

Abstract
Many RNAs are processed into biologically active transcripts, the aberrant expression of which can contribute to disease phenotypes. For example, the primary microRNA-17–92 (pri-miR-17–92) cluster contains six microRNAs (miRNAs) that collectively act in several disease settings. Herein, we used sequence-based design of structure-specific ligands to target a common structure in the Dicer processing sites of three miRNAs in the cluster, miR-17, miR-18a, and miR-20a, thereby inhibiting their biogenesis. The compound was optimized to afford a dimeric molecule that binds the Dicer processing site and an adjacent bulge, affording a 100-fold increase in potency. The dimer’s mode of action was then extended from simple binding to direct cleavage by conjugation to bleomycin A5 in a manner that imparts RNA-selective cleavage or to indirect cleavage by recruiting an endogenous nuclease, or a ribonuclease targeting chimera (RIBOTAC). Interestingly, the dimer-bleomycin conjugate cleaves the entire pri-miR-17–92 cluster and hence functionally inhibits all six miRNAs emanating from it. The compound selectively reduced levels of the cluster in three disease models: polycystic kidney disease, prostate cancer, and breast cancer, rescuing disease-associated phenotypes in the latter two. Further, the bleomycin conjugate exerted selective effects on the miRNome and proteome in prostate cancer cells. In contrast, the RIBOTAC only depleted levels of pre- and mature miR-17, −18a, and 20a, with no effect on the primary transcript, in accordance with the cocellular localization of RNase L, the pre-miRNA targets, and the compound. These studies demonstrate a strategy to tune RNA structure-targeting compounds to the cellular localization of the target.
Graphical Abstract
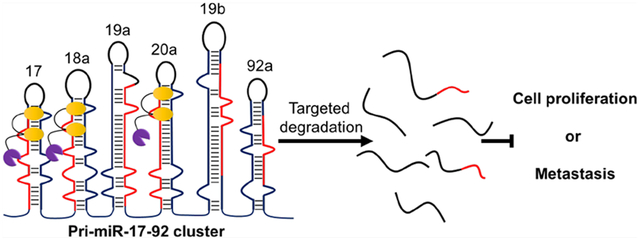
INTRODUCTION
RNA is most commonly targeted with antisense oligonucleotide-based modalities (ASOs), a strategy developed in the late 1970s by Paul Zamecnik and co-workers.1,2 Since this landmark discovery, much activity in the area has shown that RNA biology can be affected by simple binding of the ASO or by recruiting endogenous RNase H to cleave the RNA strand in the RNA-DNA hybrid.3 RNA interference (RNAi) has also emerged as an important oligonucleotide-based approach that targets an RNA for destruction; that is, triggering RNA degradation is RNAi’s only mode of action.4 Both ASOs and RNAi have achieved success in the clinic as FDA-approved medicines.5 CRISPR-based strategies to target RNA are rapidly emerging and have the potential to impact how diseases are treated.6
Each oligonucleotide-based approach recognizes RNA via sequence, and various studies have shown that unstructured regions are their ideal target sites.7 Approximately 50% of nucleotides in an RNA target are unstructured or noncanonically paired, leaving approximately half of its sequence unavailable for targeting purposes.8 Further, structured regions have been shown to regulate RNA function and processing9 and many directly mediate disease biology. Thus, a complement to the sequence-based recognition of oligonucleotides is structure-based recognition by organic ligands.10 Indeed, various studies have shown that RNA can be targeted with structure-binding small molecules, compounds that can decipher biology and can be developed into preclinical candidates.11–13 A strategy for the sequence-based design of structure-specific ligands, named Inforna, has been developed for RNA targets.14,15 This approach has been deployed successfully to rescue phenotypes associated with various diseases, to exploit known biology, to provide chemical probes to understand novel RNA biology,16 and to develop lead medicines.17–20
Herein, we used Inforna to design a ligand that targets the primary microRNA 17–92 cluster (pri-miR-17–92), a direct transcriptional target of c-MYC.21 This noncoding RNA encodes six different microRNAs (miRNAs): miR-17, −18a, −19a, −19b-1, −20a, and −92a-1. Upregulation of the miR-17–92 cluster has been observed in numerous diseases, from various cancers22–25 to fibrosis.26 Furthermore, the downstream effects are disease-dependent and linked to which members of the cluster are aberrantly expressed.27 Indeed, the miRNAs produced can act individually or synergistically to affect multiple pathways.21,28 Thus, this cluster and the miRNAs that comprise it are important targets of chemical probes and lead medicines. Our sequence-based design approach afforded a single compound that inhibits the biogenesis of three miRNAs in the cluster that share structural similarities, miR-17, −18a, and −20a. We next employed two strategies to change the compound’s mode of action from simple binding to cleavage: (i) direct, oxidative cleavage by conjugation of bleomycin A5 to our lead compound, which improved potency by ~10-fold. The compound degraded the entire miR-17–92 cluster and hence rescued miR-17–92-mediated phenotypes in prostate cancer and triple negative breast cancer (TNBC) cells; and (ii) cleavage by recruitment of an endogenous nuclease or a ribonuclease targeting chimera (RIBOTAC). Interestingly, the RIBOTAC inhibited biogenesis of miR-17, −18a, and −20a by binding and cleaving their pre-miRNAs, not the entire cluster, traced to the colocalization of the RIBOTAC, targets, and the endogenous nuclease.
RESULTS
Design, Optimization, and In Vitro Analysis of Compounds Targeting pri-miR-17–92.
Since the discovery of the miR-17–92 cluster,29 its role in disease has become increasingly apparent and diverse. Upregulation of the miR-17–92 cluster is associated with more than 14 different cancers,27 including osteosarcomas23,30 and retinoblastoma.22,31 Elevated levels of one member of the cluster, miR-92a, inhibits angiogenesis in ischemic cardiovascular endothelia,32 while its upregulation in CD4+ T cells stimulates an autoimmune response.33 Consequently, the miR-17–92 cluster is a high priority target for therapeutic intervention. We therefore focused our efforts on designing compounds that inhibit the biogenesis of the miR-17–92 cluster by inspecting the structures found at the Drosha and Dicer processing sites of each encoded miRNA. We have previously shown that binding these functional sites inhibits miRNA processing in situ and in vivo.3,15,17,18,34,35 Fortuitously, three miRNAs in the miR-17–92 cluster have the same U bulge (5′G_U/3′CUA) in their Dicer sites, miR-17, miR-18a, and miR-20a (Figure 1, yellow boxes). Both miR-17 and miR-20 have an adjacent 1-nucleotide G bulge (5′GGU/3′C_A) while miR-18a has a related purine bulge, 5′GAU/3′C_A (Figure 1, blue boxes).
Figure 1.
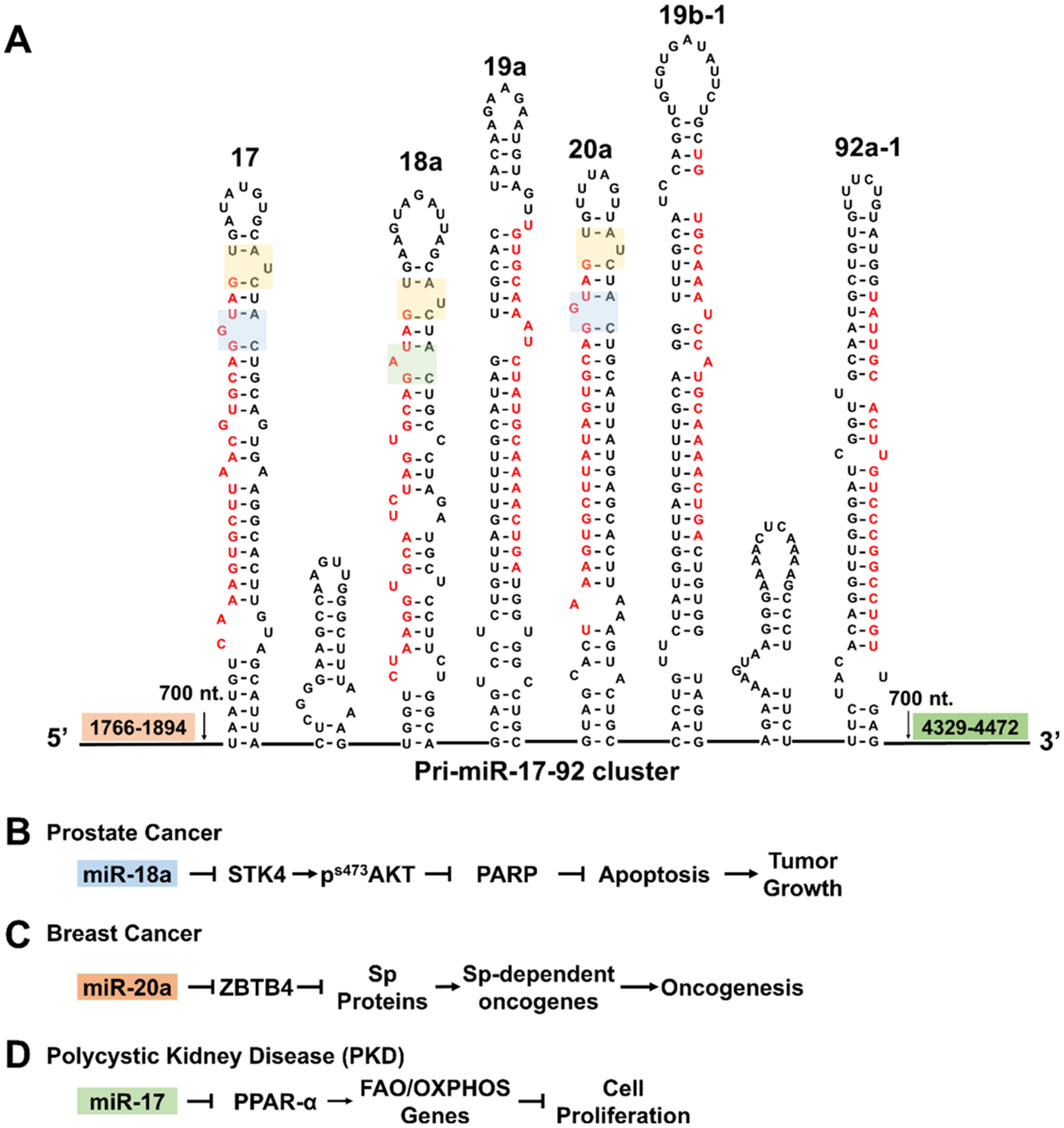
Schematic of the pri-miR-17–92 cluster and its downstream targets. (A) Schematic of the polycistronic pri-miR-17–92 cluster’s secondary structure. Interestingly, miR-17, miR-18a, and miR-20a share a common structure at their Dicer sites, the 1-nucleotide bulge 5′G_U/3′CUA targetable with 1 (yellow boxes). Adjacent targetable motifs are present in all three miRNAs, 5′GGU/3′C_A in miR-17 and miR-20a (blue box) and 5′GAU/3′C_A in miR-18a (green box). (B) In DU-145 prostate cancer cells, miR-18a represses STK4, which inhibits apoptosis. (C) In triple negative breast cancer cells, overexpression of miR-17 and miR-20a induces an invasive phenotype. (D) In polycystic kidney disease, miR-17, miR-19a, and miR-19b are upregulated, triggering cell proliferation and cyst formation.
A previous study that explored the RNA-binding capacity of various ligands showed that compound 1 (Figure 2A) preferred to bind all three of these bulges with Kd’s ranging from 30–40 μM.35 No binding was observed between 1 and RNAs in which the bulges were mutated to the corresponding GC or AU base pairs.35 These data suggested a facile route to compound optimization via dimerization of 1 to afford a single molecule that binds both bulges simultaneously.20 Fortuitously, 1 contains a chemical handle for dimerization, an azide moiety that can be conjugated to alkyne-displaying scaffolds via a Cu-catalyzed Hüisgen 1,3-dipolar cycloaddition.36,37 We conjugated 1 to peptoids displaying two alkynyl groups separated by 2–9 propylamine spacing modules, as informed by previous studies.38 (See Supporting Information for synthesis and characterization.)
Figure 2.
