

Abstract
Purpose
The bile salt export pump (BSEP), a key player in hepatic bile acid clearance, has been the center of research on drug-induced cholestasis. However, such studies focus primarily on the direct inhibition of BSEP, often overlooking the potential impact of transcriptional repression. This work aims to explore the disruption of bile acid efflux caused by drug-induced BSEP repression.
Methods
BSEP activity was analyzed in human primary hepatocytes (HPH) using a traditional biliary-clearance experiment and a modified efflux assay, which includes a 72-h pretreatment prior to efflux measurement. Relative mRNA and protein expressions were examined by RT-PCR and Western blotting, respectively.
Results
Metformin concentration-dependently repressed BSEP expression in HPH. Although metformin did not directly inhibit BSEP activity, longer metformin exposure reduced BSEP transport function in HPH by down-regulating BSEP expression. BSEP repression by metformin was found to be AMP-activated protein kinase-independent. Additional screening of 10 reported cholestatic non-BSEP inhibitors revealed that the anti-cancer drug tamoxifen also markedly repressed BSEP expression and reduced BSEP activity in HPH.
Conclusions
Repression of BSEP alone is sufficient to disrupt hepatic bile acid efflux. Metformin and tamoxifen appear to be prototypes of a class of BSEP repressors that may cause drug-induced cholestasis through gene repression instead of direct BSEP inhibition.
Keywords: BSEP, Cholestasis, Metformin, Tamoxifen, Gene regulation
INTRODUCTION
Drug-induced liver injury (DILI) is one of the most common causes of drug attrition from both drug development and post-marketing - a major concern for the pharmaceutical industry, regulatory agencies, and clinical practice (1,2). DILI can manifest in a variety of ways, mimicking a wide range of disorders associated with both acute and chronic liver diseases (3). One of the most prevalent manifestations of DILI is drug-induced cholestasis (1), characterized by interrupted bile flow from the liver to the intestines and accumulation of toxic levels of bile acids in the liver, particularly within hepatocytes (4). Physiologically, bile acids play an important role in lipid and fat-soluble vitamin emulsification and absorption from the small intestine. To facilitate their function, bile acids have distinct polar and non-polar regions that allow them to form micelles (5,6). These detergent-like micelles are necessary for lipid-trapping; however, this property also contributes to toxicity.
Typically, drug-induced cholestasis occurs soon after initiating a new drug, and is confirmed when discontinuation of the drug alleviates toxicity (7,8). While, often not life-threatening when diagnosed early, prolonged cholestasis can result in severe liver injury, liver transplantation, and death (2,4). With an increasing number of drugs reported to cause cholestasis after FDA approval, drug-induced cholestasis has become a major focus of investigation (9,10).
The bile salt export pump (BSEP, ABCB11) is the rate-limiting step of bile flow from the liver to the intestine (11,12). As the key determinant of hepatic bile acid elimination, BSEP is a major focus of drug-induced cholestasis research. Most current studies focus on disruption of BSEP function through direct inhibition of bile acid transport (13,14). Indeed, the recently issued FDA guidance for industry on transporter-based drug interactions focuses on transporter inhibition (15–17). Nevertheless, BSEP inhibition alone cannot fully explain BSEP-associated cholestasis. Genetic mutations in BSEP gene are linked to substantial attenuation or even absence of BSEP expression (e.g. progressive familial intrahepatic cholestasis type 2) (12). A group of compounds with cholestatic liability did not inhibit BSEP activity in prototypical BSEP functional assays (18). Notably, remarkable reduction of BSEP expression was observed in 8 of 12 patients with cholestatic DILI (19). Our previous study showed that several agents classified as BSEP inhibitors were also capable of repressing BSEP expression (20). Of importance, dual inhibitors and repressors of BSEP appear to be associated with severe DILI. Based on these observations, we pursued the current study wherein drug-mediated repression of BSEP expression is shown to play an important role in drug-induced cholestasis.
Metformin is one of the most commonly prescribed drugs used to treat type 2 diabetes mellitus. While generally considered a safe medication, there have been several reports of cholestasis with metformin use where discontinuation of the drug alleviated cholestatic toxicity (8,21,22). Notably, metformin does not directly inhibit BSEP (18). Accordingly, we hypothesized that instead of directly inhibiting BSEP activity, metformin represses BSEP expression, thus inducing cholestatic liver injury. Given that activation of the cellular energy sensor, AMP-activated protein kinase (AMPK), is a known mechanism through which metformin exerts many of its pharmacological actions (23), we also explored if AMPK activation is involved in the inhibitory effect of metformin on BSEP expression in HPH.
In this report, we provide experimental evidence to show that metformin exhibits potent repression of BSEP expression and disrupts BSEP-mediated bile acid efflux in HPH. Its repression of BSEP expression was independent of AMPK activation. By screening 10 cholestatic drugs that have been reported not to directly inhibit BSEP, tamoxifen was also identified as a BSEP repressor and found to disrupt BSEP efflux function in HPH. Collectively, our results indicate that a class of metformin-like compounds has the potential to repress expression of BSEP, an underappreciated mechanism that may contribute to drug-induced cholestasis.
MATERIALS AND METHODS
Reagents
Metformin, GW-4064, Compound C, D-penicillamine, nitrofurantoin, and nortriptyline were purchased from Sigma Aldrich (St. Louis, MO). Ibuprofen, sulindac, tamoxifen, trimethoprim, and verapamil were purchased from AK Scientific (Union City, CA). Carbamazepine, cimetidine, and [3H]-taurocholic acid were purchased from Perkin Elmer (Waltham, MA). PCR Primers were synthesized by Integrated DNA Technologies (Coralville, IA). High Capacity cDNA Archive Kit was obtained from Applied Biosystems (Foster City, CA). Matrigel was received from BD Biosciences (Bedford, MA). All other cell culture reagents were purchased from Thermo Fisher Scientific (Waltham, MA) or Sigma-Aldrich.
