

Abstract
Objective:
The objective of this report is to present 2 cases of cardiac paragangliomas (PGLs), and to outline the presentation, management, and associated genetic mutations.
Methods:
Case 1, a 38-year-old female, presented with a 12-month history of paroxysmal palpitations, headaches, and weight loss. Her investigations included plasma free metanephrines and urinary metanephrines, 68-gallium DOTATATE positron emission tomography/computed tomography, and cardiac imaging. Case 2, a 28-year-old male, presented with a hypertensive crisis and abdominal pain on a background of hypertension. Given his abdominal pain, he was investigated with an abdominal computed tomography (CT) scan, followed by plasma free meta-nephrines and urinary metanephrines, echocardiogram, and 123-iodine meta-iodobenzylguanidine single-photon emission CT.
Results:
Case 1 had an elevated plasma normetadrenaline of 6,750 pmol/L (reference range is <900 pmol/L) and 3-methoxytyramine of 1,845 pmol/L (reference range is <110 pmol/L). 68-gallium DOTATATE positron emission tomography/computed tomography showed an avid cardiac lesion. The lesion was resected, and histopathology confirmed PGL. Genetic studies revealed an SDHC gene mutation. For case 2, abdominal CT revealed a para-spinal mass. Workup for this lesion revealed elevated normetadrenaline of 56,000 pmol/L (reference range is <900 pmol/L). An echocardiogram, arranged for investigation of hypertension, showed an additional cardiac mass. A 123-iodine meta-iodobenzylguanidine single-photon emission CT scan confirmed that both masses were functioning. The lesions were successfully excised. He was found to have an SDHB gene mutation.
Conclusion:
Both patients had long-standing symptoms secondary to catecholamine excess, thus it is important to promptly screen patients with unexplained hypertension or paroxysmal symptoms of palpitations, headaches, and diaphoresis with plasma free metanephrines or urinary metanephrines. All patients with PGLs should be offered genetic testing due to the high incidence of genetic mutations.
INTRODUCTION
Paragangliomas (PGLs) are rare neuroendocrine tumors arising from the extra-adrenal autonomic paraganglia, which are clusters of neuroendocrine cells that produce catecholamines. Sympathetic PGLs are typically located in the abdomen. Less commonly they can occur in other sites, including the chest in <2% of cases (1,2). We report 2 interesting cases of cardiac PGLs, both presenting to a regional hospital in Queensland, Australia within a 10-year period.
CASE REPORT
Case 1
A 38-year-old female presented to a regional center in Queensland, Australia with a 12-month history of paroxysmal severe headaches and palpitations. She had measured a heart rate of 120 to 130 beats per minute during these paroxysms. She also described having unintentional weight loss of 8 kg. She had previously presented to multiple local general practitioners with these symptoms and had been incorrectly diagnosed with migraines and an anxiety disorder.
Her examination was unremarkable. She had no Cushingoid features, she was normotensive with a blood pressure of 127/83 mm Hg, and had a regular heart rate of 75 beats per minute. The patient had no past medical history, other than a gynecological procedure under general anesthesia in 2005, from which she had no intraoperative or postoperative complications. She took no regular medications and had no family history of neuroendocrine pathology, or sudden deaths.
Investigations
Initial biochemical testing revealed elevated plasma normetadrenaline of 6,750 pmol/L (reference range is <900 pmol/L) and 3-methoxytyramine of 1,845 pmol/L (reference range is <110 pmol/L). Urine metanephrine and catecholamine screens were also abnormal with elevated noradrenaline, dopamine, normetadrenaline, and 3-methoxytyramine levels (Table 1).
Table 1.
Case 1 Metanephrine and Catecholamine Measurements
| Urinary creatinine ratio | Reference range per mol creatinine | Urinary timed excretion over 24 hours | Reference range over 24 hours | |
|---|---|---|---|---|
| Noradrenaline | 353 | <55 μmol | 6,630 | <750 nmol |
| Adrenaline | 3 | <7 μmol | 56 | <80 nmol |
| Dopamine | 427 | <250 μmol | 8,040 | 200–3,500 nmol |
| Normetadrenaline | 0.82 | <0.25 mmol | 15 | <2.3 μmol |
| Metadrenaline | 0.03 | <0.10 mmol | 0.6 | <1.7 μmol |
| 3-Methoxytyramine | 1.31 | <0.15 mmol | 24.6 | <1.3 μmol |
| Plasma free metanephrines | Reference range | |||
| Normetadrenaline | 6,750 | <900 pmol/L | ||
| Metadrenaline | 183 | <500 pmol/L | ||
| 3-Methoxytyramine | 1,845 | <110 pmol/L | ||
Initial imaging included abdominal magnetic resonance imaging (MRI), which surprisingly was unremarkable. She proceeded to have a 68-gallium DOTATATE positron emission tomography/computed tomography scan, which revealed an intensely avid left atrial/interatrial septum lesion, consistent with a neuroendocrine tumor or PGL (Fig. 1). Cardiac MRI further defined the lesion and measured it to be 47 × 34 mm. An echocardiogram showed normal cardiac function with a mass noted in the interatrial groove on the apical 4-chamber view. The patient underwent preoperative assessment with a coronary angiogram, which showed 3 coronary artery tributaries feeding the tumor.
