

Abstract
Applications of adenine base editors (ABEs) have been constrained by the limited compatibility of the deoxyadenosine deaminase component with Cas homologs other than SpCas9. We evolved the deaminase component of ABE7.10 using phage-assisted non-continuous and continuous evolution (PANCE and PACE), resulting in ABE8e. ABE8e contains eight additional mutations that increase activity (kapp) 590-fold compared with ABE7.10. ABE8e offers substantially improved editing efficiencies when paired with a variety of Cas9 or Cas12 homologs. ABE8e is more processive than ABE7.10, which could benefit screening, disrupting regulatory regions and multiplex base editing applications. A modest increase in Cas9-dependent and -independent DNA off-target editing, and in transcriptome-wide RNA off-target editing can be ameliorated by introducing additional mutations in the TadA-8e domain. Finally, we show that ABE8e can efficiently edit natural mutations in a GATA1 binding site in the BCL11A enhancer or the HBG promoter in human cells, targets which were poorly edited with ABE7.10. ABE8e broadens the effectiveness and applicability of adenine base editing.
Editorial Summary
A continuously evolved adenine base editor is compatible with various Cas proteins and mediates efficient A•T-to-G•C base conversions at a wide variety of PAM sites.
Introduction
Base editors contain a catalytically impaired Cas protein fused to a DNA-modifying enzyme. The Cas9 domain directs the DNA-modifying enzyme to directly convert one base to another at a guide RNA-programmed target site1–3. Two classes of base editors have been developed to date1,2,4–6. Cytosine base editors (CBEs) convert C•G-to-T•A, and adenine base editors (ABEs) convert A•T-to-G•C. Collectively, CBEs and ABEs enable the installation and correction of all four types of transition mutations (C-to-T, G-to-A, A-to-G, and T-to-C). As half of all known disease-associated gene variants are point mutations, and transition mutations account for ~60% of known pathogenic point mutations7,8, base editors are being widely used to study and treat genetic diseases in a variety of cell types and organisms, including animal models of human genetic diseases3,9–19. ABEs are especially useful for the study and correction of pathogenic alleles, as nearly half of pathogenic point mutations in principle can be corrected by converting an A•T base pair to a G•C base pair7,8.
Base editing requires the presence of a PAM located approximately 15±2 base pairs from the target nucleotide(s) for canonical SpCas9 base editors1–3. Only about one quarter of pathogenic transition point mutations have a suitably located NGG PAM that enables compatibility with these SpCas9-derived base editors20–22. CBEs have proven to be broadly compatible with many Cas homologs including SaCas921, SaCas9-KKH21, Cas12a (Cpf1)23,24, SpCas9-NG25, and circularly permuted CP-Cas9s22, greatly expanding their targeting scope. In contrast, ABEs have shown limited compatibility with Cas homologs. Some homologs such as SaCas922, SaCas9-KKH22, SpCas9-NG25, and CP-Cas9s22 are compatible with ABEs, but editing efficiencies are substantially lower than those of the corresponding CBEs. Other homologs such as LbCas12a and enAsCas12a show virtually no activity as an ABE23,24.
We sought to broaden the applicability of ABEs by improving their compatibility with a variety of Cas domains. We hypothesized that the very low to modest efficiency of many non-SpCas9 ABEs arises from a low rate of adenine deamination (low kapp) combined with the shorter residence time on DNA (high koff) of many Cas homologs compared to that of SpCas926. The DNA-modifying enzyme in ABE is TadA-7.10, a deoxyadenosine deaminase that we previously evolved from an E. coli tRNA adenosine deaminase (TadA) to act on single-stranded DNA1. In this study, we developed and applied a new phage-assisted continuous evolution (PACE) selection system to greatly enhance the activity and compatibility of ABEs with diverse Cas homologs, including SaCas9, SaCas9-KKH, LbCas12a, enAsCas12a, SpCas9-NG, and circularly permuted CP1028-SpCas9 and CP1041-SpCas9. The resulting evolved ABE variant, ABE8e, shows substantially increased editing efficiency relative to ABE7.10. As expected, ABE8e shows increased off-target RNA and DNA editing, which we substantially reduced by introducing a V106W mutation into the TadA domain. PACE-evolved ABE variants thus expand the targeting scope, editing efficiency, and overall utility of adenine base editors.
Results
Development of a phage-assisted evolution selection circuit
PACE enables the rapid continuous evolution of biomolecules through many generations of mutation, selection, and replication per day (Fig. 1a)27–39. During PACE, host E. coli cells continuously dilute a population of bacteriophage (selection phage, SP) containing the gene of interest. The gene of interest replaces gene III, a gene required for progeny phage infectivity, in the SP. SP containing desired gene variants trigger host-cell gene III expression from an accessory plasmid (AP). Host-cell DNA plasmids encode a genetic circuit that links the desired activity of the protein encoded in the SP to the expression of gene III on the AP. Thus, SP variants containing desired genotypes can propagate, while phage encoding inactive variants do not generate infectious progeny and are rapidly diluted out of the culture vessel (‘the lagoon’, Fig. 1a). An arabinose-inducible mutagenesis plasmid (MP) controls the phage mutation rate. PACE is an ideal system for improving the kinetics of an enzyme because variant survival requires that gene III be expressed before progeny phage are packaged, and before phage are diluted out of the lagoon36. These features of PACE suggest that PACE may be ideally suited to evolve a deoxyadenosine deaminase that can mediate deamination at a rate sufficient to enable robust A•T-to-G•C base editing even when fused to Cas9 or Cas12 homologs that reside on DNA for shorter periods of time than SpCas9.
Figure 1. Phage-assisted evolution of a deoxyadenosine deaminase.
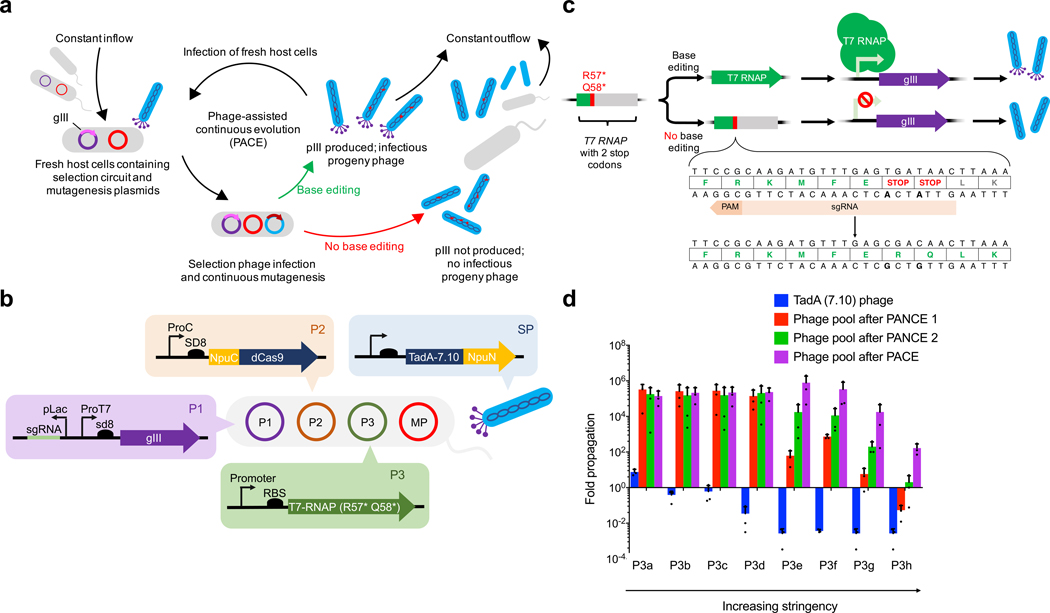
a, General PACE overview for base editor evolution37,38. E. coli host cells contain a plasmid-based genetic circuit that links expression of gene III (gIII, encoding pIII) to the activity of the base editor encoded in a modified M13 bacteriophage (blue). The production of infectious progeny phage requires expression of gene III, which only occurs in host cells infected by phage variants that encode active base editors. Phage exist in a fixed-volume vessel (the lagoon) continuously diluted with host-cell culture, so only those phage that propagate faster than the rate of dilution can persist and evolve. b, The selection circuit in PANCE or PACE for evolving the deoxyadenosine deaminase component of ABEs. Plasmid P1 (purple) contains gene III driven by a T7 promoter and an sgRNA driven by a Lac promoter. Plasmid P2 (orange) expresses an Npu C-intein fused to a catalytically dead SpCas9 (dCas9), which forms full-length base editor upon trans-intein splicing with TadA fused to a Npu N-intein (encoded on the SP, in blue). Plasmid P3 (green) expresses a T7 RNAP that contains two stop codons that can be corrected to arginine and glutamine upon adenine base editing; this editing event drives expression of gene III. We developed eight P3 variants (P3a-h) with different promoters and ribosome binding sites (RBS) to tune selection stringency. The phage genome is continuously mutated by expression of mutagenic genes from the mutagenesis plasmid (MP, red). c, The gene encoding T7 RNA polymerase (T7 RNAP), which is required for gene III expression from the T7 promoter, contains two stop codons at R57 and Q58. Deamination of both adenines by ABE converts the stop codons back to Arg and Gln, resulting in active T7 RNAP and gene III expression. d, Overnight phage propagation assays to test the activity of phage pools in host cells harboring P1, P2, and eight different variants of P3 (P3a-h) of increasing stringency. We mixed phage pools with an excess of log-phase host cells and allowed the phage to propagate overnight. The fold phage propagation is the output phage titer divided by the input titer. For all plots, dots represent individual biological replicates, bars represent mean values, and error bars represent the standard deviation of three independent biological replicates.
Following our group’s previous development of a CBE PACE selection36, we generated a new PACE selection that links ABE activity to gene III expression on the AP (plasmid P1) (Fig. 1b and Supplementary Table 1). We divided ABE into two components, each fused to half of a split intein36. We encoded TadA-7.10 fused to an Npu N-intein in the SP to focus mutagenesis and evolution on the TadA domain, and expressed catalytically dead Cas9 (dCas9) fused to an Npu C-intein from a host-cell plasmid (P2) maintained in bacteria (Fig. 1b and Supplementary Table 1)36. Phage infection followed by intein trans-splicing generates full-length base editor protein, as we previously demonstrated during the development of PACE for CBEs36. Although TadA functions natively as a dimer, we performed the selections for ABE activity using a single TadA–dCas9 fusion, as we had done previously in E. coli1, since we presumed that the TadA–dCas9 fusion is able to dimerize either with itself or with endogenous E. coli TadA.
We envisioned correcting one or more stop codons in a T7 RNA polymerase (T7 RNAP) gene on a third plasmid (P3) using ABE, thereby rescuing T7 RNAP production to drive gene III expression from a T7 promoter (Fig. 1b,c and Supplementary Table 1). We installed stop codons at amino acid positions 57 and 58 in T7 RNAP and provided a single guide RNA (sgRNA) that directs ABE to correct these stop codons on the transcription template strand back to Arg and Gln codons (Fig. 1b,c). We generated eight P3 variants that use different promoters and ribosome binding site (RBS) strengths upstream of the T7 RNAP gene to modulate selection stringency (Supplementary Table 1), and tested overnight propagation of SP encoding TadA-7.10 in host cells harboring P1, P2, and one of eight P3 variants (P3a-h). We observed phage propagation with host cells containing the least stringent P3 (P3a) as determined by measuring the number of plaque-forming units (PFU) before and after overnight incubation (Fig. 1d). These results suggest that P1+P2+P3a couples ABE activity to phage propagation, but the low rate of deamination of TadA-7.10 results in only modest gene III expression.