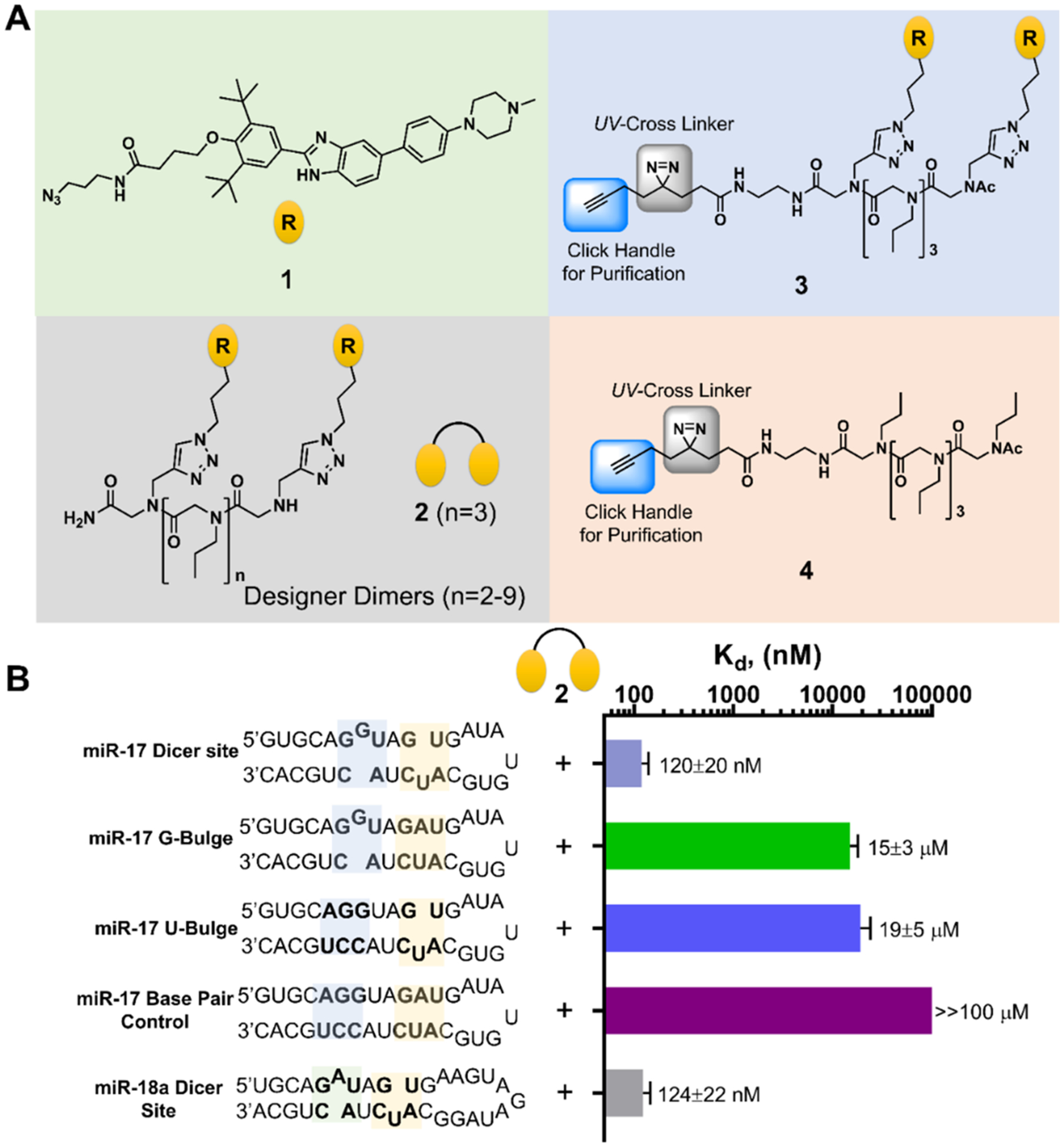
Design of a dimeric compound that binds to miR-17’s Dicer site. (A) Chemical structures of compounds used in these studies. Compound 1 is the parent monomer targeting each motif; 2 is the dimer with three propylamine spacing modules in the peptoid backbone, allowing specific binding to each miRNA’s Dicer site; 3 is a Chemical Cross-Linking and Isolation by Pull-down (Chem-CLIP) probe that contains a diazirine reactive module and click handle for conjugating biotin, enabling pull-down for assessing target engagement; 4 is the Chem-CLIP control probe that lacks the RNA-binding modules to assess nonspecific reaction of the diazirine. (B) Secondary structures of the model RNAs used in binding studies and the corresponding affinities for 2. Note that “miR-17 Dicer site” and “miR-18a Dicer site” are comprised of sequences native to the corresponding RNA. Other RNAs contain mutations that convert the bulges to base pairs.
To identify an optimal dimer that displays the RNA-binding modules that most potently targets the pre-miRNAs, the library was screened for activity in a cell-based luciferase reporter assay. It is known that peroxisome proliferator-activated receptor α (PPAR-α) mRNA is translationally repressed by miR-17.39 Thus, a construct with luciferase fused to PPAR-α’s 3′ untranslated region (UTR)39 was used to assess inhibition of miR-17 biogenesis in HEK293T cells (Figure S1). A locked nucleic acid (LNA) oligonucleotide targeting miR-17 (LNA-17) was used as a positive control, increasing luciferase activity by 1.6(±0.1)-fold; no effect was observed with a scrambled LNA oligonucleotide (Figure S1). Of the eight dimers synthesized and tested, the most potent compound contained three propylamine spacers (or compound 2; Figure 2A), which had an IC50 of ~10 μM in the luciferase-based assay (Figure S1). The number of propylamine spacing modules in the optimal compound is in agreement with previous studies for spanning three base pairs between bulges or internal loops.38
In Vitro Evaluation of 2.
As 2 showed optimal activity among the dimers, it was further characterized in vitro and in situ. We first measured the affinity of 2 for a model RNA that contains the two bulges in and adjacent to pre-miR-17’s and pre-miR-20a’s Dicer site. A 250-fold boost in affinity relative to 1 was observed, affording a Kd of 120(±20) nM (Figure 2B). No measurable binding was detected to RNAs in which the two bulges were mutated to base pairs (Figure 2B). Single mutations of the bulges, either the U bulge to an AU pair or the G bulge to a GC pair, weakened the avidity of 2 to 15(00B13) and 19(±5) μM, respectively, ~2-fold more avid than the binding of 1 which is due to stochastic effects.35,40 As expected, based on the similar avidity of 1 for the three bulges in premiR-17, −18a, and −20a, 2 exhibited similar affinity for a mimic of pre-miR-18a [124(±22) nM] while no saturable binding to the corresponding fully paired RNA was observed (Figure 2B).
Since 2 bound model constructs of pre-miR-17, pre-miR-18a, and pre-miR-20a with similar affinity, we next studied its ability to inhibit Dicer processing of all three pre-miRNAs in vitro. As expected, 2 inhibited Dicer processing of each to a similar extent, with an IC50 of ~1 μM (Figures S2, S3). To study the effect on 2’s binding site within pre-miR-17 and as a secondary assessment of selectivity (binding studies being the first), we completed a mutational analysis in which each bulge in and nearby the Dicer sites was individually and then collectively replaced with the corresponding base pair. These changes were tolerated by Dicer (Figure S2). The mutations, however, significantly reduced (in the case of the G to GC pair mutation) or ablated (in the cases of the U to AU pair mutation and the double mutant) 2’s inhibitory activity (Figure S2). Likewise, when the U bulge in pre-miR-18a was converted to an AU pair, 2 was unable to inhibit Dicer processing at the concentrations tested, up to 1 μM (Figure S3B). Further, 2 had no effect on the Dicer processing of the other three miRNAs in the cluster, pre-miR-19a, pre-miR-19b-1, or pre-miR-92a-1 (Figure S4). Altogether, these studies support that 2 is a specific RNA-binding compound, targeting the Dicer site and an adjacent bulge in pre-miR-17, pre-miR-18a, and pre-miR-20a, and that 2 does not inhibit Dicer itself.
Effect of 2 on the pri-miR-17–92 Cluster in TNBC Breast Cancer Cells.
In the TNBC cell line MDA-MB-231, miR-17 and miR-20a are highly expressed and together silence zinc finger and BTB domain containing 4 (ZBTB4) mRNA, thereby triggering an invasive phenotype (Figure 1C).24 Notably, miR-17 and miR-20a have identical seed sequences, or the sequences that form complexes with targeted mRNAs, to repress their translation (5′-AAAGUGC). Thus, both miR-17 and miR-20a inhibit ZBTB4 in TNBC cells and knockdown of both is necessary to allow robust activation of ZBTB4’s tumor suppressor functions. Loss of ZBTB4 has been correlated with shorter relapse-free survival of breast cancer patients making ZBTB4 a therapeutically important target.24 Thus, we studied if 2 could inhibit biogenesis of the targeted miRNAs and an invasive phenotype in MDA-MB-231 cells. Compound 2 selectively decreased the levels of mature miR-17, −18a, and −20a dose dependently, while not affecting the levels of the other miRNAs in the cluster (Figure 3A). The reduction in mature miRNA levels by 2 at 500 nM (~40–50%) was similar to the reduction observed for an LNA (50 nM) targeting the mature sequence of miR-20a (48(±6)%) but less than that observed for miR-17 (85(±5)%) (Figure 3B).
Figure 3.
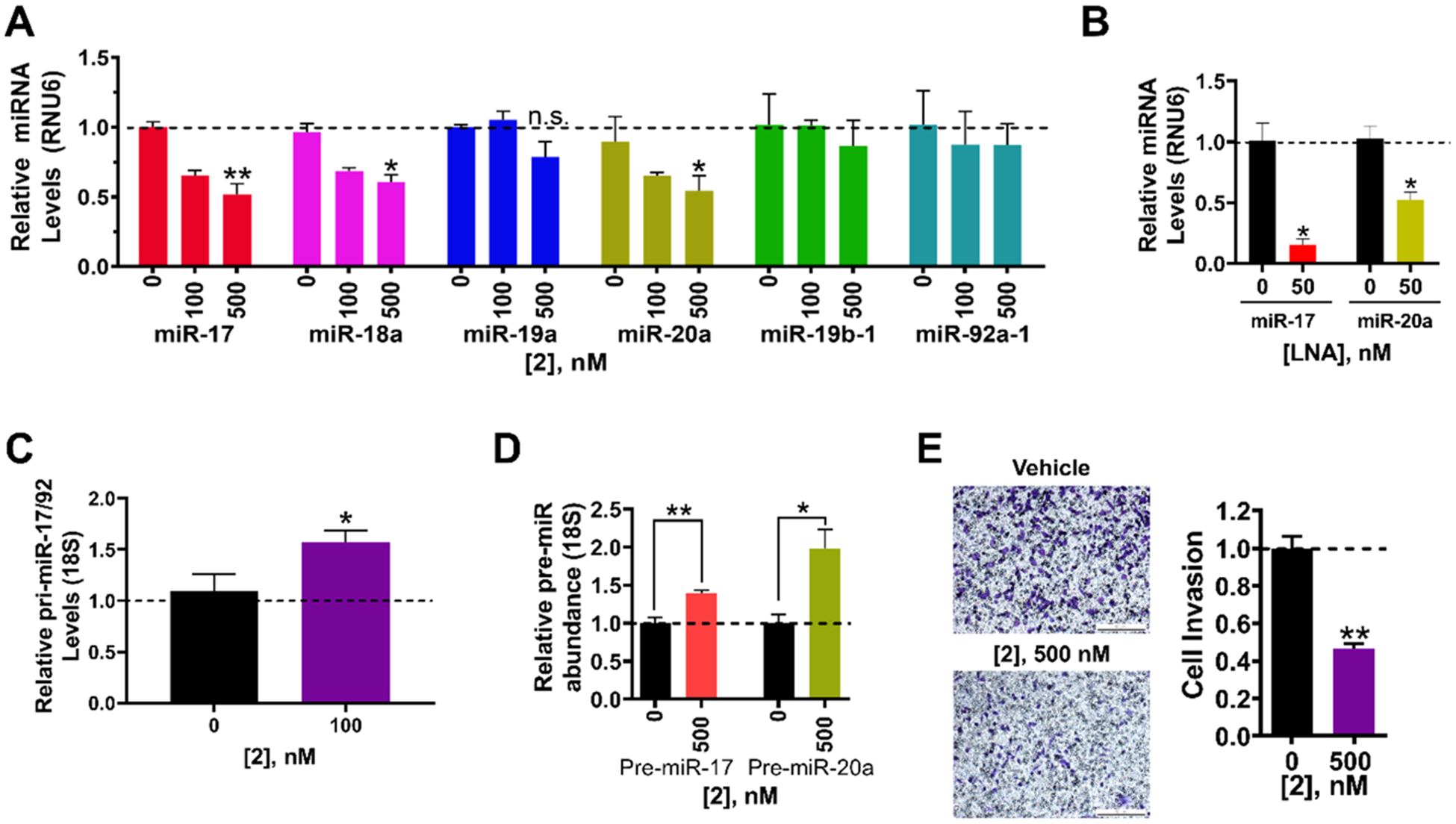
Bioactivity of dimeric binder 2 in MDA-MB-231 triple negative breast cancer cells. (A) Effect of dimeric binder 2 on the levels of mature miRNAs derived from the miR-17–92 cluster in MDA-MB-231 cells, as determined by RT-qPCR. (B) Effect of LNAs on miR-17 and miR-20a levels, as determined by RT-qPCR. (C) Effect of 2 on pri-miR-17–92 levels shows a 50% increase in its levels. (D) Effect of 2 on pri-miR-17–92, pre-miR-17, and pre-miR-20a levels, as determined by RT-qPCR. (E) Effect of 2 on the invasive properties of MDA-MB-231 cells caused by aberrant expression of miR-17 and miR-20a. Representative images of invasion assays from 2-treated and untreated MDA-MB-231 cells. Errors are reported as SEM *, p < 0.05; **, p < 0.01; ***, p < 0.001, as determined by a Student t test.