Fig. 1.
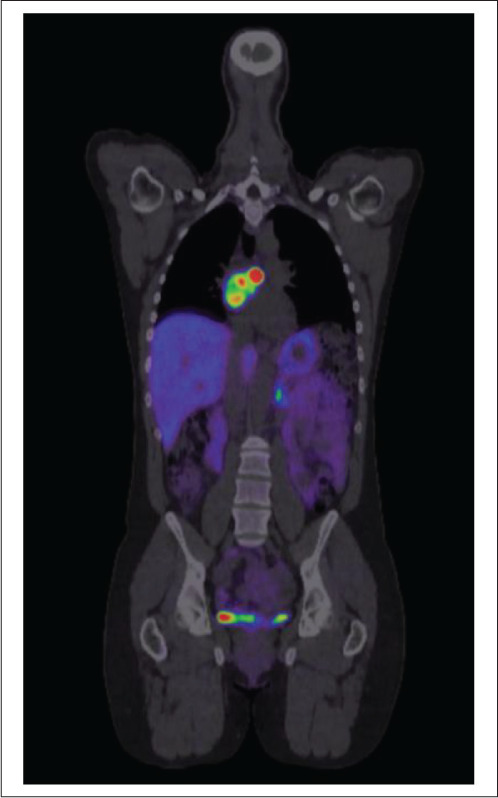
68-gallium DOTATATE positron emission tomography/computed tomography scan with intensely avid left cardiac lesion from case 1.
Management
Preoperatively, she was commenced on phenoxybenzamine at 10 mg twice daily, followed by metoprolol at 25 mg twice daily. In a combined cardiothoracic and vascular case at a tertiary center, a 70-mm tumor was resected from the intra-atrial groove (Fig. 2). The patient tolerated the procedure well and had an uncomplicated postoperative course. Histopathology confirmed PGL with no atypical mitotic figures seen, and genetic studies revealed a mutation in the succinate dehydrogenase (SDH) subunit C gene. Plasma metanephrines normalized postoperatively and her symptoms resolved.
Fig. 2.
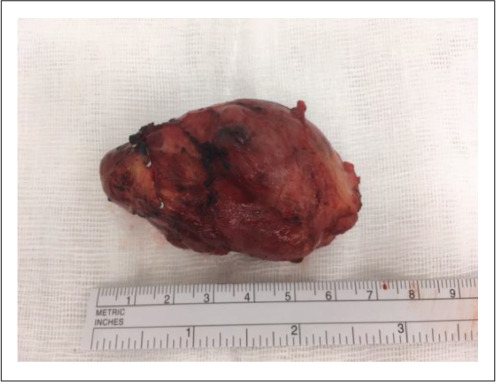
The 70-mm cardiac paraganglioma from case 1.
Case 2
Prior to case 1 by 7 years, a 28-year-old Aboriginal Australian male presented to the same center with a hypertensive crisis as well as abdominal pain, nausea, and vomiting. His symptoms had developed while playing football. On review he was hypertensive with a blood pressure of 220/110 mm Hg and tachycardic with a pulse rate of 120 beats per minute. This case was also the subject of a previous report by Marshall et al (3).
Preceding his presentation, he had a 2-year history of paroxysmal light-headedness and sweats on physical exertion. His past medical history included untreated hypertension, a previous unprovoked upper limb deep vein thrombosis, and smoking. He had no family history of neuroendocrine tumors.
Investigations
After presenting with abdominal pain, the patient was investigated with an abdominal computed tomography (CT) scan, which revealed a subdiaphragmatic paraspinal mass measuring 30 × 24 mm. Both adrenal glands were of normal radiologic appearance. Abdominal MRI further characterized and confirmed the mass. Subsequent plasma free and urinary metanephrine testing revealed markedly elevated normetadrenaline, with a plasma level of 56,000 pmol/L (Table 2).
Table 2.