Culture and Treatment of HPH
Human primary hepatocytes were obtained from BioIVT (Baltimore, MD). Hepatocytes (viability >90%) were seeded at 1.5 × 106 cells/well and 7.5 × 105 cells/well in 6-well and 12-well collagen-coated plates, respectively, and cultured in the sandwich format as described previously (24). Hepatocytes were treated 48 h after seeding with 0.1% DMSO or specified compounds for 24 h for mRNA analysis or 72 h for Western blotting and functional assays. Culture medium was replaced on a daily basis.
RT-PCR
Total RNA was isolated from HPH using the TRIzol® reagent and reverse transcribed using a High Capacity cDNA Archive Kit following the manufacturers’ instructions. Primer sequences used for PCR amplification are as follows: GAPDH, 5′-CCCATCACCATCTTCCA GGAG-3′ (forward), 5′-GTTGTCATGGATGACCTTGGC-3′ (reverse); FGF21, 5’-CTGTGGGTTTCTGTGCTGG-3′ (forward), 5’-CCGGCTTCAAGGCTTTCAG-3′ (reverse); BSEP 5′-ACATGCTT GCGAGGACCTTTA-3′ (forward), 5′-GGAGGTTCGTGCACCAGGTA-3′ (reverse). Target gene mRNA expression was normalized against that of GAPDH. Real-time PCR assays were performed in 96-well optical plates on an StepOnePlus Real-Time PCR System (Applied Biosystems) with SYBR Green PCR Master Mix as described previously (25). Data are represented as mean ± SD of three individual experiments.
Western Blotting
Antibodies against BSEP (F-6) and β-actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies against phosphor-AMPKα (Thr172) (40H9) and AMPKα were purchased from Cell Signaling Technology (Danvers, MA). Crude protein lysates from treated HPH were harvested using RIPA buffer, and protein concentrations were determined using a bicinchoninic acid (BCA) based assay from Thermo Fisher Scientific. Equal amounts of protein lysates were separated on SDS-polyacrylamide gels (4–12% bis-tris) before transferring on to polyvinylidene difluoride membranes. The membranes were subsequently probed with specific antibodies against BSEP (1:250 dilution), phosphor-AMPK (1:1000 dilution), AMPK (1:1000 dilution), or β-actin (1:30,000 dilution) overnight at 4°C, followed by incubation with horseradish peroxidase-conjugated secondary antibodies. β-actin was used as loading control. Membranes were developed with chemiluminescent reagents and the relative protein expression was estimated by densitometry analysis using NIH ImageJ (Bethesda, MD).
Biliary Efflux Assay
Human primary hepatocytes were seeded on 12-well plates as described above and cultured for 96 h after Matrigel overlay for BSEP functional assays. In a modification to the transport assay described previously (26), transport of the tritiated BSEP substrate taurocholic acid ([3H]-TCA) was monitored to assess BSEP function in the presence of selected drugs. Briefly, cells were incubated with standard or calcium (Ca2+)-free Hanks’ balanced salt solution (ST HBSS or CF HBSS) for 10 min, before HBSS (standard and Ca2+-free) containing [3H]-TCA was applied to cells for a 10-min incubation. During the inhibition assay, biliary efflux was monitored in the presence of metformin or tamoxifen at different concentrations for 10 min. Conversely, the effect of BSEP repression on its function was monitored by pretreating HPH for 72 h with metformin or tamoxifen and followed by monitoring [3H]-TCA biliary efflux for 10 min in HBSS (standard or Ca2+-free). The assay was terminated by aspirating the [3H]-TCA- containing HBSS and adding ice-cold standard HBSS. Cell lysates harvested in 0.5% Triton X-100 were analyzed by scintillation counting and normalized to protein concentration. The amount of bile acids excreted from hepatocytes, as a measurement of [3H]-TCA, was calculated into a percentage and referred to as the biliary excretion index (BEI) as described previously (26).
Statistical Analysis
Results from real-time PCR and functional efflux assays are expressed as mean ± S.D. of triplicate determinations unless otherwise indicated. Statistical analyses used one-way analysis of variance (ANOVA) and Dunnett’s posttest.
RESULTS
Metformin Represses BSEP Expression in HPH
The effects of metformin on BSEP expression were evaluated in sandwich-cultured HPH prepared from multiple liver donors. As demonstrated in Fig. 1A and B, expression of BSEP mRNA was markedly repressed by metformin treatment in a concentration-dependent manner. Maximal repression (approximately 75%) of BSEP mRNA expression was achieved with 1 mM metformin, a concentration that was frequently used in cell culture studies and is not cytotoxic in HPH (27,28). As expected, the positive control, GW-4064, robustly induced BSEP expression. western blotting analysis confirmed metformin repressed BSEP protein expression in a pattern consistent with the changes in BSEP mRNA (Fig. 1C and D). These results reveal metformin as a potent repressor of BSEP expression in HPH.
Fig. 1. Metformin represses BSEP expression in human primary hepatocytes.