TadA evolution
We attempted to evolve TadA-7.10 using this low-stringency phage propagation selection (P1+P2+P3a). Propagation was too weak to support PACE, resulting in phage washout. We then turned to phage-assisted non-continuous evolution (PANCE)20,27,35,39. PANCE uses the same genetic circuit to activate phage propagation, but instead of continuously diluting a vessel, phage are manually passaged by infecting fresh host-cell culture with an aliquot from the proceeding passage. PANCE is less stringent than PACE because there is little risk of losing a weakly active phage variant during selection, and because the effective rate of phage dilution is much lower.
We initiated PANCE of TadA-7.10 by infecting host cells harboring P1, P2, and either of the two least stringent P3 variants, P3a or P3b, with SP encoding TadA-7.10 in parallel evolutions (Supplementary Tables 1, 2). We increased genetic diversity by allowing the TadA-7.10 to mutate in the absence of selection pressure during an initial period of “genetic drift”. We provided host cells with a mutagenic drift plasmid (DP)28,37,40 that contained an anhydrotetracycline (aTc)-inducible gene III. Host cells received aTc during passages 1, 5, and 7, allowing SP to freely diversify without any selection pressure (Supplementary Table 2)28. Phage propagation levels increased in later passages, suggesting that the population had accessed more active TadA variants. We increased selection stringency by infecting the cells with fewer phage per passage (Supplementary Table 2). After 15 passages on host cells harboring P3a and P3b, the phage pool showed increased activity, propagating overnight about 100,000-fold on host cells harboring P1, P2, and the four least stringent P3 variants (P3a, P3b, P3c, or P3d) and 100- to 1,000-fold with higher stringency P3 variants P3e and P3f (Fig. 1d). These findings suggest that PANCE with intermittent genetic drift enabled the initial evolution of TadA-7.10.
To characterize TadA-7.10 mutations that contributed to increased phage propagation during PANCE, we isolated 13 individual phage clones and sequenced their TadA-7.10 genes. One mutation, T111R, was conserved in all 13 clones (Fig. 2a and Extended Data Figure 1). The mutation is predicted to lie near the active site of the enzyme, adjacent to D108N, a critical mutation that emerged early in the initial evolution of TadA-7.10 (Fig. 2b)1. The prevalence and location of this mutation suggest that T111R contributes to increased ABE activity.
Figure 2. Mutations and kinetics of TadA-8e, and editing characteristics of ABE8e in human cells.
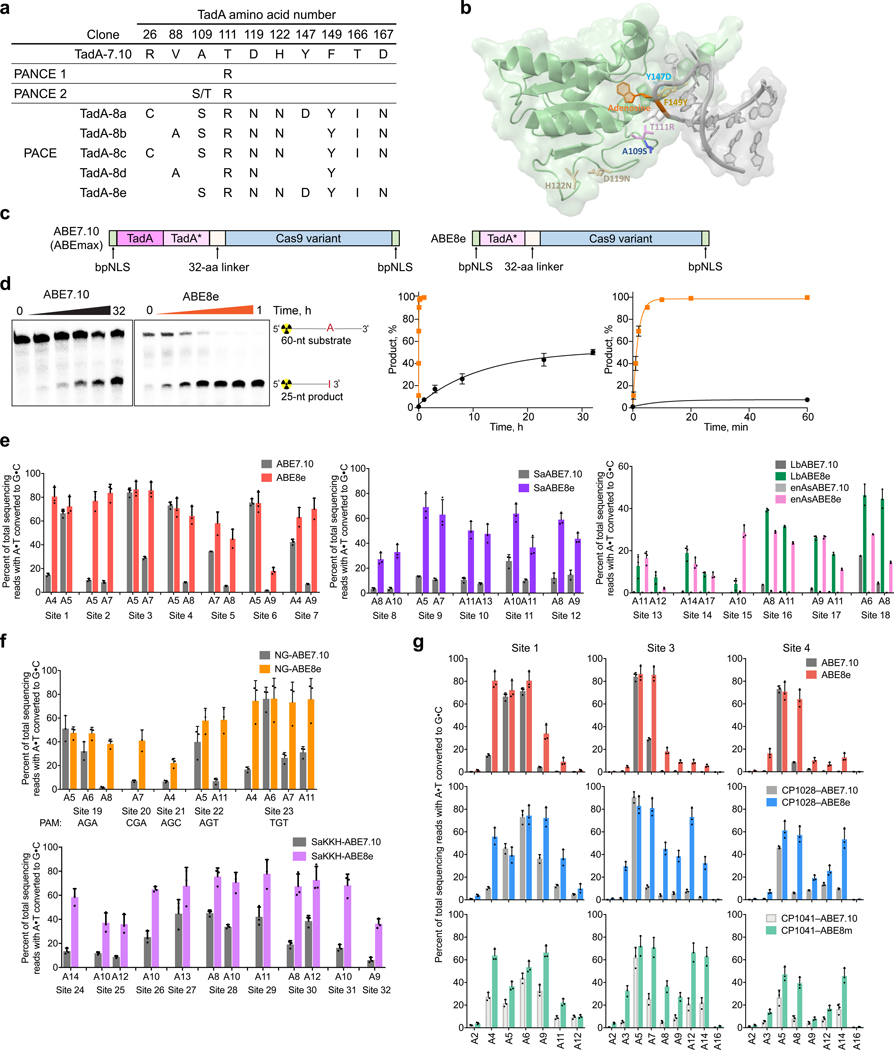
a, Conserved mutations after 25 passages of PANCE (PANCE 1 = passages 1–15; PANCE 2 = passages 11–25), and genotypes of five TadA variants emerging from 84 hours of PACE (surviving ~10174-fold total dilution). For a list of all evolved TadA genotypes, see Extended Data Figure 1. b, E. coli TadA deaminase (green, PDB 1Z3A) aligned with the structure of S. aureus TadA (not shown) complexed with tRNAArg2 (grey, PDB 2B3J). Mutations evolved during PANCE and PACE are colored to correspond to those in Extended Data Figure 1. c, The architecture of ABE7.10 (ABEmax)6 and ABE8e. d, Left: representative denaturing polyacrylamide gels of 5’-radiolabeled dsDNA deamination reactions performed with in vitro reconstituted ABE7.10 and ABE8e RNPs, followed by treatment with E. coli EndoV, which cleaves DNA 3’ of deoxyinosine. Middle: the fraction of deaminated dsDNA plotted as a function of time in hours. Right: the fraction of deaminated dsDNA plotted as a function of time in the first 60 minutes. The data were fit to a single exponential equation to extract apparent first-order deamination rate constants for ABE7.10 (black) and ABE8e (orange). Data are represented as the mean±s.d. from three independent experiments. e, Base editing in HEK293T cells by SpABE7.10 versus SpABE8e, SaABE7.10 versus SaABE8e, and LbABE7.10 and enAsABE7.10 versus LbABE8e and enAsABE8e, for the two nucleotides with the highest editing efficiency within each protospacer. Bars represent mean values, and error bars represent the standard deviation of three independent biological replicates. For editing across the entire protospacer for each site and indel frequencies, see Supplementary Figs. 4-6. f, Base editing in HEK293T cells by NG-ABE7.10 versus NG-ABE8e and SaKKH-ABE7.10 versus SaKKH-ABE8e, for the two nucleotides with the highest editing within each protospacer. For editing efficiencies across the entire protospacer and indel frequencies, see Supplementary Figs. 7, 8. g, Base editing in HEK293T cells within the protospacer by CP–ABE7.10 and CP–ABE8e variants, compared to SpABE7.10 and SpABE8e. For editing efficiencies across the entire protospacer and indel frequencies, see Supplementary Fig. 4. For all plots, bars represent mean values, and error bars represent the standard deviation of three independent biological replicates.
We continued to evolve TadA-7.10 from the phage pools that emerged from PANCE passage 15. During this second phase of PANCE experiments, we performed four evolutions in parallel, two on host cells harboring circuits P1, P2, and P3e, and two on host cells harboring P1, P2, and P3g (Supplementary Tables 1, 2). Both circuits are more stringent than those used during the initial PANCE experiments. We completed ten additional passages (Supplementary Table 2), then pooled the resulting phage and tested overnight phage propagation on host cells harboring P1, P2, and a P3 variant. Phage following passage 25 demonstrated additional fitness increases, propagating overnight ~100-fold on P1+P2+P3g host cells (Fig. 1d). We isolated 18 individual phage clones and sequenced their TadA genes. PANCE using these more stringent circuits also enriched A109S or A109T (Figs. 2a,b and Extended Data Figure 1). Given the substantial increase in phage propagation strength after 25 total PANCE passages (surviving in total ~1049-fold dilution), we decided to initiate PACE on the PANCE-derived phage population to benefit from the more stringent and efficient continuous evolution system.
We performed PACE in host cells harboring P1, P2, and either P3e or P3g in parallel lagoons with phage pools from passage 25 of PANCE. We gradually increased the lagoon flow rate from 0.5 to 2.5 lagoon vol/h over 84 hours to require that base editing of T7 RNAP occur quickly enough to support increasingly rapid phage propagation (Extended Data Figure 2). After 84 hours of PACE, we attempted to further increase the stringency by increasing the lagoon flow rate to 3.0 lagoon vol/h, but the phage could not propagate further (Extended Data Figure 2). Analysis of 21 individual SPs from 84 hours of PACE revealed enrichment of 11 consensus mutations across a variety of unique genotypes: R26G, V69A, V88A, A109S, T111R, D119N, H122N, Y147D, F149Y, T166I, D167N (Figs. 2a,b and Extended Data Figure 1). Phage emerging from PACE propagated for the first time on host cells harboring P1, P2, and P3h, which together form the most stringent circuit in this study (Fig. 1d). These observations suggest that the PANCE- and PACE-evolved TadA variants evolved greatly improved deoxyadenosine deamination activity.
Comparison of evolved TadA variants
We chose five TadA genotypes (TadA-8a, TadA-8b, TadA-8c, TadA-8d, and TadA-8e) emerging from PACE that collectively include most consensus mutations (Fig. 2a and Extended Data Figure 1) to characterize in mammalian cells. We initially assessed the activity of the new ABE variants in mammalian cell culture with the optimized architecture, codon usage, and nuclear localization signals for ABEmax6. ABEmax consists of a wild-type TadA monomer fused to the evolved TadA monomer, which in turn is fused to a Cas9 nickase domain (TadA–TadA*–Cas9 nickase, Fig. 2c)6,22. As previously demonstrated41 and discussed later in this study, the wild-type TadA monomer is not required for ABE activity. Therefore, for consistency throughout this study, an ABE8 variant containing both the wild-type TadA monomer and an evolved TadA monomer is referred to as an ABE8-dimer. For additional clarity, hereafter we refer to ABEmax6, which contains the wild-type TadA monomer, as ABE7.10 (Fig. 2c). We tested the resulting ABE8a-8e dimer variants for base editing activity in HEK293T cells along with ABE7.106, and SaABEmax6 (referred to hereafter as SaABE7.10), which uses SaCas9 nickase instead of SpCas9 nickase. We also compared TadA-8a-e variants with TadA-7.10 for levels of A•T-to-G•C base editing when tethered to catalytically dead LbCas12a (dLbCas12a). Unlike Cas9 homologs, Cas12a variants that nick the non-edited strand have not been reported due to the lack of separate active sites that cleave each DNA strand in Cas12a42.
