

Abstract
Mediated fuel cells are electrochemical devices that produce power in a manner similar to that of conventional proton exchange membrane fuel cells (PEMFCs). They differ from PEMFCs in their use of redox mediators dissolved in liquid electrolyte to conduct oxidation of the fuel or reduction of the oxidant, typically O2, in bulk solution. The mediators transport electrons (and often protons) between the electrode and the catalysts or chemical reagents in solution. This strategy can help overcome many of the challenges associated with conventional fuel cells, including managing complex multi-phase reactions (as in O2 reduction) or the use of challenging or heterogeneous fuels, such as hydrocarbons, polyols and biomass. Mediators are also commonly used in enzymatic fuel cells, where direct electron transfer from the electrode to the enzymatic active site can be slow. This review provides a comprehensive survey of historical and recent mediated fuel cell efforts, including applications using chemical and enzymatic catalysts.
Graphical Abstract

1. Introduction
1.1. Proton-exchange-membrane fuel cells (PEMFCs) and redox flow batteries (RFBs): Context for mediated fuel cells (MedFCs).
Growing worldwide energy demands continue to motivate the exploration and development of diverse energy storage1 and conversion2 technologies that could support broader use of renewable energy sources, such as solar, wind, hydroelectric, and biomass.3 Electrochemical technologies are a major focus of these efforts, with prominent examples including solid-state4–7 and redox flow batteries,8,9 supercapacitors,10,11 and (photo)electrochemical production of chemicals, ranging from hydrogen (H2) to carbon-based molecules (formic acid, syn gas, MeOH, among others).12,13 Chemicals produced from the latter processes may be stored or transported to other locations for power generation, ideally with fuel cells to leverage their higher energy efficiency relative to conventional combustion-based power generation.14 This review article surveys historical and recent advances in mediated (or “indirect”) electrochemical processes for power generation. These processes are conceptually related to proton exchange membrane fuel cells (PEMFCs) and redox flow batteries (RFBs) (Figure 1), while incorporating features distinct from these established technologies.
Figure 1.

General schematic of an A) PEM fuel cell, B) flow battery in discharge mode with [A] representing the anodic (e.g. low potential) redox couple and [C] representing the cathodic (e.g. high potential) redox couple, and C) mediated fuel cell with mediators for both anode and cathode.
PEMFCs are the most common class of fuel cells for low-temperature power generation.15 These devices convert energy from chemical bonds into electricity by oxidizing a chemical fuel at the anode and reducing an oxidant at the cathode. Conventional PEMFCs use H2 as the fuel16 and O2 as the oxidant. The electrons from H2 are transported from the electrode through the electrical circuit, while the protons are transported through a membrane electrolyte, consisting of a sulfonated fluoropolymer (e.g., Nafion®), to the cathode where they combine with O2 and the electrons to produce water (Figure 1A). A related class of fuel cells, anion exchange membrane fuel cells (AEMFCs), utilize membrane electrolytes that conduct anions, such as Cl− or OH−, and have been the focus of growing interest.17
Water management is a key challenge in conventional PEMFCs.18 Water must be present within the ionic polymer membrane to support efficient proton transport from the anode to the cathode,19 and this requirement is met by using humidified sources of H2 and O2 gases. Meanwhile, the water generated at the cathode from O2 reduction must be removed to avoid flooding of the catalyst. The issues related to water management limit the temperature range available for effective operation of PEMFCs (typical operation ≤ 100 °C). Membrane-electrode assemblies20 and bipolar plates21 have been engineered to address these challenges, while ensuring effective delivery of gases to the catalyst surface, efficient coupling of the catalytic reactions of H2 and O2 with the proton- and electron-transport processes, and precise management of water. Fuel cell technologies continue to improve, with recent advances enabling access to current and power densities of ≥ 2.5 A/cm2 and 1.2 W/cm2, respectively.22
The requirement for high loadings of Pt catalyst at the cathode to achieve optimal performance represents one of the most significant impediments to conventional PEMFCs.23 Pt catalysts are also used at the anode, but the higher catalytic rates for H2 oxidation relative to O2 reduction mean that much less catalyst is required at the anode. Advances have been made in the development of non-platinum group metal (non-PGM) catalysts for O2 reduction;24–26 however, these catalysts typically have lower intrinsic activity than Pt and, therefore, require higher catalyst loadings to achieve the similar current densities. The higher catalyst loading introduces additional limitations associated with the increased electrical resistance from the thicker catalyst layer and restricted mass transport of O2 and water through the electrode. Consequently, commercial applications of non-PGM cathode catalysts have thus far been restricted to low power-density applications, in which the complications arising from higher catalyst loadings are less problematic.27,28
Alongside the development of PEMFCs that use H2 as a fuel, considerable effort has been directed toward the development of fuel cells capable of using liquid fuels. Such devices avoid challenges associated with the storage, transport, and distribution of H2. Direct methanol fuel cells (DMFCs),29 which oxidize MeOH (supplied as a concentrated aqueous solution) to CO2 at the anode, are among the most prominent applications of liquid fuel cells. The lower rates of MeOH oxidation relative to H2 oxidation, however, leads to considerably lower power densities in DMFCs (< 120 mW/cm2).30 The use of fuels more complex than MeOH, such as sugars or other biomass-derived molecules, and limitations on the practical concentrations of these fuels in the aqueous solutions delivered to the anode, can further constrain catalytic rates and power densities in liquid fuel cells.31 Liquid fuel cells must be operated under conditions that avoid cathode flooding, which can arise from transport of water from the fuel solution through the membrane as protons are transported from the anode to the cathode. Complications of the type described here account for the limited commercial application of PEMFCs with fuels other than hydrogen.
Redox flow batteries (RFBs) represent the second class of electrochemical devices directly relevant to the subject matter of this review article. RFBs store electrical energy by transferring electrons to and from molecules or ions dissolved in solution (Figure 1B). Typical systems feature two tanks of redox couples, one containing a cathodic species that has a high reduction potential and the other with an anodic, low-potential species. Electrical energy is stored and generated by flowing the redox couple solutions through the cathode and anode compartments, which are separated by a semi-permeable membrane that enables selective transfer of protons or other ions while preventing mixing of the mediators. The underlying concepts of redox flow batteries were first demonstrated in the late 1800s,32 with significant technological development beginning in the 1960s.33 Commercial installations have emerged in recent years.34 Examples of the redox couples at the anode/cathode used in commercial systems include Cr/Fe, Zn/Br, and V/V.8 The systems using Fe, Cr, Zn and Br species benefit from the low cost of the chemicals, but the all-vanadium redox flow batteries exhibit the longest lifetimes and are the most widely used. Challenges associated with these technologies include the high toxicity and corrosivity of the electrolyte (particularly for V- and Br-based systems), the limited number of redox couples that operate near the limiting potentials for H+/H2O reduction and H2O/OH− oxidation for aqueous electrolytes, and the limited material supply and high cost of V. All-organic redox species have been the focus of much recent attention as a means to address these limitations. Examples include derivatives of quinones, nitroxyls, and viologens (Figure 2).9,35 The synthetic modularity of these molecules, which allows their redox potentials and other physical properties (e.g., solubility, stability) to be tuned, greatly expands the scope of possible compounds that may be considered. Water-soluble redox couples are especially appealing because water is low-cost solvent with inherently low solvent resistivity;36–38 however, non-aqueous RFBs have also been the focus of major research efforts.39
Figure 2.
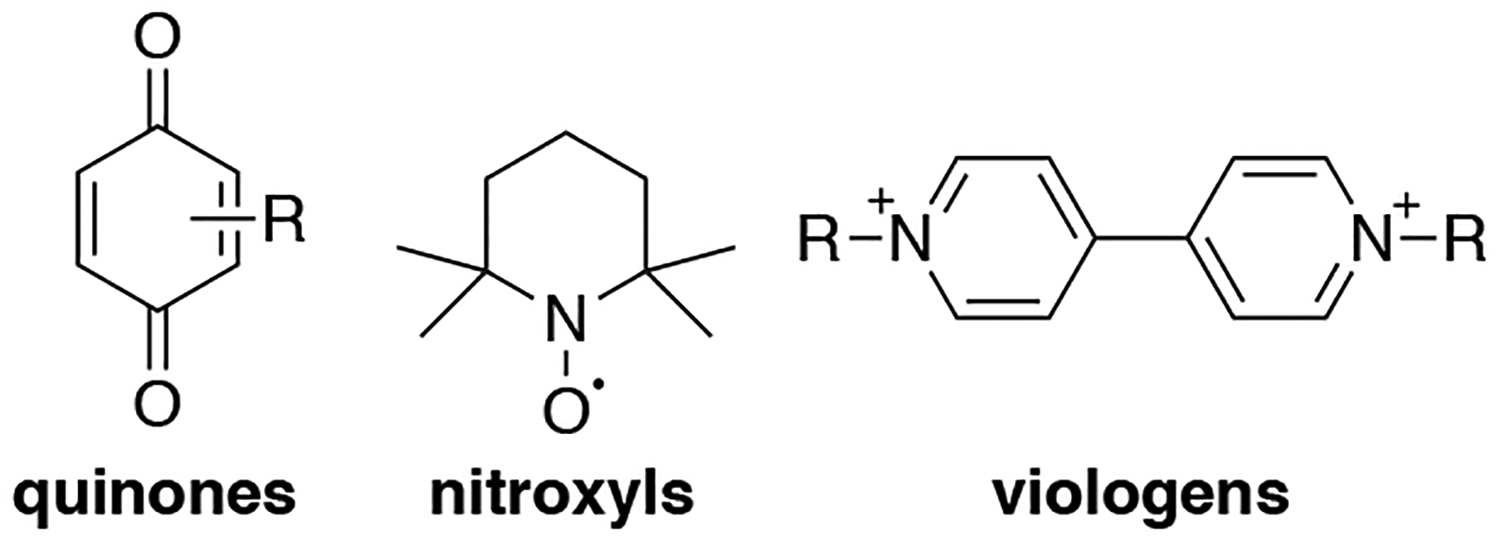
Structures of representative organic mediators examined for redox flow battery applications.
1.2. Mediated fuel cells (MedFCs): Overview and opportunities.
The above discussion of PEMFC and RFB technologies provides the relevant context for the introduction of mediated fuel cells (MedFCs, Figure 1C), which are the focus of this review article. The latter devices feature dissolved mediators analogous to the mediators used in RFBs; however, catalytic regeneration of the mediators in an off-electrode compartment enables sustained production of electricity analogous to the operation of PEMFCs. Mediator regeneration is achieved by supplying a fuel to reduce the low-potential mediator and O2 to oxidize the high potential mediator, typically employing a catalyst for both processes. Mediated fuel cell systems are distinct from RFBs because they can operate continuously, as long as the supplies of fuel and oxidant are maintained.
Several factors impact the utility of redox mediators for use in mediated fuel cells. First, the redox potential of the mediator should be as close as possible to the formal potential of the fuel or the oxidant. For a “conventional” system using H2 as the fuel and O2 as the oxidant, the anode mediator should have a redox potential near 0.0 V vs. RHE (Reversible Hydrogen Electrode; i.e., adjusting for the pH of the system), and the cathodic mediator a redox potential near 1.23 V vs. RHE. Additionally, the redox mediator should display fast electron transfer kinetics, to minimize any overpotentials caused by kinetic losses in the system, it needs to be stable in both oxidized and reduced states to enable long-term performance, and it must be highly soluble in aqueous solution for the cell to maximize the current densities.
MedFCs have several potential advantages over conventional fuel cells that warrant their consideration for further development. These features include the following:
1). Enabling the use of “challenging” fuels.
By moving fuel oxidation away from the anode, a greater number of fuel options could become available, and more diverse catalysts may be considered. For example, the use of H2 feeds containing carbon monoxide, sulfur-, or nitrogen-based contaminants has been demonstrated,40 and this advantage was one of the original motivations for investigating MedFCs.41 Another advantage is that fuels that are too complicated to oxidize at an electrode under mild conditions (<150 °C) may be considered. Examples of these alternatives include alcohols more complicated than MeOH and heterogeneous fuel sources, such coal or biomass-derived cellulose or lignin, which are challenging fuels for direct activation at an electrode.
2). Overcoming challenges with the oxygen reduction reaction (ORR).
A major motivation in previous studies of mediated fuel cells has been the replacement of Pt, which is required in relatively high loading in conventional PEM cathodes, with non-PGM alternatives.42 The high loadings of Pt are necessary to achieve sufficiently high activity within the narrow spatial dimension at the gas-diffusion electrodes. By moving the catalytic O2 reduction process into an off-electrode compartment, the ORR may be optimized independently of the electrochemistry. The lower intrinsic activity of non-PGM catalysts relative to Pt is less problematic in this configuration. For example, mass-transport restrictions are significantly reduced when the catalyst does not need to be in direct contact with the electrode and membrane, and higher mass loadings of the non-PGM catalyst may be integrated in an off-electrode reactor in a manner that does not impede system performance. The electrochemical reaction of the mediator features a simpler two-phase (liquid/solid) system at the electrode, rather than the more complicated three-phase (gas/liquid/solid) system present in conventional fuel cells. While there is still a three-phase reaction occurring at the off-electrode catalyst, many of the engineering and materials constraints are relaxed. For example, it is not essential to use a conductive catalyst support, and there are fewer limitations on the amount of catalyst that can be used, allowing higher loadings of less active and less expensive catalysts than Pt to be used.
3). Opportunities to improve overall system performance.
At first glance, mediated fuel cells appear to increase the complexity of a fuel cell stack, due to the requirement for extra plumbing and pumps to operate the mediator-regeneration loop. In fact, however, they can simplify the balance of plant accessories. PEMFCs are typically limited to operating at or below 100 °C and require humidification of the gases to avoid dehydration of the membrane. Since mediated fuel cells use aqueous mediator solutions, the membrane remains well-hydrated and gas humidification is unnecessary. Additional benefits arise from using aqueous solutions, which can absorb heat generated from the oxidation and reduction reactions, thereby facilitating temperature control of the fuel cell stack. Finally, mediated cathode systems are ideally suited for pairing with liquid fuels, including both conventional and mediated anode processes, because they are not susceptible to the flooding from water convection that can occur with gas-diffusion cathodes in PEMFCs.
4). Reduced performance losses from activation polarization.
Activation polarization is the loss in cell voltage at low current densities due to the kinetic barriers for electron transfer to the species to be oxidized or reduced. The particularly high kinetic barriers associated with catalytic reduction of O2 leads to substantial losses in the cell potential when O2 is directly reduced at an electrode. Similar issues arise at the anode, when using fuels more complex than H2, such as methanol. Redox mediators, on the other hand, may be selected to have fast electrochemical kinetics and thereby minimize activation polarization losses.
1.3. Scope of coverage and notes on nomenclature and symbolic notation used in this review.
Redox mediators have been employed in a wide range of electrochemical devices and applications, many of which have been the subject of previous review. Important examples include electrochemical synthesis of organic and inorganic chemicals,43–46 in addition to diverse energy-based technologies beyond flow batteries,8,9 including dye-sensitized solar cells,47,48 solid-state batteries (e.g., “redox targeting”),49,50 decoupled water splitting,51 electrochemical capacitors,52 and solar flow batteries.53 This review covers mediated fuel cells (MedFCs) that feature a mediated cathode for O2 reduction, a mediated anode for H2, alcohol, or biomass oxidation, or a doubly mediated system, wherein mediators are used for both half-reactions of the fuel cell. Some aspects of these reactions have been reviewed previously;41,42,54 however, significant advances in recent years, together with the partial coverage of these previous reviews, provided motivation to prepare this comprehensive survey. Only systems that are designed or intended to produce power (galvanic cells) are covered in depth, though reference is occasionally made to relevant electrolysis systems. Most of the examples consist of full fuel cells, but relevant half-cell studies are also discussed. Enzymatic fuel cells55–57 using soluble mediators are included, but microbial fuel cells58,59 are not presented. While microbial fuel cells feature certain advantages, including the capability to produce their own mediators and potential for genetic engineeringfor enhanced performance,60 their added complexity makes it is difficult to compare them directly to the other systems.
Historically, mediated fuel cells have been given the name “chemically regenerable redox fuel cells”, or CRRFCs,41,42,54,61–64 to distinguish them from “redox fuel cells”, which are now typically called redox flow batteries (RFBs). With the shift in the flow battery nomenclature, we have chosen to adopt the simplified term “mediated fuel cell” or “MedFC” for these systems (avoiding the term “MFC”, which is commonly employed for microbial fuel cells).
To simplify the notation used to describe mediated fuel cell systems, we have adopted the following notation: fuel|anode catalyst|anode mediator||cathode mediator|cathode catalyst|oxidant, whereby the double bar (||) refers to the membrane separating the anode and anode compartments and the single bar (|) separates the different components within the mediated anode or cathode systems. Certain studies only investigated one of the half-cell systems, or a system was conducted in the absence of a mediator or catalyst. In such cases, certain components are omitted from the symbolic notation. The distinction between a “mediator” and a “catalyst” is not always well defined in multi-component electrochemical mediator systems. We have attempted to be consistent in use of the term “mediator” for the major redox-active species that undergoes electron-transfer at the electrode, and the term “catalyst” for the species that undergoes the redox reaction with the fuel or oxidant in a redox cascade. Ambiguities can arise, for example, in single-component mediator systems in which the “mediator” also serves as the “catalyst”. A table containing information for all reported mediated fuel cells using chemical catalysts is given in Appendix I. All reports of mediated fuel cells using enzymatic catalysts is provided in Section 3.2.
2. The Cathode: Oxygen Reduction
The reduction of O2 is the predominant cathodic reaction for fuel cell technology, owing to the availability of O2 and formation of water as a benign product. State-of-the-art catalysts in PEMFCs and DMFCs are composed of Pt or Pt alloys that exhibit the best combination of high activity, low overpotential, long-term stability, and selectivity for 4 e− reduction of O2 to H2O.65,66 While optimal performance is achieved with these Pt-based catalysts, their high costs continues to be a limiting factor in PEMFC commercialization. The cost of Pt has been forecasted to account for over 40% of the stack cost.67 Eliminating or decreasing the loading of Pt or Pt-group metals is a high priority, and heterogeneous electrocatalysts such as Pt-alloys and non-PGM electrocatalysts, including metal-free carbon-based materials, have been developed.68,69 The first commercial PEMFC system utilizing non-PGM catalysts was announced in 2017.28 Nonetheless, no catalysts have thus far achieved the activity and stability metrics of Pt or Pt alloys, and alternate solutions continue to be the focus of active study. Mediated O2 reduction was the focus of considerable historical investigation, and new advances have emerged in recent years. The following sections survey the different mediators that have been reported for mediated ORR, including historical and recent examples, and systems implemented in fuel cell devices and fundamental studies.
2.1. Br−/Br2 mediator
The bromide/bromine redox couple has been widely used in flow battery research due to its high redox potential (1.06 V vs. NHE), fast electrode kinetics, and high solubility of the relevant species in different redox states. The potential is within 200 mV of the thermodynamic potential for O2 reduction to H2O and, therefore, could provide a low-overpotential strategy for mediated ORR. Fuel cells reported with bromine-based mediators for the ORR are shown in Table 1.
Table 1.
Reported systems using Br mediators for O2 reduction
The first report of a MedFC utilizing bromine-based cathode mediators was published in 1955 by Posner.70 In this work, other cathodic mediators were examined, but they exhibited low cell potentials. A variety of catalysts and mediators were screened, and the use of bromine in combination with NO2 showed promise (Figure 3). The anode contained chars, coke, or coal as the fuel and a variety of metal ions were tested as anodic mediators. Tests were also performed without NOx/O2-based regeneration, and the results showed that Br2 at the cathode shows minimal polarization (<0.05 V) up to a cell current density of >50 A/ft2 (i.e., ~54 mA/cm2) with 1.2 N Br2 and 0.7 N HBr in 3 N HCl. Challenges with long-term stability of the cathode system was acknowledged, but >8 h of sustained activity was demonstrated.
Figure 3.

