoni R., Sudati F., Savi A., Pepe A., Masiello V., Todde S., Gianolli L., Messa C., Fazio F., Appl. Radiat. Isot. 2010, 68, 5; [DOI] [PubMed] [Google Scholar]; d) Hoberück S., Wunderlich G., Michler E., Hölscher T., Walther M., Seppelt D., Platzek I., Zöphel K., Kotzerke J., J. Labelled Compd. Radiopharm. 2019, 62, 523; [DOI] [PubMed] [Google Scholar]; e) Kreller M., Pietzsch H. J., Walther M., Tietze H., Kaever P., Knieß T., Füchtner F., Steinbach J., Preusche S., Instruments 2019, 3, 9. [Google Scholar]
- 12.a) Obata A., Kasamatsu S., Lewis J. S., Furukawa T., Takamatsu S., Toyohara J., Asai T., Welch M. J., Adams S. G., Saji H., Yonekura Y., Fujibayashi Y., Nucl. Med. Biol. 2005, 32, 21; [DOI] [PubMed] [Google Scholar]; b) Yoshii Y., Furukawa T., Matsumoto H., Yoshimoto M., Kiyono Y., Zhang M. R., Fujibayashi Y., Saga T., Cancer Lett. 2016, 376, 74; [DOI] [PubMed] [Google Scholar]; c) Yoshii Y., Matsumoto H., Yoshimoto M., Zhang M. R., Oe Y., Kurihara H., Narita Y., Jin Z. H., Tsuji A. B., Yoshinaga K., Fujibayashi Y., Higashi T., Transl. Oncol. 2018, 11, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chakravarty R., Goel S., Dash A., Cai W. B., Q. J. Nucl. Med. Mol. Imaging 2017, 61, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.a) Sun X. L., Huang X. L., Guo J. X., Zhu W. L., Ding Y., Niu G., Wang A., Kiesewetter D. O., Wang Z. L., Sun S. H., Chen X. Y., J. Am. Chem. Soc. 2014, 136, 1706; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Sun X. L., Huang X. L., Yan X. F., Wang Y., Guo J. X., Jacobson O., Liu D. B., Szajek L. P., Zhu W. L., Niu G., Kiesewetter D. O., Sun S. H., Chen X. Y., ACS Nano 2014, 8, 8438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.a) Zhou M., Zhang R., Huang M. A., Lu W., Song S. L., Melancon M. P., Tian M., Liang D., Li C., J. Am. Chem. Soc. 2010, 132, 15351; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Pang B., Zhao Y., Luehmann H., Yang X., Detering L., You M., Zhang C., Zhang L., Li Z. Y., Ren Q., Liu Y., Xia Y., ACS Nano 2016, 10, 3121. [DOI] [PubMed] [Google Scholar]
- 16. Zhou M., Chen Y. Y., Adachi M., Wen X. X., Erwin B., Mawlawi O., Lai S. Y., Li C., Biomaterials 2015, 57, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.a) Zhou M., Li J., Liang S., Sood A. K., Liang D., Li C., ACS Nano 2015, 9, 7085; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhou M., Zhao J., Tian M., Song S., Zhang R., Gupta S., Tan D., Shen H., Ferrari M., Li C., Nanoscale 2015, 7, 19438. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 18.a) Schipper M. L., Cheng Z., Lee S. W., Bentolila L. A., Iyer G., Rao J. H., Chen X. Y., Wul A. M., Weiss S., Gambhirl S. S., J. Nucl. Med. 2007, 48, 1511; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jarrett B. R., Gustafsson B., Kukis D. L., Louie A. Y., Bioconjugate Chem. 2008, 19, 1496; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Hong H., Zhang Y., Sun J. T., Cai W. B., Nano Today 2009, 4, 399; [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Xie J., Chen K., Huang J., Lee S., Wang J., Gao J., Li X., Chen X., Biomaterials 2010, 31, 3016; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Barreto J. A., Matterna M., Graham B., Stephan H., Spiccia L., New J. Chem. 2011, 35, 2705. [Google Scholar]
