

Abstract
Nucleic acids are often covalently modified with fluorescent reporter molecules to create a hybridization state-dependent optical signal. Designing such a nucleic acid reporter involves selecting a fluorophore, quencher, and fluorescence quenching design. This report outlines the effect that these choices have on the DNA hybridization characteristics by examining six fluorophores in four quenching schemes: a quencher molecule offset from the fluorophore by 0, 5, or 10 bases, and nucleotide quenching. The similar binding characteristics of left-handed L-DNA were evaluated in comparison with right-handed DNA to quantify the effect of each quenching scheme. These results were applied to the Adaptive PCR method, which monitors fluorescently-labeled L-DNA as a sentinel for analogous unlabeled D-DNA in the reaction. All of the tested fluorophores and quenching schemes increased the annealing temperature of the oligonucleotide pairs by values ranging from 0.5 to 8.5 °C relative to unlabeled oligonucleotides. The design with the smallest increase (0.5 °C) was a sense strand with a FAM fluorophore and an anti-sense strand with Black Hole Quencher 2 offset by 10 bases from the FAM. An identical design that did not offset the quencher molecules resulted in a shift in annealing temperature of 5 °C. PCR was performed using temperature switching based on each of these L-DNA designs, and efficiency was significantly increased for the 10-base offset design, which had the smallest shift in annealing temperature. These results highlight the importance of selecting an appropriate fluorescence quenching scheme for nucleic acid optical signals.
Graphical Abstract
Introduction
Optical detection of nucleic acid products and interactions allows for highly specific and sensitive detection of a variety of targets.1–4 Due to the range of applications including genomic analysis, detection of analytes, and amplification monitoring during PCR, different nucleic acid fluorescent biosensor formats have been engineered.5, 6 For example, fluorescent signals may be generated with intercalating dyes which bind in the major or minor groove of double-stranded DNA.7 Other more specific formats rely on the fluorescent signal from a fluorophore which is modulated by proximity to a quenching molecule.8–10 In order to monitor hybridization of a specific oligonucleotide, the nucleic acids of interest are often covalently modified with these fluorophores and quenchers. Binding events are revealed by either bringing the donor and acceptor fluorophores together to permit energy transfer, or separating them to cease energy transfer.
Quencher molecules reduce fluorescence through fluorescence resonance energy transfer (FRET) when fluorescent energy from the donor molecule is transferred to the acceptor quenching molecule.3, 9 FRET is most effective when the donor and acceptor molecules are within a radius of 2-10 nm. In terms of nucleic acid fluorescent labels, a quenched fluorescent signal indicates that the two labeled strands are annealed. Quenching can also occur through direct contact when the donor and acceptor are very close, diminishing the fluorescence of both molecules and instead releasing energy primarily as heat.9 This type of quenching is known as contact-mediated quenching or contact quenching. Both contact-mediated and FRET-mediated quenching mechanisms may be facilitated with a variety of quenching molecules, including dyes such as Dabcyl and Black Hole Quencher, other particles such as gold nanoparticles and carbon nanotubes, and even single nucleotides.9, 11, 12
Chiral molecules are found in one of two enantiomeric conformations: often denoted as the left-handed and right-handed forms. Perhaps the most attention is given to enantiomeric conformation in the pharmaceutical industry, where drug administration authorities often require enantiomerically pure drug synthesis.13 Additionally, enantiomers play an important role in organic synthesis and materials components.14 Left-handed DNA (L-DNA) is the enantiomer of naturally-occurring right-handed DNA (D-DNA), and was first reported in 1984.15 L-DNA exhibits identical solubility, duplex stability, and selectivity as its right-handed enantiomer, but does not base pair or hybridize with D-DNA and is resistant to enzymatic degradation.16, 17 These properties have led to L-DNA being utilized for molecular beacons and DNA microarrays with enhanced specificity, as well as therapeutic aptamers with enhanced stability.17–21 For applications where an optical readout is desired, L-DNA may be fluorescently labeled with fluorophores and quenchers. Previous studies have shown that these fluorescent labels affect the thermodynamic properties of the DNA duplex.22–24 We have recently utilized optical L-DNA sensing for the development of a hybridization-controlled Adaptive PCR instrument, which offers a suitable application to study this effect. Adaptive PCR monitors the fluorescence of L-DNA hybridization to dynamically switch between heating and cooling phases instead of monitoring temperature.25 The underlying assumption is that the observed hybridization state of fluorescently-labeled L-DNA accurately reflects that of the D-DNA in the reaction. Matching the thermodynamic hybridization characteristics of fluorescently-labeled L-DNA analogs to those of unlabeled D-DNA PCR primers and targets is important for optimal thermal cycling.