Redox sequence for bromine-mediated cathode with a NOx-based catalyst (||Br2|NOx|O2). For relevant studies, see references 40, 70–73.
In the same year, Posner and Merton-Bingham studied the mechanism of NOx-catalyzed autoxidation of HBr.71 The authors proposed a 6-step sequence (Figure 4A), with the slow step involving the reaction of NOBr in solution with an unknown, gas-phase species resulting from the reaction of HNO2 and HBr (species “X” in Figure 4A). Later studies by Reneke noted reproducibility problems with the analytical techniques for bromine quantification employed by Posner and Merton-Bingham.72 Reneke also suggested that the mechanism of Posner and Merton-Bingham was unnecessarily complex and proposed an alternative 3-step mechanism (Figure 4B). Calculations from Reneke’s report propose that a 10-kW fuel cell using this system to regenerate Br2 would require a regeneration tower over 130 ft in height and 1.5 ft in diameter. These dimensions likely prevent large-scale implementation of this technology; however, the report from Posner, as well as subsequent studies, show that fuel cell operation is possible.
Figure 4.

Proposed mechanisms for HBr oxidation to Br2 by NOx catalysts.
In 1959, Carson and Feldman from the General Electric Company described a 100 W fuel cell stack, using seven cells connected in series.40 The cathodic mediator was Br2/Br−, again using nitric oxide to promote aerobic regeneration of Br2. The fuel was H2, which reduced a TiIV mediator to TiIII (i.e., TiO2+/Ti3+) using Pd-black as an off-electrode catalyst (H2|Pt|TiO2+||Br2|NOx|O2) (see Section 3.1.2 for discussion of the anode performance). The cells were operated at 80 °C, and an open-circuit potential of around 0.95 V was obtained. Continuous operation of the system was demonstrated for over 16 days, but the regeneration of the mediators was not sufficient to operate the system at its peak activity for longer than a few minutes. Additionally, the overall efficiency was low, due to high internal resistance of the cell, and the overall device was quite large, due to a complicated liquid delivery system to each cell. Carson and Feldman noted that, while this system demonstrated technical feasibility, several improvements would be necessary for commercialization, including the use of non-volatile mediators/catalysts on the cathode.
In 1964, Juda, at Ionics, Inc., patented a fuel cell using bromine-based mediators.73 H2 oxidation at a heterogeneous Pt electrode was used as the anodic reaction. The cathode compartment contained a solution of KBr, HBr and Br2 with NO2 as the catalyst. An open circuit potential of 0.98 V and an operating potential of 0.7 V at 10 mA/cm2 at room temperature were obtained. Juda compared these results to a system using similar loadings of KI, HI, and I2 without a catalyst, which generated an open circuit potential of 0.5 V and an operating potential of 0.3 V at a current density of 10 mA/cm2. No sustained operation of these cells was reported.
2.2. Vanadyl and vanadium-containing polyoxometalate cathodic mediators
The VO2+/VO2+ redox couple is the most widely studied cathodic mediator in mediated fuel cells. Some major advantages of this system include the high redox potential (~1.0 V vs. NHE), its non-volatile nature, good stability, and reasonably high solubility (> 2 M). These features underlie the widespread implementation of the same mediator system for redox flow battery applications.74 A catalyst is required to increase the rate of VO2+ reoxidation by O2. Both NOx-based catalysts and heteropolyacids (HPAs; also called polyoxometalates, POMs) have been used for this purpose. Table 2 lists the various implementations reported for cathodic vanadium mediators.
Table 2.
Reported fuel cell systems using V-based mediators for O2 reduction.
| fuel | catalyst | mediator | mediator | catalyst | ref |
|---|---|---|---|---|---|
| H2 | Pt | – | VO2+/VO2+ | HCl | 75 |
| – | – | V2+/V3+ | NOx | 79 | |
| H2 | Pt | Ti3+/TiO2+ | 80 | ||
| CH4, ethylene glycol, coal | Pt | Fe2+/Fe3+ | 82–84 | ||
| biomass | – | Fe2+/Fe3+ | 85, 86 | ||
| sugars | Pt | V3+/VO2+ | Pt | 81 | |
| H2 | Pt | H4SiW12O40 | POM | 76–78 | |
| Pt | – | V-containing POM | 63, 95–102,104 | ||
| Pt/C | AQ | 105 | |||
| sugars | POM | Fe2+/Fe3+ | 110 | ||
| biomass | POM | 107, 111, 112 | |||
| alcohols | 108 | ||||
| lignin | 113 | ||||
| coal | 109 | ||||
The first example of VO2+/VO2+ mediators for electrocatalytic O2 reduction was patented by Schaefer and Kordesch at Union Carbide Corporation in 1966.75 While details of the invention are somewhat limited, they described a cell using an H2 gas anode and NaVO3 dissolved in a strong HCl electrolyte (19–39% by weight). When the cathode was supplied with O2, it was operated for over 24 h at 80 °C and sustained a potential between 1.5 and 1.7 V vs. cadmium (conversion to a standardized reference was not given) at a current density of 50 mA/cm2. An HCl electrolyte was necessary for regeneration; when H2SO4 was used as the electrolyte, addition of HBr and HNO3 was necessary to achieve sustained activity.
Oei, working at Ford, reported the use of a VO2+/VO2+ mediated cathode. The initial report investigated the performance of various electrolytes in a RFB context (i.e., no regeneration of either redox couple by O2 or a fuel): VO2+/VO2+ as the cathodic redox couple and Sn2+/Sn4+, Fe2+/Fe3+, or Cu/Cu2+ as the anodic redox couples.63 Initial tests optimized the flow-field configuration and electrode materials. The different electrode materials displayed similar performance for the vanadium redox process, but substantially different activity for the Sn2+/Sn4+ redox couple. Later in the same year, Kummer and Oei reported a mediated cathode system with VO2+ and POMs as the mediator/catalyst composition (POM = H3PMo12O40 or H5PMo10V2O40). A silicon-containing POM was used as the anodic mediator (H2|Pt|Si-POM||VO2+|POM|O2; see Section 3.1.4 for discussion of anodic performance).76–78 Polarization curves were obtained using 0.8 M VOSO4 and 0.28 M H3PMo12O40 at a pH 0.75 (Figure 5). The highest power density obtained was ~38 mW/cm2. Reasonable iR-free voltages were obtained, but the internal resistance of the cell contributed to significant losses in cell potential (0.83 V with iR-correction vs. 0.55 V without iR-correction at ~80 mA/cm2). Slow regeneration of the cathode mediator system caused the potential of the cathode compartment to drop from 468 mV to 330 mV over 900 minutes of operation. A pH change was observed in the cathode compartment, which was attributed to vanadium migration through the membrane. The authors commented that long-term performance would be determined by the size of the regenerators, and to achieve reasonable rates, the regenerator would be too large for commercial vehicle use. For example, a 20-kW engine would weigh over 5,000 pounds.
Figure 5.