We next studied the effect of 2 on pri-miR-17–92, pre-miR-17, and pre-miR-20a levels. Depending on its cellular localization, 2 could engage the pri-miRNA or the pre-miRNAs to reduce mature miRNA levels. It is known that the 17–92 cluster folds into a compact tertiary structure and that alterations in this structure affects the processing of the primiR-17–92 transcript.41,42 Although 2 localized mainly to the cytoplasm, fluorescence was also detected in the nucleus, suggesting that it could inhibit processing of the pri-miRNA as well as pre-miR-17, −18a, and 20a (Figure S5). Indeed, 2 increased pri-miR-17–92 levels by 50(±6)%, as determined by RT-qPCR using primers upstream of all six miRNAs (p < 0.05; Figure 3C). [Notably, direct engagement of pri-miR-17–92 by 2 was confirmed via a competition experiment between 2 and a 2-bleomycin conjugate in which 2 restored pri-miR-17–92 levels (vide infra).] Thus, the increase in pri-miR-17–92 levels observed is due to engagement of the primary transcript in the nucleus, likely affecting its overall structure and its global processing.
If 2 only engaged pri-miR-17–92 and inhibited its biogenesis, then a decrease in pre-miRNA levels is expected. If, however, 2 directly binds and inhibits processing of both the pri- and pre-transcripts, two outcomes are possible: (i) no change in pre-miRNA levels are observed. That is, the reduction in pre-miRNA levels due to inhibition of pri-miRNA processing and the boost in pre-miRNA levels due to inhibition of Dicer processing are similar and thus cancel each other out; or (ii) an increase in pre-miRNA levels is observed because Dicer processing is inhibited to a greater extent than Drosha processing. In accordance with the last possibility and its presence in the cytoplasm (Figure S5), 2 increased levels of pre-miR-17 by 1.4(±0.04)-fold (p < 0.01) and pre-miR-20a by 2.0(±0.2)-fold (p < 0.05) (as determined by RT-qPCR using primers specific for each pre-miRNA; Figure 3D). [Note: RT-qPCR primers that amplify pre-miRNAs cannot distinguish between the processed, cytoplasmic pre-miRNA and the pre-miRNA in the context of the pri-miRNA; thus, the increase observed is a combined effect of changes in the levels of the cluster and the pre-miRNA of interest.] These results were confirmed by absolute quantification of mature, pre- and primiR-17–92 levels (Figure S6A).
We next investigated 2’s effect on miR-17 and −20a’s downstream target ZBTB4.24 Indeed, 2 increased Zbtb4 mRNA levels by 1.4(±0.2)-fold at 500 nM (p < 0.05) (Figure S6B) and ZBTB4 protein levels by 1.7(±0.2)-fold (p < 0.05) at 500 nM, similar to the effect observed upon treatment with 100 nM of LNA-17 (Figure S6C). Previous studies have shown that derepression of ZBTB4 via inhibition of miR-17 and miR-20a decreases the invasive properties of breast cancer cells.24 We therefore studied if 2 could rescue this phenotype in TNBC cells. Similar to LNA-17 treatment (Figure S6D), 2 inhibited the invasive characteristics of MDA-MB-231 cells (Figure 3E). Interestingly, this effect can be traced to inhibition of miR-17 and miR-20a biogenesis by 2 as levels of other miRNAs predicted to regulate expression ZBTB4 by TargetScan were unaffected (Figure S6E).43 These data support that the observed rescue of phenotype is via 2’s inhibition of the miR-17/20a-ZBTB4 circuit.
Effect of 2 on the pri-miR-17–92 Cluster in Autosomal Dominant Polycystic Kidney Disease (ADPKD).
Autosomal dominant polycystic kidney disease is the most common genetically defined kidney disease, ultimately leading to renal failure.39 Recent studies have shown that miR-17, −19a, −19b-1, and −20a from the miR-17–92 cluster are upregulated in cystic kidneys, which in turn repress PPAR-α and aggravate cyst growth (Figure 1D).39 Thus, the effect of 2 was evaluated in WT 9–12 cells, an immortalized cell line generated from renal epithelial cell from proximal and distal tubules of ADPKD patients.44 Similar to our studies in DU-145 (vide infra) and MDA-MB-231 TNBC cells, 2 decreased expression of its cognate targets, miR-17, miR-18a, and miR-20a, dose dependently (by 55(±6)%, 52(±9)%, and 59(±13)%, respectively, at 500 nM), with no effect on miR-19a, miR-19b-1, and miR-92a-1 (Figure S7A). As expected, reduction of mature miR-17 levels by 2 derepressed PPAR-α expression, increasing protein levels by 2.7-fold at 500 nM (Figure S7B). Importantly, these studies show that reduction of miR-17 and miR-20a levels alone are sufficient to derepress PPAR-α, as levels of miR-19a and −19b-1, which also regulate PPAR-α,39 were unaffected by 2 treatment (Figure S7A).
Effect of 2 on the pri-miR-17–92 Cluster in the Prostate Cancer Cell Line DU-145.
The pri-miR-17–92 cluster, particularly by the overexpression of miR-18a, promotes prostate cancer.45 Thus, we evaluated the inhibitory effect of 2 on the biogenesis of the miR-17–92 cluster in the prostate cancer cell line DU-145. As expected, based on its in vitro binding affinity and activity, application of 2 inhibited the biogenesis of miR-17, −18a, and −20a, decreasing mature miRNA levels of each (Figure 4A). As little as 100 nM of 2 was sufficient to significantly inhibit production of the mature miRNAs: by 49(±9)% for miR-17 (p < 0.05); by 49(±8) for miR-20a (p < 0.05); and by 58(±11)% for miR-18a (p < 0.01) (Figure 4A). Importantly, 2 did not affect levels of miR-19a, −19b-1, and −92a-1, which it does not bind. LNA-18a reduced miR-18a levels by 27(±5)% (Figure 4B). We previously studied 1, the monomer from which 2 is derived, for reducing miR-18a levels in DU-145 cells.35 The reduction in mature miR-18a levels observed upon treatment with 100 nM of 2 is similar to that upon treatment with 10 μM of 1,35 or a 100-fold increase in potency.
Figure 4.
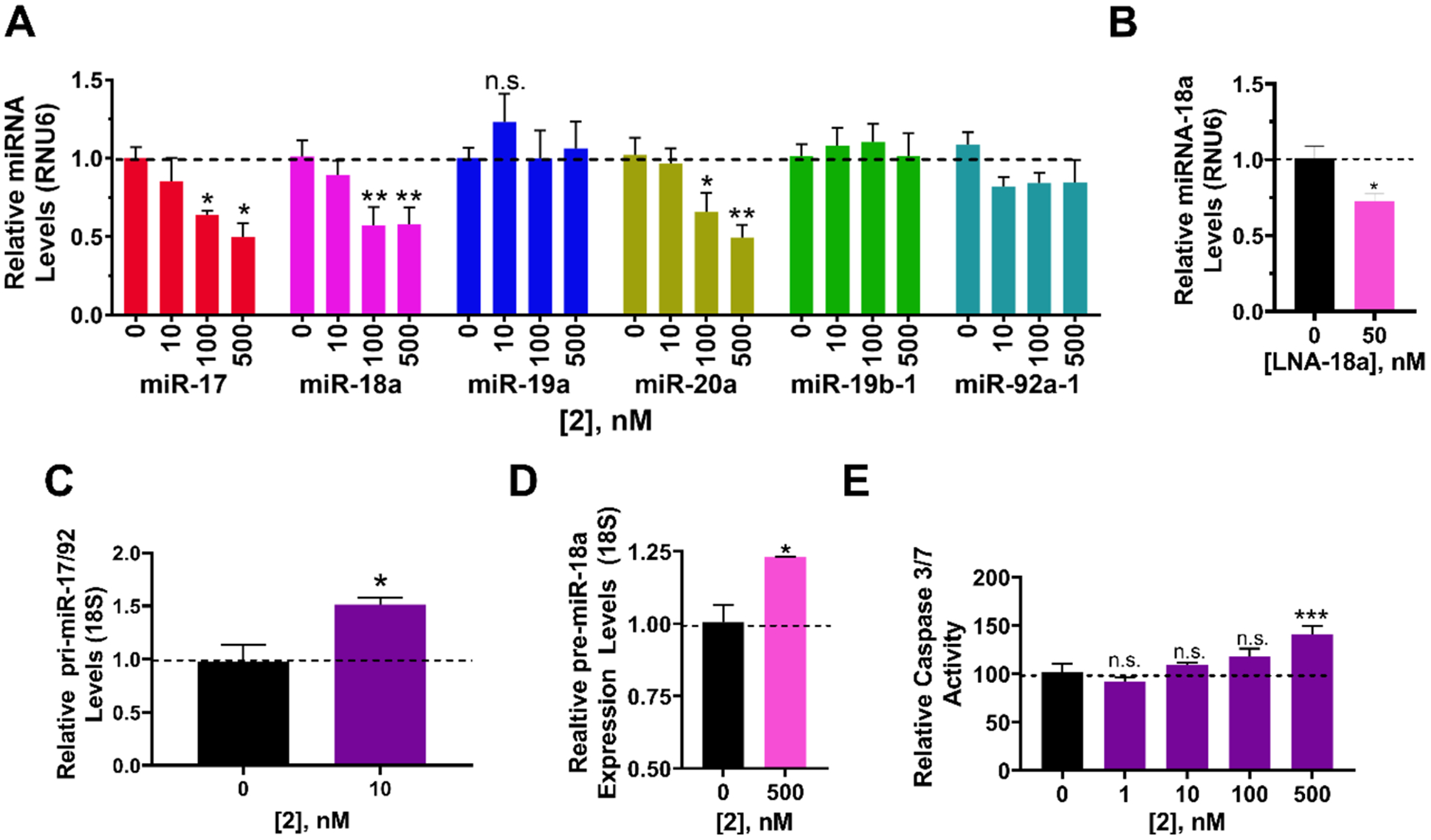
Bioactivity of dimer binder 2 in DU-145 prostate cancer cells. (A) Effect of 2 on the levels of mature miRNAs derived from the miR-17–92 cluster in DU-145 cells, as determined by RT-qPCR. (B) RT-qPCR of an LNA targeting miR-18a shows a modest effect on miR-18a levels. (C) Effect of 2 on pri-miR-17–92 levels shows a 50% increase in pri levels. (D) Effect of 2 on pre-miR-18a levels, as determined by RT-qPCR. (E) Effect of 2 (500 nM) on phenotype (apoptosis), as measured by Caspase 3/7 activity. Errors are reported as SEM *, p < 0.05; **, p < 0.01; ***, p <0.001, as determined by a Student t test.