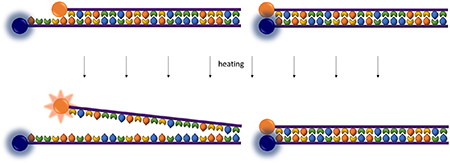
Here we investigate the effects of six fluorophores on DNA annealing temperature. For each of these fluorophores we investigate four quenching schemes: quenching with Black Hole Quencher 2 offset from the fluorophore by 0, 5, and 10 bases, and quenching with guanine (Figure 1). These quenching formats were outlined by Marras et al in their investigation of quenching efficiencies.9 Additionally, we examine the effect of these different fluorescence quenching schemes on our Adaptive PCR application. Using this information, we outline the rational design of fluorescently-labelled L-DNA oligomers which closely mirror the hybridization characteristics of natural, unmodified D-DNA.
Figure 1.

Optical methods for detecting hybridization. A. Unlabeled DNA is measured with SYTO 82 intercalating dye. B, C, D. Quenching with Black Hole Quencher offset from the fluorophore by 0, 5, and 10 bases, respectively. E. Guanine quenching.
Materials and Methods
Hybridization studies oligonucleotides
Analysis of DNA hybridization temperature was performed using primers from an established PCR detection protocol for malarial parasite Plasmodium falciparum.26 Primers and the complementary portion of the target DNA were purchased from Integrated DNA Technologies (Skokie, IL). Identical left-handed oligonucleotides were purchased from Biomers.net (Germany). A complete list of oligonucleotide sequences is given in supporting information Table S1. For studies using fluorescently labelled oligomers, sequences were purchased with Black Hole Quencher 2 (BHQ2) covalently attached to the 3’ end of the target strand and fluorophores attached to the 5’ end of the primer strand. Fluorophores included FAM, HEX, Texas Red, ROX, ATTO Rho101, and Cy5. Tests using an offset between the quencher and fluorophore had either a 5 or 10-base offset on the 3’ end of the quenching strand only. The extra bases to create the offset were taken from the neighboring P. falciparum genetic code. Tests involving guanine quenching replaced the 3’ BHQ2 with a single guanine base (Figure 1, scheme E).
PCR studies oligonucleotides
PCR experiments were performed using previously described P. falciparum target DNA, corresponding primers, and hydrolysis probe.27 L-DNA oligonucleotides with an identical sequence as the forward primers were purchased from Biomers.net with 5’ FAM fluorophores. Complementary nucleotides were purchased with 3’ BHQ2 with no offset, 3’ BHQ2 with a 10-base offset, and 3’ guanine. A complete list of oligonucleotide sequences is given in supporting information Table S1.
Hybridization temperature experiments
Annealing curves were obtained using a Qiagen Rotor-Gene Q. Samples were 25 µL in volume and contained 100 nM fluorophore-labeled oligonucleotides and 100 nM complementary quencher-labeled oligonucleotides in 1X VersaTaq PCR buffer. The samples were raised to an initial temperature of 90 °C and allowed to reach thermal equilibrium. The temperature was then lowered by one °C/minute to a final temperature of 50 °C. Fluorescence was measured at each temperature and the derivative with respect to temperature was calculated. Using Matlab, the derivative data was fitted to a Gaussian curve (Figure S1), and the temperature value corresponding to the peak was taken as the annealing temperature. Each sample was tested in triplicate to obtain a mean and 95% confidence interval. Quenching efficiencies were also calculated by comparing samples with no quencher to those with quenchers. A two-sample t-test was used to determine if quenching method affected quenching efficiency.