To select a TadA-8 variant for further studies, we transfected each base editor into HEK293T cells with a sgRNA targeting a site with a cognate PAM for SpCas9 (NGG), SaCas9 (NNGRRT), or LbCas12a (TTTV) and sequenced target loci after three days. Strikingly, all five TadA-8 variants demonstrated large improvements in A•T-to-G•C base editing efficiency, up to 9.4-, 12-, and 24-fold when tethered to SpCas9, SaCas9, and dLbCas12a, respectively, without any evident changes to the very low indel formation levels of ABE7.10 (Supplementary Figs. 1–3). Most notably, we observed efficient A•T-to-G•C base editing activity with dLbCas12a for the first time, despite the lack of a suitable Cas12a nickase to nick the non-edited strand to enhance editing efficiencies2. Because ABEs containing TadA-8e showed consistently high editing activity and low indel formation, particularly when fused with dLbCas12a, we chose base editors containing TadA-8e for subsequent analyses.
As noted above, although early ABE variants required a heterodimeric TadA containing an N-terminal wild-type TadA monomer for maximal activity1, Joung and coworkers showed that later ABE variants have comparable activity with and without the wild-type TadA monomer41. We confirmed that eliminating the wild-type TadA from the wtTadA–TadA-8e–Cas fusion did not affect base editing activity with SpCas9, SaCas9, or dLbCas12a (Supplementary Figs. 4-6). We performed a dose-titration experiment comparing ABE8e and ABE8e-dimer at eight plasmid doses and observed no difference in A•T-to-G•C base conversion activity at three separate genomic sites in HEK293T cells (Extended Data Figure 3). Additional comparisons of ABE8 variants with ABE8-dimer variants yielded no apparent differences in activity; therefore, we performed all subsequent experiments with monomeric ABE8e (Fig. 2c, Supplementary Figs. 4-8).
Characterization of ABE8e
To determine if PANCE and PACE resulted in improved deamination kinetics, we compared the DNA adenine deamination kinetics of ABE7.10 and ABE8e in vitro. We purified recombinant ABE7.10 and ABE8e through His-tag column chromatography, high-resolution heparin-affinity chromatography, and size-exclusion chromatography. We performed single-turnover DNA deamination assays by measuring A-to-I conversion in vitro to determine their apparent first-order kinetics. deamination rate constants (kapp). Remarkably, kapp of deoxyadenosine deamination is 590-fold higher for ABE8e than for ABE7.10 (0.59±0.034 min−1 for ABE8e versus 0.0010±0.00030 min−1 for ABE7.10) (Fig. 2d). The dramatically increased rate of deoxyadenosine deamination by ABE8e compared to ABE7.10 suggests that TadA-8e may be fast enough to yield efficient DNA adenine deamination even when coupled to non-SpCas9 Cas effectors that have decreased residence times on DNA substrates. These results establish that PANCE and PACE of TadA-7.10 resulted in a highly evolved TadA variant that mediates very efficient and fast deoxyadenosine deamination.
To further characterize the expanded targeting capabilities of adenine base editing with the evolved TadA-8e, we treated HEK293T cells with SpABE8e, SaABE8e, LbABE8e, enAsABE8e, or the corresponding ABE7.10 variants targeting several endogenous genomic sites each and measured A•T-to-G•C base conversion efficiency after three days (Fig. 2e and Supplementary Figs. 4-6). With all four Cas homologs, we observed substantially enhanced editing with the ABE8e variant compared to the ABE7.10 variant. For SpABE variants, the increased editing efficiency of SpABE8e editors was most evident when examining the editing levels at the second-most efficiently edited A within each protospacer (Fig. 2e). With SpABE7.10, editing levels at the second-most edited A ranged from 1.7% to 20%, while with SpABE8e editing levels ranged from 18%–86%, improvements of 3.0- to 11-fold.
ABE8e variants using SaCas9 and two Cas12a homologs showed large increases in editing levels. For SaABE7.10, A•T-to-G•C conversion at the highest edited position ranged from 3.6%–26% at the five genomic loci tested, while for SaABE8e, A•T-to-G•C conversion at the highest edited position ranged from 33%–69% (Fig. 2e). For Cas12a homologs, no nickase for the unedited strand is known. Therefore, we analyzed base editors with dead LbCas12a and enAsCas12a (engineered AsCas12a23). Strikingly, average LbCas12a-mediated adenine base editing increased from 2.9% average editing for LbABE7.10 to 24% average editing (and as high as 46±5.0%) for LbABE8e, and average enAsCas12a-mediated editing increased 59-fold from 0.31% with enAsABE7.10 to 18% (and as high as 29±0.51%) for enAsABE8e at the mostly highly edited position within each protospacer (Fig. 2e)23.
Next we examined the base editing window of ABE8e variants. The editing windows for SpCas9-derived ABE7.10 are slightly narrower (typically, protospacer positions 4–7, counting the PAM as positions 21–23) than the editing windows for canonical SpCas9 CBEs36. Editing windows of ABE8e variants are now consistent with those of the corresponding CBEs: positions 4–8 for SpABE8e, 3–14 for SaABE8e, and 8–14 for both LbABE8e and enAsABE8e (Supplementary Figs. 4-6). These results demonstrate that ABE8e variants can efficiently edit positions that were previously challenging to target with ABE7.10.
To further expand the targeting scope of ABEs caused by modest compatibility with alternative PAM Cas9 variants, we generated ABE8e variants with Cas9 variants previously engineered or evolved to be compatible with alternative PAM sequences, including SpCas9-NG (PAM = NGN)25 and SaCas9-KKH (PAM = NNNRRT)22. We tested each variant in HEK293T cells at five or more endogenous genomic sites each and measured A•T-to-G•C conversion after three days (Fig. 2f and Supplementary Figs. 5, 7, 8). Compared to the corresponding ABE7.10 variants, NG-ABE8e and SaKKH-ABE8e demonstrated increased editing efficiencies (Fig. 2f and Supplementary Figs. 5, 7, 8). For NG-ABEs, we analyzed the 11 most highly edited adenines across five genomic sites. While A•T-to-G•C conversion levels were similar between NG-ABE7.10 and NG-ABE8e at four of the 11 target adenines, at the other seven target adenines, NG-ABE8e was substantially (up to 28-fold) more efficient (Fig. 2f). Similarly, SaKKH-ABE8e supported increased editing efficiencies (37–78% at the most highly edited position) at all tested adenines at each of the nine loci tested compared with SaKKH-ABE7.10 (12–45% at the most highly edited position) (Fig. 2f). These data collectively show that TadA-8e is broadly compatible with diverse Cas9 and Cas12 homologs, greatly expanding the targeting scope of adenine base editors.
The use of circularly permuted Cas9 variants in base editors alters the position of base editing activity windows22. We previously demonstrated that the editing windows of ABEs using circularly permuted Cas9 variants are broadened from protospacer positions 4–7 for canonical ABE7.10 to positions 4–11 for the circularly permuted variants. We constructed circularly permuted CP1028-ABE8e and CP1041-ABE8e, and observed further expansion of the editing window to protospacer positions 3–14 (Fig. 2g and Supplementary Figs. 4). We also observed increased editing at the boundaries of the editing window. For protospacer positions 9–14, which lie outside the canonical editing window of ABEs, base editing efficiency across seven tested genomic loci increased an average of 2.9- and 2.5-fold with CP1028-ABE8e and CP1041-ABE8e, respectively, compared to the corresponding ABE7.10 variants, without substantial changes in indel frequencies (Fig. 2g and Supplementary Figs. 4). Together, circularly permuted ABE8e variants and non-canonical PAM ABE8e variants expand the targeting scope of efficient adenine base editing.
Next, we sought to understand differences in the processivity between ABE8e and ABE7.10. Enhanced processivity is useful for some applications where multiple edits within a given editing window are desired such as the disruption of regulatory regions or genetic screens driven by base editing. We assessed the processivity of ABEs at genomic sites 5, 6, and 7 by calculating the number of alleles containing multiple A•T-to-G•C edits within the editing window. At these three sites, the frequency of alleles with multiple base conversions increase 11- to 17-fold for ABE8e relative to ABE7.10. (Supplementary Fig. 9). This increased processivity could be caused by the overall increased editing activity of ABEs, or by an evolved response to the need for the deaminase to edit two target bases within the same protospacer in the selection circuit. These data suggest that ABE8e is particularly useful when multiple A•T-to-G•C conversions at a single locus are desired or when ABE7.10 is inefficient at editing a desired site.
Off-target analysis of ABE8e
We analyzed off-target activity of ABE8e in HEK293T cells at previously reported Cas9-dependent off-target sites43,44. We tested the top three known ABE off-target sites for site 5 (HBG) and site 6 (VEGFA3) as identified by EndoV-Seq43, and for EMX1 as identified by GUIDE-seq44. We observed an increase in editing at six of the nine off-target sites when comparing ABE8e to ABE7.10, but the ratio of on-target to off-target editing was comparable at all but two sites (Fig. 3a). At the top three EMX1 off-targets sites44, we observed similar levels of ABE8e mediated A•T-to-G•C editing (averaging 74±3.0%, 58±4.9%, and 5.9±4.8% at the top three off-target sites, respectively) and Cas9 mediated indel formation (averaging 81±2.2%, 48±4.9%, and 6.0±1.0%, respectively) (Supplementary Fig. 10). These data demonstrate that the off-target editing levels of ABE8e are higher than the off-target editing levels of ABE7.10 and are consistent with the off-target editing levels of Cas9 nuclease. Given our previous observations that off-target base editing is greatly reduced when delivering base editors as protein-RNA (RNP) complexes45, we examined on-target and off-target base editing with ABE8e RNP delivery. RNP delivery of ABE8e purified from E. coli resulted in similar levels of on-target editing, but much lower editing levels at the nine off-target sites, and thus a dramatic increase in on-target:off-target editing ratios (up to 1,300-fold) at every off-target site analyzed (Fig. 3a). For applications in which off-target editing must be minimized, we recommend the use of ABE8e RNP delivery.
Figure 3. Off-target analysis of ABE8e.