Redox sequence proposed for V-mediated cathode with POM catalyst for O2 reduction (||VO2+|POM|O2). For relevant systems using this mediated cathode, see references 76–78.
In 1985, Kummer and Oei reported further optimization of their mediated fuel cell design.79 To avoid the substantial performance decreases observed in VO2+ oxidation catalyzed by POMs, they shifted to the use of NOx gases as a catalyst (Figure 6A). Over a 12 h experiment, the cathodic performance remained nearly constant (Figure 6B), showing more stable performance than previous reports.63 To prevent loss of NOx species, a static pressure of O2 was used without bubbling gas into the cathode solution. When using a V2+/V3+ anode solution, power densities of 73.5 mW/cm2 were achievable.
Figure 6.

(A) Redox sequence for vanadium-mediated cathode with NOx catalyst for O2 reduction (||VO2+|NOx|O2). For systems featuring this mediated cathode, see references 79–86. (B) Sustained performance curve of a mediated cathode utilizing a VO2+/VO2+ redox mediator, reoxidized with NOx-based catalysts. The anode solution contained a V2+/V3+ redox mediator which was not regenerated during the experiment (V3+||VO2+|NOx|O2). Cell was discharged through a 5-ohm external load with a flow rate of 230 mL/min. Adapted from ref. 79. Copyright 1985, Chapman and Hall Ltd.
The VO2+/VO2+ redox couple has been adopted by other groups in various fuel cell studies focused on anodic reaction chemistry. Folkesson constructed a cell using a VO2+/VO2+ redox mediator regenerated aerobically using NOx as a catalyst with a variety of anode redox mediators for H2 oxidation (see Sections 3.1.2, 3.1.3, and 3.1.7 for further discussion of the anodic chemistry).80 Folkesson demonstrated reasonably stable cell performance generating around 15 mW/cm2 using the VO2+/VO2+ and Ti3+/TiO2+ redox couples. A later report mentions the aerobic oxidation of VO2+ catalyzed by Pt for a fuel cell using a V3+/VO2+ anode for the oxidation of sugars in a fuel cell stack capable of producing 10 kW of power; however, no evidence for the aerobic oxidation of VO2+ was presented.81 Whitesides and coworkers have used vanadyl-based mediators paired with NOx catalysts in a variety of applications (c.f. Figure 6A). Their initial report focused on the use of methane as a fuel, mediated by Fe2+/Fe3+ and catalyzed by Pt black (CH4|Pt|Fe2+||VO2+|NOx|O2; see Section 3.1.3 for further discussion of the anode).82 Methane oxidation occurred under forcing conditions in a high-pressure reactor (120 °C, 54 atm CH4), and the Fe2+ in the resulting aqueous reaction mixture was oxidized at the anode (Figure 7). Polarization curves were obtained, but the performance of the overall system was low, due to engineering difficulties in cell construction and the large potential losses associated with use of the Fe2+/Fe3+ redox couple as the anodic redox mediator (~600 mV difference between thermodynamic potential of methane oxidation to CO2 and the Fe2+/Fe3+ redox potential). This approach was later extended by Whitesides and coworkers to the use of ethylene glycol as a fuel, generating a power density of 9.9 mW/cm3,83 and to the use of solid carbonaceous fuels, including coal (see Section 3.1.3 for further discussion).84
Figure 7.
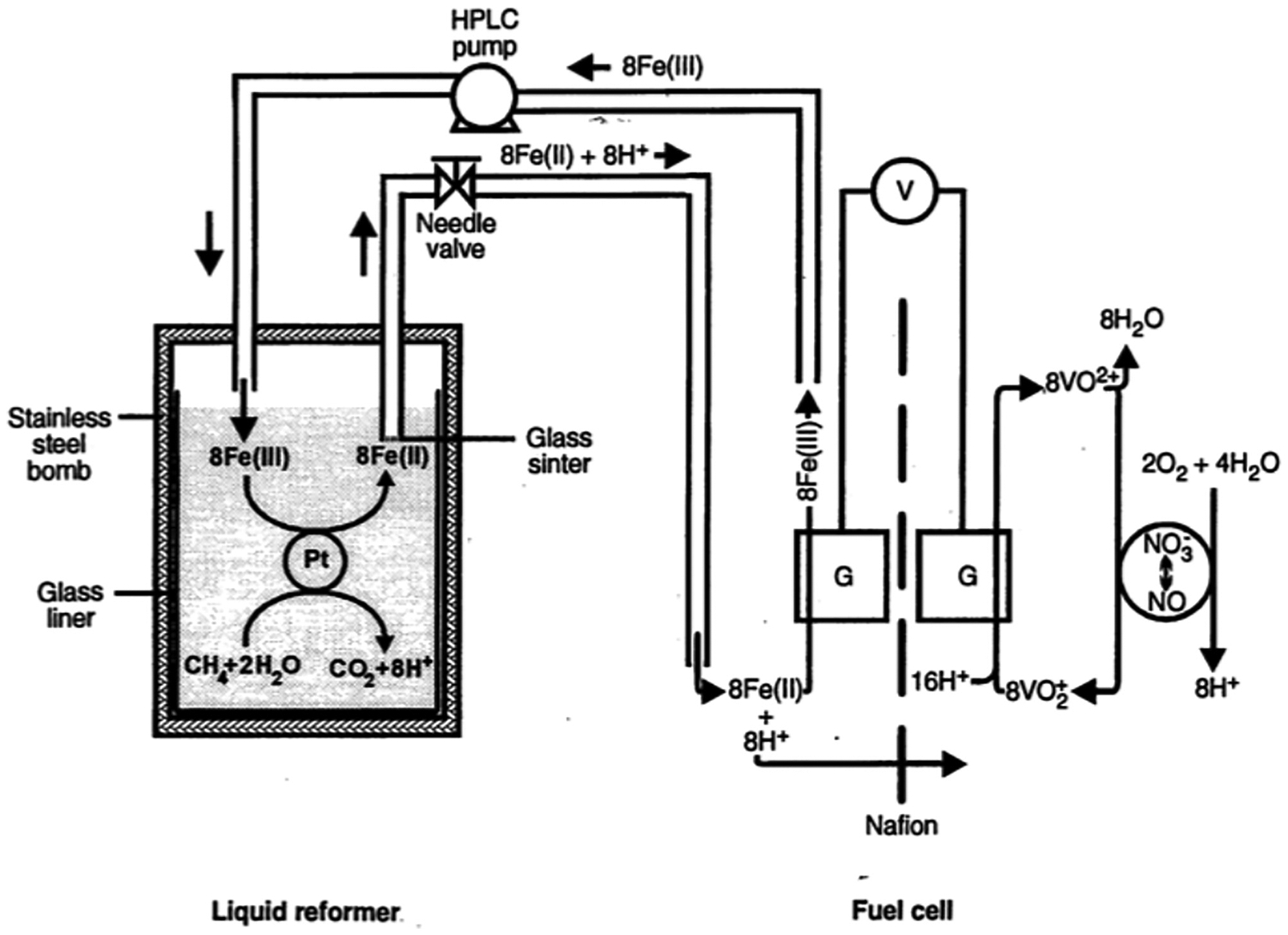
Schematic of doubly mediated fuel cell (CH4|Pt|Fe2+||VO2+|NOx|O2) using methane as fuel developed by Whitesides and coworkers. Adapted from ref. 82. Copyright 1994, American Association for the Advancement of Science.
Deng and coworkers reported a fuel cell using VO2+/VO2+-based mediators and an HNO3 catalyst to mediate O2 reduction on the cathode paired with an Fe2+/Fe3+ anodic mediator for biomass (wheat straw) oxidation.85 Power densities of over 100 mW/cm2 were achieved with 10 g of wheat straw in 100 mL of electrolyte (10 mL HCl and 30 g FeCl3 in H2O), and a stable current density of 320 mA/cm2 was achieved at a voltage of 0.3 V for over 12 h (see Section 3.1.3 for discussion of the anode). Further studies by Deng and coworkers compared the performance of a mediated cathode utilizing a VO2+/VO2+ mediator with HNO3 as a catalyst with the performance of a mediated cathode using an H12P3Mo18V7O85 POM mediator (see section 3.1.4 for discussion of the relevant anode).86 Glucose and other biomass sources were used as the fuel with an FeCl3 anodic mediator (glucose|Fe3+||VO2+|NOx|O2 vs. glucose|Fe3+||POM|O2). The cell utilizing the vanadium/NOx system achieved a maximum power density of approximately 90 mW/cm2, whereas the POM-mediated cell only achieved a maximum power density of approximately 35 mW/cm2. This difference was attributed to the higher open circuit potential achieved by the vanadium-based system compared to the POM-based system.
As discussed above, Oei demonstrated that mixtures of POMs and VO2+ undergo aerobic oxidation. POM/VO2+ reactions with O2 have been intensely studied for decades, particularly for Mo/V-containing POMs.87–94 These reactions are quite complex, due to the complicated speciation of POMs in acidic solutions. This complexity can be observed in the 31P and 51V NMR spectra of H12P3Mo18V7O85, in which a multitude of different POM isomers and structures are observed (Figure 8).
Figure 8.

31P and 51V NMR spectra of 0.25 M solution of H12P3Mo18V7O85. Numerals 1, 2, 3, and 4 correspond to collection of lines of HPA-x Keggin POMs H3+xPMo12-xVxO40, where x = 1–4, see reference 87. Adapted from ref. 91. Copyright 2008, Elsevier B.V.
Starting in the mid-2000s, Acal Energy Ltd., a company based in the UK, filed numerous patents describing the use of POMs as combined mediator/catalysts for the electrocatalytic ORR for fuel cells. In contrast to the Oei work discussed above, Acal used a conventional Pt/C H2 anode. POMs have a variety of promising characteristics, including their inexpensive components, ability to operate at high potentials, and thermodynamic stability.95,96 Several POM species with promising activities were identified. Higher vanadium incorporation into the vanadium-substituted phosphomolybdate (H3+xPMo12-xVxO40, 0<x<6) led to enhanced activity, as did the inclusion of sodium ions (NaxH3PMo12-xVxO40, 0<x,6).97 Addition of 0.15 M VO2+ (as V2O4) as an additional mediator to a solution of 0.3 M H3Na3PMo9V3O40 gave higher sustained voltages when the fuel cell was run at 400 mA/cm2. A cell voltage of ~0.5 V was observed with no added V2O4 and ~0.58 V with added V2O4.98 In a later patent application, Acal disclosed that a mediated fuel cell with 0.3 M Na4H3PMo8V4O40, operated at 80 °C and an ambient pressure, achieved sustained performance over 40 min at approximately 0.4 V and 1.5 A/cm2.99 In 2019, Knuckey, Creeth, and coworkers disclosed the role of elevated temperatures and air pressures on POM reoxidation.100 Using H7PMo8V4O40, they tested the rate of open-circuit potential (OCP) recovery after cell discharge at 0.8 V. At a regenerator temperature of 80 °C and atmospheric pressure using air, it took over 4000 s for the OCP to increase to 0.88 V. In contrast, at 120 °C and 2 barg (air), the same OCP was reached in under within 300 s.
Building on the work of Acal, Matsui, Eguchi, and coworkers published a systematic study of three POMs as fuel cell cathode mediators: H3PMo12O40, H5PMo10V2O40, and H6PMo9V3O40.101 Polarization curves examining the influence of POM identity, concentration (0.01–0.3 M), and cell temperature (20, 40, or 80 °C) were reported using a conventional Pt/C H2 anode (Figure 9). The vanadium-containing POMs displayed higher activity than the all-molybdenum POM, and increased performance was observed at higher mediator concentrations and higher temperatures. The aerobic regeneration activity of the three POMs during fuel cell operation was examined. Using 0.01 M solutions at either 25 °C or 80 °C and purged with either Ar or O2, the voltage of the cell was monitored as a function of time at a current density of 5.36 mA/cm2. The voltage using the all-Mo H3PMo12O40 mediator decayed identically under O2 or Ar, indicating that the aerobic oxidation of the reduced mediator was slow. H5PV2Mo10O40 showed similar behavior under O2 and Ar at 25 °C, indicating slow aerobic reoxidation; however, steady currents under O2 at 80 °C indicated that the POM was oxidized at a sufficient rate for sustained activity at elevated temperature. H6PV3Mo9O40 is oxidized at both temperatures, yielding stable voltages over the course of the experiment. The maximum power density obtained (measured by polarization curves) was approximately 40 mW/cm2, obtained with 0.3 M H6PV3Mo9O40 at 20 °C or with 0.1 M H6PV3Mo9O40 at 80 °C.
Figure 9.