Samples containing L-DNA oligomers labeled with Texas Red and complements labeled with BHQ2 with no offset (Figure 1, scheme B) were compared to identical D-DNA samples to confirm that the handedness of DNA does not affect the annealing temperature. The other fluorescence quenching schemes were not compared between enantiomeric structures due to the relative expense of L-DNA in the small quantities required. Based on the initial results of matched enantiomeric hybridization, as well as similar findings in literature,16, 17, 25 we assume that the hybridization characteristics of L-DNA and D-DNA are identical for all quenching schemes. The annealing temperatures of oligomers labeled with six fluorophores were then determined using the same procedure for D-DNA only. The fluorophores tested were FAM, HEX, Texas Red, ROX, ATTO Rho101, and Cy5. For each of these fluorescent labels, four quenching schemes were tested: BHQ2 quenching with 0, 5, and 10-base offsets, and guanine quenching with no offset. These designs are depicted in Figure 1. To estimate the annealing temperature of unlabeled DNA, unmodified sequences were also analyzed using SYTO 82 intercalating dye at 10 µM. This dye has been shown to be thermodynamically stable, shifting the annealing temperature <0.4 °C at 10 µM concentration.28
Effect of fluorescent labels on DNA hybridization during rapid thermal cycling
Samples containing Texas Red-labeled L-DNA and FAM-labeled D-DNA together in one tube were used to study the difference in hybridization caused by the fluorescent labels. Both oligonucleotides were quenched by their respective complements in the 0-base offset quenching format (Figure 1, scheme B). Samples were 25 µL volume in 1X VersaTaq PCR buffer. The samples were thermally cycled in the Adaptive PCR machine between 50 and 90 °C at heating and cooling rates of 1.00 and 0.49 °C/second, respectively. Temperature was monitored by inserting a thermocouple (Omega, 5TC-TT-K-36-36) into the reaction tube. Fluorescence of each oligonucleotide was monitored by the Adaptive PCR instrument. Three heating/cooling cycles were performed for each sample, and each sample was run in triplicate. An example waveform is shown in Figure S2, where the annealing points for the second thermal cycle are circled. This process was then repeated with swapped L-DNA and D-DNA labels to ensure that the handedness of the DNA played no part in any observed differences in hybridization. Fluorescence data as a function of time was imported into Matlab and analyzed to plot the derivative with respect to time. These derivative plots were examined to find the annealing timepoints.
Effect of fluorescence quenching scheme on Adaptive PCR efficiency
Adaptive PCR was performed using previously described methods (i.e., switching between heating and cooling is based not on temperature but on the fluorescence of L-DNA primer-analogs).27 The L-DNA oligomers were labeled with 5’ FAM and included in the reaction at equal concentrations as the D-DNA primers. Two different designs were used for the complementary quenching L-DNA: 3’ BHQ2 in the 0-base offset quenching format and in the 10-base offset format (Figure 1, schemes B and D). Adaptive PCR was then performed for 40 cycles for each design, and tested in triplicate. PCR reaction efficiency was calculated for each sample using the LinRegPCR software package (available at https://www.medischebiologie.nl/files/). Samples were baseline corrected for fluorescence and then windowed based on the linear portion of the log increase of fluorescence to calculate the efficiency.29
Results and Discussion
L-DNA vs D-DNA annealing analysis
L-DNA has previously been shown to exhibit identical hybridization properties as natural D-DNA.16, 17, 25 This key assumption was verified by performing side-by-side annealing analyses on both left and right-handed DNA with BHQ2 quenching and no offset (Figure 1, scheme B). The selectivity of sequence binding occurs during the annealing phase of PCR because strands of varying length bind at distinct temperatures during cooling, differentiating the temperature at which polymerase can efficiently extend primers to create new target complements. Too high a temperature and primers will not bind; too low and binding will be nonspecific. Therefore, we focused primarily on the annealing events. Analysis of the annealing curves for identical L-DNA and D-DNA sequences labeled with Texas Red revealed annealing temperatures of 70.2 ± 0.1 and 70.1 ± 0.1 °C, respectively, reinforcing that D-DNA and L-DNA exhibit identical annealing behavior.
Annealing temperatures of each fluorophore-quencher pair
The annealing temperatures determined for each of the six fluorophores in each of the four described quenching schemes are shown in Figure 2. In all cases regardless of the quenching scheme, the presence of fluorescent labels raised the observed annealing temperature compared to that of unmodified DNA measured with SYTO 82 intercalating dye. Regardless of the offset between fluorophore and quencher, quenching efficiencies for BHQ2 quenching were >89% (Table S2). The efficiency dropped slightly as the quencher offset grew, but this difference was found to be statistically insignificant (p>0.05).
Figure 2.