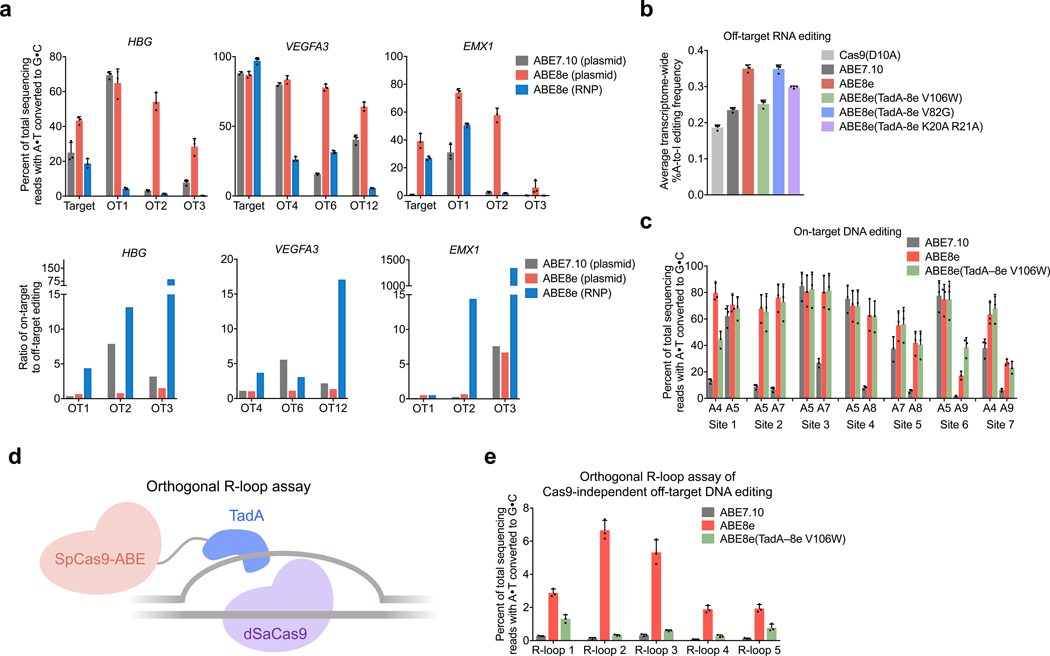
a, DNA off-target analysis comparing ABE7.10 plasmid delivery, ABE8e plasmid delivery, and ABE8e RNP delivery at site 5 (HBG), site 6 (VEGFA3), and EMX1. Editing efficiencies and on-target:off-target editing ratios are shown. b, Off-target transcriptome-wide A-to-I conversion analysis in cellular RNA. c, DNA editing comparing ABE7.10, ABE8e, and ABE8e(TadA-8e V106W) at seven genomic sites in HEK293T cells. d, Orthogonal R-loop assay overview. e, Cas9-independent off-target A•T to G•C editing frequencies detected by the orthogonal R-loop assay at each R-loop site with dSaCas9 and a SaCas9 sgRNA. Each R-loop was performed by cotransfection of ABE7.10, ABE8e, or ABE8e (TadA–8e V106W), and a SpCas9 sgRNA targeting site 3 with dSaCas9 and a SaCas9 sgRNA targeting R-loops 1–5, respectively. For all plots, bars represent mean values, and error bars represent the standard deviation of three independent biological replicates.
ABE7.10 has previously been shown to cause a low-level increase in deamination of adenines in cellular RNA, which can be minimized by introducing mutations in the TadA domains that decrease RNA editing but preserve DNA editing41,46–48. To measure the extent of cellular RNA editing by ABE8e, we treated HEK293T cells with plasmids encoding ABE7.10, ABE8e, or Cas9 (D10A) nickase and an sgRNA targeting LDLR, then measured the A-to-I mutation frequency across the transcriptome. Consistent with previous reports41,46,47, we detected a modest increase in transcriptome-wide RNA editing with ABE7.10 compared to the Cas9 (D10A) control, with A-to-I conversions increasing from 0.19±0.017% for Cas9 (D10A) to 0.24±0.0066% for ABE7.10. ABE8e induced additional transcriptome-wide A-to-I conversion to 0.35±0.010% (Fig. 3b). To reduce RNA editing by ABE8e, we installed mutations previously reported to minimize RNA editing of ABEs41,46,47, and measured transcriptome-wide A-to-I RNA editing levels. We tested three ABE8e mutants, including ABE8e(TadA-8e V106W)47, ABE8e(TadA-8e V82G)41, and ABE8e(TadA-8e K20A R21A)41. ABE8e(TadA-8e V106W) resulted in the greatest reduction in RNA editing levels, decreasing the transcriptome-wide A-to-I conversion level to 0.25±0.011% (Fig. 3b). On-target DNA editing was similar among all of these tested mutants to that of ABE8e (Supplementary Fig. 11). We also characterized on-target DNA editing of SpCas9, SaCas9, and LbCas12a versions of ABE8e(TadA-8e V106W) and observed similar editing levels to that of ABE8e for all adenines across all protospacers (Fig. 3c,Extended Data Figure 4, and Supplementary Figs 12-14). We therefore recommend the use of ABE8e(TadA-8e V106W) for applications that require minimizing off-target RNA editing.
Next we characterized the ability of ABE8e to mediate Cas9-independent off-target DNA editing. We used a recently developed orthogonal R-loop assay49 to detect the propensity of base editors to edit single-stranded DNA regions unrelated to their target loci with much greater sensitivity and lower cost than assays that require whole-genome sequencing (Fig. 3d). We co-transfected HEK293T cells with plasmids encoding an SpABE variant and an on-target sgRNA for SpABE, along with a catalytically inactive SaCas9 (dSaCas9) and an SaCas9 sgRNA targeting a genomic locus unrelated to the SpABE on-target site. We previously correlated base editing levels within these dSaCas9-generated R-loops with the propensity for Cas9-independent off-target editing in mammalian cells49.
Using this assay, we compared A•T-to-G•C base conversion levels in five dSaCas9 R-loops for ABE7.10 and ABE8e, observing an increase in off-target editing at these orthogonal R-loops, ranging from 0.079%–0.32% with ABE7.10 to 1.9%–6.7% with ABE8e (Fig. 3e, Supplementary Fig. 15). To mitigate Cas9-independent off-target DNA editing, we performed the same analysis with ABE8e(TadA–8e V106W), and observed substantial decrease in editing at these orthogonal R-loops, now ranging from 0.32%–1.3% (Fig. 3e, Supplementary Fig. 15). These data indicate that Cas9-independent off-target editing increases with ABE8e as expected given its higher activity, but can be ameliorated by introducing the V106W mutation into TadA-8e, which likely reduces affinity of TadA-8e for both DNA and RNA, thereby increasing its dependence on Cas9 for substrate engagement.
Application of ABE8e to disease-relevant loci
We targeted two sites to install mutations known to induce the upregulation of fetal hemoglobin, a promising approach to treat hemoglobinopathies such as sickle cell disease and β-thalassemia50. We first assessed ABE8e editing at an erythroid enhancer of BCL11A, which encodes a transcriptional repressor that silences fetal hemoglobin in adult erythroid cells. Mutations in a GATA1 binding site at the +58 BCL11A erythroid enhancer lead to decreased BCL11A expression and concomitant fetal hemoglobin induction51–53. We compared the ability of ABE7.10 and ABE8e to install two A•T-to-G•C edits in the GATA1 binding site at positions 4 and 7 within a single protospacer in HEK293T cells. We observed simultaneous editing of both target adenines in 54.4±12.5% of alleles following cellular treatment with ABE8e compared to only 7.9±3.4% of alleles following treatment with SpABE7.10 (Fig. 4a and Supplementary Fig. 16).
Figure 4. Adenine base editing with ABE8e at disease-relevant loci in human cells.
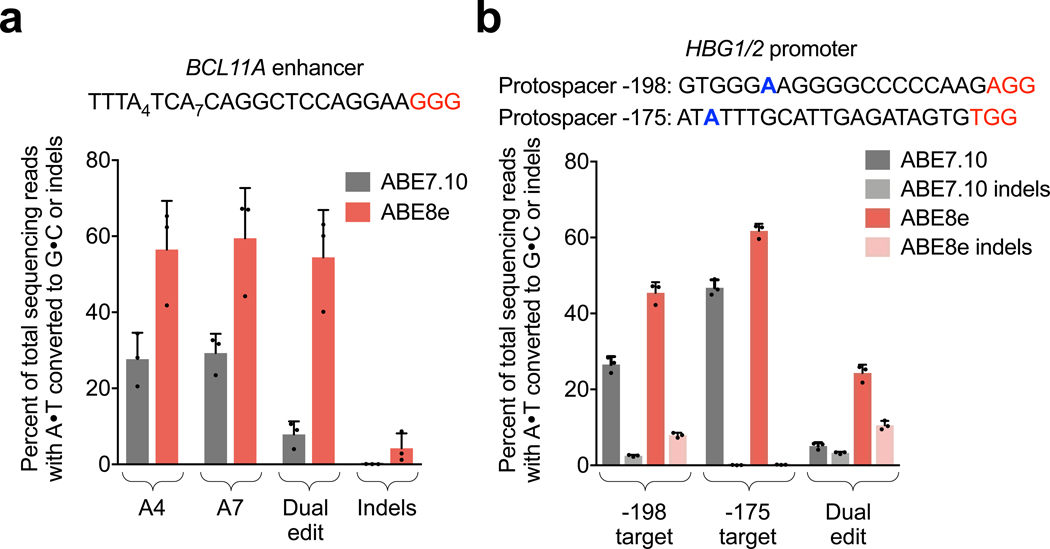
a, Base editing efficiency and indel frequencies in HEK293T cells at a GATA1 binding site of the BCL11A enhancer by ABE7.10 and ABE8e. Editing efficiencies at each adenine in the protospacer individually and efficiencies of editing both adenines within the same allele. b, Base editing efficiencies and indel frequencies in HEK293T cells of the HBG1/2 promoter with ABE7.10 and ABE8e. Data are shown for each sgRNA provided individually, and for dual editing of both target sites within a single allele when both sgRNAs are provided simultaneously. Protospacers are named based on the position of the target adenine relative to the HBG transcription start site. For all plots, bars represent mean values, and error bars represent the standard deviation of three independent biological replicates.
We also applied ABE8e to install two naturally-occurring A•T-to-G•C single-nucleotide polymorphisms (SNPs) present in the promoters of the HBG1 and HBG2 fetal hemoglobin genes50. Each of these SNPs confers a hereditary persistence of fetal hemoglobin, which may alleviate symptoms of hemoglobinopathies54. The target adenines are located at HBG promoter positions −198 and −175, and their base editing requires two guide RNAs since they are separated by 23 nucleotides. We treated HEK293T cells with ABE7.10 or ABE8e and each guide RNA separately, and observed 1.3- to 1.7-fold increases in editing at each site with ABE8e (Fig. 4b). Although each SNP individually increases the expression of fetal hemoglobin, editing both SNPs simultaneously might further augment fetal hemoglobin levels. When we delivered both guide RNAs simultaneously with ABE7.10, we observed combined editing at both HBG promotor positions of only 5.1±0.9% within a single allele (Fig. 4b and Supplementary Fig. 17). In contrast, we observed 24±2.2% of HBG alleles containing both edits with ABE8e, a 4.8-fold increase (Fig. 4b and Supplementary Fig. 17). These results suggest that ABE8e is especially well suited for multiplexed base editing applications.
Discussion
Collectively, these results demonstrate the ability of PANCE and PACE to evolve an adenine base editor with 590-fold improved deamination kinetics that supports efficient base editing using a much larger set of Cas9 and Cas12a homologs than was previously possible. The newly evolved TadA-8e supports efficient adenine base editing with every Cas protein tested in this study—SpCas9, SaCas9, LbCas12a, enAsCas12a, SpCas9-NG, SaCas9-KKH, CP1028-SpCas9, and CP1041-SpCas9—to mediate efficient A•T-to-G•C base conversions.
The improved kinetics, editing efficiency, and targeting scope of ABE8e variants enabled efficient simultaneous base editing to install two mutations in a GATA1 binding site of a BCL11A enhancer, or two mutations in the promoter of HBG genes, which are known to increase fetal hemoglobin expression.
The dramatic increase in deamination kinetics, together with compatibility with previously uncooperative Cas effectors, suggest the value of a high-resolution structure of ABE8e that illuminates the molecular basis of these features. ABE8e substantially advances the capabilities of adenine base editors by expanding their targeting scope, efficiency, and suitability for base editing applications.