Consistent with studies in MDA-MB-231 TNBC cells, 2 increased levels of pri-miR-17–92 by 51(±6)% and pre-miR-18a by 23(±0.05)% (Figure 4C,D). These results, in conjunction with the decreased levels of mature miR-18a, support the hypothesis that 2 inhibits processing of the priand pre-miRNA. To validate these observations, we measured the effects of compounds treatment on mature and pre-miR-17, −18a, and −20a was well as pri-miR-17–92 levels by absolute quantification, which is in agreement with relative quantification data (Figure S8A).
Downstream Effects of 2 in DU-145 Prostate Cancer Cells.
In DU-145 cells, miR-18a translationally represses serine/threonine-protein kinase 4 (STK4).45 A previous study showed that an antagomir directed against miR-18a increased STK4 protein levels and triggered apoptosis via STK4-mediated dephosphorylation of protein kinase B [also known as AKT serine/threonine kinase (AKT)]. Treatment of DU-145 cells with 2 increased levels of Stk4 mRNA by 22(±7)% (Figure S8B) and STK4 protein by 65(±7)% (Figure S8C). To determine whether 2 can trigger apoptosis, Caspase 3/7 levels were measured upon 2 treatment. Indeed, 2 triggered apoptosis, increasing Caspase 3/7 activity by 41(±8)% at 500 nM (Figure 4E). An LNA targeting miR-18a increased Caspase 3/7 activity by 24(±8)% while no effect was observed for a scrambled control (Figure S8D).
To confirm that 2 triggered apoptosis by the miR-18a-STK4 circuit, we (i) overexpressed pri-miR-17–92 via a plasmid and (ii) knocked down STK4 via an shRNA. As expected, both overexpression of the cluster and knock down of its downstream target (STK4) reduced 2’s ability to trigger apoptosis (Figure S8E). The activity of 2 was unaffected upon transfection of a control plasmid or scrambled shRNA (Figure S8E). These observations support the hypothesis that 2 induced apoptosis in a miR-17–92 cluster- and STK4-dependent manner.
Collectively, these studies show that 2’s inhibition of the processing of three pre-miRNAs derived from the pri-miR-17–92 derepressed their downstream targets to alleviate oncogenic phenotypes in two different cellular models of disease, prostate cancer and breast cancer. Compound 2 also shows promising activity in an ADPKD model and will be the subject of further investigation.
Compound 2 Directly Engages the pri-miR-17–92 Cluster In Situ, as Determined by Chem-CLIP.
A previously developed method named Chemical Cross-Linking and Isolation by Pull-down (Chem-CLIP) was used to assess direct target engagement of 2 (Figure 5A).35,46,47 To enable these studies, 2 was equipped with a diazirine module, a photo cross-linking group that reacts with nucleic acids upon irradiation, and a terminal alkyne that can react with biotinazide, used for isolation and purification (compound 3, Figure 2A). A control compound, 4, which lacks the RNA-binding modules but retains the reactive diazirine and alkyne on the peptoid backbone, was also synthesized (Figure 2A).
Figure 5.
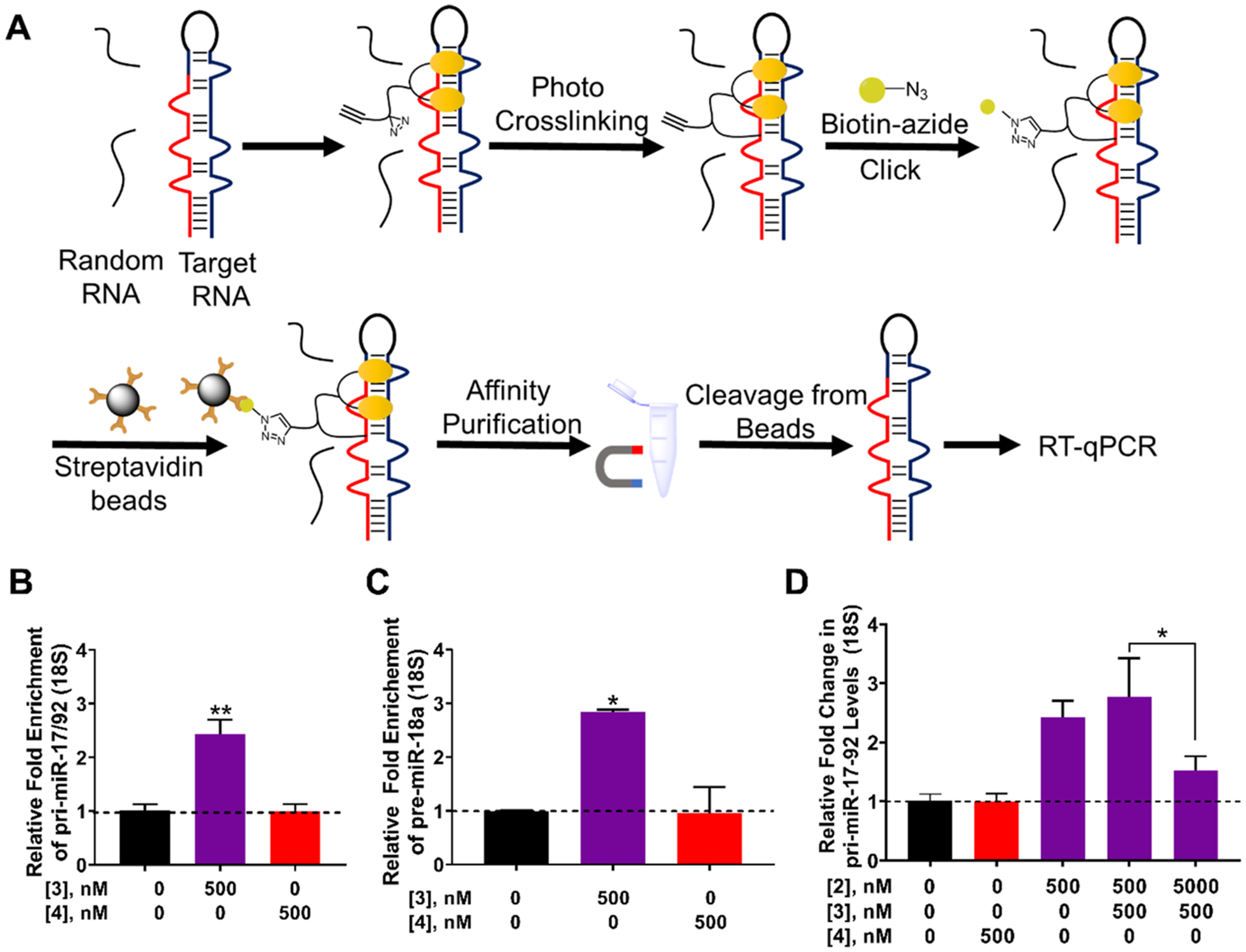
Studying target engagement via Chemical Cross-Linking and Isolation by Pull-down (Chem-CLIP) and Competitive-Chem-CLIP (CChem-CLIP) in DU-145 cells. (A) Schematic of the Chem-CLIP target engagement methodology. (B) Enrichment of the pri-miR17–92 transcript by 3 in Chem-CLIP studies, as compared to the lysate prior to pull-down. (C) Pull-down of pre-miR-18a by 3, as compared to the lysate prior to pull-down. (D) C-Chem-CLIP studies in which DU-145 cells were cotreated with 3 and increasing concentrations of 2, which dose-dependently reduced the amount of pri-miR-17–92 pulled down. Errors are reported as SEM *, p < 0.05; **, p < 0.01; ***, p < 0.001, as determined by a Student t test.
DU-145 cells were treated with 500 nM of 3, followed by isolation of 3-RNA adducts. A 2.4-fold enrichment of pri-miR-17–92 was observed, as compared to its levels in the lysate prior to pull-down, while no significant enrichment was observed for cells treated with 4 (Figure 5B). Interestingly, these results suggest that 3 directly engaged the primary transcript in the nucleus. We also measured enrichment using RT-qPCR primers complementary to pre-miR-18a, as overexpression of miR-18a represses apoptosis in prostate cancer cells via STK4. Indeed, 3 enriched pre-miR-18a by 2.8-fold in Chem-CLIP studies; no significant enrichment of pre-miR-18a was observed with 4 (Figure 5C). Finally, to further confirm that the parent compound 2 engages the same sites as the Chem-CLIP probe 3, a competition experiment (C-Chem-CLIP) was completed by pretreating DU-145 cells with varying concentrations of 2, followed by dosing with 3. Indeed, these studies showed that 2 reduced the amount of pri-miR-17–92 pulled down by 3 in a dose-dependent manner, demonstrating that the two compounds bind the same sites (Figure 5D).
Targeted Cleavage of the pri-miR-17–92 Cluster.
On the basis of cellular localization, RT-qPCR, and Chem-CLIP analyses, 2 inhibited the biogenesis of only those miRNAs that it bound, namely miR-17, miR-18a, and miR-20a. Since all members of the miR-17–92 cluster are implicated in disease, it would be desirable to have a compound that inhibits generation of all mature miRNAs in the cluster (Figure 6A).27 We therefore studied destruction of the precursor and primary transcripts by equipping 2 with a bleomycin A5 cleaving module. We have shown that conjugation of bleomycin A5 to an RNA-binding compound allows for the programmable cleavage of the desired RNA target in cells and in an animal model.19,48,49 That is, this approach can direct cleavage selectively to the RNA target with which the RNA-binding compound interacts while directing cleavage away from DNA, bleomycin’s canonical target. RNA-selective cleavage is afforded by conjugation of bleomycin A5′s free amine, which contributes to the recognition of DNA.19,49,50 Thus, we synthesized a 2-bleomycin A5 conjugate, 5, and its control compound 6, which lacks RNA-binding modules (Figure 6B).
Figure 6.