Voltage time-courses of POM-mediated cathodes (H2|Pt||POM|O2) at a current density of 5.36 mA/cm2 at either 25 °C or 80 °C under O2 or Ar for 0.01 M solutions of (a) H3PMo12O40, (b) H5PV2Mo10O40, and (c) H6PV3Mo9O40. Adapted from ref. 101. Copyright 2016, American Chemical Society.
Davies and coworkers published a separate study of the use of a H6PV3Mo9O40 POM (HV3) solution as the cathodic mediator, again utilizing a conventional Pt/C H2 anode (Figure 10).63 Many of the reaction parameters of the cathode were examined. A primary focus in this report was the influence of the concentration of reduced vanadium (VO2+) on the performance of the cell and examination of the “regenerator efficiency” to probe how well the reduced mediator undergoes oxidation by O2. The authors reported the performance of the cell at various VO2+ fractions (i.e., the amount of reduced vanadium relative to total vanadium): 0.05 (almost fully oxidized), 0.25, 0.45, 0.65, and 0.85 (mostly reduced). As expected, the highest OCP values were obtained with the most oxidized solution (0.05 VO2+), and a maximum power density of nearly 1 W/cm2 was achieved with 95% oxidized 0.3 M H6PV3Mo9O40 at 80 °C. Regeneration experiments, however, indicated that only approximately half of the VIV can be aerobically oxidized to VV, which will limit overall performance. Based on their regeneration data and the polarization curves, the authors predicted the steady-state performance of the combined system, and the results indicated that the steady-state performance will be similar to the results observed with the 0.85 VO2+ (mostly reduced) fraction. This result indicated that the regeneration ability of their system was too low to reach optimal power densities. The authors compared their calculated steady state results, as well as their data at 0.05 and 0.65 VO2+ fractions, to state-of-the-art conventional PEMFCs and to other mediated cathode fuel cells (Table 3). Their result at 0.05 VO2+ fraction was above that for state-of-art PEMFCs and far above those reported for other cathode-mediated systems, while the 0.65 VO2+ fraction and the calculated steady state performance were below that of traditional PEMFCs, with a maximum sustainable power density estimated to be 0.38 W/cm2.
Figure 10.

Schematic of POM-mediated cathode fuel cell (H2|Pt||POM|O2) reported by Davies and coworkers. Adapted from ref. 63. Copyright 2017, Elsevier B.V.
Table 3.
Performance comparison of a reported conventional PEM fuel cell, previously reported mediated cathode fuel cell systems, and results from Davies and coworkers.63
| System | Cathode material | Cathode reaction | Liquid Electrolyte | OCP (V) | Cell voltage @ 0.2 A/cm2 (V) | Cell voltage @ 1 A/cm2 (V) | Max power (W/cm2) | Ref. |
|---|---|---|---|---|---|---|---|---|
| Conventional PEMFC | Pt/C | O2 + 4 e− + 4 H+ → 2 H2O | None | 0.96 | 0.81 | 0.67 | ≥ 0.83 | 23 |
| Mediated cathode (HNO3) | N-doped carbon | NO3− + 2 e− + 2 H+ → NO2− + H2O | 5 M HNO3 | 1.04 | 0.95 | 0.46 | 0.51 | 129 |
| Mediated cathode (POM) | carbon | VO2+ + e− → VO2+ | 0.45 M POMa | 0.83 | 0.68 | 0.46 | 0.51 | 96 |
| Mediated cathode (POM) | carbon | VO2+ + e− → VO2+ | 0.3 M HV3 @ 0.05 VIV | 0.99 | 0.9 | 0.72 | 1 | 63 |
| Mediated cathode (POM) | carbon | VO2+ + e− → VO2+ | 0.3 M HV3 @ 0.65 VIV | 0.78 | 0.72 | 0.5 | 0.58 | 63 |
| Mediated cathode (POM) | carbon | VO2+ + e− → VO2+ | 0.3 M HV3 @ steady stateb | 0.81 | 0.73 | – | 0.36 | 63, 102 |
| Mediated cathode (POM) | carbon | VO2+ + e− → VO2+ | 0.3 M NaV4 @ steady stateb | 0.80 | 0.75 | 0.53 | 0.58 | 63, 102 |
POM identity was not disclosed
Steady-state data was calculated from regeneration rates and cell data
In a follow up report, Davies and coworkers examined four different POM species in the same manner described above,102 with the four POM solutions labeled HV3 (empirical formula H6PV3Mo9O40), HV4 (empirical formula H7PV4Mo8O40), NaV3 (empirical formula Na3H3PV3Mo9O40), and NaV4 (empirical formula Na4H3PV4Mo8O40). The “protonic” HV3 and HV4 POMs displayed higher open circuit voltages at all VIV fractions relative to the sodiumcontaining solutions, due to the lower pH of the protonic solutions. Polarization curves using almost fully oxidized HV3 or HV4 (5% VIV) displayed power densities of over 1 W/cm2. However, the HV3 composition underwent oxidative regeneration much more slowly than the other POM compositions, leading to poor extrapolated steady-state performance. The authors predicted the steady-state performance for each POM identity based on the polarization curves at different VIV fractions and the regeneration data, and they then projected the expected steady-state polarization curves for each POM (Figure 11). Due to its poor reoxidation, HV3 had the lowest estimated performance at steady-state. The two V4 POMs had nearly identical performance, with NaV4 displaying slightly higher maximum power density (578 mW/cm2; see also, last entry in Table 3). The authors pointed out that the regenerator performance continued to be a limiting factor in achieving maximal power densities and cite an Acal patent describing the formation of “microbubbles” to enhance POM oxidation.103 A steady-state durability test was performed using the NaV4 POM at a current density of 0.4 A/cm2, and no loss in activity was detected over a 200 h run.
Figure 11.

Projected (a) i-V and (b) power density curves generated from simulated steady state performance for HV3, HV4, NaV3, and NaV4 POM solutions (H2|Pt||POM|O2). Adapted from ref. 102. Copyright 2017, Elsevier B.V.
In 2018, Ward and Davies published a study investigating the role of temperature and catholyte concentration of a POM-mediated cathode.104 Using both HV4 and NaV4 POM compositions, the temperatures tested ranged from 40–90 °C and catholyte concentrations ranged from 0.2–0.45 M. Increases in temperature led to a slight decrease in the redox potential of the catholyte, while an increase in concentration led to larger increases in the catholyte redox potential. The authors also examined the role of temperature and concentration on the pH and conductivity of the catholyte and observed that increasing temperature and concentration both led to decreases in the pH and increases to the conductivity. Polarization curves with different concentrations of HV4 (95% oxidized) showed a 25% increase in maximum power density when shifting from 0.2 M POM to 0.3 M POM at 80 °C, but only a 6% increase when shifting from 0.3 M POM to 0.45 M POM. The minimal improvement at high concentrations was attributed to an increase in cell ohmic resistance as the catholyte concentration increased, perhaps due to VO2+ and VO2+ incorporation into the membrane. For NaV4 catholyte solutions, the best results at 80 °C were obtained using a 0.3 M concentration of the POM, which also corresponded to the lowest observed ohmic cell resistance. Increasing the temperature of both 0.3 M HV4 and NaV4 solutions resulted in enhanced performance at low current densities due to decreased activation losses, as expected; however, detrimental effects were observed at 90 °C at high current densities. The authors suggested that the poorer performance arose from anode effects, e.g., a decrease in electrochemical Pt surface area and decreased exchange current densities. POM regeneration data, collected using the same procedure as in previous reports, indicated that NaV4 has an increased rate of reoxidation relative to HV4. Concentration had only a minor impact on the rate of regeneration. Increasing the temperature typically led to higher reoxidation rates, with an exception observed upon increasing from 80 °C to 90 °C with highly reduced (>60 % reduced) NaV4 samples. The authors attributed a decreased reoxidation rate in this case to decreased O2 solubility and mass transport constraints. Steady-state experiments for each condition were performed at 1 A/cm2, and the highest voltage (0.47 V) was obtained with 0.45 M NaV4 at 90 °C. Very similar performance, however, was obtained using 0.3 M NaV4 at 80 °C, with only a 10 mV drop in cell voltage relative to the more forcing conditions. The authors concluded that the optimal conditions for fuel cell performance are 80 °C with 0.3 M NaV4 and estimated that a steady state peak power density of approximately 510 mW/cm2 could be achieved.
Stahl and coworkers reported a double MedFC using Na4H3PMo8V4O40 as a cathodic mediator paired with a mediated anode consisting of a Pt/C catalyst and anthraquinone mediator to mediate H2 oxidation.105 In initial tests using the mediated anode with a conventional cathode consisting of a Pt/C electrocatalyst, low power densities were obtained due to flooding of the cathode. Significantly higher power densities (up to 528 mW/cm2 with iR-correction) were obtained when a POM-mediated cathode was used instead (see Section 3.1.6 for further discussion of the mediated anode).
Recently, analogous vanadium-containing POMs have been used as cathodic mediators for ORR in various biomass-based fuel cells. A different set of lower-potential POMs were used as mediators for anodic biomass oxidation.106 The relevant anodic chemistry is discussed in Section 3.1.4. The selection of POMs used in the cathode was expanded beyond the Keggin-type POMs typically used by Acal and others described above, inspired by the fundamental POM/O2 reactivity reported by Zhizhina and Odyakov.91
The first example was reported by Deng and coworkers in 2014,107 describing a cell capable of using fresh grass, switchgrass, starch, or cellulose as the anodic fuels (Figure 12). A 0.3 M solution of H12P3Mo18V7O85 (or the di- or tri-sodium salt thereof) was used as the cathodic mediator solution, and H3PW11MoO40 was used as the anodic mediator. Stable power densities of 30 mW/cm2 were obtained for 10 h of operation at 80 °C. Consistent with the high loading of vanadium in the cathodic POM mediator, a very high potential (> 1.0 V vs. NHE) was achieved, even with multiple reduced vanadium centers in the POM. The influence of concentration and degree of reduction on the rate of aerobic oxidation was investigated. The degree of reduction was defined as the average number of electrons each POM complex had collected, and the highest rate of oxidation was reached at a high degree of reduction (>1.8 e−/POM) and at intermediate concentrations (~0.15 M). Analyses of anodic reactions are presented in Section 3.1.4.
Figure 12.