HPH prepared from multiple liver donors were treated with vehicle control (0.1% DMSO), metformin (0.1, 0.5, and 1 mM), or GW-4064 (2 μM) as a positive control as detailed in Materials and Methods. mRNA expression of BSEP was measured using RT-PCR in HL#66 (A) and #111 (B). Data are expressed as mean ± S.D. (n = 3). The expression of BSEP protein (C & D) was measured using Western blotting in HL #111 & #120. Densitometry was determined using the NIH ImageJ software
Metformin Alters BSEP Function by Repressing its Expression
To investigate whether metformin alters BSEP function, two transport assays were performed in HPH. The effects of metformin on hepatic bile acid efflux were tested in a prototypical inhibition assay with 10 min drug exposure (Fig. 2A) or in a modified inhibition assay condition where a 72-h drug pretreatment precedes the bile acid efflux measurement (Fig. 2B). Our results demonstrated that under typical inhibitory conditions, metformin did not alter bile acid efflux compared to the vehicle control (0.1% DMSO) (Fig. 2C and E). As a positive control, a known cholestatic BSEP-inhibitor, troglitazone, potently inhibited bile acid transport through BSEP. Importantly, when HPH were pretreated for 72 h with metformin (0.1, 0.5, and 1 mM), [3H]-TCA efflux was markedly decreased (Fig. 2D and F). Under typical inhibition assay conditions (10-min incubation), metformin (1 mM) did not alter the clearance of [3H]-TCA (82.45% for metformin vs. 81.72% for vehicle control) whereas in the 72-h repression assay bile acid clearance in the presence of metformin was reduced to 19.22% compared to the control value of 81.52% (Table 1). These results indicate that metformin does not inhibit BSEP through direct substrate competition; instead, longer exposure to metformin repressed BSEP expression, thereby decreasing bile acid clearance capacity at 72 h.
Fig. 2. Metformin represses BSEP function in human primary hepatocytes.

The prototypical inhibition assay with short substrate incubation of 10 min (A) and a modified repression assay including a 72-h drug pretreatment (B) were schematically depicted. BSEP inhibition assay measured via biliary excretion of [3H]-TCA in two HPH donors, HL #90 (C) & #91 (E), upon 10-min exposure to vehicle control (0.1% DMSO), positive control (troglitazone 100 μM), or metformin (0.1, 0.5, and 1 mM). BSEP repression was measured via biliary excretion of [3H]-TCA in two human primary hepatocytes donors, HL #90 (D) & 91(F), upon 72-h pre-treatment with vehicle control (0.1% DMSO) or metformin (0.1, 0.5, and 1 mM). Solid bars represent dpm/mg protein of [3H]-TCA that accumulated in the cells + bile canaliculi, while open bars represent dpm/mg protein [3H]-TCA in the cells only
Table 1.
Biliary excretion index (BEI) of metformin treatments in inhibition and repression assays
| No Pre-treatment (Inhibition) | 72 h Pre-treatment (Repression) | |
|---|---|---|
| DMSO 0.1% | 81.72 ± 6.75 | 81.52 ± 4.83 |
| MET 0.1 mM | 82.71 ± 8.53 | 83.50 ± 1.75 |
| MET 0.5 mM | 84.18 ± 2.14 | 68.95 ± 11.77 |
| MET 1 mM | 82.45 ± 1.08 | 19.22 ± 16.22*** |
P < 0.001
Repression of BSEP Expression by Metformin Is Independent of AMPK Activation in HPH
AMPK activation is a known mediator of metformin actions. As shown in Fig. 3A and B, metformin markedly increased AMPK phosphorylation in HPH, without altering the expression of total AMPK. Hence, the role of AMPK activation in BSEP repression by metformin was further investigated by co-treatment with compound C, a widely used AMPK inhibitor. To confirm the inhibitory effect of compound C on AMPK in HPH, the mRNA expression of FGF21, whose expression is correlated with AMPK activation (29), was measured after treatment with metformin alone or metformin and compound C at different concentrations (5, 10, 20 μM). As expected, metformin at 1 mM significantly increased FGF21 mRNA expression, which was attenuated by compound C in a concentration-dependent manner, indicating the successful inhibition of AMPK activation by compound C in HPH (Fig. 3C and D). Notably, co-treatment with compound C did not reverse the repression of BSEP by metformin (Fig. 3E and F). These results suggest that repression of BSEP expression by metformin is independent of AMPK activation in HPH.
Fig. 3. Repression of BSEP by metformin in HPH is AMPK-independent.