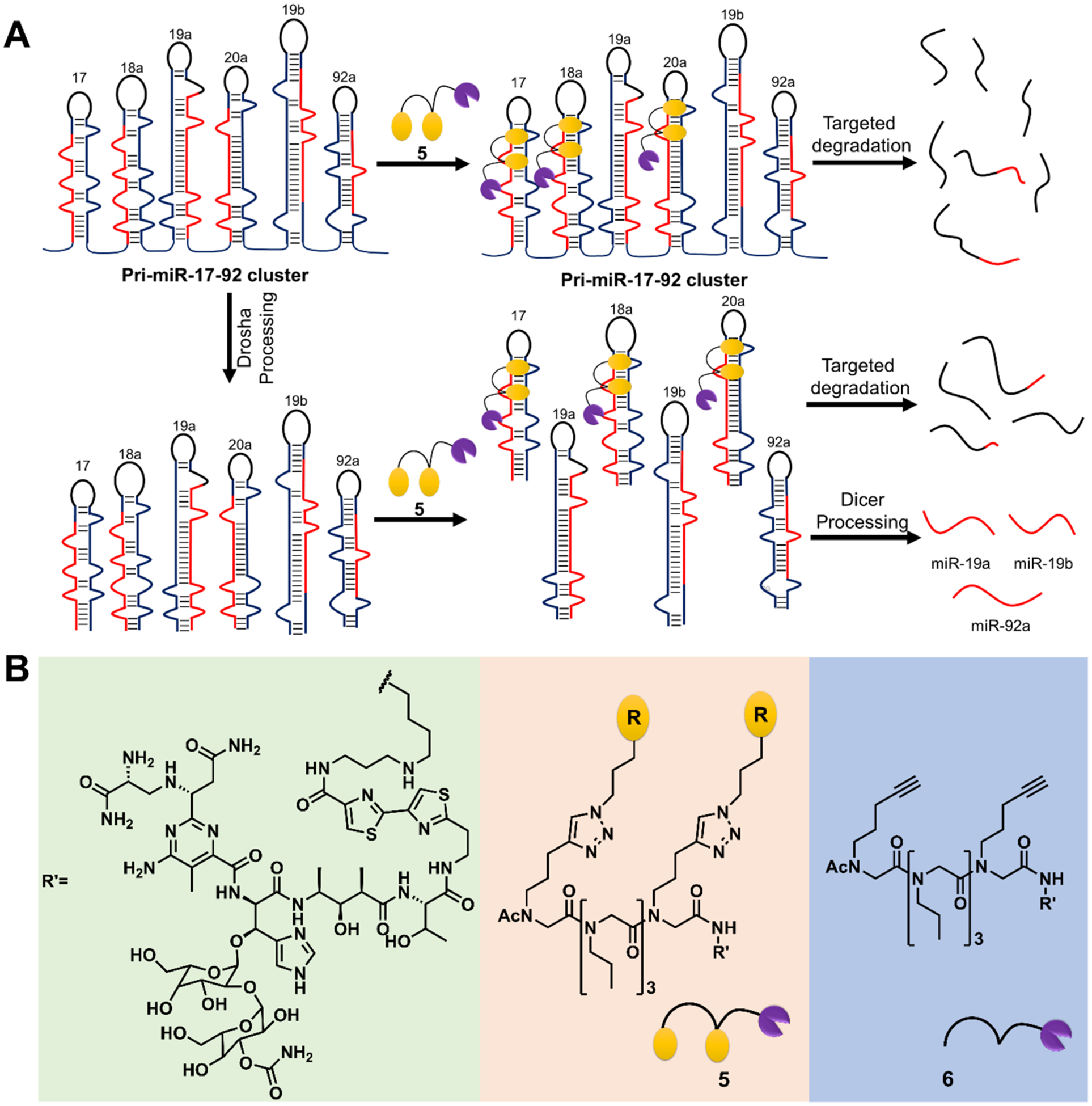
Designing a small molecule to directly cleave pri-miR-17–92. (A) Schematic of targeted degradation of the cluster with a cleaving compound. (B) Structures of the 2-bleomycin A5 conjugate, 5, and control compound 6 that lacks the RNA-binding modules.
Compound 5 was assessed for its ability to cleave pre-miR-17 in vitro. As expected, based on our in vitro binding and Dicer processing studies (Figures 2B and S2), 5 specifically cleaved pre-miR-17 nearby its Dicer processing site in a dose dependent manner with an IC50 of ~500 nM. (Figure S9A). In contrast, no dose-dependent cleavage was observed with control compound 6, which lacks RNA binding modules (Figure S9A). When the U bulge at the Dicer site and the adjacent G bulge were mutated to AU and GC base pairs, respectively, no cleavage due to the compound was detected (Figure S9B). These results collectively demonstrate specific binding of 5 at the designed site, the Dicer processing site and adjacent bulge. Although conjugation of bleomycin A5′s amine to RNA-binding molecules has been previously shown to reduce DNA cleavage overall and ablate cleavage at active concentrations that cleave the desired RNA target,19,49,50 we assessed whether 5 or 6 cleaved plasmid DNA to a greater extent than bleomycin A5 itself. Bleomycin A5 significantly cleaved DNA with as little as 100 nM of compound (Figure S9C, blue line), while at the same concentration 5 did not significantly cleave the DNA (Figure S9C, red line). Compound 6 had attenuated DNA cleavage capacity relative to parent bleomycin A5 at 100 nM concentration (Figure S9C, green line). Thus, 5 is an RNA-specific cleaver in vitro.
Cleavage of the pri-miR-17–92 Cluster by 5 in MDA-MB-231 TNBC Cells.
Compound 5 was delivered to MDA-MB-231 cells to assess its ability to cleave both pri-miR-17–92 and its cognate pre-miRNAs. Notably, cellular localization studies showed that 5 can be found throughout the cell, with enhanced fluorescence observed in the perinuclear space (Figure S5). Indeed, 5 cleaved pri-miR-17–92, reducing its levels by 33(±8)% at 100 nM, with control compound 6 having no effect (Figure 7A). We next completed a competitive cleavage experiment between 2 and 5. If the two compounds bind the same site within pri-miR-17–92 then increasing concentrations of 2 should diminish cleavage in this competition experiment. Indeed, 2 restored pri-miR-17–92 levels in a dose-dependent fashion (Figure 7B). Mirroring these results, 5 (500 nM) diminished pre-miR-17 levels by 33(±7)%, pre-miR-20a by 46(±6)% (Figure 7C), and all six mature miRNAs from 40(±7) to 25(±15)% (Figure 7D). We corroborated our relative analysis by conducting absolute quantification of mature, pre- and pri-miRNA levels (Figure S10B).
Figure 7.
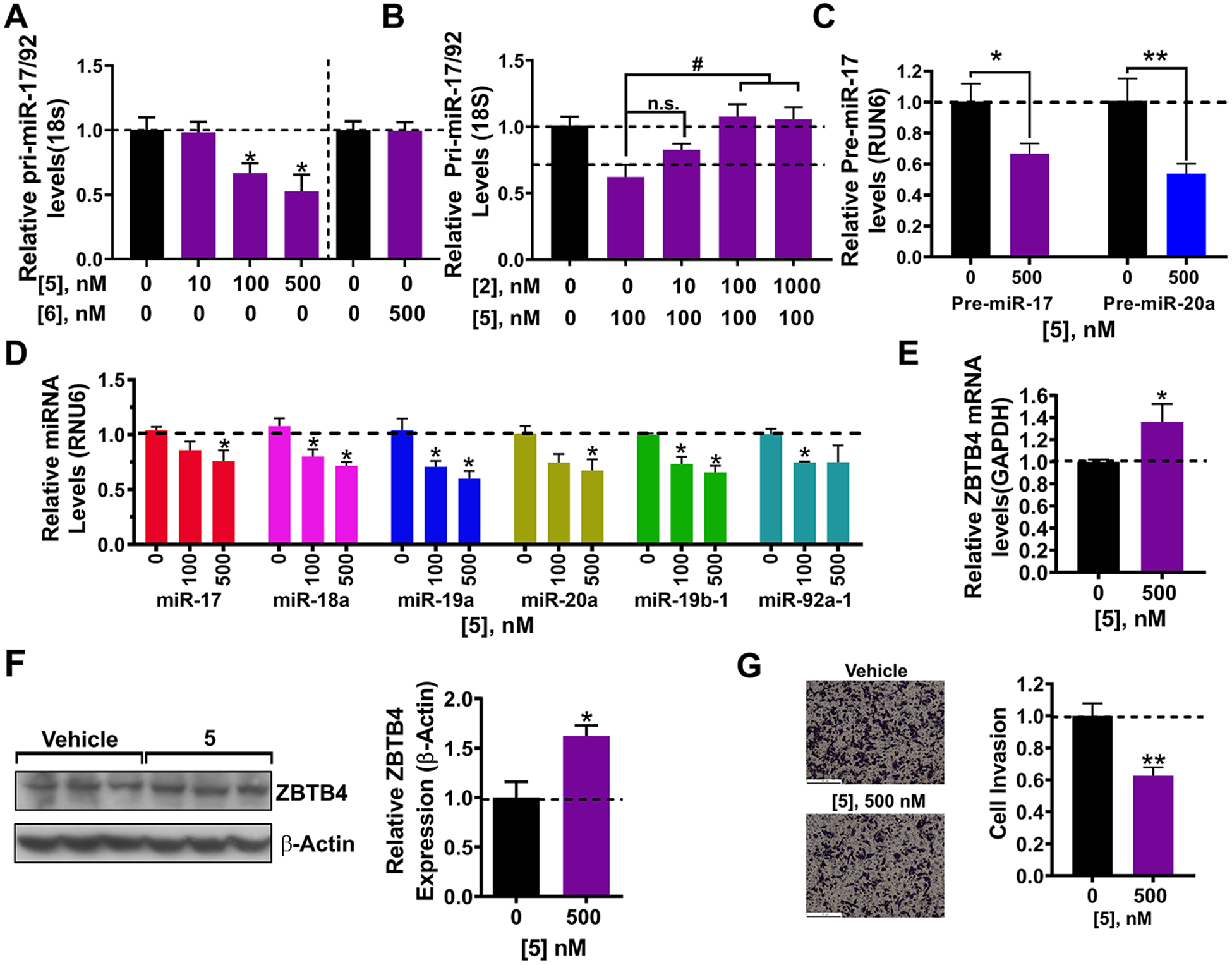
Bioactivity of dimer-bleomycin conjugate 5 in MDA-MB-231 triple negative breast cancer cells. (A) Effect of cleaving compound 5 and negative control compound 6 on pri-miR-17–92 levels in MDA-MB-231 cells, as determined by RT-qPCR. (B) Competition experiment between dimer binder 2 and 5 and their effect of pri-miR-17–92 levels, as determined by RT-qPCR. (C) Effect of 5 on pre-miR-17 levels, as determined by RT-qPCR. (D) Effect of 5 on the levels of mature miRNAs derived from the miR-17–92 cluster, as determined by RT-qPCR. (E) Effect of 5 on Zbtb4 mRNA levels, a direct target of miR-17, as determined by RT-qPCR. (F) Effect of 5 on ZBTB4 expression in MDA-MB-231 cells. (G) Effect of 5 on the invasive characteristics of MDA-MB-231 cells, due to repression of ZBTB4. Errors are reported as SEM *, p < 0.05; **, p < 0.01; ***, p < 0.001, as determined by a Student t test.
Reduction of miR-17–92 levels derepressed both ZBTB4 mRNA and protein, by 36(±16)% and 62(±11)%, respectively, (500 nM of 5, Figure 7E,F). Further, 5 diminished the invasive characteristics of MDA-MB-231 cells (Figure 7G). As described for 2, we completed studies to investigate if 5 indeed acts along the miR-17–92-ZBTB4 axis by overexpressing primiR-17–92 or ablating ZBTB4 with an shRNA. Indeed, both types of experiments ablated 5’s ability to derepress ZBTB4 mRNA and its anti-invasive phenotype (Figure S10C–G).
To assess 5’s selectivity, we identified miRNAs that contain the same motifs as those targeted by 2 in the miR-17–92 cluster and those predicted by Inforna to bind 2 [dubbed RNA isoforms (n = 30); Table S2]. All 30 miRNAs only contain a single binding site for 1, four of which are located in a Dicer site and one in a Drosha site (Table S2). Treatment of 5 did not affect the levels of any of these miRNA isoforms, underscoring 5’s selectivity for the cluster (Figure S10H). The well-known oncogenic miR-2151 contains an A bulge in its Dicer site and a U bulge four base pairs downstream; three base pairs separate the bulges in pre-miR-18a and they are in different orientations (same side of the helix in pre-miR-21 and opposite sides in pre-miR-18a (Table S2). Further the A and U bulges in miR-21 have different closing base pairs than premiR-18a and are predicted by Inforna to bind 1 weakly. Indeed, 5 had no effect on mature miR-21 levels (Figure S10H). Collectively, these data support that (i) 5 directly cleaved the pri-miR-17–92 transcript and induced an anti-invasive phenotype that is dependent on the downregulation of the cluster and ZBTB4 derepression and (ii) its selectivity is due to precise display of the RNA-binding modules at a distance that mimics the distance between the bulges, three base pairs.
Cleavage of the pri-miR-17–92 Cluster by 5 in DU-145 Prostate Cancer Cells.
Treatment of DU-145 cells with 5 decreased the levels of pri-miR-17–92 dose-dependently, with significant cleavage observed with as little as 10 nM of compound. No change in pri-miR-17–92 levels were observed with control compound 6 up to the highest concentration tested (500 nM), indicating selective cleavage of the cluster by 5 (Figure 8A). Competitive cleavage studies (completed at a constant concentration of 10 nM of 5, the approximate IC50) showed that 2 restored pri-miR-17–92 levels in a dose dependent fashion, validating that they both bind the same site within the target (Figure 8B). Furthermore, 5 also diminished pre-miR-18a levels by 40(±4)% at 100 nM (Figure 8C) and levels of all miRNAs within the cluster with treatment with as little as 10 nM compound, except for miR-92a-1 where inhibitory effects were significant at 100 and 500 nM concentrations (Figure 8D). In agreement with studies of the pri-miRNA, 6 did not affect levels of mature miRNAs within the cluster except miR-19a, which decreased by ~50% at 500 nM (Figure S11A), consistent with results obtained from absolute quantification (Figure S11B). This is likely due to the stretches of AU pairs in its hairpin precursor, which were shown previously to be a target of bleomycin A5 itself.50
Figure 8.