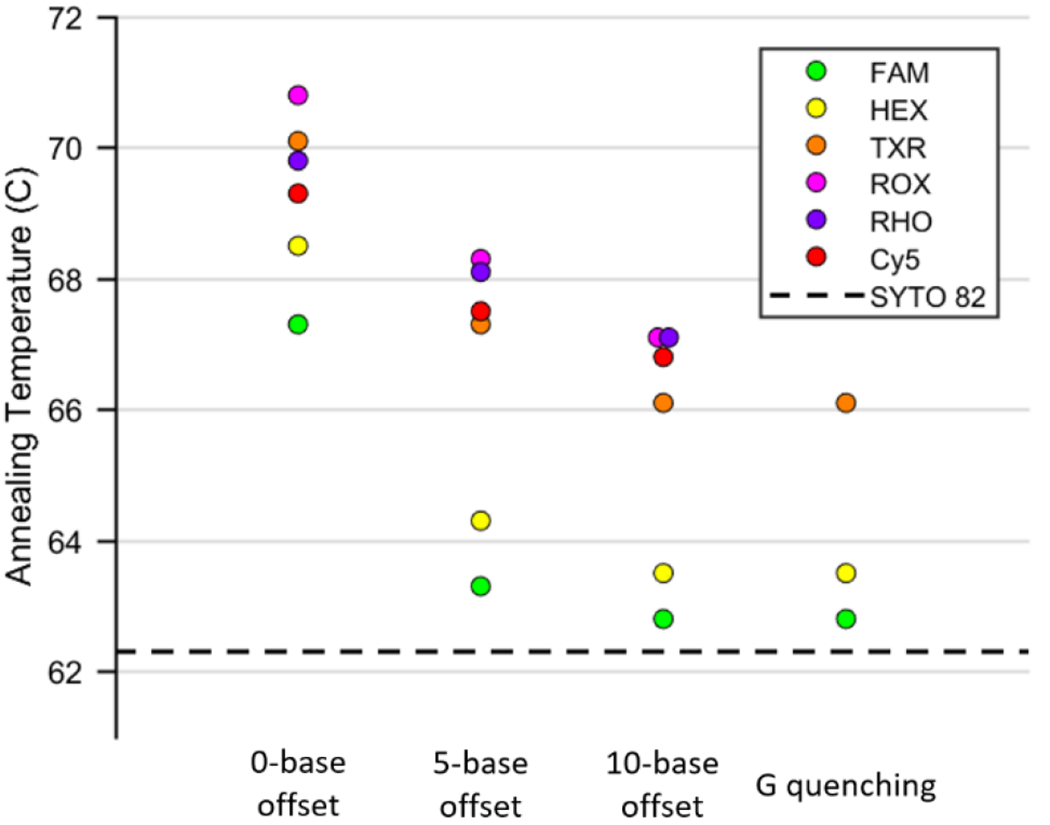
Plot of annealing temperatures for each fluorophore quenched by BHQ2 with 0, 5, and 10-base offsets, as well as quenched by guanine with no offset. The horizontal dashed line represents the annealing temperature of unmodified DNA measured with SYTO 82 intercalating dye.
The observed increase in annealing temperature was largest for 0-base offset quenching, where it ranged from 4.0-7.5 °C depending on the fluorophore (Table S3). Quenching with an offset of 5 bases resulted in a smaller increase in annealing temperature for all fluorophores, and a 10-base offset lowered that increase even further. It appears that as the distance between fluorophore and quencher grows and the quenching mechanism shifts from contact-mediated quenching to FRET quenching, the stabilizing effect on the DNA duplex decreases. Guanine quenching showed almost identical results as 10-base offset quenching, although it only quenched sufficiently to make out an annealing curve for FAM, HEX, and Texas Red. The similarity of results between 10-base offset BHQ2 quenching and guanine quenching suggests that the quencher molecule has minimal effect on duplex stability when separated from the fluorophore by 10 bases. Separating the fluorophore and quencher by a distance of 5 bases likely decreases but does not eliminate the contact between the two. The current protocol for Adaptive PCR utilizes quenchers with no offset; these results suggest not only that the fluorescently-labelled L-DNA will anneal earlier than their unlabeled analogs in the cooling phase of PCR, but also that the difference should depend on the fluorophore/quencher pair used.