A) Schematic for the doubly mediated fuel cell using POM-I as mediator for biomass oxidation and POM-II as mediator for O2 reduction (biomass|POM-I||POM-II|O2). B) Further details showing electrode reactions and regeneration reactions of POM-I and POM-II. Adapted from ref. 107. Copyright 2014 Wiley-VCH Verlag GmbH & Co.
Several additional studies have employed cathodes with V-containing POM mediators or mediated systems using both VO2+ and POMs in efforts primarily focused on processes at the anode (i.e., thorough analysis of the cathode performance was not conducted). These studies will be elaborated in Section 3, but are briefly noted here. Deng and coworkers described a fuel cell using alcohol-based fuels, such as methanol, glycerol, or glucose, using H12P3Mo18V7O85 as a cathodic POM mediator. Maximum power densities of over 30 mW/cm2 with sorbitol as the alcohol, based on single-scan polarization curves with a fully oxidized mediator at the cathode (i.e., without steady-state POM regeneration by O2).108 The same group later reported a fuel cell system utilizing the same POM mediator in the cathode and using low-rank coal as the fuel.109 The same cathodic POM mediator was subsequently utilized by Li and coworkers in a fuel cell utilizing glucose, cellulose, or starch as a fuel.110 Deng and coworkers utilized sewage sludge as the fuel in their system using H3PMo12O40 as the anodic mediator and H12P3Mo18V7O85 as the cathodic mediator.111 Chao, Deng, and coworkers reported a biomass fuel cell using sugars treated photochemically and thermally with H3PMo12O40 at the anode and H12P3Mo18V7O85 at the cathode.112
A fuel cell using lignin as a fuel and H3PMo12O40 as the anodic mediator was reported by Zhao and Zhu,113 with several cathode variations. Examples included (a) a traditional Pt/C gas-diffusion cathode with O2 as the oxidant, (b) carbon electrodes with PMo12O40 or K5PV2Mo10O40 as POM mediators with O2, and (c) a carbon electrode with KMnO4 as a stoichiometric oxidant. Open circuit potentials and maximum power densities were measured at 90 °C and are summarized in Table 4. In general, the power densities were higher with solution-phase oxidants versus a conventional O2 cathode. Continuous operation of the cell at 90 °C with each oxidant was examined. The cathodes using O2 at Pt/C (current density ~ 0.4 mA/cm2) and the mediated H3PMo12O40|O2 system (current density ~ 0.5 mA/cm2) displayed reasonably stable behavior, while the current density decreased substantially for cathodes utilizing KMnO4 or K5PV2Mo10O40|O2.
Table 4.
Performance of various cathode chemistries on fuel cell performance using lignin as fuel.
| Oxidanta | OCP (mV) | Maximum Power Density (mW/cm2) |
|---|---|---|
| O2 @ Pt/C | 350 | 0.96 |
| O2 + 0.1 M H3PMo12O40 | 690 | 5 |
| 0.2 M KMnO4 | 900 | 45.1 |
| O2 + 0.1 M K5PV2Mo10O40 | 330 | 0.41 |
: 1 atm O2
2.3. Fe2+/Fe3+ mediator
The Fe2+/Fe3+ redox couples, including those from aqueous Fe salts, [Fe(CN)6]4/3−, and ferrocene derivatives, have been the subject of substantial interest from the flow battery community.8 The electrode kinetics can be rapid, and these mediators have low cost and toxicity. On the other hand, the redox potential of free Fe2+/Fe3+ (~0.7 V) is relatively low compared to the thermodynamic potential for O2, even while ancillary ligands may be used to access higher potentials. The various mediated cathode systems using this mediator for O2 reduction are summarized in Table 5.
Table 5.
Reported systems using iron-based mediators for O2 reduction.
In 1931, Lamb and Elder studied the aerobic oxidation of FeSO4 in acidic solutions catalyzed by various substances, with the goal of utilizing this redox mediator in a fuel cell application.114 The rate of oxidation of Fe2+ was monitored by measuring the potential of the solution during the oxidation. The reaction rate was found to be first order in pO2, second order in [FeSO4], and independent of [H2SO4] above 0.23 M, but the reaction rate increased rapidly below this H2SO4 concentration. CuSO4 was found to accelerate the oxidation, and it was noted that in the presence of CuSO4 the oxidation of FeSO4 accelerated with increasing acid concentration. Additionally, charcoal accelerated the reaction rate with a roughly first order dependence, and Pt black accelerated the reaction rate in a non-linear fashion, which the authors attributed to catalyst poisoning. The authors then constructed a working cathode solution with a Zn/ZnSO4 anode to study the behavior of the mediated cathode under electrochemical conditions. Comparison of O2 consumption in their kinetic studies with data obtained from the electrochemical studies show good agreement. In the electrochemical studies, the authors measured a current density of 21.5 mA/cm2 while operating at a potential of 0.606 V.
In 1953, Posner published a pair of papers examining the oxidation of Fe2+ in concentrated HCl solutions with no catalyst and in dilute HCl solutions catalyzed by charcoal.115,116 At high concentrations of HCl (> 4.0 N), Fe2+ was rapidly autoxidized, while below this value, the reaction was slow, consistent with the previous observations of Lamb and Elder. At the high concentrations of acid, the rate of Fe2+ oxidation was first order in [Fe2+], approximately first order in [HCl] between 6–8 N, and first order in pO2. Posner proposed that the rate of oxidation increases at high [HCl] due to the formation of an Fe2+-HCl complex, the oxidation of which bypasses formation of superoxide and proceeds directly to HO2•. When H2SO4 is used as the acid, no acceleration of the autoxidation rate is observed at high concentrations, suggesting that the active complex is not formed with this acid. At lower acid loadings, with charcoal as a catalyst, the autoxidation rate was first order in the catalyst loading, pO2, and [H+], and proportional to [Fe2+]/([Fe2+]+[Fe3+]). The use of iron as a mediator for the ORR was not pursued further, however, due to its low potential.70 Efforts were made to identify ligands that could increase the Fe2+/Fe3+ potential.117 No suitable ligands were identified, however, and use of Fe2+/Fe3+ as ORR cathode mediators were abandoned for many years.
Iron-based mediators reemerged in the mid- to late-2000s, when Acal Energy Ltd. published a series of patents using various iron-based mediators in fuel cell cathodes. A patent application118 described a MedFC using a conventional Pt/C H2 anode paired with a mediated cathode system containing K3Fe(CN)6 and KOH. The use of both a cationic- and anionic-selective membrane was required for optimal performance. In a separate experiment, they disclosed the aerobic regeneration of K3Fe(CN)6 from K4Fe(CN)6 using a solution containing 0.02 M EDTA and 0.01 M MnSO4. However, there was no integration between the aerobic regeneration studies and the fuel cell experiments. In subsequent patents, ferrocene (Fc) species were described as mediators using iron complexes to catalyze aerobic oxidation of the reduced mediator.119–122 Examples include the use of Fc(CH2SO3H)2 as a cathodic mediator and [Fe(trilen-(SO3Na)3)]2+ as the catalyst at pH 2 (Figure 13). A fuel cell was constructed incorporating a conventional Pt/C H2 anode (i.e., H2|Pt/C||Fc(CH2SO3H)2|[Fe(trilen-SO3Na)3)]2+|O2), and the mediator was partially oxidized by bubbling O2 through a solution of the mediator and catalyst (10 mM mediator and 1.0 mM catalyst). A polarization curve was measured with this cell. This system exhibited an open-circuit potential of approximately 0.78 V, but the potential decreased rapidly with increasing current, resulting in a current density of only 16 mA/cm2 at a cell voltage of 0.2 V. The authors confirmed that the mediator undergoes regeneration during fuel cell operation by monitoring the open-circuit potential vs. time. Fc derivatives with the cyclopentadienyl rings linked together by an alkyl tether exhibit increased stability relative to unlinked derivatives at pH 2.5.123 In spite of these improvements, subsequent efforts by Acal emphasized polyoxometalate mediators (cf. Section 2.2).
Figure 13.

Ligand used with Fe2+ (trilen-SO3Na)3 and to oxidize Fc(CH2SO3H)2 mediator.
In 2016, Park and coworkers reported the use of iron phthalocyanine, Fe(pc), as a catalyst for the aerobic oxidation of Fe2+ in the context of a MedFC.61 They studied the kinetics of Fe2+ [as Fe(SO4)] oxidation by O2 (1 atm) in 1 M H2SO4 catalyzed by 0.25 mM Fe(pc) or [FeIII(tetra(4-OMephenyl)porphyrin)]Cl. Fe(pc) was a more effective catalyst with >90% conversion of Fe2+ to Fe3+ after 5 h. This reaction was proposed to proceed via O2 binding to the iron macrocycle in the FeII oxidation state and subsequent reduction of O2 to water with H2SO4 and aqueous Fe2+ ions as the source of protons and electrons (Figure 14A). The authors then measured polarization curves for the three oxidized solutions (0.5 M FeSO4 plus 1.0 M H2SO4 and (i) no catalyst, (ii) Fe(pc), or (iii) [FeIII(tetra(4-MeOphenyl)porphyrin)]Cl). They found the best performance for the solution containing Fe(pc), stemming from the much higher conversion of Fe2+ to Fe3+ (Figure 14B). In this study, the cell was held at 80 °C and H2 was supplied as the fuel to a Pt/C anode (H2|Pt/C||Fe2+|Fe(pc)|O2). The solution containing Fe(pc) as the catalyst led to a peak power density of ~250 mW/cm2, which was over three-fold higher than the other two solutions (65 mW/cm2 for the uncatalyzed solution and 76.8 mW/cm2 for the solution containing the Fe-porphyrin catalyst). However, no sustained electrolysis data was included to provide evidence for efficient on-stream regeneration of the mediator during fuel cell operation. The authors noted that further adoption of mediated cathode systems would require higher potential mediators, as well as more active catalysts for the mediator oxidation.
Figure 14.
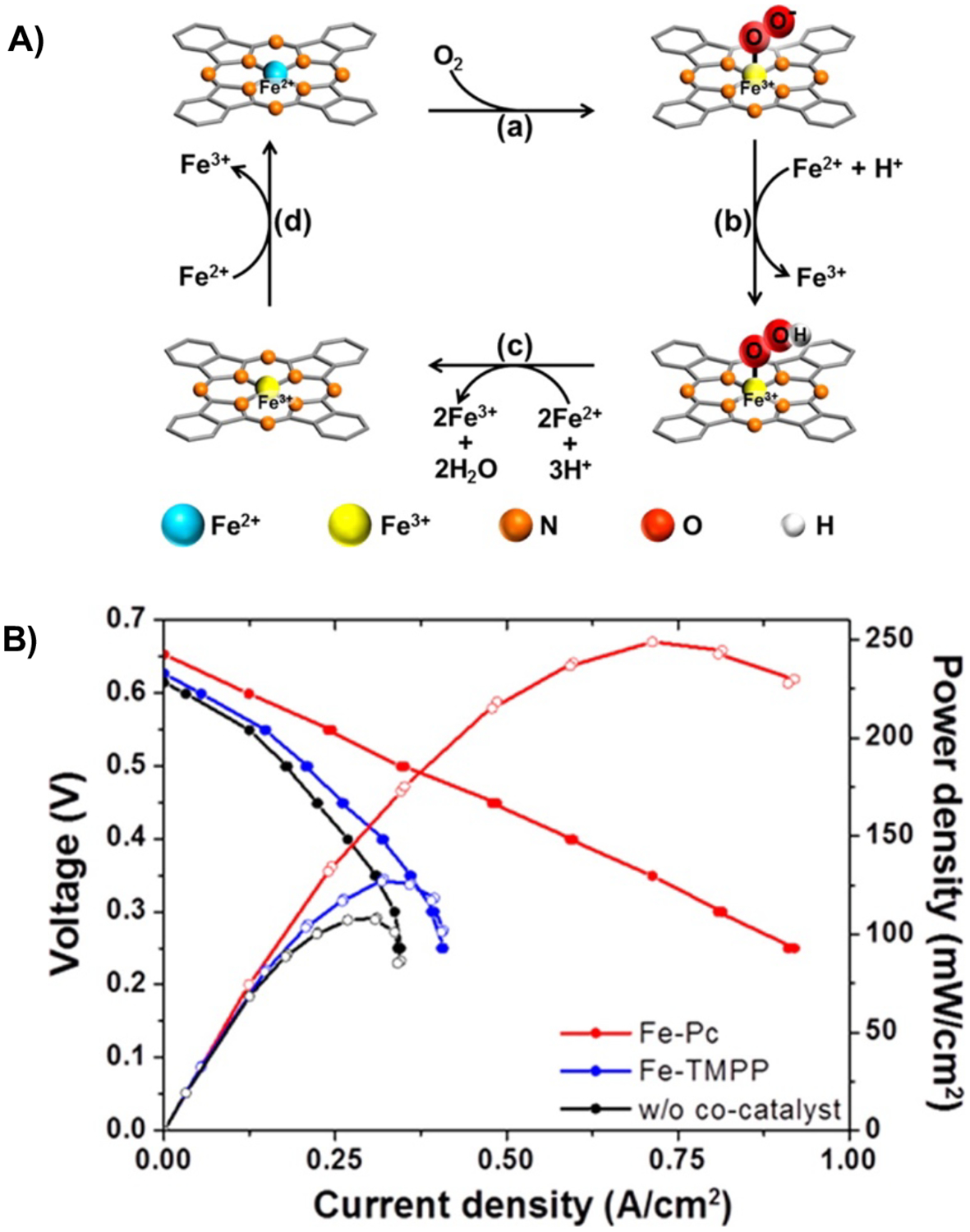
(A) Proposed mechanism for Fe2+ oxidation by O2 catalyzed by Fe(pc). (B) Polarization curves measured using 0.5 M FeSO4 + 1.0 M H2SO4 solutions after oxidation at 80 °C with O2 for 5 h in the presence of no catalyst (black traces), [FeIII((4-OMephenyl)porphyrin)]Cl (Fe-TMPP) as catalyst (blue traces), or Fe(Pc) as catalyst (red traces). H2 was used as the fuel at a Pt/C electrode (H2|Pt/C||Fe2+|Fe(pc)|O2). Adapted from ref. 61. Copyright 2016 American Chemical Society.
In order to study Fe complexes similar to those reported by Acal, Metz and coworkers used computational methods to calculate the reduction potential of a variety of different Fe complexes bearing chelating nitrogen ligands (Figure 15).62 Using experimental E1/2 values for [Fe(II)trilen (R = H/Cl/SO3Na)-OH2]2+ and Fe(II)N4Py(R = H/CH3)-OH2]2+, the authors calculated the difference between potential energy surfaces by the self-consistent field method, ΔE(SCF), and found good agreement between these values. The authors used these benchmarks to estimate the redox potential of [Fe(II)trilen (R = CH3/CF3/CN)-OH2]2+ and [Fe(II)TFM-trilen(R = H/SO3Na)-OH2]2+. The complex [Fe(II)TFM-trilen(R = SO3Na)-OH2]2+ was expected to have a redox potential of nearly 1 V, which would increase the open-circuit potential by ~25% relative to the highest previous Fe complex studied experimentally. A synthetic route to this complex was proposed, but it wasn’t prepared. Thus, experimental validation of these results, including assessment of mediator stability and aerobic regeneration, awaits further study.
Figure 15.