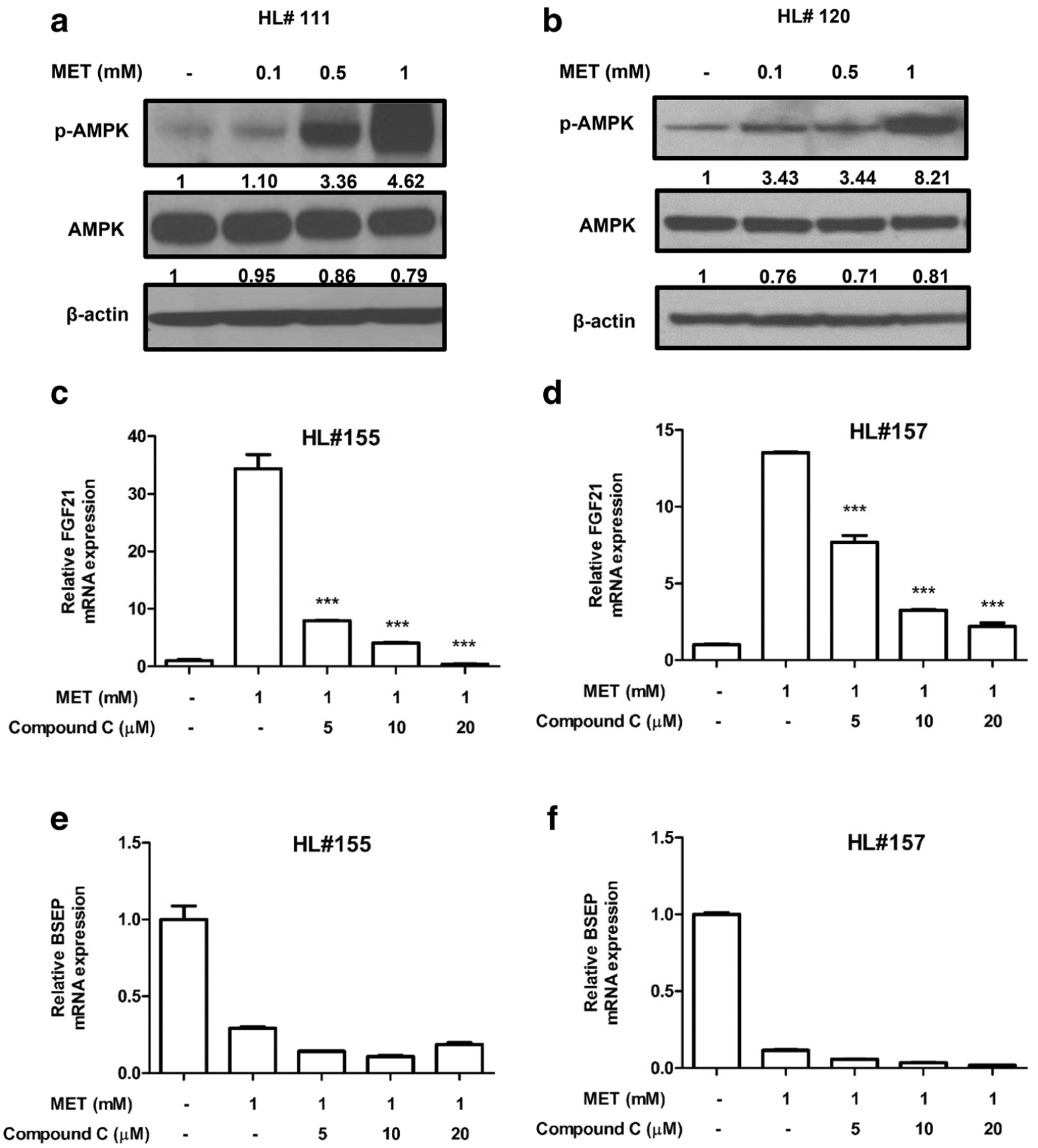
HPH were treated with vehicle control (0.1% DMSO), metformin (0.1, 0.5, and 1 mM) or metformin (1 mM) and compound C (5, 10, and 20 μM) as outlined in Materials and Methods. AMPK phosphorylation (A & B) was measured using Western blotting in HL#111 & #120. RT-PCR was used to measure the mRNA expression of FGF21 and BSEP in HPH from donors HL #155 (C & E) and #157 (D & F). Data were expressed as mean ± S.D. (n = 3). Densitometry was determined using the NIH ImageJ software
Drug-Induced Repression of BSEP Expression Is Not Unique to Metformin
In addition to metformin, which we used as a prototype compound to study BSEP repression, other drugs are reported to cause cholestasis without inhibiting BSEP (18). We evaluated 10 such drugs for their effects on BSEP expression in HPH. The concentrations of these drugs were selected based on literature search and all the compounds at the concentrations selected showed no HPH cytotoxicity during the treatments. As shown in Fig. 4A, tamoxifen, an estrogen receptor (ER) antagonist widely used to treat ER-positive breast cancer, potently repressed BSEP gene expression in HPH at 20 μM, a concentration at which this compound exhibited no cytotoxicity (data not shown). Concentration-dependent repression of BSEP by tamoxifen was confirmed in hepatocytes from additional liver donors at both the mRNA (Fig. 4B and C) and protein (Fig. 4D and E) levels. Like metformin, tamoxifen showed no effects on the [3H]-TCA efflux in a prototypical inhibition assay in HPH (Fig. 5A and C), in line with its classification as a non-inhibitor of BSEP. However, in hepatocytes pre-treated with 10 μM and 20 μM tamoxifen for 72 h, [3H]-TCA efflux was significantly reduced to 52.38% and 16.61%, respectively, compared with 71.75% in control cells (Fig. 5B, D and Table 2). Such alterations in biliary excretion index are most likely caused by the repression of BSEP expression. These results confirm disruption of BSEP efflux function in HPH by some cholestatic BSEP non-inhibitors occurs via gene repression rather than competitive inhibition.
Fig. 4. Cholestatic non-BSEP-inhibitor tamoxifen represses BSEP expression.

(A) mRNA expression of BSEP was measured using RT-PCR in HPH from donors HL #109, #130, & #131. Cells were treated with vehicle control DMSO (0.1%), tamoxifen (20 μM, TAM), ibuprofen (50 μM, IBUP), trimethoprim (50 μM, TRIM), D-penicillamine (250 μM, D-PEN), sulindac (10 μM, SULI), carbamazepine (200 μM, CARB), verapamil (100 μM, VERA), nitrofurantoin (10 μM, NITR), nortriptyline (10 μM, NORT), and cimetidine (1 mM, CIME). Data are expressed as mean ± S.D. (n = 3). The top and bottom dotted lines indicate 20% and 60% reduction in the BSEP mRNA expression, respectively. Concentration-dependent mRNA and protein expression of BSEP was measured in HPH treated with vehicle control (0.1% DMSO), positive control (GW-4064 2 μM), or tamoxifen (5, 20 μM). mRNA expression was measure using RT-PCR in HPH from donors HL#113 (B) & #127 (C). Data are expressed as mean ± S.D. (n = 3). ***P < 0.001; **P < 0.01. Protein expression of BSEP was measured using Western blotting in HPH from donors HL #120 (D) & #123 (E). Densitometry was determined using the NIH ImageJ software
Fig. 5. Tamoxifen disrupts BSEP function in human primary hepatocytes.