Effect of fluorescent labels on DNA hybridization during rapid thermal cycling
The annealing temperatures previously measured were obtained at slow rates of thermal change, ensuring that the samples reach thermodynamic equilibrium at each temperature. We next sought to investigate the effect fluorescent labels would have on hybridization observed at the comparably rapid rates of cooling seen in PCR. FAM-labeled DNA was found to anneal approximately 7.2 seconds later than Texas Red-labeled DNA (Table S4). The annealing temperatures of the FAM-labeled and Texas Red-labeled samples measured on the Rotor-Gene Q were 69.3 and 65.9 °C, respectively (difference of 3.4 °C). Note that these values are slightly lower than those given in Figure 2 because the DNA is at a lower concentration.
Since the cooling rate of our system is known, the difference in annealing timepoints should be predictable using the following equation: Here, ΔT is difference in annealing temperatures in °C, Δt is difference in annealing timepoints in seconds, and is the cooling rate of the solution. With a cooling rate of 0.49 °C/second and a difference in annealing temperatures of 3.4 °C, we predict a difference in annealing timepoints of 6.9 seconds. This is close to the observed difference of 7.2 seconds (Table S4), which suggests that the difference in annealing time caused by fluorescent labels can be predicted using the cooling rate of the system and the known annealing temperatures.
To test this hypothesis, the experiment was repeated using a different combination of fluorescent markers: L-DNA labeled with FAM and D-DNA labeled with HEX. The annealing temperature of HEX-labelled oligomers was found to be only 0.8 °C higher than that of FAM-labelled oligomers. If the difference in annealing points seen on the Adaptive PCR machine is truly determined only by the difference in annealing temperatures, then the FAM-labeled L-DNA should anneal approximately 1.6 seconds after the HEX-labeled DNA. The hypothesis was confirmed: the difference in annealing was measured as 1.2 ± 0.4 seconds. Assuming total DNA concentration is equal, the difference in hybridization temperature due to the presence of fluorescent labels appears to be predictable based on annealing temperature measurements.
Effect of fluorescence quenching schemes on Adaptive PCR efficiency
We next investigated the effect of the fluorescence quenching design on PCR product formation using two different L-DNA designs to switch between heating and cooling. The first is the design which led to the smallest rise in annealing temperature: FAM fluorophore and BHQ2 quencher with a 10-base offset (Figure 1, scheme D). The other design used FAM and BHQ2 with no offset (scheme B). A guanine quenching design could not be tested because left-handed guanine does not exhibit the same quenching properties as right-handed guanine (data not shown). The results in Figure 3 show the mean amplification curves obtained using each design. The mean reaction efficiency for no-offset and 10 base-offset quenching were 75.0 ± 7.9% and 90.4 ± 9.9%, respectively. As a reference, PCR was also performed on the Rotor-Gene using the same procedure as previously described,27 and the efficiency was 66.8 ± 19.8%. Statistical analysis using a one-tailed t-test assuming unequal variance showed that the 10-base offset quenching design led to a statistically significant increase in efficiency compared to the 0-base offset design. (p = .04).