Ligands studied computationally by Metz and coworkers to determine the redox potentials of the Fe complexes.62 Experimentally determined redox potentials are shown in red, while the calculated redox potentials are shown in black.
2.4. NOx mediators
In previous sections, NOx species have been utilized as catalysts for aerobic oxidation of a mediator. HNO3 has also been utilized as a single-component mediator/catalyst system. Both HNO3 and HNO2 have high reduction potentials to NO, 0.957 V and 1.028 V vs. NHE, respectively. These values are close to the formal reduction potential of O2 to H2O, 1.23 V, and it is possible to access high concentrations of HNO3 that support high current densities. The systems using NOx species derived from HNO3 as cathode mediator/catalysts for O2 reduction are given in Table 6.
Table 6.
Reported systems using NOx species as mediators for O2 reduction.
In the early 1960s, research into the use of HNO3-mediated ORR was conducted by researchers at Esso Research and Engineering Company in New Jersey.124 Alcohols, such as methanol or ethylene glycol, were targeted as the fuels for this system. In the initial report, low concentrations of HNO3 (approximately 1 wt% in H2SO4) gave substantially higher currents than O2 alone. The researchers studied the mechanism of electroreduction of HNO3 and proposed an autocatalytic mechanism with a rate-limiting step involving the reaction of HNO3 with nitric oxide (NO) to form nitrous acid (HNO2).125 The latter was proposed as the electroactive species. Engineering of the external regenerator allowed up to 30 cycles of HNO3 turnover, measured by comparing the total charge passed to stoichiometric redox equivalents available from HNO3.126 A challenge with this system was the loss of NO or other NOx species to the gas phase, but further development of the MeOH-HNO3/O2 fuel cell enabled power densities of 44 mW/cm2 at 0.34 V using a new Pt/C-based catalyst at the cathode. A maximum power density of 19 mW/cm2 was obtained during continuous operation for over 180 h using a standard Pt mesh cathode.127
The use of NOx-based mediators for fuel cell cathodes was revisited in 2011, when Park and coworkers reported a fuel cell using the NO2−/NO3− as a cathodic redox mediator using a nitrogen-doped carbon-felt electrode.128 The reduction of NO3− to NO2− has a formal potential of 0.94 V, and aerobic oxidation of NO2− is facile. The authors prepared an Fe-N/C catalyst by treatment of iron nitrate and melamine with carbon black and annealing of the resultant powder at 700 °C under N2. This electrode showed enhanced activity for nitrate reduction relative to commercial Pt/C catalysts. Specifically, polarization curves using a conventional Pt/C H2 anode and 5 M HNO3 as the oxidant in the cathode compartment showed higher current densities with Fe-N/C than with Pt/C cathodes (325 mA/cm2 vs. 250 mA/cm2, respectively) at 0.9 V. To monitor the stability of the cathode materials, the power density at 0.8 V was measured over the course of 100 h with an operating temperature of 80 °C. The Fe-N/C electrode showed minimal drop in activity, while the performance of the Pt/C cathode dropped dramatically. This decrease in activity was attributed to Pt dissolution. The selectivity of NO3− reduction using the Fe-N/C electrode was measured, and a 98.3% selectivity for formation of NO2− was observed, with the remainder forming N2 and NO. No data describing aerobic regeneration of NO3− was presented, though the authors suggested that this should be achievable.
In 2017, Park and coworkers reported an H2/O2 fuel cell using a Pt/C anode and a heteroatom-doped cathode with an HNO3/NO mediator (Figure 16).129 Nitrogen-doped carbon felt, prepared by treatment of commercial carbon felt at 700 °C under an atmosphere of NH3, proved to be an efficient electrode material to facilitate the reduction of NO3− to NO. Polarization curves using 5 M HNO3 were collected at a variety of temperatures, and at 80 °C a power density of over 0.5 W/cm2 was achieved at a potential of 0.8 V. To examine the long-term activity of this system, the fuel cell performance was monitored by holding the cell at a potential of 0.8 V for 30 days with circulation of the 5 M HNO3 solution supplied with 1 atm O2 (H2|Pt/C||HNO3|O2). A slow linear decline in performance was observed during the experiment, falling to approximately 0.3 W/cm2 after 30 days of operation (Figure 17); however, replacement of the cathode solution with fresh HNO3 caused the cell performance to return to its original values. The authors calculated a “regeneration efficiency” of approximately 93%. With no O2 supply, the system was capable of producing power for only 4 days, reflecting the redox capacity of the HNO3 in the cathode solution.
Figure 16.
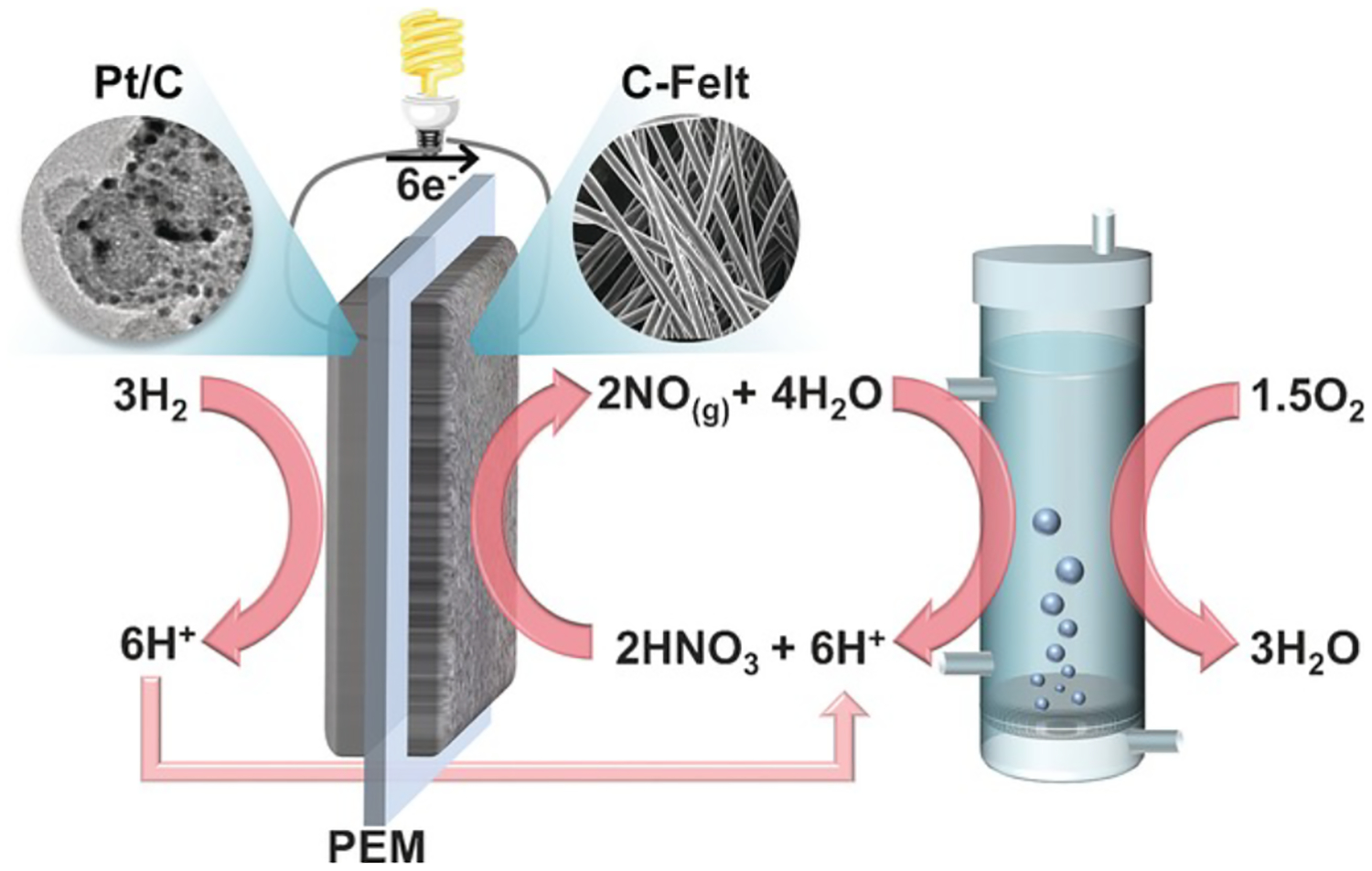
Schematic of a mediated fuel cell using the NO3−/NO redox mediator to mediate O2 reduction. Adapted from ref. 129. Copyright 2017 Wiley-VCH Verlag GmbH & Co. KGaA.
Figure 17.

Long term stability test of HNO3-mediated cathode with nitrogen-doped carbon-felt electrode. Cell run at 80 °C with H2 anode. Adapted from ref. 129. Copyright 2017 Wiley-VCH Verlag GmbH & Co. KGaA.
A direct ethanol fuel cell utilizing a PtSn alloy anodic catalyst and a HNO3-based catholyte mediator was reported in 2018 by Park and coworkers (Figure 18).130 The system achieved a maximum power density of 68 mW/cm2 and an open-circuit potential of ~0.85 V. In contrast, a system using direct O2 reduction on a commercial Pt/C catalyst on the cathode achieved a maximum power density of 34 mW/cm2 and an open-circuit potential of ~0.65 V. No NO2 regeneration data was presented in this work.
Figure 18.

Schematic of a direct ethanol fuel cell using an HNO3 mediated cathode. Adapted from ref. 130. Copyright 2018 Hydrogen Energy Publications LLC.
2.5. Nitroxyl/Oxoammonium mediators
Organic aminoxyls, such as TEMPO (2,2,6,6-tetramethylpiperidine N-oxyl), are organic radicals that can undergo 1 e− oxidation to an oxoammonium species. They have been used as mediators in a wide variety of alcohol oxidation reactions, including under electrochemical46,131 and aerobic132,133 reaction conditions. The use of NOx co-catalysts in the latter reactions provided the starting point for a fundamental study by Gerken and Stahl to investigate the ORR with an aminoxyl/NOx mediator/catalyst system (Figure 19A).134 Cyclic voltammetry and controlled-potential electrolysis experiments highlighted the cooperative redox behavior of this system. A series of different nitroxyl mediators (TEMPO, ACT, 3-CARP, and ABNO; Figure 19B) were examined. TEMPO/TEMPO+ electrochemistry was facile, but TEMPO did not react efficiently with O2 (Figure 19C, blue trace). NO, generated by addition of acid to a solution of NaNO2, reacted rapidly with O2, but the NO2 product exhibited poor electrochemical behavior (Figure 19C, black trace). The combination of both mediators led to a significant and sustained increase in the observed currents (Figure 19C, red trace). Other aminoxyl mediators (ABNO, ACT, and 3-CARP) yielded similar results (Figure 19D), and the operating potential correlates with the potential (vs. Fc+/0) of the aminoxyl/oxoammonium redox couple.
Figure 19.