Methods
General methods and molecular cloning
Antibiotics were used at the following working concentrations: carbenicillin, 50 μg/mL; spectinomycin, 50 μg/mL; chloramphenicol, 40 μg/mL; and kanamycin, 30 μg/mL. Nuclease-free water (ThermoFisher Scientific) was used for PCR reactions and cloning. For all other experiments, water was purified using a MilliQ purification system (Millipore). Phusion U Green Multiplex PCR Master Mix (ThermoFisher Scientifc) was used for all PCRs.
Plasmids were cloned by uracil-specific excision reagent (USER) assembly, Golden Gate assembly, or KLD cloning following manufacturer’s instructions. For USER cloning, 42–60 °C melt temperature junctions were used, and constructs were assembled by digesting at 37 °C for 45 min followed by transformation into chemically competent cells. For Golden Gate assembly, LguI (SapI isoschizomer, Life Technologies) was used as the type IIS restriction enzyme along with T4 DNA ligase (New England BioLabs). Typical assemblies contained final concentrations of ~0.5–2 ng per kb per μL plasmids, with a ~2:1 ratio of donor to acceptor plasmids. Assemblies were incubated at 37 °C for between 1 h and 18 h followed by transformation into chemically competent cells. Guide RNA plasmids were assembled following the manufacturer’s instructions with KLD enzyme mix (New England BioLabs).
Codon-optimized sequences for human cell expression were obtained from Genscript. Plasmids were cloned and amplified using Mach1 T1R competent cells (ThermoFisher Scientific). Plasmid DNA was isolated using the Qiagen Spin Miniprep Kit and Qiagen Midiprep Kit according to the manufacturer’s instructions. All constructs assembled using PCR were fully sequence-verified using Sanger sequencing (Quintara Biosciences), while constructs assembled using Golden Gate cloning were sequence-verified across all assembly junctions. Protospacer sequences for guide RNA plasmids are described in Supplementary Table 3. The amino acid sequences for codon-optimized, bis-bpNLS base editor variants are listed in Supplementary Sequence 1.
Bacteriophage cloning
Phage were cloned with the second generation backbone using Golden Gate assembly as previously described36, with minor modifications. Briefly, the phage genome was split between two donor plasmids (pBT114-splitC and pBT29-splitD) and the desired phage insert was supplied on a third donor plasmid (pBT100.164). The donor plasmid (pBT100.164) contains TadA-7.10 fused to an Npu C-intein. pBT114-splitC differs from the second-generation donor plasmid used previously (pBT29-splitC). pBT29-splitC contains a small portion of the C-terminal end of gene III, which serves as the promoter for gene VI36. Due to problems with gene III recombination events into the phage, leading to a “cheater” phenotype in which base editing was not required for phage propagation, the C-terminal end of gene III was removed from the phage backbone and replaced by an artificial promoter for gene VI in pBT114-splitC.
Phage were cloned with Golden Gate assembly as described above with LguI (SapI isoschizomer, Life Technologies) used as the type IIS restriction enzyme. Following Golden Gate assembly, phage were transformed into chemicompetent S206031 E. coli host cells containing plasmid pJC175e, which enables activity-independent phage propagation, and grown overnight at 37 °C with shaking in Davis Rich Medium (DRM). Bacteria were then centrifuged for 5 min at 15,000 g, and plaqued as described below. Individual phage plaques were grown in 2xYT media until the bacteria reached late growth phase. Bacteria were centrifuged as before, and the supernatants containing phage were purified with a 0.2 micron filter to remove residual bacteria. Finally, phage were sequenced to ensure proper cloning.
Preparation and transformation of chemically competent cells
Strain S206031 was used in all experiments, including phage propagation tests, PANCE, and PACE. Chemically competent cells were prepared as described36, unless otherwise noted. Briefly, an overnight culture was diluted 50-fold into 2xYT media and grown at 37 °C with shaking at 230 r.p.m. to an optical density (OD600) of around 0.4–0.5. Cells were cooled on ice and pelleted by centrifugation at 4,000 g for 10 min at 4 °C. The cell pellet was then resuspended by gentle stirring in ice-cold TSS solution (LB media supplemented with 5% v/v DMSO, 10% w/v PEG 3350, and 20 mM MgCl2). The cell suspension was mixed thoroughly, aliquoted and frozen in a dry ice/acetone bath, then stored at −80 °C until use. To transform cells, 100 μl of competent cells thawed on ice was added to a plasmid(s) and 100 μl KCM solution (100 mM KCl, 30 mM CaCl2, and 50 mM MgCl2 in water). The mixture was heat shocked at 42 °C for 60 s and SOC media (200 μL) was added. Cells were allowed to recover at 37 °C with shaking at 230 r.p.m. for 1 h, then spread on LB media with 1.5% agar (United States Biologicals) plates containing the appropriate antibiotic(s) and incubated at 37 °C for 16–18 h.
Plaque assays for phage titer quantification and phage cloning
Phage were plaqued on S206031 E. coli host cells containing plasmid pJC175e (activity-independent propagation)38 or plasmid pT7-AP13 (to check for the presence of T7 RNAP recombinants)38. To prepare a cell stock for plaquing, overnight culture of host cells (fresh or stored at 4 °C for up to ~1 week) was diluted 50-fold in 2xYT media containing appropriate antibiotic(s) and grown at 37 °C to an OD600 of 0.5–0.8. Serial dilutions of phage (ten-fold) were made in PBS buffer (pH 7.4) or water. To prepare plates, molten 2xYT medium agar (1.5% agar, 55 °C) was mixed with Bluo-gal (10% w/v in DMSO) to a final concentration of 0.04% Bluo-gal. The molten agar mixture was pipetted into quadrants of quartered Petri dishes (1.5 mL per quadrant) or wells of a 12-well plate (~1 mL per well) and allowed to set. To prepare top agar, a 2:1 mixture of 2xYT media and molten 2xYT medium agar (1.5%, 0.5% agar final) was prepared. Top agar was maintained tightly capped at 55 °C for up to 1 week. To plaque, cell stock (50–100 μL) and phage (10 μL) were mixed in 2 mL library tubes (VWR International), and 55 °C top agar added (400 or 1,000 μL for 12-well plate or Petri dish, respectively) and mixed one time by pipetting up and down, and then the mixture was immediately pipetted onto the solid agar medium in one well of a 12-well plate or one quadrant of a quartered Petri dish. Top agar was allowed to set undisturbed (10 min at room temperature), then plates or dishes were incubated (without inverting) at 37 °C overnight. Phage titer were determined by quantifying blue plaques.
Phage propagation assays
S2060 cells containing plasmids of interest were prepared as described above and inoculated in Davis Rich Medium (DRM) (prepared from US Biological CS050H-001/CS050H-003). Host cells from an overnight culture in DRM were diluted 50-fold into fresh DRM and grown for ~1.5 h at 37 °C. Previously titered phage stocks were added to 2 mL of bacterial culture at a final concentration of 105 plaque forming units mL−1. The cultures were grown overnight with shaking at 37 °C and then centrifuged (3,600 g, 10 min) to remove cells. The supernatants were titered by plaquing as described above. Fold enrichment was calculated by dividing the titer of phage propagated on host cells by the titer of phage at the same input concentration shaken overnight in DRM without host cells.
PANCE experiments
Chemically competent host cells were transformed with DP640 and plated on 2xYT agar containing 0.5% glucose (w/v) along with appropriate concentrations of antibiotics. Five colonies were diluted in DRM with the appropriate antibiotics, grown to OD600 0.5–0.6, and treated with 40 mM arabinose to induce mutagenesis and the desired amount of anhydrotetracycline for a given passage (0 or 40 ng/mL). Treated cultures were split into the desired number of either 2 mL cultures in single culture tubes or 500 μL cultures in a 96-well plate and infected with selection phage. Infected cultures were grown overnight at 37 °C and harvested the next day via centrifugation (3000 g for 10 min). Supernatant containing evolved phage was isolated and stored at 4 °C. Isolated phage were then used to infect the next passage and the process repeated for the desired number of selection passages for the selection. For phage dilutions, see Supplementary Table 2. Phage titers were determined by plaquing as described above. Phage genotypes were assessed from pool samples or single plaques by diagnostic PCR using primers BT-52F (5′-GTCGGCGCAACTATCGGTATCAAGCTG) and BT-52R2 (5′-AGTAAGCAGATAGCCGA ACAAAGTTACCAGAAGGAAAC), and the PCR products were assessed by Sanger sequencing.
PACE experiments
Unless otherwise noted, PACE apparatus, including lagoons, chemostats, pumps and media, were prepared and used as previously described39. Host cells were prepared as described for PANCE above. Five colonies were diluted into 5 mL DRM with the appropriate antibiotics and grown to OD600 0.4–0.8, which was then used to inoculate a chemostat (60 mL), which was maintained under continuous dilution with fresh DRM at 1–1.5 volumes per h to keep cell density roughly constant. Lagoons were initially filled with DRM, then continuously diluted with chemostat culture for at least 2 h before seeding with phage.
Stock solution of arabinose (1 M) was pumped directly into lagoons (10 mM final) as previously described39 for 1 h before the addition of phage. For the first 12 h after phage inoculation, anhydrotetracycline (aTc) was present in the stock solution (3.3 μg/mL). Syringes containing aTc solution were covered in aluminum foil, and work was conducted to minimize light exposure of tubing and lagoons.
Lagoons were seeded at a starting titer of ~107 pfu per mL. Dilution rate was adjusted by modulating lagoon volume (5–20 mL) and/or culture inflow rate (10–20 mL/h). Lagoons were sampled at indicated times (usually every 24 h) by removal of culture (500 μL) by syringe through the waste needle. Samples were centrifuged at 13,500 g for 2 min and the supernatant removed and stored at 4 °C. Titers were evaluated by plaquing as described above. The presence of T7 RNAP or gene III recombinant phage was monitored by plaquing on S2060 cells containing pT7-AP and no plasmid. Phage genotypes were assessed from single plaques by diagnostic PCR as described in the PANCE section.
Cell culture
HEK293T cells (ATCC CRL-3216) were cultured in Dulbecco’s modified Eagle’s medium (Corning) supplemented with 10% fetal bovine serum (ThermoFisher Scientific) and maintained at 37 °C with 5% CO2.
Transfections
HEK293T cells were seeded at 50,000 cells per well on 48-well poly-D-lysine plates (Corning) in the same culture medium. Cells were transfected 24–30 h after plating with 1.5 μL Lipofectamine 2000 (ThermoFisher Scientific) using 750 ng base editor plasmid, 250 ng guide RNA plasmid and 20 ng green fluorescent protein as a transfection control following the manufacturer’s instructions. Titration experiments were performed as previously reported6. When targeting HBG1/2 and BCL11A sites with plasmid DNA, 150 ng of base editor plasmid and 50 ng of guide RNA plasmid was complexed with 1 μL of Lipofectamine 3000. For all transfection experiments unless otherwise noted, cells were cultured for 3 d, then washed with 1x PBS (ThermoFisher Scientific), followed by genomic DNA extraction by addition of 100 μL freshly prepared lysis buffer (10 mM Tris-HCl, pH 7.5, 0.05% SDS, 25 μg/mL proteinase K (ThermoFisher Scientific)) directly into each transfected well. The mixture was incubated at 37 °C for 1 h then heat inactivated at 80 °C for 30 min. Genomic DNA lysate was subsequently used immediately for high-throughput sequencing (HTS).