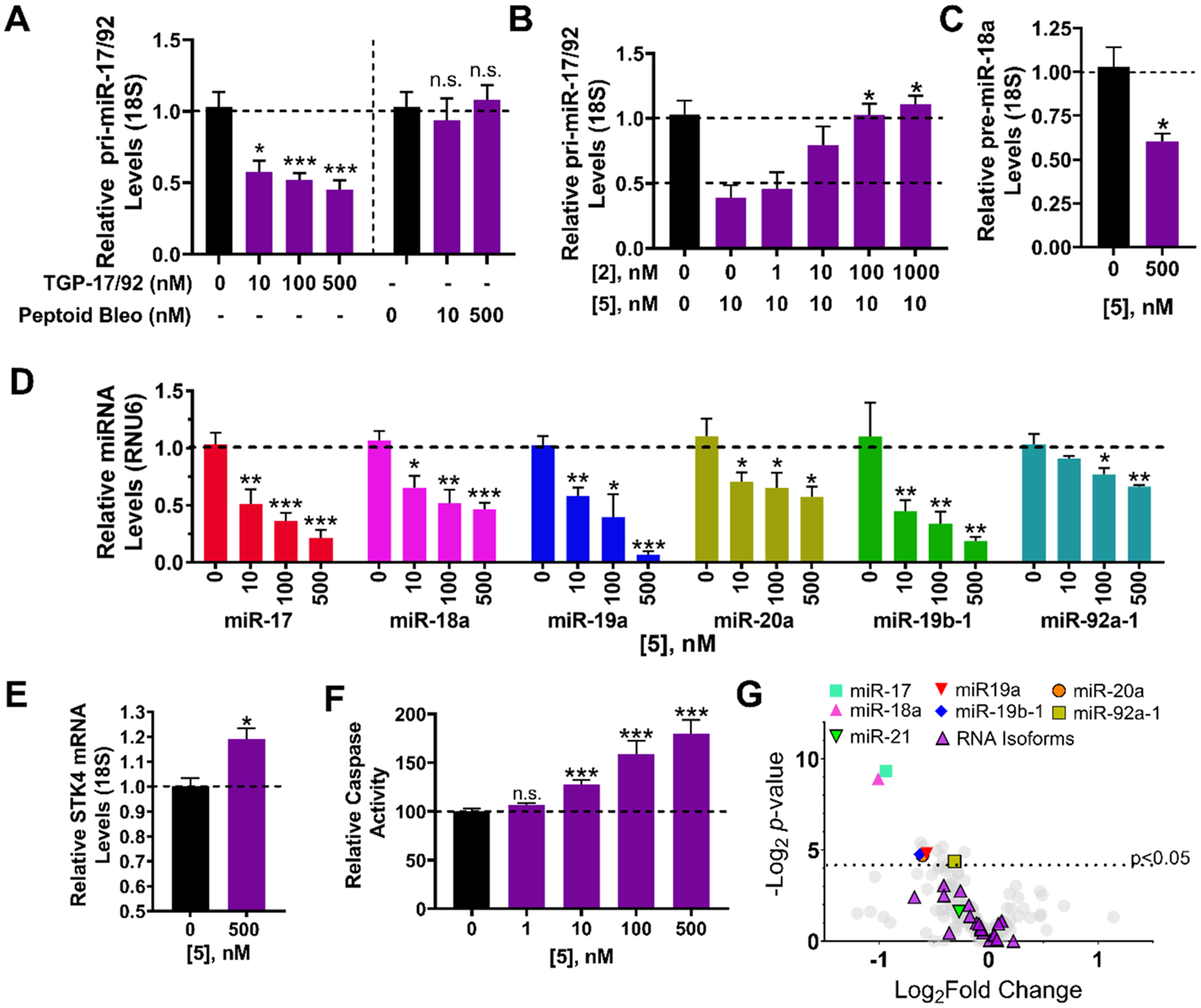
Bioactivity of the dimer-bleomycin conjugate 5 in DU-145 prostate cancer cells. (A) Effect of cleaving compound 5 and negative control compound 6 on pri-miR-17–92 levels in DU-145 cells, as determined by RT-qPCR. (B) Competition experiment between dimer binder 2 and 5 and their effect of pri-miR-17–92 levels, as determined by RT-qPCR. (C) Effect of 5 on pre-miR-17 levels, as determined by RT-qPCR. (D) Effect of 5 on the levels of mature miRNAs derived from the miR-17–92 cluster, as determined by RT-qPCR. (E) Effect of 5 on Stk4 mRNA levels, a direct target of miR-18a, as determined by RT-qPCR. (F) Effect of 5 on Caspase 3/7 activity, an indicator of apoptosis which is impeded in DU-145 cells due to repression of STK4. (G) Profiling of 373 miRNAs in DU-145 cells shows that only mature miRNAs derived from the 17–92 cluster are significantly affected, with miR-17 and −18a being the most significantly affected. Note that profiling was completed after a 6 h treatment period to minimize downstream effects as the compound induces apoptosis. All other data were collected after at 24 h treatment period. Errors are reported as SEM *, p < 0.05; **, p < 0.01; ***, p < 0.001, as determined by a Student t test.
The effect of the 5 on the levels of miR-18a’s downstream target (STK4) in DU-145 cells and on phenotype was also assessed. Indeed, application of 5 (500 nM) increased levels of Stk4 mRNA by 19(±4)% (Figure 8E) and protein levels by 2.6-fold (Figure S11C). Further, the compound triggered apoptosis in DU-145 cells, as measured via induction of Caspase 3/7, with statistically significant effects observed with as little as 10 nM compound (Figure 8F), consistent with its effect on pri-miR-17–92 levels (Figure 8A). Control compound 6 did not induce apoptosis, as expected (Figure S11D). We compared the IC50’s of 2 and 5 for knockdown of mature miR-18a or induction of apoptosis, enabling small molecules to cleave the desired target increased potency by ~10-fold (Figures 4A vs 8D and 4E vs 8F).
Similar to the studies completed for 2 (Figure S8C), we studied whether 5’s rescue of phenotype is due to inhibiting the miR-18a-STK4 circuit via cleavage of the primary transcript. Indeed, overexpression of the cluster from a plasmid attenuated 5’s activity, as assessed by cleavage of pri-miR-17–92, derepression of STK4 mRNA, and activation of Caspase 3/7 (Figure S11E–G). Interestingly, while the STK4 shRNA had no effect on 5’s ability to cleave pri-miR-17–92, it ablated derepression of Stk4 mRNA and activation of Caspase 3/7 (Figure S11H–J). Taken together, these data support that 5’s activity is a direct result of downregulating the pri-miR-17–92 cluster and that induction of apoptosis is dependent on derepression of STK4.
Selectivity of 5 in DU-145 Cells.
To assess the selectivity of 5 cleavage on the miRNome, the levels of all expressed miRNAs in DU-145 cells (n = 373) were measured upon treatment with 500 nM of 5 by RT-qPCR. As shown in Figures 8G and S11K, the most affected miRNAs were those derived from the cluster, with no statistically significant effects on noncluster miRNAs, RNA isoforms, or miR-21. As mentioned above, none of these RNAs has two bulges separated by the same distance as in pre-miR-17, −18a, or −20a. Collectively, these studies suggest that 5 is a selective cleaver of the miR-17–92 cluster among the miRNome.
We next assessed whether 5 has selective effects on the proteome. Global proteomics analysis showed that only 9/3730 (0.24%) proteins were significantly affected by 5 treatment (Figure S12A), and only four were correlated with changes in mRNA levels (Figure S12B). Among down-regulated proteins, zinc finger CCCH domain-containing protein 7A (ZC3H7A) was found to be abnormally expressed in pancreatic ductal adenocarcinoma (PDAC) and mutation of ZC3H7A is related to metastasis of PDAC.52 Interestingly, programmed cell death 1 ligand 1 (PDL1), a known to be a target of miR-17,53,54 was upregulated, although its cell surface expression was not affected, as assessed by immunohistochemical staining and flow cytometry (Figure S12C–E). The BRI3 binding protein (BRI3BP), a pro-apoptotic protein that increases drug-induced apoptosis by increasing cytochrome c release and improving Caspase-3 activity,55,56 was also upregulated. Please see the Supporting Information for a detailed discussion of this proteomics analysis (Figure S12).
Design and Evaluation of a miR-17-, miR-18a-, and miR-20a-Targeting RIBOTAC.
We developed an alternative strategy to target members of the miR-17–92 cluster for destruction by recruiting an endogenous nuclease via a ribonuclease targeting chimera (RIBOTAC).3,57,58 A RIBOTAC comprises an RNA-binding small molecule, in this case 2, conjugated to an RNase L-recruiting module, in this case, a recently discovered heterocycle, affording RIBOTAC 7 (Figure 9A).57 RNase L functions in innate immunity and is expressed at minute levels in all cells as an inactive monomer. It is activated and dimerized upon viral infection and has inherent substrate specificity, preferring 5′UA and 5′UU steps.59,60 RIBOTACs locally recruit RNase L to the desired target to effect selective cleavage.3,57,58
Figure 9.
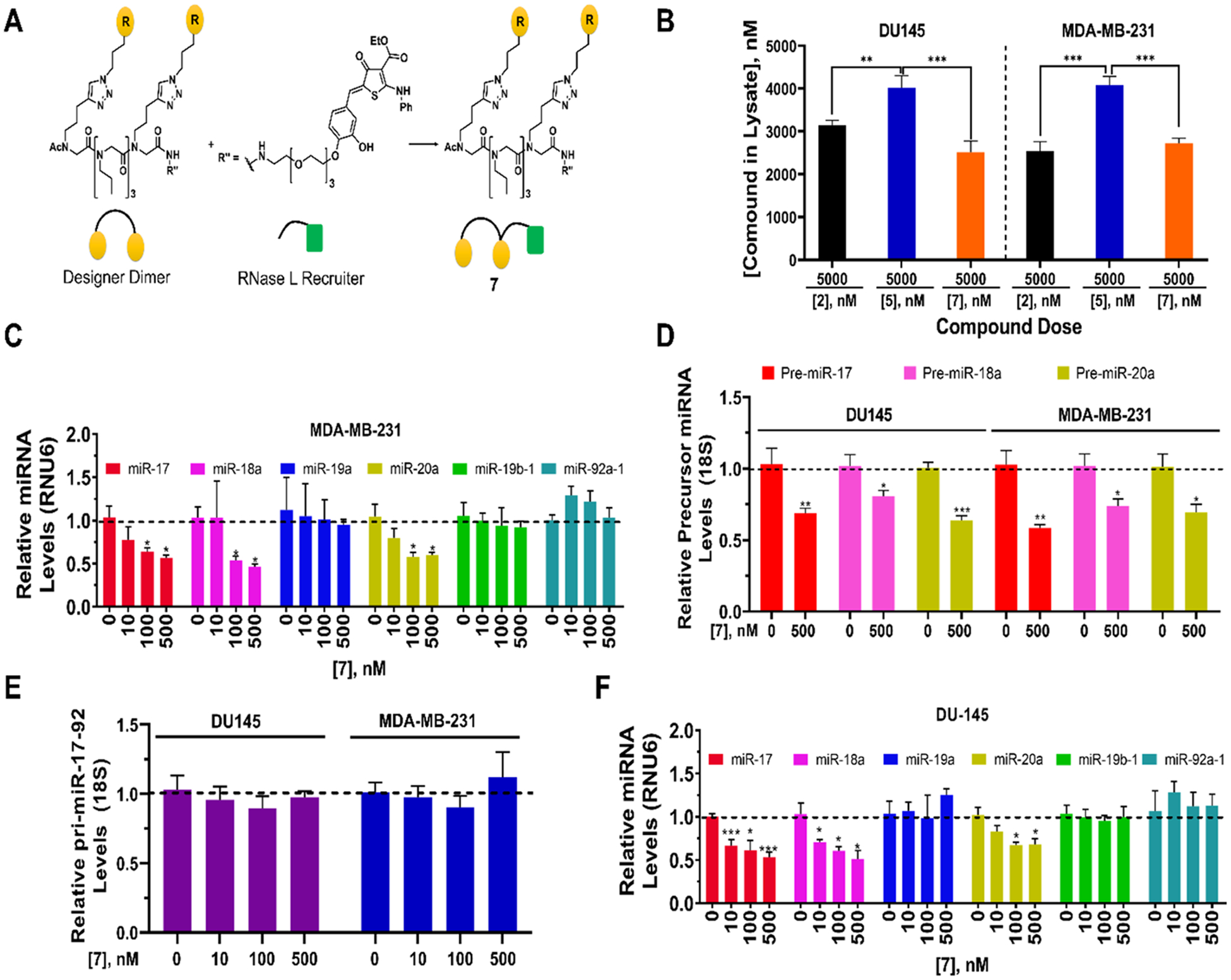
Bioactivity of RIBOTAC 7 in MDA-MB-231 TNBC and DU-145 prostate cancer cells. (A) Structure of RIBOTAC 7, generated by coupling dimer binder 2 with a small molecule that recruits RNase L discovered previously.57 (B) Cellular permeability of 2 (dimer binder), 5 (dimer-bleomycin conjugate) and RIBOTAC 7 at 5 μM. (C) Effect of 7 on the levels of mature miRNAs from the 17–92 cluster in MDA-MB-231 TNBC cells, as determined by RT-qPCR. (D) Effect of 7 on pre-miR-17, −18a, and −20a levels in MDA-MB-231 and DU-145 cells, as determined by RT-qPCR. (E) Effect of 7 on pri-miR-17–92 in MDA-MB-231 TNBC and DU-145 prostate cancer cells, as determined by RT-qPCR. (F) Effect of 7 on the levels of mature miRNAs from the 17–92 cluster in DU-145 prostate cancer cells, as determined by RT-qPCR. *, p < 0.05, **, p < 0.01, ***, p < 0.001 by a Student t test. All errors are reported as SEM.