Figure 3.
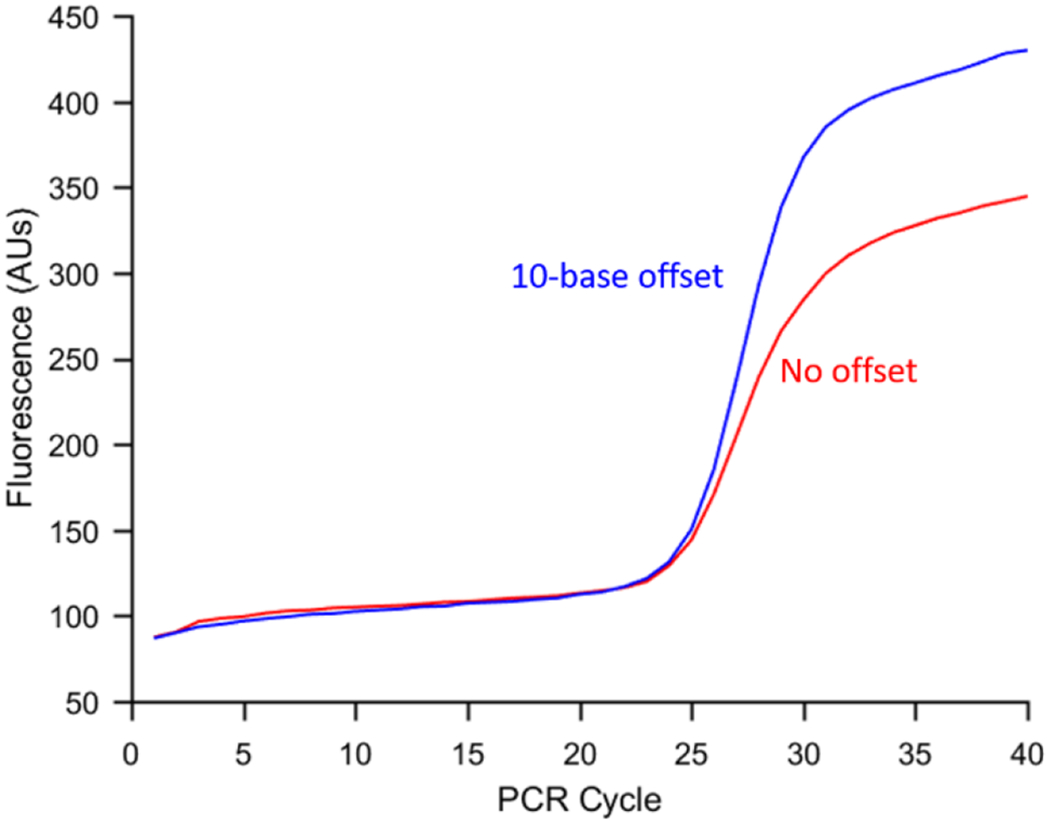
Mean amplification curve (n=3) for Adaptive PCR with 0-base offset quenching of FAM (red) and 10-base offset quenching of FAM (blue). Separating the quencher and fluorophore increased the mean reaction amplification efficiency approximately 15%.
The low efficiency of this reaction can be attributed to its original use in single nucleotide polymorphism (SNP) detection, which greatly limits the choice of primer sequences. This makes the reaction an excellent candidate to study the effect of fluorescent labels on amplification efficiency, although we still expect to see the same trends in other more efficient reactions.
Conclusions
We have shown that nucleic acids covalently modified with various fluorophore-quencher pairs do not exhibit the same hybridization characteristics as their unmodified enantiomers. Contact-mediated quenching, in which the fluorophore and quencher are in physical contact, caused the largest shift in annealing temperature. Preventing the interaction between fluorophore and quencher, whether it be by offsetting them or replacing the quencher with a guanine, led to a much smaller effect. FAM showed the smallest shift in hybridization temperature of the fluorophores tested (0.5-5.0 °C), and ROX showed the largest (4.8-8.5 °C). It is worth noting that the extent of this stabilizing effect is very likely sequence-dependent; however, we expect that the general trends seen here remain true. The quenching scheme and choice of fluorophore are both important determinants of the stability of the duplex, and its use for predicting the equivalent enantiomeric hybridization state. As an example, we applied this strategy to hybridization-controlled Adaptive PCR and, as predicted, the L-DNA designed to have an annealing temperature closer to that of the unmodified D-DNA primer resulted in a more efficient PCR reaction.
Previous work regarding Adaptive PCR has demonstrated high efficiency amplification despite utilizing L-DNA which is quenched with no offset. While this may seem contradictory with the findings presented here, each reaction required optimization of the parameters controlling the switch between heating and cooling. In this way, an imperfect L-DNA design was worked around using the instrument settings. The findings presented here offer guidelines which should eliminate the need to individually optimize each reaction, as done in the past, because the L-DNA hybridization will mirror that of the D-DNA in the reaction. This example highlights the importance of selecting an appropriate fluorescence quenching design for nucleic acid optical signals.
Supplementary Material
Acknowledgements
This work was supported in part by the National Institutes of Health grant R42HG009470.
Footnotes
Electronic Supplementary Information (ESI) available: [details of any supplementary information available should be included here]. See DOI: 10.1039/x0xx00000x
Conflicts of interest
There are no conflicts to declare.