Orthogonal R-loop assay
Orthogonal R-loop assays to measure off-target editing were performed as previously described49, with minor modifications. Under standard conditions, 200 ng of SpCas9 sgRNA plasmid, 200 ng of SaCas9 sgRNA plasmid, 300 ng of base editor plasmid, and 300 ng of dSaCas9 plasmid were co-transfected into HEK293T cells using 1.5 μL of Lipofectamine 2000. For these transfection experiments, cells were cultured for 3 d, then washed with 1x PBS (ThermoFisher Scientific), followed by genomic DNA extraction by addition of 100 μL freshly prepared lysis buffer (10 mM Tris-HCl, pH 7.5, 0.05% SDS, 25 μg/mL proteinase K (ThermoFisher Scientific)) directly into each transfected well. The mixture was incubated at 37 °C for 1 h then heat inactivated at 80 °C for 30 min. Genomic DNA lysate was used immediately for high-throughput sequencing (HTS).
Protein purification for in vitro studies
SpABE7.10 and SpABE8e, which were used in in vitro deamination assays, were cloned into a pBR322 plasmid. The cloned constructs contain an N-terminal His6-tag. The proteins were expressed in E. coli strain BL21 Rosetta 2 (DE3) (EMD Biosciences) and purified as described previously55 with a few alterations. The cells were lysed via sonication in 30 mM HEPES pH 8.0, 1 M KCl, 2 mM TCEP, 10 % (v/v) glycerol, 0.5% Triton X-100, and 10 mM imidazole buffer supplemented with protease inhibitor cocktail (Roche). The proteins were purified in several steps. First, clarified lysates were loaded onto a HisTrap FF column (GE Healthcare), and proteins were eluted with a gradient of lysis buffer supplemented with up to 300 mM imidazole. Next, the eluted proteins were separated from non-specific nucleic acids using heparin HiTrap column (GE Healthcare) and eluted with a linear gradient of 100 mM to 1.2 M KCl. Finally, the proteins were further purified by size-exclusion chromatography using a Superdex 200 16/60 column and 20 mM HEPES pH 7.5, 400 mM KCl, 2 mM TCEP, 10 % (v/v) glycerol buffer. The eluted proteins were concentrated to ~10 mg/mL.
Single turnover in vitro deamination assay
A single guide RNA (5’-GUUCACCUUUCUUUGUCUGUGUUUUAGAGCUAGAAAUAGCAAGUUAAAAUAAGGCUAGUCCGUUAUCAACUUGAAAAAGUG-3’) was transcribed in vitro using synthetic DNA oligonucleotides (IDT) containing a T7 promoter sequence. The sgRNA was purified by 12 % polyacrylamide gel electrophoresis (PAGE), then extracted from the gel using the crush-and-soak method followed by ethanol precipitation and dephosphorylated using rSAP (New England Biolabs).
For in vitro deamination assays, 10 pmol of DNA oligonucleotide (non-target strand which contains a single adenine 5’-GTTCGGTGGCTCCGTCCGTGTTCACCTTTCTTTGTCTGTGGGCGTTTTGGTTGCTCTTCG-3’) was 5’-radiolabeled using [γ−32P] ATP (PerkinElmer) and 5 units of T4 polynucleotide kinase (New England Biolabs) in 1× T4 PNK buffer (New England Biolabs) at 37 °C for 30 min. The labeling reaction was purified using an illustra™ MicroSpin G-25 column (GE Life Sciences) to remove free nucleotides. The dsDNA substrates were prepared by annealing the 5’-radiolabeled non-target strand and an excess of unlabeled target strand (5’-CGAAGAGCAACCAAAACGCCCACAGACAAAGAAAGGTGAACACGGACGGAGCCACCGAAG-3’).
The SpABE7.10 and sgRNA and SpABE8e and sgRNA RNP complexes were prepared by mixing the sgRNA and appropriate SpABE in a 1.5:1 molar ratio in RNP assembly buffer (20 mM Tris-HCl pH 7.5, 200 mM KCl, 5 % (v/v) glycerol, 5 mM MgCl2, 2 mM DTT) and incubating at room temperature for 20 min.
The deamination reactions containing 1 μM RNPs in deamination buffer (20 mM Tris-HCl pH 7.5, 100 mM KCl, 5 % (v/v) glycerol, 2.5 mM MgSO4, 2 mM DTT) were initiated by adding 5’-radiolabeled dsDNA to a final concentration of 1 nM, then incubated at 37 °C. 20 μL aliquots were removed at the 0, 1, 3, 8, 23, and 32 hours in case of SpABE7.10 and at the 0, 1, 2, 5, 10, 20, and 60 min in case of SpABE8e. The reactions were quenched by mixing them with 30 μL of hot water and incubating them at 95 °C for 2 min. The reactions were then purified using Monarch PCR & DNA Cleanup kit (New England Biolabs). To detect adenosine deamination, eluted DNA was incubated with 20 units of E. coli Endonuclease V (EndoV) in 1× NEB4 buffer at 37 °C for 1 h. EndoV specifically recognizes deaminated adenosine (inosine) in dsDNA and ssDNA and cleaves DNA one nucleotide downstream of the modification site43,56,57. After cleavage by EndoV, the samples were mixed with an equal volume of formamide gel loading buffer (95% formamide, 100 mM EDTA, 0.025% SDS, and 0.025% (w/v) bromophenol blue), heated at 95 °C for 2 min, resolved on 15% denaturing polyacrylamide gel, and visualized by phosphorimaging. Assays were performed in three independent replicates, and the intensities of the uncleaved and cleaved DNA were analyzed using ImageQuant TL Software (GE Healthcare). Apparent rates were calculated by a fit to a single exponential decay (Prism7, GraphPad).
Protein purification for RNP delivery to mammalian cells
ABE8e was codon-optimized for bacterial expression and cloned into the protein expression plasmid pD881-SR (Atum, Cat. No. FPB-27E-269). The expression plasmid was transformed into BL21 Star DE3 competent cells (ThermoFisher, Cat. No. C601003). Colonies were picked for overnight growth in Terrific Broth (TB) with 25ug/mL kanamycin and grown overnight with shaking at 37 °C. The next day, 2 L of pre-warmed TB were inoculated with overnight culture at a starting OD600 of 0.05. Cells were shaken at 37 °C for about 2.5 h until the OD600 was ~1.5. Cultures were cold shocked in an ice-water slurry for 1 h, after which L-rhamnose was added to a final concentration of 0.8% (w/v). Cultures were then incubated at 18 °C with shaking for 24 h to induce. Following induction, cells were pelleted and flash-frozen in liquid nitrogen and stored at −80 °C. The next day, cells were resuspended in 30 mL cold lysis buffer (1 M NaCl, 100 mM Tris-HCl pH 7.0, 5 mM TCEP, 20% glycerol, with 5 tablets of complete, EDTA-free protease inhibitor cocktail tablets (Millipore Sigma, Cat. No. 4693132001)). Cells were passed 3 times through a homogenizer (Avestin Emulsiflex-C3) at ~18,000 psi for lysis. Cell debris was pelleted for 20 min using a 20,000 g centrifugation at 4 °C. Supernatant was collected and spiked with 40 mM imidazole, followed by a 1 h incubation at 4 °C with 1 mL of Ni-NTA resin slurry (G Bioscience Cat. No. 786–940, prewashed once with lysis buffer). Protein-bound resin was washed twice with 12 mL of lysis buffer in a gravity column at 4 °C. Protein was eluted in 3 mL of elution buffer (300 mM imidazole, 500 mM NaCl, 100 mM Tris-HCl pH 7.0, 5 mM TCEP, 20% glycerol). Eluted protein was diluted in 40 mL of low-salt buffer (100 mM Tris-HCl, pH 7.0, 5 mM TCEP, 20% glycerol) just before loading into a 50 mL Akta Superloop for ion exchange purification on the Akta Pure25 FPLC. Ion exchange chromatography was conducted on a 5 mL GE Healthcare HiTrap SP HP pre-packed column (Cat. No. 17115201). After washing the column with low salt buffer, the diluted protein was flowed through the column to bind. The column was then washed in 15 mL of low salt buffer before being subjected to an increasing gradient to a maximum of 80% high-salt buffer (1 M NaCl, 100 mM Tris-HCl, pH 7.0, 5 mM TCEP, 20% glycerol) over the course of 50 mL, at a flow rate of 5 mL/min. 1 mL fractions were collected during this ramp to high-salt buffer. Peaks were assessed by SDS-PAGE to identify fractions containing the desired protein, which were concentrated first using an Amicon Ultra 15mL centrifugal filter (100-kDa cutoff, Cat. No. UFC910024), followed by a 0.5 mL 100 kDa cutoff Pierce concentrator (Cat. No. 88503). Concentrated protein was quantified using a BCA assay (ThermoFisher, Cat. No. 23227); the stock concentration was 3.8 mg/mL.
Protein nucleofection
Nucleofection was conducted as per manufacturer’s recommendations using a Lonza 4D nucleofector and the Lonza SF Cell Line 4D-Nucleofector X Kit S (Cat No. V4XC-2032). After formulating the SF nucleofection buffer, 200,000 cells were resuspended in 5 μL of buffer per nucleofection. In the remaining 15 μL of buffer per nucleofection, 20 pmol of chemically modified sgRNA from Synthego was combined with 18 pmol of ABE8e protein and incubated 5 min at room temperature to complex. Cells were added to the 20 μL nucleofection cuvettes, followed by protein solution, pipetting up and down to mix. Cells were nucleofected with program CM-130, immediately after which 80 μL of warmed media was added to each well for recovery. After 5 min, 25 μL from each sample was added to 250 μL of fresh media in a 48-well poly-D-lysine plate (Corning). Cells were then treated the same way as lipofected cells above for genomic DNA extraction after 3 more days of culture.
HTS of genomic DNA samples
HTS of genomic DNA from HEK293T cells was performed as previously described2. Primers for PCR 1 of target genomic site amplification are listed in Supplementary Table 4, and amplicons for analyses are listed in Supplementary Table 5. Following Illumina barcoding, PCR products were pooled and purified by electrophoresis with a 2% agarose gel using a Monarch DNA Gel Extraction Kit (New England Biolabs), eluting with 30 μl H2O. DNA concentration was quantified with a Qubit dsDNA High Sensitivity Assay Kit (ThermoFisher Scientific) and sequenced on an Illumina MiSeq instrument (paired-end read, R1: 250–280 cycles, R2: 0 cycles) according to the manufacturer’s protocols.
HTS data analysis
Sequencing reads were demultiplexed using the MiSeq Reporter (Illumina) and FASTQ files were analyzed using CRISPResso258, as previously described22. Dual editing in individual alleles were analyzed by a Python script provided in Supplementary Note 1. Base-editing values are representative of n = 3 independent biological replicates collected by different researchers, with the mean ± s.d. shown. Base-editing values are reported as a percentage of the number of reads with adenine mutagenesis over the total aligned reads. All raw FASTQ files generated are available from the NCBI SRA under BioProject PRJNA589228.
Preparation of RNA libraries for RNA-Seq
HEK293T cells were treated as before via transfection with base editor and guide plasmids. After two days, cells were washed with 1x PBS, lysed with TRIzol (Thermo Fisher), and cleaned using RNeasy mini kit (Qiagen) with on-column DNAseI treatment following the manufacturer’s protocols. Polyadenylated mRNA was enriched from 1 μg of total RNA using the PolyA mRNA isolation kit (Takara) on the Apollo 324 instrument. Stranded RNA-seq libraries were generated from these samples using the PrepX mRNA 48 kit (Takara) on the Apollo 324 followed by barcoding and amplification (12 cycles). Following PCR and bead cleanup with AmpureXP beads (Beckman Coulter), libraries were visualized on a 2200 TapeStation (Agilent) and quantified using a Library Quantification Kit (KAPA Biosystems) for multiplexing. Libraries were sequenced on a NextSeq high-throughput flowcell (Illumina) as 75 bp paired-end reads. All raw FASTQ files generated are available from the NCBI SRA under BioProject PRJNA589228.