- 19. Choi H. S., Liu W., Misra P., Tanaka E., Zimmer J. P., Ipe B. I., Bawendi M. G., Frangioni J. V., Nat. Biotechnol. 2007, 25, 1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.a) Imhof A., Brunner P., Marincek N., Briel M., Schindler C., Rasch H., Macke H. R., Rochlitz C., Muller‐Brand J., Walter M. A., J. Clin. Oncol. 2011, 29, 2416; [DOI] [PubMed] [Google Scholar]; b) Jin Z. H., Furukawa T., Sogawa C., Claron M., Aung W., Tsuji A. B., Wakizaka H., Zhang M. R., Boturyn D., Dumy P., Fujibayashi Y., Saga T., Eur. J. Pharm. Biopharm. 2014, 86, 478. [DOI] [PubMed] [Google Scholar]
- 21. Cai W. B., Chen K., Li Z. B., Gambhir S. S., Chen X. Y., J. Nucl. Med. 2007, 48, 1862. [DOI] [PubMed] [Google Scholar]
- 22. Kreyling W. G., Abdelmonem A. M., Ali Z., Alves F., Geiser M., Haberl N., Hartmann R., Hirn S., de Aberasturi D. J., Kantner K., Khadem‐Saba G., Montenegro J. M., Rejman J., Rojo T., de Larramendi I. R., Ufartes R., Wenk A., Parak W. J., Nat. Nanotechnol. 2015, 10, 619. [DOI] [PubMed] [Google Scholar]
- 23. Goel S., Chen F., Ehlerding E. B., Cai W. B., Small 2014, 10, 3825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.a) Wong R. M., Gilbert D. A., Liu K., Louie A. Y., ACS Nano 2012, 6, 3461; [DOI] [PubMed] [Google Scholar]; b) Zhao Y. F., Sultan D., Detering L., Luehmann H., Liu Y. J., Nanoscale 2014, 6, 13501; [DOI] [PubMed] [Google Scholar]; c) Chakravarty R., Chakraborty S., Ningthoujam R. S., Nair K. V. V., Sharma K. S., Ballal A., Guleria A., Kunwar A., Sarma H. D., Vatsa R. K., Dash A., Ind. Eng. Chem. Res. 2016, 55, 12407. [Google Scholar]
- 25. Riedinger A., Avellini T., Curcio A., Asti M., Xie Y., Tu R. Y., Marras S., Lorenzon A., Rubagotti S., Iori M., Capponi P. C., Versari A., Manna L., Seregn E., Pellegrino T., J. Am. Chem. Soc. 2015, 137, 15145. [DOI] [PubMed] [Google Scholar]
- 26.a) Joo J., Na H. B., Yu T., Yu J. H., Kim Y. W., Wu F., Zhang J. Z., Hyeon T., J. Am. Chem. Soc. 2003, 125, 11100; [DOI] [PubMed] [Google Scholar]; b) Ghosh S., Avellini T., Petrelli A., Kriegel I., Gaspari R., Almeida G., Bertoni G., Cavalli A., Scotognella F., Pellegrino T., Manna L., Chem. Mater. 2016, 28, 4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mei B. C., Susumu K., Medintz I. L., Mattoussi H., Nat. Protoc. 2009, 4, 412. [DOI] [PubMed] [Google Scholar]
- 28. Wang W. T., Ji X., Kapur A., Zhang C. Q., Mattoussi H., J. Am. Chem. Soc. 2015, 137, 14158. [DOI] [PubMed] [Google Scholar]
- 29. Wang W. T., Ji X., Na H. B., Safi M., Smith A., Palui G., Perez J. M., Mattoussi H., Langmuir 2014, 30, 6197. [DOI] [PubMed] [Google Scholar]
- 30. Li H., Brescia R., Krahne R., Bertoni G., Alcocer M. J., D'Andrea C., Scotognella F., Tassone F., Zanella M., De Giorgi M., ACS Nano 2012, 6, 1637. [DOI] [PubMed] [Google Scholar]
- 31.a) Dorfs D., Hartling T., Miszta K., Bigall N. C., Kim M. R., Genovese A., Falqui A., Povia M., Manna L., J. Am. Chem. Soc. 2011, 133, 11175; [DOI] [PubMed] [Google Scholar]; b) Comin A., Manna L., Chem. Soc. Rev. 2014, 43, 3957. [DOI] [PubMed] [Google Scholar]
- 32. Gaspari R., Della Valle G., Ghosh S., Kriegel I., Scotognella F., Cavalli A., Manna L., Nano Lett. 2017, 17, 7691. [DOI] [PubMed] [Google Scholar]
- 33. Cai W. B., Chen K., Li Z. B., Gambhir S. S., Chen X. Y., J. Nucl. Med. 2007, 48, 1862. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information