Case 2 Metanephrine and Catecholamine Measurements
| Urinary creatinine ratio | Reference range per mol creatinine | Urinary timed excretion over 24 hours | Reference range over 24 hours | |
|---|---|---|---|---|
| Noradrenaline | 298 | <55 μmol | 6,050 | <750 nmol |
| Adrenaline | 1.8 | <7.0 μmol | 35 | <80 nmol |
| Dopamine | 128 | <250 μmol | 2,600 | 200–3,500 nmol |
| Normetadrenaline | 1.3 | <0.25 mmol | 27 | <2.3 μmol |
| Metadrenaline | 0.04 | <0.10 mmol | 0.71 | <1.7 μmol |
| 3-Methoxytyramine | 0.22 | <0.15 mmol | 4.4 | <1.3 μmol |
| Plasma free metanephrines | Reference range | |||
| Normetadrenaline | 56,000 | <900 pmol/L | ||
| Metadrenaline | 450 | <500 pmol/L | ||
An echocardiogram was arranged given his hyper-tensive crisis which unexpectedly revealed a right atrial mass. There was also left ventricular septal hypertrophy and a near full thickness left ventricular apical infarct with akinesis. A cardiac MRI scan further characterized this mass as a well-defined, uniformly enhancing mass centered on the right atrioventricular groove measuring 40 × 32 mm. Coronary angiogram showed no obstructive coronary artery disease and confirmed that the cardiac mass was supplied by branches of the right coronary artery. A 123-iodine meta-iodobenzylguanidine single-photon emission CT scan confirmed that both the intraabdominal and cardiac masses were associated with a large focus of activity.
Management
The patient was commenced on phenoxybenzamine, which was titrated to 30 mg twice daily, and subsequently metoprolol at 12.5 mg daily, with good clinical effect. At a tertiary center, his cardiac mass was resected en bloc with the involved right coronary artery, and a saphenous vein graft was performed for his right coronary artery repair. He also had a laparoscopic excision of his retrocaval mass. His postoperative plasma free metanephrines normalized and he ceased his antihypertensives.
Histopathology confirmed that both masses were PGLs and displayed cellular atypia with no evidence of necrosis or mitotic figures. Genetic testing revealed a SDH subunit B gene mutation. Unfortunately, the patient was lost to follow up, so appropriate screening in light of his genetic mutation has not been undertaken.
DISCUSSION
Here we report 2 cases of cardiac PGLs which manifested as paroxysmal symptoms due to catecholamine excess. For both of these patients, diagnosis was delayed due to a lack of clinical suspicion, despite typical symptoms, which is consistent with the literature. Diagnosis was delayed by a mean of 9.5 years in a cohort of patients with cardiac PGLs described by Liu et al (4).
Sympathetic PGLs are similar to pheochromocytomas, both clinically and at a cellular level. They produce catecholamines (adrenalin, noradrenalin, dopamine) which may cause symptoms of hypertension, episodic flushing, headache, sweating, and tachycardia. These tumors are typically located in the sympathetic trunk of the abdomen, although they can less commonly occur in the base of skull (5%), bladder and prostate (10%), and chest in <2% of cases (1,2). Parasympathetic PGLs differ in that they are typically located in the head and neck and do not produce catecholamines. PGLs have a higher rate of malignancy than pheochromocytomas, with rates estimated to be 14 to 50% (2).
The recommended screening tests for pheochromocytomas and PGLs are plasma free or urinary metanephrines, which are the inactive metabolites of catecholamines. Tumors related to SDH subunit mutations preferentially secrete high levels of normetadrenaline and 3-methoxytyramine (5). Values >2 times the upper limit of normal indicate a high probability of pheochromocytomas and PGLs, and imaging to localize disease is recommended. CT and MRI are highly sensitive in localizing tumors, although for patients with sporadic PGLs, or SDH subunit mutation-related tumors, additional whole-body imaging with 68-gallium DOTATATE positron emission tomography/computed tomography is recommended to exclude additional tumors or metastases (5).
Cardiac PGLs are rare. A review by Wang et al (1) in 2015 described 158 cases in the literature. In this review, typical catecholamine-induced symptoms at the time of diagnosis were present in 77.3% of patients. The incidence of malignancy, defined as the presence of a definite metastatic lesion, was reported to be 5.9%, with other case series reporting an incidence of 2.4 to 14%.
Up to 47% of PGLs are related to inherited syndromes, particularly in the genes for SDH subunits B, C, and D. This holds even in patients with no family history (6), which is consistent with our 2 cases. A prospective study of patients with PGLs found an SDH subunit gene mutation in 242 (54.4%) patients (7). Of these, a SDH subunit C mutation was found in only 16 (6.6%) patients and 14 of these patients had head and neck PGLs. The other 2 patients had thoracic PGLs. Another series of 8 patients with SDH subunit C mutations and PGLs found that 7 had head and neck tumors while 3 had thoracic tumors (2 had synchronous head and neck tumors) (8). In our study, case 1 was found to have a thoracic PGL and a SDH subunit C mutation, which is consistent with the observation that people with SDH subunit C mutations have tumors most commonly located in the head and neck, followed by the thorax (7).