The prototypical inhibition assay and a modified repression assay including a 72-h drug pretreatment were described in Materials and Methods. BSEP inhibition assay was measured via biliary excretion of [3H]-TCA in two HPH donors, HL #158 (A) & #159 (C), upon 10-min exposure to vehicle control (0.1% DMSO), positive control (troglitazone 100 μM), or tamoxifen (5, 10, and 20 μM). BSEP repression assay was measured via biliary excretion of [3H]-TCA in two human primary hepatocytes donors, HL #158 (B) & #159 (D), upon 72-h pre-treatment with vehicle control (0.1% DMSO) or tamoxifen (5, 10, and 20 μM). Solid bars represent dpm/mg protein of [3H]-TCA that accumulated in the cells + bile canaliculi, while open bars represent dpm/mg protein [3H]-TCA in the cells only
Table 2.
Biliary excretion index (BEI) of tamoxifen treatments in inhibition and repression assays
| No Pre-treatment (Inhibition) | 72 h Pre-treatment (Repression) | |
|---|---|---|
| DMSO 0.1% | 65.99 ± 2.52 | 71.75 ± 5.14 |
| Tamoxifen 5 μM | 62.86 ± 3.95 | 58.94 ± 5.49 |
| Tamoxifen 10 μM | 65.60 ± 6.98 | 52.38 ± 4.75* |
| Tamoxifen 20 μM | 52.30 ± 7.43 | 16.61 ± 9.32*** |
P < 0.05;
P < 0.001
DISCUSSION
Drug-induced cholestasis is a leading cause of drug attrition, prompting a growing research effort to understand the underlying mechanisms (30,31). Until recent years, the primary focus has been the ability of drugs to inhibit bile acid efflux from the liver by interaction with the bile salt export pump BSEP (13,14,32). Although this strategy has elucidated the mechanism of action for many cholestatic drugs, some chemicals with cholestatic liability do not directly inhibit BSEP activity (18), suggesting alternate mechanisms may contribute to drug-induced cholestasis. Our previous screen of 30 known BSEP inhibitors identified five potent (> 60% repression) and 10 moderate (20–60% repression) repressors of BSEP expression (20). Importantly, drugs exhibiting both potent repression of BSEP expression and inhibition of its activity were associated with the greatest DILI concern, i.e. black box warnings and withdrawal from the market (33). Using metformin as a model compound, the present study unequivocally shows that drug-induced BSEP repression alone is sufficient to affect bile acid clearance from HPH and, thus, may represent an overlooked mechanism leading to drug-induced cholestatic liver injury.
As the first-line medication for the treatment of type 2 diabetes, metformin exhibits several metabolic benefits and is generally well-tolerated with few side effects. Nonetheless, several case reports revealed the association of metformin with intrahepatic cholestasis and liver injury in type 2 diabetics (8,21,22). In mice, metformin treatment increased bile acid accumulation in both the liver and blood (34). The underlying mechanisms of these observations however remained elusive. Using membrane vesicles from Sf9 cells overexpressing human BSEP, Pedersen et al., screened 250 compounds for BSEP inhibition and identified metformin as a non-inhibitor of BSEP (35). In the current study, we found that metformin markedly repressed BSEP expression in sandwich-cultured HPH. These results suggest metformin may represent a pure repressor of human BSEP that could influence bile acid clearance without direct substrate competition.
Transport activity of membrane proteins has often been evaluated using insect membrane vesicles, immortalized cell lines ectopically expressing particular transporters, or sandwich-cultured hepatocytes that maintain functional endogenous transporters (14,26,36,37). Typical inhibition assays involve a short-term (10 to 30 min) incubation of test compounds with substrate probes in membrane vesicles or cultured cells (14,38). Utilizing this traditional approach, we showed that metformin does not directly inhibit BSEP-mediated efflux of taurocholic acid in HPH, which is consistent with earlier observations in inverted insect membrane vesicles (35). Nevertheless, this methodology fails to evaluate a drug’s effects on BSEP after an extended period of exposure. In our modified experiments as depicted in Fig. 2B, the 72-h pretreatment prior to bile acid efflux measurements in HPH allowed a transport assay to coordinate effects of gene expression on transporter function, and confirmed that repression of BSEP expression alone can alter BSEP-mediated bile acid transport.
Despite the fact that AMPK activation is a known mechanism by which metformin exerts many of its pharmacological effects, in our study the repression of BSEP expression in HPH by metformin was independent of AMPK activation. To date, accumulating evidence has demonstrated the AMPK-independent effects of metformin, such as its inhibitory effects on hepatic gluconeogenesis and adipogenesis (39,40). Nevertheless, how metformin down-regulates BSEP expression in HPH needs further investigation. In contrast, previous studies in mice demonstrated that activation of AMPK by metformin enhanced BSEP expression (41), however this was paradoxically accompanied by increased cholestatic toxicity (34,42). Although the mechanisms underlying these contradictory observations are largely unknown, as increasingly recognized, species differences between mice and humans contribute to the relatively low pre-dictability of preclinical models for toxicological evaluation during drug development (43–45).