RNA-Seq data analysis
Analysis of the transcriptome-wide editing RNA sequencing data was performed as follows. Prior to the analysis described below, FASTQ files were generated using Bcl2fastq2, then trimmed using Trimmomatic version 0.32 to remove adaptor sequences, unpaired sequences, and low-quality bases. We created sam alignments using HISAT2 to align paired reads from each of three biological replicates to the hg19 human reference genome (UCSC). Precomputed HISAT2 indexes where obtained from https://ccb.jhu.edu/software/hisat2/index.shtml. The resulting sam files were sorted and indexed using the samtools software package.
To calculate the average % of A-to-I editing amongst adenosines sequenced in transcriptome-wide sequencing analysis, we used REDItools v1.3 to quantify the % A-to-I editing in each sample. We removed all nucleotides except adenosines from our analysis, and then removed all adenosines with a read coverage less than 10 to avoid errors due to low sampling; additionally, we removed positions with a mapping or read quality score below 25. Next, we calculated the number of adenosines converted to an inosine in each sample and divided this by the total number of adenosines in our dataset after filtering to obtain a percentage of adenosines edited to inosine in the transcriptome. Calculation of s.e.m. was generated by comparison of 3 biological replicates.
Reproducibility
Biologically independent experiments reported here were performed by different researchers using independent splits of the mammalian cell type used.
Extended Data
Extended Data Figure 1. Mutation table of variants from PANCE and PACE.
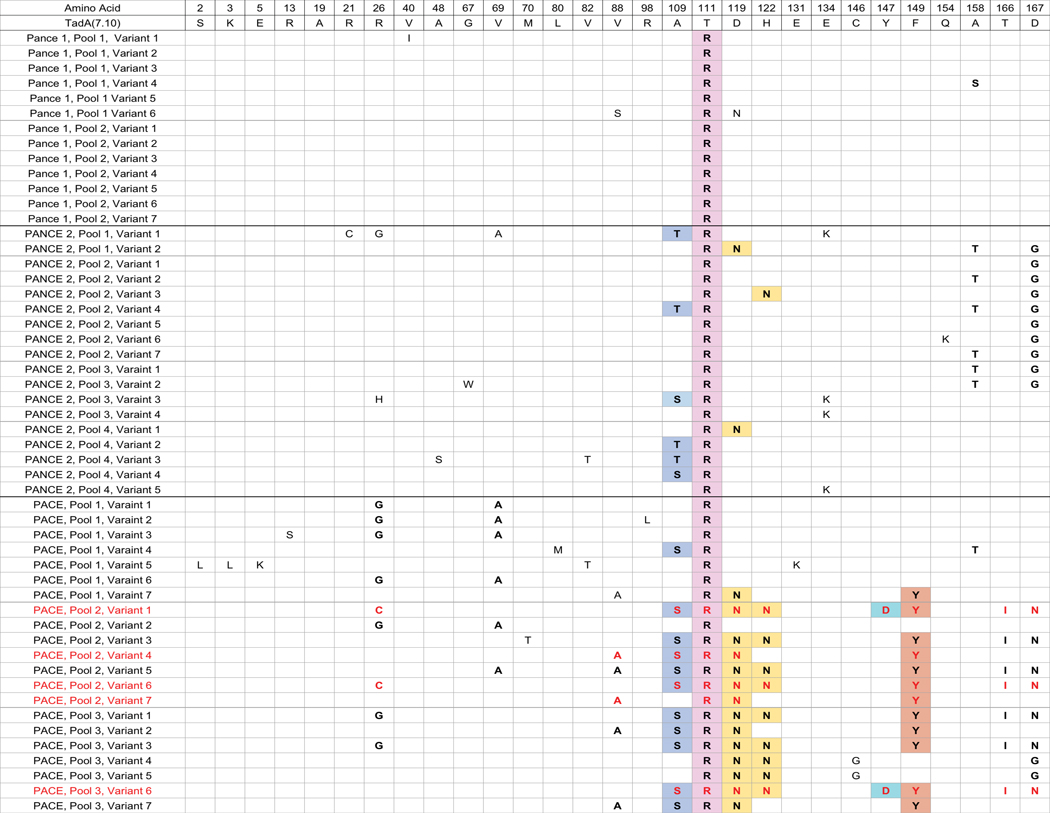
Data were obtained by sequencing individual plaques. Conserved mutations are bolded. Mutations that are highlighted in the structure in Fig. 2b are highlighted to match the amino acid positions in the structure. Genotypes in red were tested for base editing activity in mammalian cells.
Extended Data Figure 2. PACE schedule for deoxyadenosine deaminase evolution.
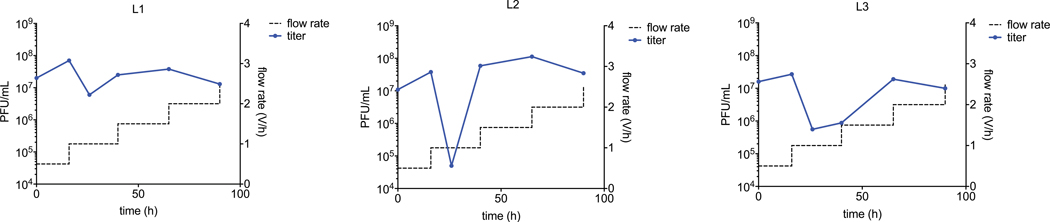
Lagoon L1 contains host cells harboring P1, P2, and P3e. Lagoons L2 and L3 contain host cells harboring P1, P2, and P3g, which form a more stringent selection circuit than the circuit in lagoon L1. For details on plasmids, see Supplementary Table 1. The stringency of the ABE selection was further modulated by increasing the lagoon flow rate (dashed lines). For the first 12 hours, gene III was expressed by the addition of anhydrotetracycline to enable genetic drift in the absence of selection pressure12,13.
Extended Data Figure 3. Titration data at eight editor doses comparing base editing efficiencies for ABE8e and ABE8e-dimer at three sites in HEK293T cells.
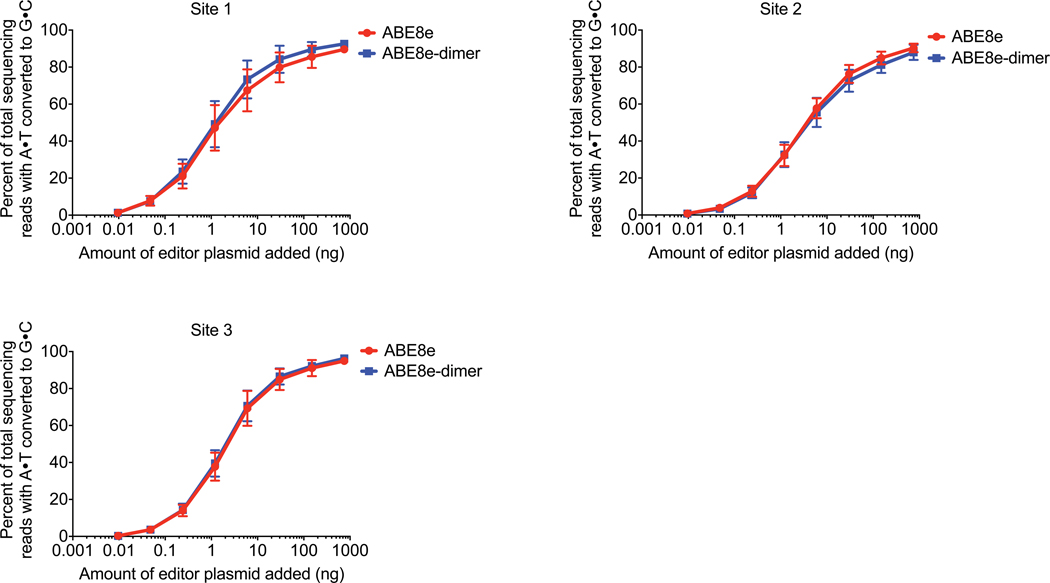
Base editing with ABE8e and ABE8e-dimer in HEK293T cells at three genomic sites in HEK293T cells. Transfections were performed with constant amount of sgRNA plasmid but eight varying doses of ABE plasmid. For all plots, dots represent individual biological replicates and bars represent mean±s.d. of three independent biological replicates.
Extended Data Figure 4. TadA-8e V106W analysis for SaCas9 and LbCas12a.
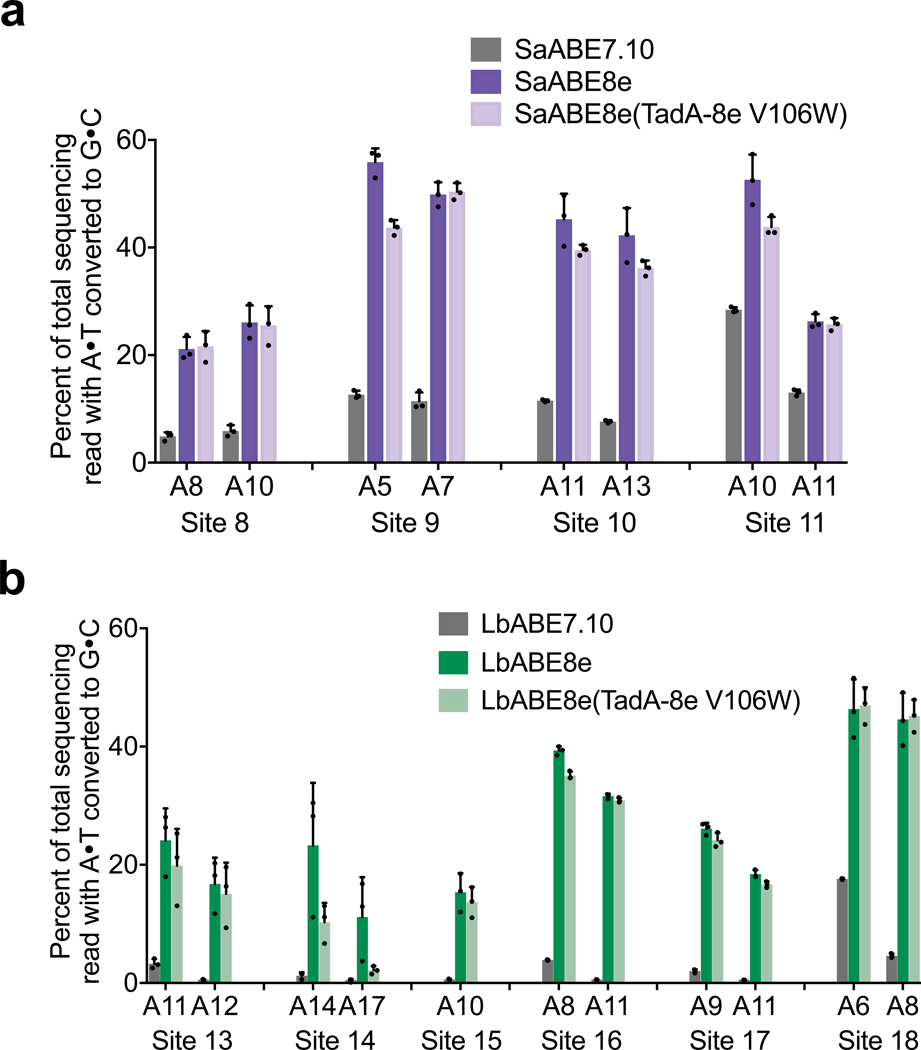
a, DNA editing comparing SaABE7.10, SaABE8e, and SaABE8e(TadA-8e V106W) at four genomic sites in HEK293T cells. b, DNA editing comparing LbABE7.10, LbABE8e, and LbABE8e(TadA-8e V106W) at six genomic sites in HEK293T cells. For all plots, dots represent individual biological replicates and bars represent mean±s.d. of three independent biological replicates.