SDH subunit B mutations, as detected in case 2, are mostly associated with thoraco-abdominal PGLs and have a higher association with malignancy and poor prognosis (7). The younger the patient is at diagnosis, the more likely they are to have familial disease and it is recommended that all patients with PGLs are tested for genetic mutations (9).
The treatment of cardiac PGLs is surgical resection; however complete resection is often difficult as these tumors are highly vascular and tend to involve the coronary arteries (1). A review by Liu et al (4) found that all 17 patients in their case series had cardiac PGLs supplied by coronary arteries. Thus, it is imperative that patients have a preoperative coronary angiogram to determine the coronary supply. Operative outcomes are generally favorable. In the cohort of 17 patients described by Liu et al (4), 1 patient died postoperatively, 2 patients developed tumor recurrence, and 14 remained symptom-free with normal urine catecholamines during a mean follow-up period of 6.5 years.
CONCLUSION
To have 2 cases of cardiac PGLs presenting to a regional center within a 10-year period is exceptionally rare. These cases highlight the importance of timely screening of individuals with unexplained hypertension or paroxysmal symptoms of palpitations, headaches, and diaphoresis with plasma free metanephrines or urinary metanephrines. Both patients had SDH subunit gene mutations, which is common in PGL cases, although SDH subunit C mutations are rare. All patients with a PGL should be offered genetic testing.
ACKNOWLEDGMENT
We thank Dr. Marshall and colleagues who previously published on case 2.
Abbreviations
- CT
computed tomography
- MRI
magnetic resonance imaging
- PGL
paraganglioma
- SDH
succi-nate dehydrogenase
Footnotes
DISCLOSURE
The authors have no multiplicity of interest to disclose.
REFERENCES
- 1.Wang JG, Han J, Jiang T, Li YJ. Cardiac paragangliomas. J Card Surg. 2015;30:55–60. doi: 10.1111/jocs.12455. [DOI] [PubMed] [Google Scholar]
- 2.Lee JA, Duh QY. Sporadic paraganglioma. World J Surg. 2008;32:683–687. doi: 10.1007/s00268-007-9360-4. [DOI] [PubMed] [Google Scholar]
- 3.Marshall L, Shah P, Yeung S, Mundy J. Synchronous presentation of cardiac and abdominal paragangliomas. Ann Thorac Surg. 2012;93:e115–e117. doi: 10.1016/j.athoracsur.2011.11.067. [DOI] [PubMed] [Google Scholar]
- 4.Liu XP, Miao Q, Liu XR, Zhang CJ, Ma GT, Liu JZ. Outcomes of surgery for functional cardiac paragangliomas: a single-center experience of 17 patients. J Thorac Cardiovasc Surg. 2019;157:1556–1564. doi: 10.1016/j.jtcvs.2018.09.013. [DOI] [PubMed] [Google Scholar]
- 5.Nölting S, Ullrich M, Pietzsch J et al. Current management of pheochromocytoma/paraganglioma: a guide for the practicing clinician in the era of precision medicine. Cancers (Basel) 2019;11:1505. doi: 10.3390/cancers11101505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fishbein L, Merrill S, Fraker DL, Cohen DL, Nathanson KL. Inherited mutations in pheochromocytoma and paraganglioma: why all patients should be offered genetic testing. Ann Surg Oncol. 2013;20:1444–1450. doi: 10.1245/s10434-013-2942-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Burnichon N, Rohmer V, Amar L et al. The succinate dehydrogenase genetic testing in a large prospective series of patients with paragangliomas. J Clin Endocrinol Metab. 2009;94:2817–2827. doi: 10.1210/jc.2008-2504. [DOI] [PubMed] [Google Scholar]
- 8.Else T, Marvin ML, Everett JN et al. The clinical phenotype of SDHC-associated hereditary paraganglioma syndrome (PGL3) J Clin Endocrinol Metab. 2014;99:E1482–E1486. doi: 10.1210/jc.2013-3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lenders JW, Duh QY, Eisenhofer G et al. Pheochromocytoma and paraganglioma: an Endocrine Society Clinical Practice Guideline. J Clin Endocrinol Metab. 2014;99:1915–1942. doi: 10.1210/jc.2014-1498. [DOI] [PubMed] [Google Scholar]