Recently, a list of drugs labeled “cholestatic BSEP non-inhibitors” were reported to induce cholestatic toxicity without direct inhibition of BSEP activity (18). We speculated a plausible explanation for the cholestatic effects of these drugs might be repression of BSEP gene expression. Of 10 drugs investigated in our study, tamoxifen potently repressed BSEP expression and disrupted BSEP-mediated bile acid efflux in HPH, while the rest had moderate or no effects. Notably, tamoxifen is classified as a selective estrogen receptor modulator that can act as either an agonist or antagonist in a tissue-specific manner (46). Interestingly, previous reports have demonstrated that estrogen receptor alpha (ERα) exhibits transrepressive effects on BSEP expression, and estradiol-activated ERα contributes to intrahepatic cholestasis of pregnancy (47,48). As an ERα agonist in the liver (49), it is possible that, like estradiol, tamoxifen induces ERα-mediated transrepressive effects on BSEP, which merits further investigation (47). It is worth noting that while repression of BSEP expression alone is sufficient to reduce bile acid elimination in the liver and potentially leads to drug-induced cholestasis, it cannot be generalized as the mechanism for all cholestatic BSEP non-inhibitors. Given that the overall bile acid homeostasis in the liver is coordinated by its tightly controlled bio-synthesis and transport, the cholestatic effects of these BSEP non-inhibitors could be potentially contributed by compensatory mechanisms involving bile acid elimination in the liver, such as canalicular efflux transporters MRP2 and P-gp as well as sinusoidal efflux transporters MRP3, MRP4 and OSTα-OSTβ (50).
CONCLUSION
In conclusion, BSEP inhibition cannot fully explain drug-induced cholestasis; hence, we directed our work at identifying alternate mechanisms. In the current study, we found that metformin and tamoxifen, two cholestatic BSEP non-inhibitors, alter BSEP function through repression of BSEP expression without directly inhibiting its activity, leading to decreased bile acid efflux from the liver. Our results suggest transcriptional BSEP repression represents an overlooked mechanism underlying drug-induced cholestasis.
Acknowledgements and Disclosures.
The study was partly supported by NIH grants R01 GM121550 and F31 DK105750, and the Food and Drug Administration (FDA) Maryland’s Center of Excellence in Regulatory Science and Innovation (M-CERSI) initiative (1U01FD005946). Tao Hu was supported by the Oak Ridge Institute for Science and Education (ORISE) postdoctoral fellowship, FDA. This work was supported in part by Merit Review Award # BX002129 from the United States (U.S.) Department of Veterans Affairs Biomedical Laboratory Research and Development Program.
ABBREVIATIONS
- AMPK
AMP-activated protein kinase
- BEI
Biliary excretion index.
- BSEP
Bile salt export pump
- DILI
Drug-induced liver injury
- DIC
Drug-induced cholestasis
- HPH
Human primary hepatocytes
- MRP
Multidrug resistance-associated protein
- OST
Organic solute transporter
- P-gp
P-glycoprotein
Footnotes
The authors disclose that there is no potential conflict of interest. The contents do not represent the views of the FDA, the U.S. Department of Veterans Affairs or the United States Government.
Publisher’s Note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
REFERENCES
- 1.Oorts M, Baze A, Bachellier P, Heyd B, Zacharias T, Annaert P, Richert L. Drug-induced cholestasis risk assessment in sandwich-cultured human hepatocytes. Toxicol In Vitro. 2016. [DOI] [PubMed] [Google Scholar]
- 2.Vatakuti S, Pennings JL, Gore E, Olinga P, Groothuis GM. Classification of Cholestatic and necrotic Hepatotoxicants using Transcriptomics on human precision-cut liver slices. Chem Res Toxicol. 2016;29(3):342–51. [DOI] [PubMed] [Google Scholar]
- 3.Schadt HS, Wolf A, Pognan F, Chibout SD, Merz M, Kullak-Ublick GA. Bile acids in drug induced liver injury: key players and surrogate markers. Clin Res Hepatol Gastroenterol 2016. [DOI] [PubMed] [Google Scholar]
- 4.Noor F A shift in paradigm towards human biology-based systems for cholestatic-liver diseases. J Physiol. 2015;593(23):5043–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qiu X, Zhang Y, Liu T, Shen H, Xiao Y, Bourner MJ, et al. Disruption of BSEP function in HepaRG cells alters bile acid disposition and is a susceptive factor to drug-induced Cholestatic injury. Mol Pharm. 2016;13(4):1206–16. [DOI] [PubMed] [Google Scholar]
- 6.Chatterjee S, Richert L, Augustijns P, Annaert P. Hepatocyte-based in vitro model for assessment of drug-induced cholestasis. Toxicol Appl Pharmacol. 2014;274(1):124–36. [DOI] [PubMed] [Google Scholar]
- 7.Namas R, Marquardt A. Case report and literature review: Quinacrine-induced Cholestatic hepatitis in undifferentiated connective tissue disease. J Rheumatol. 2015;42(7):1354–5. [DOI] [PubMed] [Google Scholar]