References
- 1.Livak KJ, Flood S, Marmaro J, Giusti W, Deetz K, Genome Research, 1995, 4 (6), 357–362. [DOI] [PubMed] [Google Scholar]
- 2.Cardullo RA, Agrawal S, Flores C, Zamecnik PC, Wolf DE, Proceedings of the National Academy of Sciences, 1988, 85 (23), 8790–8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qiu X, Guo J, Jin Z, Petreto A, Medintz IL, Hildebrandt N, Small, 2017, 13 (25), 1700332. [DOI] [PubMed] [Google Scholar]
- 4.Navani NK, Li Y, Current Opinion in Chemical Biology, 2006, 10 (3), 272–281. [DOI] [PubMed] [Google Scholar]
- 5.Juskowiak B, Analytical and Bioanalytical Chemistry, 2011, 399 (9), 3157–3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Marras SA, Tyagi S, Kramer FR, Clinica Chimica Acta, 2006, 363 (1–2), 48–60. [DOI] [PubMed] [Google Scholar]
- 7.Qiu J, Wilson A, El-Sagheer AH, Brown T, Nucleic Acids Research, 2016, 44 (17), e138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ogawa M, Kosaka N, Longmire MR, Urano Y, Choyke P, Kobayashi H, Molecular Pharmaceutics, 2009, 6 (2), 386–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Marras SA, R Kramer F, Tyagi S, Nucleic Acids Research, 2002, 30 (21), e122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng J, Yang R, Shi M, Wu C, Fang X, Li Y, Li J, Tan W, Chemical Society Reviews, 2015, 44 (10), 3036–3055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang R, Jin J, Chen Y, Shao N, Kang H, Xiao Z, Tang Z, Wu Y, Zhu Z, Tan W, Journal of the American Chemical Society, 2008, 130 (26), 8351–8358. [DOI] [PubMed] [Google Scholar]
- 12.Wang W, Chen C, Qian MX, Zhao XS, Sensors and Actuators B: Chemical, 2008, 129 (1), 211–217. [Google Scholar]
- 13.Shen J, Okamoto Y, Chemical Reviews, 2015, 116 (3), 1094–1138. [DOI] [PubMed] [Google Scholar]
- 14.Pu L, Accounts of Chemical Research, 2017, 50 (4), 1032–1040. [DOI] [PubMed] [Google Scholar]
- 15.Anderson DJ, Reischer RJ, Taylor AJ, Wechter WJ, Nucleosides & Nucleotides, 1984, 3 (5), 499–512. [Google Scholar]
- 16.Urata H, Shinohara K, Ogura E, Ueda Y, Akagi M, Journal of the American Chemical Society, 1991, 113 (21), 8174–8175. [Google Scholar]
- 17.Hauser NC, Martinez R, Jacob A, Rupp S, Hoheisel JD, Matysiak S, Nucleic Acids Research, 2006, 34 (18), 5101–5111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ke G, Wang C, Ge Y, Zheng N, Zhu Z, Yang CJ, Journal of the American Chemical Society, 2012, 134 (46), 18908–18911. [DOI] [PubMed] [Google Scholar]
- 19.Lee AY, Kim KR, Yu JH, Ahn DR, Elsevier: Journal of Industrial and Engineering Chemistry, In Press, 2019. [Google Scholar]
- 20.Yatime L, Maasch C, Hoehlig K, Klussmann S, Andersen GR, Vater A, Nature Communications, 2015, 6, 6481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams KP, Liu XH, Schumacher TN, Lin HY, Ausiello DA, Kim PS, Bartel DP, Proceedings of the National Academy of Sciences, 1997, 94 (21), 11285–11290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.You Y, Tataurov AV, Owczarzy R, Biopolymers, 2011, 95 (7), 472–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Moreira BG, You Y, Owczarzy R, Biophysical Chemistry, 2015, 198, 36–44. [DOI] [PubMed] [Google Scholar]
- 24.Telser J, Cruickshank KA, Morrison LE, Netzel TL, Journal of the American Chemical Society, 1989, 111 (18), 6966–6976. [Google Scholar]
- 25.Adams NM, Gabella WE, Hardcastle AN, Haselton FR, Analytical Chemistry, 2016, 89 (1), 728–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bitting AL, Bordelon H, Baglia ML, Davis KM, Creecy AE, Short PA, Albert LE, Karhade AV, Wright DW, Haselton FR, Journal of Laboratory Automation, 2016, 21 (6), 732–742. [DOI] [PubMed] [Google Scholar]
- 27.Leelawong M, Adams NM, Gabella WE, Wright DW, Haselton FR, Journal of Molecular Diagnostics, In Press, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gudnason H, Dufva M, Bang DD, Wolff A, Nucleic Acids Research, 2007, 35 (19), e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ruijter J, Ramakers C, Hoogaars W, Karlen Y, Bakker O, Van den Hoff M, Moorman A, Nucleic Acids Research, 2009, 37 (6), e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu WT, Wu JH, Y Li ES, S Selamat E, Applied Environmental Microbiology, 2005, 71 (10), 6453–6457. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.