A) Simplified scheme for TEMPO-mediated O2 reduction using a NOx-based catalyst B) Different nitroxyls examined in the study. C) Controlled-potential electrolysis traces in the presence and absence of mediator and/or catalyst (TEMPO and NOx, respectively). D) Controlled-potential electrolysis traces in the presence of NOx and various nitroxyl mediators (red = TEMPO; black = ACT; blue = ABNO; green = 3-CARP). Adapted from ref. 134. Copyright 2015 American Chemical Society.
The thermodynamic potential for O2 reduction under these reaction conditions was determined by measuring the H+/H2 open-circuit potential under the non-aqueous conditions and applying the relevant corrections.135,136 The highest potential mediator, ACT, was shown to feature an ORR overpotential of roughly 300 mV, similar to overpotentials associated with the onset of ORR with conventional Pt catalysts. This system was not well suited for practical application (e.g., due to the use of an organic solvent and loss of NOx species to the gas phase), but it provides some of the most thorough insights to date into the mechanisms of electrochemical O2 reduction with NOx and mixed aminoxyl/NOx mediator systems.
Park and coworkers reported a MedFC using aqueous acidic conditions with TEMPO as a mediator in the absence of a redox catalyst (Figure 20A).137 The authors report the redox potential of TEMPO to be 0.76 V and used a conventional Pt/C H2 anode (H2|Pt/C||TEMPO|O2). A fuel cell test with a cathode flow rate of 2 mL/min produced a maximum power density of 90 mW/cm2 (Figure 20B). A constant potential experiment was conducted at 0.4 V, and the current was observed to go to zero in approximately 190 minutes (Figure 20C). A rough analysis of the total charge passed suggests that no regeneration of TEMPO occurs with O2, consistent with previous observations (e.g., Figure 20B). The reduced TEMPO mediator will be present as TEMPOH2+ under these conditions and will not undergo facile reactions with O2.138
Figure 20.

A) Schematic of mediated cathode fuel cell using TEMPO mediator. B) Polarization curve of the mediated fuel cell using TEMPO as a cathodic mediator and a conventional H2 anode. Conditions: 0.3 M TEMPO and 1 M H2SO4 in the cathode, fuel cell temperature 30 °C. C) Time-on-stream test using a TEMPO-mediated cathode. Inset shows original color of TEMPO solution (left) and solution color after time-on-stream experiment (right). Adapted from ref. 137. Copyright 2018, Elsevier B.V.
2.6. Quinone/Hydroquinone mediators
Quinones have a rich redox chemistry. They undergo both electron-transfer or proton-coupled electron transfer, they are used in dehydrogenation and other oxidation reactions with organic molecules, and they serve as cocatalysts in aerobic oxidation reactions.139,140 The last of these roles has direct relevance to the ORR because hydroquinone (H2Q) reacts with activated oxygen species in the reduction of O2 with a molecular Co(salophen) catalyst.141 Stahl and Anson showed that the use of H2Q as an “electron-proton transfer mediator” led to a change in ORR product selectivity from H2O2 (two-electron reduction) to H2O (four electron reduction) during electrocatalytic O2 reduction.142 Cyclic voltammetry and bulk electrolysis experiments further showed that H2Q led to an enhanced rates and lower overpotentials for the Co(salophen)-catalyzed ORR in organic solvent (N,N-dimethylformamide, with acetic acid as the proton source; Figure 21). The conditions for the Co(salophen)/H2Q study reflected those used in the prior aerobic oxidation reactions, but the results have implications for more-conventional ORR effort by showing that a mediator could participate in a cooperative, inner-sphere mechanism and thereby influence the rate and/or selectivity of the reaction.
Figure 21.

Simplified scheme for quinone-mediated O2 reduction utilizing Co(salophen) as a soluble catalyst. Adapted from ref. 142. Copyright 2017, American Chemical Society.
The first demonstration of hydroquinone-mediated ORR under PEMFC-like conditions was reported by Stahl and coworkers in 2018, using a water-soluble quinone mediator paired with an off-electrode heterogeneous Co-N/C catalyst.143 Water-soluble high-potential quinones containing sulfonate groups have been utilized in flow-battery applications144 (Compounds A-C, Figure 22), but the previously reported materials rapidly decomposed and had inadequate stability for fuel cell operation. To address this limitation, a new tetra-substituted quinone bearing four alkylsulfide-linked sulfonate groups was designed and synthesized (Compound D, Figure 22). This mediator exhibited substantially improved stability with a half-life over >5000 h. Various M-N/C catalysts were tested for their ability to oxidize the corresponding hydroquinone to the quinone under fuel cell relevant conditions (1 M H2SO4 and 60 °C), and a Co-N/C catalyst using 1,10-phenanthroline as the nitrogen source during the pyrolytic catalyst preparation was identified as the most effective catalyst. A fully integrated fuel cell was constructed using a conventional Pt/C H2 anode (H2|Pt/C||quinone|Co-N/C|O2) and a Nafion membrane between the anode and cathode (Figure 23A). The cathodic solution contained 0.1 M quinone in 1 M H2SO4. This solution was circulated between the fuel cell cathode, where the oxidized mediator was reduced, and a packed-bed reactor, where the mediator was oxidized by O2 using the Co-N/C catalyst. An iR-corrected peak power density of 251 mW/cm2 (161 mW/cm2 uncorrected) was obtained (Figure 23B). Under steady-state conditions, a stable current output was observed for over 8 h at a constant potential of 0.5 V (Figure 23C). Control experiments showed that a decrease in current density occurred when O2 flow was shut off to the packed bed reactor, and the current density returned to its previous value when O2 flow was resumed. It was noted that development of stable higher potential mediators would enable the fuel cell performance to match or exceed that of conventional fuel cells.
Figure 22.

Stability of quinone structures previously tested for flow battery applications, A-C, and new tetrasubstituted quinone D. The stability was assessed by 1H NMR, and the half-lives were calculated by assessing the first-order rate constant for quinone decomposition. Conditions: 0.1 M quinone concentration in 0.5 M D2SO4 + 0.05 M CH3SO3H in D2O at 60 °C. Adapted from ref. 143. Copyright 2018, Elsevier B.V.
Figure 23.

A) Schematic of mediated cathode utilizing quinone mediator and Co-Phen/AC catalyst, H2|Pt/C||quinone|Co-phen/AC|O2. B) Polarization curves and C) time-on-stream performance for quinone-mediated fuel cell cathode with a Co-N/C catalyst. Conditions: 0.1 M quinone mediator, 1 M H2SO4, 60 °C, H2 fuel with Pt/C anode, MWCNT-coated carbon paper cathode, Nafion 117 membrane, 5 g Co-N/C catalyst in packed bed reactor. Time-on-stream experiment conducted at a constant potential of 0.5 V. Adapted from ref. 143. Copyright 2018, Elsevier B.V.
2.7. 2,2’-Azino-bis(3-ethylbenzothiazoline-6-sulphonate) mediator
2,2’-Azino-bis(3-ethylbenzothiazoline-6-sulfonate), ABTS2−, is a water-soluble, redox-active organic molecule that has been used as a mediator for O2 reduction with various enzymes at the cathode of enzymatic fuel cells (Figure 24). Mediators have found widespread use in enzymatic fuel cells to facilitate electron transfer between the electrode and the active site of an enzyme, which can be buried within the enzyme. A more detailed discussion of the role of mediators in enzymatic fuel cells is provided in Section 3.2 in the context of anodic reactions, and a review of O2 reduction in enzymatic biofuel cells, including both mediated and direct electrochemical approaches, was recently published.145 In general, mediated fuel cells employing enzymatic catalysts achieve power densities that are much lower (often, by two or three orders of magnitude) than those described in the previous sections.
Figure 24.

(A) Structure of ABTS2− and (B) redox sequence for ABTS2−-mediated cathode with enzymes as catalyst. For systems using this mediated cathode, see references 146–154.
The first use of ABTS2− in this context was reported by Palmore and Kim in 1999, using fungal laccase from Pyricularia oryzae as the enzyme catalyst.146 Many laccases reduce O2 at very high potentials, though prior reports using direct electrochemical methods displayed very low current densities. The outer-sphere electron-transfer reactivity of laccase accommodates a wide array of mediators. The authors considered mediators with an array of redox potentials, with the hope of identifying one close to the formal potential of the enzyme (E°’ = 0.539 V vs. SCE at pH 4.0, at which the formal potential for the reduction of O2 to H2O is 0.723 V vs. SCE). ABTS2− was selected due to its high potential (E°’ = 0.440 V vs. SCE at pH 4.0), good solubility and reasonable stability.
The authors investigated the aerobic oxidation of ABTS2− to ABTS•− catalyzed by laccase. Lower pH values favored higher laccase activity, and the stoichiometry of ABTS2− oxidized to O2 reduced was found to be 4:1, consistent with full reduction of O2 to water. A fuel cell employing a pH 4, 0.2 M acetate buffer was constructed with a Pt-gauze anode under 1 atm H2 and a glassy carbon or Pt-foil cathode containing both laccase and ABTS2− under 1 atm O2 (H2|Pt||ABTS2− |laccase|O2). The anode and cathode compartments were separated by a Nafion membrane. Polarization curves were obtained, and this system showed both a higher open-circuit potential and lower polarization relative to a Pt/O2 or carbon/O2 cathode system (Figure 25A). For example, at a current density of 25 μA/cm2 the biocathode exhibits a potential of 0.50 V vs. SCE, while the Pt/O2 and carbon/O2 system have potentials of 0.04 V and −0.29 V, respectively. Measured power densities were substantially higher for the biocathode, with the mediator system achieving a power density of approximately 40 μW/cm2, compared to ~15 μW/cm2 for a Pt cathode and ~3 μW/cm2 for a carbon cathode (Figure 25B). The performance of mediated laccase cathodes decreases at pH > 5. A biofuel cell pairing ABTS2− and fungal laccase at the cathode was reported to achieve a max power density of 99.8 μW/cm2 at a pH 4.0, with a decrease to 2.0 μW/cm2 at a pH 7.147
Figure 25.

A) Polarization curves of a fuel cell constructed with a laccase/ABTS2− biocathode (dotted line with open circles), Pt cathode (solid line with filled triangles), or carbon cathode (solid line with filled circles). B) Power density for the mediated fuel cell (dotted line with open circles) compared with a Pt cathode (solid line with filled triangles) and a carbon cathode (solid line with filled circles). Adapted from ref. 146. Copyright 1999 Elsevier Science S.A.
A pair of studies investigating the use of ABTS2− as a mediator at pH 7 were published by Ikeda and coworkers in 2001. The initial study explored electrocatalytic reduction of O2 with bilirubin oxidase from M. verrucaria as the catalyst and ABTS2− as the mediator.148 Cyclic voltammetry studies indicated catalytic O2 reduction occurred under these conditions, with a half-wave potential of 490 mV vs. Ag/AgCl, which is close to the thermodynamic potential for O2 reduction to H2O (615 mV vs. Ag/AgCl at pH 7.0; Figure 26). The mediator was found to decompose via disproportionation of ABTS•− to ABTS2− and ABTS0, which is unstable under these conditions. In a subsequent study, a biofuel cell with an ABTS2−/bilirubin oxidase cathode system was paired with Desulfovibrio vulgaris bacteria and methyl viologen for H2 oxidation at the anode.149 A pH of 7 was maintained in each compartment, which were separated by an anion-exchange membrane. Results from this study showed that the cathode compartment exhibited good activity and stability, but the overall power output was limited by the anodic chemistry.
Figure 26.

Cyclic voltammagrams of: A) pH 7.0 phosphate buffer, B) pH 7 phosphate buffer and 0.25 mM ABTS2–, and C) pH 7 phosphate buffer, 0.25 mM ABTS2–, and 0.11 mM BOD. Scan rate was 10 mV/s, working electrode was glassy carbon, and a Pt disk was used as the counter electrode Adapted from ref. 148. Copyright 2001 Elsevier Science B.V.
In 2007, Dunn and coworkers reported a fuel cell using bilirubin oxidase immobilized in a sol-gel with ABTS2− as a mediator for O2 reduction.150 The sol-gel substantially decreased diffusion of ABTS2− to the electrode. Using an anode consisting of a sol-gel immobilized glucose oxidase enzyme as the catalyst, ferrocene methanol as a soluble mediator, and glucose as the fuel (glucose|glucose oxidase|FcCH2OH||ABTS2−|bilirubin oxidase|O2), an open circuit potential of 480 mV and a peak power density of approximately 0.12 mW/cm2 were obtained. When air was used instead of O2, the power density dropped to 0.086 mW/cm2. Similar work was published by Xu and coworkers, using ABTS2− as a mediator with laccase at the cathode and glucose oxidase with either a ferrocene derivative or phenazine methylsulfate as anodic mediators.151 A maximum power density of 5.8 μW/cm2 was obtained in this case.
An enzymatic biofuel cell with a maximum power density of ~2 mW/cm2 was reported by Zhu and coworkers.152 At the cathode, laccase was bound to a graphene-gold nanoparticle (AuNP-graphene) hybrid electrode, and ABTS2− was used as a mediator. Glucose oxidase was anchored to the same electrode material at the anode, and direct electron-transfer was possible (Figure 27A–B). Using 0.5 mM ABTS2− as a cathode mediator and 50 mM glucose as fuel for the anode, an open-circuit potential of 1.16 V and a maximum power density of 1.96 mW/cm2 were achieved (Figure 27B). This system also displayed good stability; after 70 days of operation the OCP retained 80% of its original value (Figure 27C) and the maximum power of the cell decreased to 1.30 mW/cm2, corresponding to 66% of the original value.
Figure 27.