We therefore assessed whether the pri-miRNA, pre-miRNAs, or both are targeted for degradation by RIBOTAC 7. Interestingly, fluorescence microscopy showed that 7 was dispersed throughout the cell but notably in the cytoplasm where the pre-miRNA targets and RNase L also reside (Figure S5). These results suggest that 7 could trigger cleavage of premiR-17, −18a, and −20a, which bind 2, but not the cleavage of pri-miR-17–92 as RNase L is not detectable in the nucleus. That is, cleavage only occurs in the cytoplasm where all three components of the ternary complex are present, i.e., 7, its RNA targets, and RNase L. Notably, 7 was taken up to a similar extent as 2 in both DU-145 and MDA-MB-231 cells (Figure 9B).
To study if this is indeed the case, we measured the levels of mature miRNAs, pre-miRNAs, and the primary transcript of the 17–92 cluster in MDA-MB-231 TNBC cells upon treatment with 7. Indeed, 7 reduced the levels of mature miR-17, miR-18a, and miR-20a dose dependently, with no effect on the levels of the other three mature miRNAs in the cluster (Figure 9C). Consistent with our hypothesis, 7 (500 nM) decreased levels of pre-miR-17, pre-miR-18a, and premiR-20a by 31(±3)%, 19(±4)%, and 36(±3)%, respectively (Figure 9D), while pri-miR-17/92 levels were unchanged (Figure 9E). To corroborate these data, the effect of 7 on levels of the cluster, whether mature, pre-, or pri-miRNAs, was also measured in DU-145 cells. The same trends were observed; that is, only the pre-miRNAs that bind 2 were degraded, as evidenced by reduced levels of their mature (Figure 9F) and pre-miRNAs (Figure 9D). No effect on pri-miR-17/92 levels was observed (Figure 9E). The changes observed in both cell lines were verified by absolute quantification (Figure S13).
DISCUSSION
The most common way to target an RNA is the binding of ASOs to unfolded or unstructured regions.7,61 However, much of the biology of an RNA is due to its structure.62–64 Indeed, we have used sequence-based design to afford a simple binding compound that is structure-specific. The compound’s mode of action was then extended to cleave the desired target selectively either directly by conjugation to bleomycin A5 or indirectly by conjugation to an RNase L-recruiting module, or RIBOTAC. Application of the bleomycin cleavage strategy to the targeting of the miR-17–92 cluster enabled cleavage of the nuclear primary transcript, thereby reducing levels of all encoded mature miRNAs. These results, taken together with the application of this strategy to cleave r(CUG) repeat expansions in vivo,19 demonstrate the generalizability of this method to enhance the bioactivity of RNA targeting ligands. Given that ~25% of all miRNAs are embedded in clusters and their roles in disease,28 developing strategies to eliminate a cluster in its entirety could have far reaching effects. Further, we demonstrate that cleavage can be tuned to a cellular compartment, such that only a single or a few miRNAs in a cluster can be marked for decay. RIBOTAC 7 selectively cleaved pre-miR-17, pre-miR-18a, and pre-miR-20a, those that bind 2 from which it is derived, reducing levels of only their encoded mature miRNAs. That is, no effect was observed on the three other members in the pri-miR-17–92 cluster or the primary transcript itself. Collectively, our studies suggest that the cleaving module can be customized for each target based on its cellular localization and the site adjacent to the RNA binder’s binding site, as both bleomycin50 and RNase L59,60 have inherent substrate preferences. Importantly, both the RIBOTAC and bleomycin approach remove the requirement of molecular recognition of a structure located at a functional site (such as a Dicer or Drosha processing site). Rather, any structure-specific ligand can be imbued with the ability to cleave the target, thereby inhibiting its downstream function.
Many ligands developed to target RNA are likely to interact with sites that will not affect the biology of the target. That is, although interactions at these sites may be selective, they will not elicit a biological effect. The ability to imbue compounds with the capacity to degrade an RNA target will likely enhance the number of bioactive ligands as triggering decay will affect the biology of a target, regardless of whether the compound binds to a functional or nonfunctional site.
There is much to be learned about designing small molecules that bind to RNA targets in cells and modulate biology. The observation that the targeted degradation can selectively modulate disease pathways and on-target phenotypes in several disease settings suggests that these approaches can be used to broadly provide chemical probes. The development of these probes is a first and important step in developing precision medicines to leverage known biology into therapeutic modalities.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by the National Institutes of Health (R01 GM097455 and R01 CA249180 to MDD). We thank Prof. Vishal Patel, M.D. (Department of Internal Medicine, University of Texas Southwestern Medical Center) for the PPARα-3′ UTR luciferase reporter. We also thank Ms. Alicia Angelbello for her assistance in acquiring confocal microscopy images.
All raw data associated that support the findings of this study are available from the corresponding author upon reasonable request.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.9b13159.
Experimental methods, discussion of proteomics analysis, synthetic methods and characterization of all compounds (PDF)
The authors declare the following competing financial interest(s): Matthew Disney is a founder of Expansion Therapeutics.
REFERENCES
- (1).Agrawal S; Goodchild J; Civeira MP; Thornton AH; Sarin PS; Zamecnik PC Oligodeoxynucleoside phosphoramidates and phosphorothioates as inhibitors of human immunodeficiency virus. Proc. Natl. Acad. Sci. U. S. A 1988, 85 (19), 7079–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Zamecnik PC; Stephenson ML Inhibition of Rous sarcoma virus replication and cell transformation by a specific oligodeoxynucleotide. Proc. Natl. Acad. Sci. U. S. A 1978, 75 (1), 280–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Costales MG; Matsumoto Y; Velagapudi SP; Disney MD Small molecule targeted recruitment of a nuclease to RNA. J. Am. Chem. Soc 2018, 140 (22), 6741–6744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Davis ME; Zuckerman JE; Choi CH; Seligson D; Tolcher A; Alabi CA; Yen Y; Heidel JD; Ribas A Evidence of RNAi in humans from systemically administered siRNA via targeted nanoparticles. Nature 2010, 464 (7291), 1067–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Stein CA; Castanotto D FDA-approved oligonucleotide therapies in 2017. Mol. Ther 2017, 25 (5), 1069–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Abudayyeh OO; Gootenberg JS; Essletzbichler P; Han S; Joung J; Belanto JJ; Verdine V; Cox DBT; Kellner MJ; Regev A; Lander ES; Voytas DF; Ting AY; Zhang F RNA targeting with CRISPR-Cas13. Nature 2017, 550 (7675), 280–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Lima WF; Vickers TA; Nichols J; Li C; Crooke ST Defining the factors that contribute to on-target specificity of antisense oligonucleotides. PLoS One 2014, 9 (7), No. e101752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Stombaugh J; Zirbel CL; Westhof E; Leontis NB Frequency and isostericity of RNA base pairs. Nucleic Acids Res. 2009, 37 (7), 2294–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Crews LA; Balaian L; Delos Santos NP; Leu HS; Court AC; Lazzari E; Sadarangani A; Zipeto MA; La Clair JJ; Villa R; Kulidjian A; Storb R; Morris SR; Ball ED; Burkart MD; Jamieson CHM RNA splicing modulation selectively impairs leukemia stem cell maintenance in secondary human AML. Cell Stem Cell 2016, 19 (5), 599–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Tor Y Targeting RNA with small molecules. ChemBioChem 2003, 4 (10), 998–1007. [DOI] [PubMed] [Google Scholar]
- (11).Disney MD Targeting RNA with small molecules to capture opportunities at the intersection of chemistry, biology, and medicine. J. Am. Chem. Soc 2019, 141 (17), 6776–6790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Connelly CM; Moon MH; Schneekloth JS Jr. The emerging role of RNA as a therapeutic target for small molecules. Cell Chem. Biol 2016, 23 (9), 1077–1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Leon B; Kashyap MK; Chan WC; Krug KA; Castro JE; La Clair JJ; Burkart MD A challenging pie to splice: drugging the spliceosome. Angew. Chem., Int. Ed 2017, 56 (40), 12052–12063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Disney MD; Winkelsas AM; Velagapudi SP; Southern M; Fallahi M; Childs-Disney JL Inforna 2.0: A platform for the sequence-based design of small molecules targeting structured RNAs. ACS Chem. Biol 2016, 11 (6), 1720–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Velagapudi SP; Gallo SM; Disney MD Sequence-based design of bioactive small molecules that target precursor microRNAs. Nat. Chem. Biol 2014, 10 (4), 291–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Wang ZF; Ursu A; Childs-Disney JL; Guertler R; Yang WY; Bernat V; Rzuczek SG; Fuerst R; Zhang YJ; Gendron TF; Yildirim I; Dwyer BG; Rice JE; Petrucelli L; Disney MD The Hairpin Form of r(G4C2)exp in c9ALS/FTD Is Repeat-Associated Non-ATG Translated and a Target for Bioactive Small Molecules. Cell Chem. Biol 2019, 26 (2), 179–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Costales MG; Haga CL; Velagapudi SP; Childs-Disney JL; Phinney DG; Disney MD Small molecule inhibition of microRNA-210 reprograms an oncogenic hypoxic circuit. J. Am. Chem. Soc 2017, 139 (9), 3446–3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Velagapudi SP; Costales MG; Vummidi BR; Nakai Y; Angelbello AJ; Tran T; Haniff HS; Matsumoto Y; Wang ZF; Chatterjee AK; Childs-Disney JL; Disney MD Approved anti-cancer drugs target oncogenic non-coding RNAs. Cell Chem. Biol 2018, 25 (9), 1086–1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Angelbello AJ; Rzuczek SG; McKee KK; Chen JL; Olafson H; Cameron MD; Moss WN; Wang ET; Disney MD Precise small-molecule cleavage of an r(CUG) repeat expansion in a myotonic dystrophy mouse model. Proc. Natl. Acad. Sci. U. S. A 2019, 116 (16), 7799–7804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Velagapudi SP; Cameron MD; Haga CL; Rosenberg LH; Lafitte M; Duckett DR; Phinney DG; Disney MD Design of a small molecule against an oncogenic noncoding RNA. Proc. Natl. Acad. Sci. U. S. A 2016, 113 (21), 5898–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Mu P; Han YC; Betel D; Yao E; Squatrito M; Ogrodowski P; de Stanchina E; D’Andrea A; Sander C; Ventura A Genetic dissection of the miR-17~92 cluster of microRNAs in Myc-induced B-cell lymphomas. Genes Dev. 2009, 23 (24), 2806–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Conkrite K; Sundby M; Mukai S; Thomson JM; Mu D; Hammond SM; MacPherson D miR-17~92 cooperates with RB pathway mutations to promote retinoblastoma. Genes Dev. 2011, 25 (16), 1734–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Huang G; Nishimoto K; Zhou Z; Hughes D; Kleinerman ES miR-20a encoded by the miR-17–92 cluster increases the metastatic potential of osteosarcoma cells by regulating Fas expression. Cancer Res. 2012, 72 (4), 908–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Kim K; Chadalapaka G; Lee SO; Yamada D; Sastre-Garau X; Defossez PA; Park YY; Lee JS; Safe S Identification of oncogenic microRNA-17–92/ZBTB4/specificity protein axis in breast cancer. Oncogene 2012, 31 (8), 1034–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Li Y; Chen M; Liu J; Li L; Yang X; Zhao J; Wu M; Ye M Upregulation of microRNA 18b contributes to the development of colorectal cancer by inhibiting CDKN2B. Mol. Cell. Biol 2017, 37 (22), e00391–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Topkara VK; Mann DL Role of microRNAs in cardiac remodeling and heart failure. Cardiovasc. Drugs Ther 2011, 25 (2), 171–82. [DOI] [PubMed] [Google Scholar]