- 8.Saadi T, Waterman M, Yassin H, Baruch Y. Metformin-induced mixed hepatocellular and cholestatic hepatic injury: case report and literature review. Int J Gen Med. 2013;6:703–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kotsampasakou E, Ecker GF. Predicting drug-induced cholestasis with the help of hepatic transporters-an in Silico modeling approach. J Chem Inf Model. 2017;57(3):608–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parmentier C, Couttet P, Wolf A, Zaccharias T, Heyd B, Bachellier P, Uteng M, Richert L. Evaluation of transcriptomic signature as a valuable tool to study drug-induced cholestasis in primary human hepatocytes. Arch Toxicol. 2017. [DOI] [PubMed] [Google Scholar]
- 11.Gerloff T, Stieger B, Hagenbuch B, Madon J, Landmann L, Roth J, et al. The sister of P-glycoprotein represents the canalicular bile salt export pump of mammalian liver. J Biol Chem. 1998;273(16): 10046–50. [DOI] [PubMed] [Google Scholar]
- 12.Strautnieks SS, Bull LN, Knisely AS, Kocoshis SA, Dahl N, Arnell H, et al. A gene encoding a liver-specific ABC transporter is mutated in progressive familial intrahepatic cholestasis. Nat Genet. 1998;20(3):233–8. [DOI] [PubMed] [Google Scholar]
- 13.Marroquin LD, Bonin PD, Keefer J, Schroeter T. Assessment of Bile Salt Export Pump (BSEP) Inhibition in Membrane Vesicles Using Radioactive and LC/MS-Based Detection Methods. Curr Protoc Toxicol. 2017;71:14 14 11–14 14 20. [DOI] [PubMed] [Google Scholar]
- 14.Montanari F, Pinto M, Khunweeraphong N, Wlcek K, Sohail MI, Noeske T, et al. Flagging drugs that inhibit the bile Salt export pump. Mol Pharm. 2016;13(1):163–71. [DOI] [PubMed] [Google Scholar]
- 15.Giacomini KM, Huang SM. Transporters in drug development and clinical pharmacology. Clin Pharmacol Ther. 2013;94(1):3–9. [DOI] [PubMed] [Google Scholar]
- 16.Tweedie D, Polli JW, Berglund EG, Huang SM, Zhang L, Poirier A, et al. Transporter studies in drug development: experience to date and follow-up on decision trees from the international transporter consortium. Clin Pharmacol Ther. 2013;94(1):113–25. [DOI] [PubMed] [Google Scholar]
- 17.FDA. Drug Interaction Studies- study design, data analysis, implications for dosing, and labeling recommendations In: Clinical Pharmacology CfDEaR, FDA, U.S: Department of Health and Human Services, editor.; 2012. [Google Scholar]
- 18.Kock K, Ferslew BC, Netterberg I, Yang K, Urban TJ, Swaan PW, et al. Risk factors for development of cholestatic drug-induced liver injury: inhibition of hepatic basolateral bile acid transporters multidrug resistance-associated proteins 3 and 4. Drug Metab Dispos. 2014;42(4):665–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zollner G, Thueringer A, Lackner C, Fickert P, Trauner M. Alterations of canalicular ATP-binding cassette transporter expression in drug-induced liver injury. Digestion. 2014;90(2):81–8. [DOI] [PubMed] [Google Scholar]
- 20.Garzel B, Yang H, Zhang L, Huang SM, Polli JE, Wang H. The role of bile salt export pump gene repression in drug-induced cholestatic liver toxicity. Drug Metab Dispos. 2014;42(3):318–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Desilets DJ, Shorr AF, Moran KA, Holtzmuller KC. Cholestatic jaundice associated with the use of metformin. Am J Gastroenterol. 2001;96(7):2257–8. [DOI] [PubMed] [Google Scholar]
- 22.Nammour FE, Fayad NF, Peikin SR. Metformin-induced cholestatic hepatitis. Endocr Pract. 2003;9(4):307–9. [DOI] [PubMed] [Google Scholar]
- 23.Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, et al. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 2001;108(8):1167–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Faucette SR, Sueyoshi T, Smith CM, Negishi M, Lecluyse EL, Wang H. Differential regulation of hepatic CYP2B6 and CYP3A4 genes by constitutive androstane receptor but not pregnane X receptor. J Pharmacol Exp Ther. 2006;317(3):1200–9. [DOI] [PubMed] [Google Scholar]
- 25.Li L, Chen T, Stanton JD, Sueyoshi T, Negishi M, Wang H. The peripheral benzodiazepine receptor ligand 1-(2-Chlorophenyl-methylpropyl)-3-isoquinoline-carboxamide is a novel antagonist of human constitutive Androstane receptor. Mol Pharmacol. 2008;74(2):443–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Swift B, Pfeifer ND, Brouwer KLR. Sandwich-cultured hepatocytes: an in vitro model to evaluate hepatobiliary transporter-based drug interactions and hepatotoxicity. Drug Metab Rev. 2010;42(3):446–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Nygaard EB, Vienberg SG, Orskov C, Hansen HS, Andersen B. Metformin stimulates FGF21 expression in primary hepatocytes. Exp Diabetes Res. 2012;2012:465282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wanninger J, Neumeier M, Weigert J, Liebisch G, Weiss TS, S c h a f f l e r A , e t a l . M e t f o r m i n r e d u c e s c e l l u l a r lysophosphatidylcholine and thereby may lower apolipoprotein B secretion in primary human hepatocytes. Biochim Biophys Acta. 2008;1781(6–7):321–5. [DOI] [PubMed] [Google Scholar]
- 29.Zarei M, Barroso E, Leiva R, Barniol-Xicota M, Pujol E, Escolano C, et al. Heme-regulated eIF2alpha kinase modulates hepatic FGF21 and is activated by PPARbeta/delta deficiency. Diabetes. 2016;65(10):3185–99. [DOI] [PubMed] [Google Scholar]
- 30.Chen M, Hong H, Fang H, Kelly R, Zhou G, Borlak J, et al. Quantitative structure-activity relationship models for predicting drug-induced liver injury based on FDA-approved drug labeling annotation and using a large collection of drugs. Toxicol Sci. 2013;136(1):242–9. [DOI] [PubMed] [Google Scholar]