Supplementary Material
Acknowledgements
This work was supported by US NIH U01 AI142756, RM1 HG009490, R01 EB022376, and R35 GM118062, St. Jude Collaborative Research Consortium, the Bill and Melinda Gates Foundation, and the Howard Hughes Medical Institute. M.F.R. was supported by an HHMI Hanna Gray Fellowship. K.T.Z. was supported by Harvard Chemical Biology Training Grant (T32 GM095450). G.A.N was supported by the Helen Hay Whitney Fellowship. C.W. was supported as a Marlon Abbe Fellow of the Damon Runyon Cancer Research Foundation (DRG-2343–18). L.W.K. was supported by an NSF GRFP. D.E.B. was supported by NHLBI (P01HL053749), Burroughs Wellcome Fund and the St. Jude Children’s Research Hospital Collaborative Research Consortium. We thank Shannon Miller and Tina Wang for providing sgRNA plasmids. We thank Aditya Raguram for help with computational analyses. We thank Jordan Doman and Aditya Raguram for plasmids used during the orthogonal R-loop assay.
Footnotes
Competing interests
The authors declare competing financial interests. D.R.L. is a consultant and co-founder of Editas Medicine, Pairwise Plants, Beam Therapeutics, and Prime Medicine, companies that use genome editing. The authors have filed patent applications on evolved ABEs. The Regents of the University of California have patents issued and pending for CRISPR technologies on which J.A.D. is an inventor. J.A.D. is a co-founder of Caribou Biosciences, Editas Medicine, Scribe Therapeutics, and Mammoth Biosciences, and a scientific advisory board member of Caribou Biosciences, Intellia Therapeutics, eFFECTOR Therapeutics, Scribe Therapeutics, Mammoth Biosciences, Synthego, and Inari; she is a director at Johnson & Johnson. The authors declare no competing non-financial interests.
Data availability. High-throughput sequencing data have been deposited in the NCBI Sequence Read Archive database (PRJNA589228). All plasmids encoding ABE8e variants in this study will be available through Addgene. A subset of selection plasmids used in this study will be available through Addgene. Other materials are available upon reasonable request.
Code availability. Custom scripts used to analyze processivity is available in Supplementary Note 1.
References
- 1.Gaudelli NM et al. Programmable base editing of A*T to G*C in genomic DNA without DNA cleavage. Nature 551, 464–471 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Komor AC, Kim YB, Packer MS, Zuris JA & Liu DR Programmable editing of a target base in genomic DNA without double-stranded DNA cleavage. Nature 533, 420–424 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rees HA & Liu DR Base editing: precision chemistry on the genome and transcriptome of living cells. Nat. Rev. Genet 19, 770–788 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Nishida K. et al. Targeted nucleotide editing using hybrid prokaryotic and vertebrate adaptive immune systems. Science 353 (2016). [DOI] [PubMed] [Google Scholar]
- 5.Komor AC et al. Improved base excision repair inhibition and bacteriophage Mu Gam protein yields C:G-to-T:A base editors with higher efficiency and product purity. Sci. Adv 3, eaao4774 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koblan LW et al. Improving cytidine and adenine base editors by expression optimization and ancestral reconstruction. Nat. Biotechnol 36, 843–846 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Landrum MJ et al. ClinVar: public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 44, D862–868 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Landrum MJ et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res. 42, D980–985 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Komor AC, Badran AH & Liu DR Editing the genome without double-stranded DNA breaks. ACS Chem. Biol 13, 383–388 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Villiger L. et al. Treatment of a metabolic liver disease by in vivo genome base editing in adult mice. Nat. Med 24, 1519–1525 (2018). [DOI] [PubMed] [Google Scholar]
- 11.Ryu SM et al. Adenine base editing in mouse embryos and an adult mouse model of Duchenne muscular dystrophy. Nat. Biotechnol 36, 536–539 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Yeh WH, Chiang H, Rees HA, Edge ASB & Liu DR In vivo base editing of post-mitotic sensory cells. Nat. Commun 9, 2184 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tanaka S. et al. In vivo targeted single-nucleotide editing in zebrafish. Sci. Rep 8, 11423 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y. et al. Programmable base editing of zebrafish genome using a modified CRISPR-Cas9 system. Nat. Commun 8, 118 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ma Y. et al. Highly efficient and precise base editing by engineered dCas9-guide tRNA adenosine deaminase in rats. Cell Discov. 4, 39 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zafra MP et al. Optimized base editors enable efficient editing in cells, organoids and mice. Nat. Biotechnol 36, 888–893 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Z. et al. Efficient generation of mouse models of human diseases via ABE- and BE-mediated base editing. Nat. Commun 9, 2338 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Z. et al. Highly efficient RNA-guided base editing in rabbit. Nat. Commun 9, 2717 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Song C-Q et al. Adenine base editing in an adult mouse model of tyrosinaemia. Nat. Biomed. Eng (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hu JH et al. Evolved Cas9 variants with broad PAM compatibility and high DNA specificity. Nature 556, 57–63 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kim YB et al. Increasing the genome-targeting scope and precision of base editing with engineered Cas9-cytidine deaminase fusions. Nat. Biotechnol 35, 371–376 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Huang TP et al. Circularly permuted and PAM-modified Cas9 variants broaden the targeting scope of base editors. Nat. Biotechnol 37, 626–631 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kleinstiver BP et al. Engineered CRISPR-Cas12a variants with increased activities and improved targeting ranges for gene, epigenetic and base editing. Nat. Biotechnol 37, 276–282 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Li X. et al. Base editing with a Cpf1-cytidine deaminase fusion. Nat. Biotechnol 36, 324–327 (2018). [DOI] [PubMed] [Google Scholar]
- 25.Nishimasu H. et al. Engineered CRISPR-Cas9 nuclease with expanded targeting space. Science 361, 1259–1262 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yourik P, Fuchs RT, Mabuchi M, Curcuru JL & Robb GB Staphylococcus aureus Cas9 is a multiple-turnover enzyme. RNA 25, 35–44 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bryson DI et al. Continuous directed evolution of aminoacyl-tRNA synthetases. Nat Chem Biol 13, 1253–1260 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Carlson JC, Badran AH, Guggiana-Nilo DA & Liu DR Negative selection and stringency modulation in phage-assisted continuous evolution. Nat. Chem. Biol 10, 216–222 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dickinson BC, Leconte AM, Allen B, Esvelt KM & Liu DR Experimental interrogation of the path dependence and stochasticity of protein evolution using phage-assisted continuous evolution. Proc. Natl. Acad. Sci. U.S.A 110, 9007–9012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dickinson BC, Packer MS, Badran AH & Liu DR A system for the continuous directed evolution of proteases rapidly reveals drug-resistance mutations. Nat. Commun 5, 5352 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hubbard BP et al. Continuous directed evolution of DNA-binding proteins to improve TALEN specificity. Nat. Methods 12, 939–942 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Leconte AM et al. A population-based experimental model for protein evolution: effects of mutation rate and selection stringency on evolutionary outcomes. Biochemistry 52, 1490–1499 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Packer MS, Rees HA & Liu DR Phage-assisted continuous evolution of proteases with altered substrate specificity. Nat. Commun 8, 956 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang T, Badran AH, Huang TP & Liu DR Continuous directed evolution of proteins with improved soluble expression. Nat. Chem. Biol 14, 972–980 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Roth TB, Woolston BM, Stephanopoulos G. & Liu DR Phage-Assisted Evolution of Bacillus methanolicus Methanol Dehydrogenase 2. ACS Synth. Biol 8, 796–806 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Thuronyi BW et al. Continuous evolution of base editors with expanded target compatibility and improved activity. Nat. Biotechnol, 1070–1079 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Badran AH & Liu DR In vivo continuous directed evolution. Curr. Opin. Chem. Biol 24, 1–10 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Esvelt KM, Carlson JC & Liu DR A system for the continuous directed evolution of biomolecules. Nature 472, 499–503 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Badran AH et al. Continuous evolution of Bacillus thuringiensis toxins overcomes insect resistance. Nature 533, 58–63 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Badran AH & Liu DR Development of potent in vivo mutagenesis plasmids with broad mutational spectra. Nat. Commun 6, 8425 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Grunewald J. et al. CRISPR DNA base editors with reduced RNA off-target and self-editing activities. Nat. Biotechnol 37, 1041–1048 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Swarts DC, van der Oost J. & Jinek M. Structural Basis for Guide RNA Processing and Seed-Dependent DNA Targeting by CRISPR-Cas12a. Mol. Cell 66, 221–233 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liang P. et al. Genome-wide profiling of adenine base editor specificity by EndoV-seq. Nat. Commun 10, 67 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tsai SQ et al. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol 33, 187–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rees HA et al. Improving the DNA specificity and applicability of base editing through protein engineering and protein delivery. Nat. Commun 8, 15790 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Grunewald J. et al. Transcriptome-wide off-target RNA editing induced by CRISPR-guided DNA base editors. Nature 569, 433–437 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rees HA, Wilson C, Doman JL & Liu DR Analysis and minimization of cellular RNA editing by DNA adenine base editors. Sci. Adv 5, eaax5717 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zhou C. et al. Off-target RNA mutation induced by DNA base editing and its elimination by mutagenesis. Nature 571, 275–278 (2019). [DOI] [PubMed] [Google Scholar]
- 49.Doman JL A. R.; Newby GA; Liu DR Evaluation and minimization of Cas9-Independent off-target DNA editing by cytosine base editors. Nat. Biotechnol in press (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Amato A. et al. Interpreting elevated fetal hemoglobin in pathology and health at the basic laboratory level: new and known gamma- gene mutations associated with hereditary persistence of fetal hemoglobin. Int. J. Lab. Hematol 36, 13–19 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Canver MC et al. BCL11A enhancer dissection by Cas9-mediated in situ saturating mutagenesis. Nature 527, 192–197 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu Y. et al. Highly efficient therapeutic gene editing of human hematopoietic stem cells. Nat. Med 25, 776–783 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Vierstra J. et al. Functional footprinting of regulatory DNA. Nat. Methods 12, 927–930 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Powars DR, Weiss JN, Chan LS & Schroeder WA Is there a threshold level of fetal hemoglobin that ameliorates morbidity in sickle cell anemia? Blood 63, 921–926 (1984). [PubMed] [Google Scholar]
- 55.Jinek M. et al. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yao M, Hatahet Z, Melamede RJ & Kow YW Purification and characterization of a novel deoxyinosine-specific enzyme, deoxyinosine 3’ endonuclease, from Escherichia coli. J. Biol. Chem 269, 16260–16268 (1994). [PubMed] [Google Scholar]
- 57.Dalhus B. et al. Structures of endonuclease V with DNA reveal initiation of deaminated adenine repair. Nat. Struct. Mol. Biol 16, 138–143 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Clement K. et al. CRISPResso2 provides accurate and rapid genome editing sequence analysis. Nat. Biotechnol 37, 224–226 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.