- (27).Mogilyansky E; Rigoutsos I The miR-17/92 cluster: a comprehensive update on its genomics, genetics, functions and increasingly important and numerous roles in health and disease. Cell Death Differ. 2013, 20 (12), 1603–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Kabekkodu SP; Shukla V; Varghese VK; J, D. S.; Chakrabarty, S.; Satyamoorthy, K. Clustered miRNAs and their role in biological functions and diseases. Biol. Rev. Camb. Philos. Soc 2018, 93 (4), 1955–1986. [DOI] [PubMed] [Google Scholar]
- (29).He L; Thomson JM; Hemann MT; Hernando-Monge E; Mu D; Goodson S; Powers S; Cordon-Cardo C; Lowe SW; Hannon GJ; Hammond SM A microRNA polycistron as a potential human oncogene. Nature 2005, 435 (7043), 828–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Thayanithy V; Sarver AL; Kartha RV; Li L; Angstadt AY; Breen M; Steer CJ; Modiano JF; Subramanian S Perturbation of 14q32 miRNAs-cMYC gene network in osteosarcoma. Bone 2012, 50 (1), 171–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Sage J; Ventura A miR than meets the eye. Genes Dev. 2011, 25 (16), 1663–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Bonauer A; Carmona G; Iwasaki M; Mione M; Koyanagi M; Fischer A; Burchfield J; Fox H; Doebele C; Ohtani K; Chavakis E; Potente M; Tjwa M; Urbich C; Zeiher AM; Dimmeler S MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science 2009, 324 (5935), 1710–3. [DOI] [PubMed] [Google Scholar]
- (33).Tsitsiou E; Lindsay MA microRNAs and the immune response. Curr. Opin. Pharmacol 2009, 9 (4), 514–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Velagapudi SP; Disney MD Two-dimensional combinatorial screening enables the bottom-up design of a micro-RNA-10b inhibitor. Chem. Commun 2014, 50 (23), 3027–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Velagapudi SP; Luo Y; Tran T; Haniff HS; Nakai Y; Fallahi M; Martinez GJ; Childs-Disney JL; Disney MD Defining RNA-small molecule affinity landscapes enables design of a small molecule inhibitor of an oncogenic noncoding RNA. ACS Cent. Sci 2017, 3 (3), 205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Chan TR; Hilgraf R; Sharpless KB; Fokin VV Polytriazoles as copper(I)-stabilizing ligands in catalysis. Org. Lett 2004, 6 (17), 2853–5. [DOI] [PubMed] [Google Scholar]
- (37).Kolb HC; Finn MG; Sharpless KB Click chemistry: diverse chemical function from a few good reactions. Angew. Chem., Int. Ed 2001, 40 (11), 2004–2021. [DOI] [PubMed] [Google Scholar]
- (38).Childs-Disney JL; Tsitovich PB; Disney MD Using modularly assembled ligands to bind RNA internal loops separated by different distances. ChemBioChem 2011, 12 (14), 2143–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Hajarnis S; Lakhia R; Yheskel M; Williams D; Sorourian M; Liu XQ; Aboudehen K; Zhang SR; Kersjes K; Galasso R; Li J; Kaimal V; Lockton S; Davis S; Flaten A; Johnson JA; Holland WL; Kusminski CM; Scherer PE; Harris PC; Trudel M; Wallace DP; Igarashi P; Lee EC; Androsavich JR; Patel V MicroRNA-17 family promotes polycystic kidney disease progression through modulation of mitochondrial metabolism. Nat. Commun 2017, 8, 14395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).McGhee JD; von Hippel PH Theoretical aspects of DNA-protein interactions: co-operative and non-co-operative binding of large ligands to a one-dimensional homogeneous lattice. J. Mol. Biol 1974, 86 (2), 469–89. [DOI] [PubMed] [Google Scholar]
- (41).Chakraborty S; Mehtab S; Patwardhan A; Krishnan Y PrimiR-17–92a transcript folds into a tertiary structure and autoregulates its processing. RNA 2012, 18 (5), 1014–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Chaulk SG; Thede GL; Kent OA; Xu Z; Gesner EM; Veldhoen RA; Khanna SK; Goping IS; MacMillan AM; Mendell JT; Young HS; Fahlman RP; Glover JN Role of pri-miRNA tertiary structure in miR-17~92 miRNA biogenesis. RNA Biol. 2011, 8 (6), 1105–14. [DOI] [PubMed] [Google Scholar]
- (43).Lewis BP; Burge CB; Bartel DP Conserved seed pairing, often flanked by adenosines, indicates that thousands of human genes are microRNA targets. Cell 2005, 120 (1), 15–20. [DOI] [PubMed] [Google Scholar]
- (44).Loghman-Adham M; Nauli SM; Soto CE; Kariuki B; Zhou J Immortalized epithelial cells from human autosomal dominant polycystic kidney cysts. Am. J. Physiol. Renal. Physiol 2003, 285 (3), F397–412. [DOI] [PubMed] [Google Scholar]
- (45).Hsu TI; Hsu CH; Lee KH; Lin JT; Chen CS; Chang KC; Su CY; Hsiao M; Lu PJ MicroRNA-18a is elevated in prostate cancer and promotes tumorigenesis through suppressing STK4 in vitro and in vivo. Oncogenesis 2014, 3, e99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Guan L; Disney MD Covalent small-molecule-RNA complex formation enables cellular profiling of small-molecule-RNA interactions. Angew. Chem., Int. Ed 2013, 52 (38), 10010–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Su Z; Zhang Y; Gendron TF; Bauer PO; Chew J; Yang WY; Fostvedt E; Jansen-West K; Belzil VV; Desaro P; Johnston A; Overstreet K; Oh SY; Todd PK; Berry JD; Cudkowicz ME; Boeve BF; Dickson D; Floeter MK; Traynor BJ; Morelli C; Ratti A; Silani V; Rademakers R; Brown RH; Rothstein JD; Boylan KB; Petrucelli L; Disney MD Discovery of a biomarker and lead small molecules to target r(GGGGCC)-associated defects in c9FTD/ALS. Neuron 2014, 83 (5), 1043–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Rzuczek SG; Colgan LA; Nakai Y; Cameron MD; Furling D; Yasuda R; Disney MD Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nat. Chem. Biol 2017, 13 (2), 188–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Li Y; Disney MD Precise small molecule degradation of a noncoding RNA identifies cellular binding sites and modulates an oncogenic phenotype. ACS Chem. Biol 2018, 13 (11), 3065–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Angelbello AJ; Disney MD, Bleomycin can cleave an oncogenic noncoding RNA. 2018, 19 (1), 43–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Krichevsky AM; Gabriely G miR-21: a small multi-faceted RNA. J. Cell. Mol. Med 2009, 13 (1), 39–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Zhou B; Irwanto A; Guo YM; Bei JX; Wu Q; Chen G; Zhang TP; Lei JJ; Feng QS; Chen LZ; Liu J; Zhao YP Exome sequencing and digital PCR analyses reveal novel mutated genes related to the metastasis of pancreatic ductal adenocarcinoma. Cancer Biol. Ther 2012, 13 (10), 871–9. [DOI] [PubMed] [Google Scholar]
- (53).Casey SC; Tong L; Li Y; Do R; Walz S; Fitzgerald KN; Gouw AM; Baylot V; Gütgemann I; Eilers M; Felsher DW MYC regulates the antitumor immune response through CD47 and PD-L1. Science 2016, 352 (6282), 227–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (54).Audrito V; Serra S; Stingi A; Orso F; Gaudino F; Bologna C; Neri F; Garaffo G; Nassini R; Baroni G; Rulli E; Massi D; Oliviero S; Piva R; Taverna D; Mandala M; Deaglio S PD-L1 up-regulation in melanoma increases disease aggressiveness and is mediated through miR-17–5p. Oncotarget 2017, 8 (9), 15894–15911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Yamazaki T; Sasaki N; Nishi M; Yamazaki D; Ikeda A; Okuno Y; Komazaki S; Takeshima H Augmentation of drug-induced cell death by ER protein BRI3BP. Biochem. Biophys. Res. Commun 2007, 362 (4), 971–5. [DOI] [PubMed] [Google Scholar]
- (56).Mendes M; Perez-Hernandez D; Vazquez J; Coelho AV; Cunha C Proteomic changes in HEK-293 cells induced by hepatitis delta virus replication. J. Proteomics 2013, 89, 24–38. [DOI] [PubMed] [Google Scholar]
- (57).Costales MG; Aikawa H; Li Y; Childs-Disney JL; Abegg D; Hoch DG; Pradeep Velagapudi S; Nakai Y; Khan T; Wang KW; Yildirim I; Adibekian A; Wang ET; Disney MD Small-molecule targeted recruitment of a nuclease to cleave an oncogenic RNA in a mouse model of metastatic cancer. Proc. Natl. Acad. Sci. U. S. A 2020, 117 (5), 201914286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (58).Costales MG; Suresh B; Vishnu K; Disney MD Targeted degradation of a hypoxia-associated non-coding RNA enhances the selectivity of a small molecule interacting with RNA. Cell Chem. Biol 2019, 26 (8), 1180–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Floyd-Smith G; Slattery E; Lengyel P Interferon action: RNA cleavage pattern of a (2’−5′)oligoadenylate–dependent endonuclease. Science 1981, 212 (4498), 1030–2. [DOI] [PubMed] [Google Scholar]
- (60).Wreschner DH; McCauley JW; Skehel JJ; Kerr IM Interferon action–sequence specificity of the ppp(A2’p)nA-dependent ribonuclease. Nature 1981, 289 (5796), 414–7. [DOI] [PubMed] [Google Scholar]
- (61).Lima WF; Monia BP; Ecker DJ; Freier SM Implication of RNA structure on antisense oligonucleotide hybridization kinetics. Biochemistry 1992, 31 (48), 12055–61. [DOI] [PubMed] [Google Scholar]
- (62).Zaug A; Cech T The intervening sequence RNA of Tetrahymena is an enzyme. Science 1986, 231 (4737), 470–475. [DOI] [PubMed] [Google Scholar]
- (63).Doudna JA Structural genomics of RNA. Nat. Struct. Biol 2000, 7 (11), 954–6. [DOI] [PubMed] [Google Scholar]
- (64).Ding Y; Tang Y; Kwok CK; Zhang Y; Bevilacqua PC; Assmann SM In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features. Nature 2014, 505 (7485), 696–700. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.