- 31.Ritschel T, Hermans SM, Schreurs M, van den Heuvel JJ, Koenderink JB, Greupink R, et al. In silico identification and in vitro validation of potential cholestatic compounds through 3D ligand-based pharmacophore modeling of BSEP inhibitors. Chem Res Toxicol. 2014;27(5):873–81. [DOI] [PubMed] [Google Scholar]
- 32.Morgan RE, Trauner M, van Staden CJ, Lee PH, Ramachandran B, Eschenberg M, et al. Interference with bile salt export pump function is a susceptibility factor for human liver injury in drug development. Toxicol Sci. 2010;118(2):485–500. [DOI] [PubMed] [Google Scholar]
- 33.Chen M, Vijay V, Shi Q, Liu Z, Fang H, Tong W. FDA-approved drug labeling for the study of drug-induced liver injury. Drug Discov Today. 2011;16(15–16):697–703. [DOI] [PubMed] [Google Scholar]
- 34.Chen Q, Yang X, Zhang H, Kong X, Yao L, Cui X, et al. Metformin impairs systemic bile acid homeostasis through regulating SIRT1 protein levels. Biochim Biophys Acta. 2017;1864(1): 101–12. [DOI] [PubMed] [Google Scholar]
- 35.Pedersen JM, Matsson P, Bergstrom CA, Hoogstraate J, Noren A, LeCluyse EL, et al. Early identification of clinically relevant drug interactions with the human bile salt export pump (BSEP/ABCB11). Toxicol Sci. 2013;136(2):328–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kis E, Ioja E, Nagy T, Szente L, Heredi-Szabo K, Krajcsi P. Effect of membrane cholesterol on BSEP/Bsep activity: species specificity studies for substrates and inhibitors. Drug Metab Dispos. 2009;37(9):1878–86. [DOI] [PubMed] [Google Scholar]
- 37.Mita S, Suzuki H, Akita H, Hayashi H, Onuki R, Hofmann AF, et al. Vectorial transport of unconjugated and conjugated bile salts by monolayers of LLC-PK1 cells doubly transfected with human NTCP and BSEP or with rat Ntcp and Bsep. Am J Physiol Gastrointest Liver Physiol. 2006;290(3):G550–6. [DOI] [PubMed] [Google Scholar]
- 38.Wolf KK, Vora S, Webster LO, Generaux GT, Polli JW, Brouwer KL. Use of cassette dosing in sandwich-cultured rat and human hepatocytes to identify drugs that inhibit bile acid transport. Toxicol in Vitro. 2010;24(1):297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Foretz M, Hebrard S, Leclerc J, Zarrinpashneh E, Soty M, Mithieux G, et al. Metformin inhibits hepatic gluconeogenesis in mice independently of the LKB1/AMPK pathway via a decrease in hepatic energy state. J Clin Invest. 2010;120(7):2355–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chen SC, Brooks R, Houskeeper J, Bremner SK, Dunlop J, Viollet B, et al. Metformin suppresses adipogenesis through both AMP-activated protein kinase (AMPK)-dependent and AMPK-independent mechanisms. Mol Cell Endocrinol. 2017;440:57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Molusky MM, Hsieh J, Lee SX, Ramakrishnan R, Tascau L, Haeusler RA, et al. Metformin and AMP kinase activation increase expression of the sterol transporters ABCG5/8 (ATP-binding cassette transporter G5/G8) with potential Antiatherogenic consequences. Arterioscler Thromb Vasc Biol. 2018;38(7):1493–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chopra AR, Kommagani R, Saha P, Louet JF, Salazar C, Song J, et al. Cellular energy depletion resets whole-body energy by promoting coactivator-mediated dietary fuel absorption. Cell Metab. 2011;13(1):35–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yang K, Pfeifer ND, Kock K, Brouwer KL. Species differences in hepatobiliary disposition of taurocholic acid in human and rat sandwich-cultured hepatocytes: implications for drug-induced liver injury. J Pharmacol Exp Ther. 2015;353(2):415–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Song X, Chen Y, Valanejad L, Kaimal R, Yan B, Stoner M, et al. Mechanistic insights into isoform-dependent and species-specific regulation of bile salt export pump by farnesoid X receptor. J Lipid Res. 2013;54(11):3030–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kostrubsky VE, Strom SC, Hanson J, Urda E, Rose K, Burliegh J, et al. Evaluation of hepatotoxic potential of drugs by inhibition of bile-acid transport in cultured primary human hepatocytes and intact rats. Toxicol Sci. 2003;76(1):220–8. [DOI] [PubMed] [Google Scholar]
- 46.Goodsell DS. The molecular perspective: tamoxifen and the estrogen receptor. Oncologist. 2002;7(2):163–4. [DOI] [PubMed] [Google Scholar]
- 47.Chen Y, Vasilenko A, Song X, Valanejad L, Verma R, You S, et al. Estrogen and estrogen receptor-alpha-mediated Transrepression of bile Salt export pump. Mol Endocrinol. 2015;29(4):613–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song X, Vasilenko A, Chen Y, Valanejad L, Verma R, Yan B, et al. Transcriptional dynamics of bile salt export pump during pregnancy: mechanisms and implications in intrahepatic cholestasis of pregnancy. Hepatology. 2014;60(6):1993–2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yoshikawa Y, Miyashita T, Higuchi S, Tsuneyama K, Endo S, Tsukui T, et al. Mechanisms of the hepatoprotective effects of tamoxifen against drug-induced and chemical-induced acute liver injuries. Toxicol Appl Pharmacol. 2012;264(1):42–50. [DOI] [PubMed] [Google Scholar]
- 50.Dawson PA, Lan T, Rao A. Bile acid transporters. J Lipid Res. 2009;50(12):2340–57. [DOI] [PMC free article] [PubMed] [Google Scholar]