A) General schematic of the mediated fuel cell system using ABTS2− as a mediator for O2 reduction in the cathode. B) Formal redox potentials (vs. SHE) for each redox couple in the mediated fuel cell (GOD = glucose oxidase and HABTS = protonated ABTS2−). C) Power curve of the assembled fuel cell. D) Open-circuit potential measurements of the cell over 70 days of operation. Adapted from ref. 152. Copyright 2015, The Royal Society of Chemistry.
Chen, Hou, and coworkers reported a single-enzyme biofuel cell that utilizes immobilized laccase enzymes at both the anode and the cathode.153 Bisphenol A (BPA), was oxidized at the anode and O2 was reduced at the cathode. While the unmediated system was capable of producing power, enhanced performance was obtained when ABTS2− was introduced as a mediator for the cathode, with an approximately 3-fold increase in voltage after addition of ABTS (Figure 28).
Figure 28.
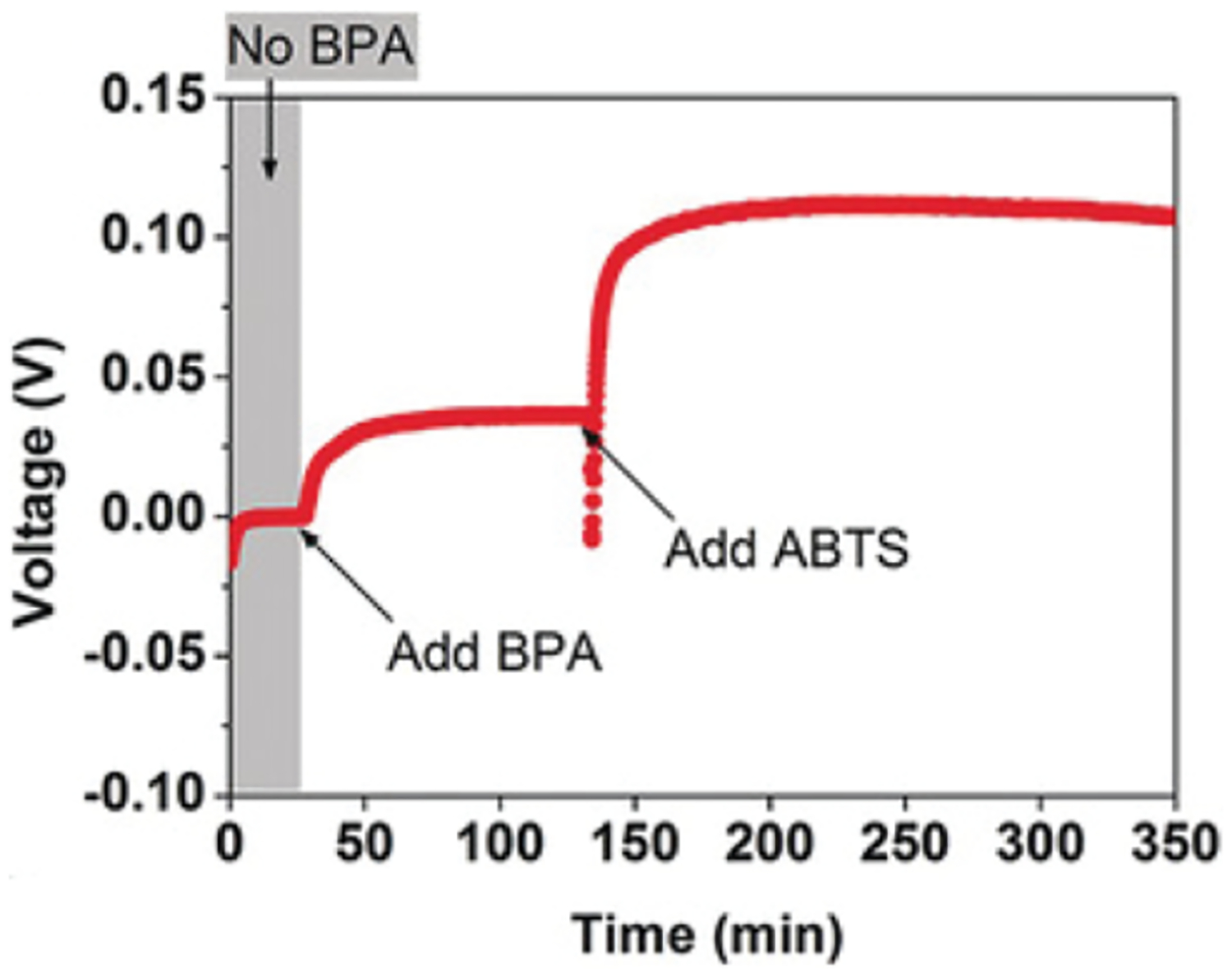
Change in cell potential after addition of bisphenol A (BPA) and ABTS2− using immobilized laccase enzyme on both anode and cathode. Adapted from ref. 153. Copyright 2017, Wiley-VCH Verlag GmbH & Co.
Cosnier and coworkers entrapped P2ABTS2− (bis-pyrene-ABTS2−) in a glyconanoparticle and paired this mediator with solubilized bilirubin oxidase to mediate O2 reduction in a “solubilized enzymatic fuel cell.”154 This entrapment strategy facilitates the use of higher concentrations of hydrophobic mediators, and the increased mediator size allows for the use of size-exclusion membranes to limit mediator and enzyme crossover. The cathode containing the entrapped P2ABTS2− (PS-bCD-P2ABTS) glyconanoparticle and bilibrubin oxidase was paired with an anode containing a 9,10-phenanthrenequinone (PS-bCD-PQ) glyconanoparticle and fungal flavin adenine dinucleotide-dependent glucose dehydrogenase (Figure 29). The glyconanoparticles were prepared by self-assembly of amphiphilic b-cyclodextrin modified polystyrene polymer, which was synthesized through click chemistry of functionalized cyclodextrin and polystyrene blocks,155 and the resulting PS-bCD-P2ABTS glyconanoparticles were approximately 43 nm in size. A 3-chamber fuel cell was designed, with the central chamber connected to a peristaltic pump and separated from the anode and cathode compartments by dialysis membranes with a cutoff value of 25 kDa. Fresh solution containing pH 7.0 McIlvaine buffer, ambient O2 concentration, and 5 mM glucose was flowed into the central compartment at a rate of 80 μL/min. The assembled fuel cell achieved a maximum power of 116 ± 21 μW. Charge-discharge experiments were conducted over 10 days. After seven days of testing, the peak power dropped to 85 ± 3 μW, for a power loss of approximately 26%. The spent fuel solution was tested and no evidence was found for the presence of enzymes or mediator. In contrast, a system without encapsulation of the mediators failed to charge after only 20 h, due to leaching of the mediators out of the cathode and anode compartments.
Figure 29.

Anode and cathode designs for “solubilized enzymatic fuel cell” using glyconanoparticle-entrapped 9,10-phenanthrenequinone and P2ABTS2− mediators. The cathodic enzyme used was bilirubin oxidase (Box) and the anodic enzyme used was fungal flavin adenine dinucleotide-dependent glucose dehydrogenase (FAD-GDH). Adapted from ref. 154. Copyright 2018, American Chemical Society.
3. The Anode: Hydrogen, Alcohol, and Biomass Oxidation
A variety of fuels have been examined in PEMFCs. Hydrogen is the most widely used and is the current fuel for commercial fuel cell systems; however, challenges associated with the transportation and storage of H2 continue to motivate efforts to identify alternative fuels. Methanol (MeOH) is the most common alternate fuel for low-temperature (<120 °C) fuel cells, in part, because methanol can undergo electrochemical oxidation to CO2 without the need to break carbon-carbon bonds. Mediated approaches to electrochemical oxidation of H2 and MeOH have been demonstrated in fuel cell applications, but the opportunities for MedFCs may be more significant for applications with challenging liquids, solids or other complex fuels that do not undergo facile direct electrochemical oxidation. In such cases, the redox chemistry with the fuel can occur off-electrode with a dissolved mediator. This approach allows for consideration of a wider variety of catalysts, since they do not need to be conductive or have other properties required of direct electrocatalysts. Results of mediated electrochemistry at the anode of fuel cells, including examples with chemical and enzymatic catalysts, are summarized below.
3.1. Fuel oxidations using chemical catalysts
3.1.1. Introduction
Early studies of MedFCs included studies of mediated anodes with H2 as the fuel, but major advances in the performance of gas-diffusion electrodes and the ability to use low Pt catalyst loadings diminished interest and effort in such applications. Nonetheless, the historical efforts provide important context for mediator selection for oxidation reactions with more complex fuels, such as alcohols and biomass. An overview of the various mediated systems using chemical catalysts for anodic reactions is given in Table 7, each of which will be discussed in the following sections.
Table 7.
Mediated anode systems utilizing chemical catalysts. In all cases, the oxidant used is O2.
| fuel | catalyst | mediator | mediator | catalyst | ref |
|---|---|---|---|---|---|
| H2 | Pd | Ti3+/TiO2+ | Br−/Br2 | NOx | 40 |
| Pt/Al2O3 | VO2+/VO2+ | 80 | |||
| Pd/Al2O3 | Fe(EDTA)2−/Fe(EDTA)− | 80 | |||
| Pt/Al2O3 | Mo3+/Mo4+ | 79 | |||
| H4SiW12O40 | 79 | ||||
| POM | 63 | ||||
| Pt/C | AQ | POM | 105 | ||
| alcohols, sugars or lignin | – | – | – | 166–168 | |
| sugars | POM | Fe2+/Fe3+ | POM | 110 | |
| methane, alcohols | Pt | VO2+/VO2+ | NOx | 82–83 | |
| biomass | – | 84–86 | |||
| sugars | Pt | V3+/VO2+ | Pt | 81, 169 | |
| sugars | – | viologen | – | Co | 171–175, 177 |
| biomass | POM | – | Pt/C | 113, 159 | |
| alcohols | POM | 107 | |||
| coal | 109 | ||||
| biomass | 111, 112 | ||||
Two different MedFC configurations have been employed for the oxidation of biomass or related “challenging” fuels with mediators (Figure 30). The first features a one-stage process in which the fuel oxidation and electrochemical reduction of the mediator are coupled within a single continuous process (Figure 30A). The second features a two-stage process in which the fuel is oxidized by the mediator in an independent process, and electrochemical regeneration of the reduced mediator occurs in a separate process (Figure 30B). This latter strategy allows the chemical and electrochemical redox process to be conducted under different conditions (e.g., temperatures, time), while retaining the ability to translate chemical redox processes into electrical power.
Figure 30.

Depiction the anode sequence for A) one-stage versus B) two-stage mediated biomass oxidation processes.
3.1.2. TiO2+/Ti3+ mediator
The first use of mediators to facilitate H2 oxidation was reported in 1959 by Carson and Feldman working at General Electric (see Section 2.1 for discussion on their cathode system).40 They used an on-line reformer without gas purification, meaning that this mediator system must tolerate CO2 and H2S impurities. Pd black was used as the catalyst for TiO2+ reduction by H2 in 6 N HCl (Figure 31). The activity of the catalyst increased over time, due to solubilization/precipitation of the Pd catalyst, arising from interconversion between insoluble Pd black (Pd0) and Pd2+ ions. The reaction of Pd2+ ions with H2 generated Pd0, and the freshly deposited Pd black was found to be more active. The Ti3+/TiO2+ redox couple has an E° of 0.1 V vs. NHE and, therefore, is well-positioned thermodynamically to mediate H2 oxidation.
Figure 31.

Redox scheme for TiO2+-mediated anode using H2 fuel with a Pd0/Pd2+ catalyst. For relevant studies, see refs. 40 and 80.
A kinetic study of this system was performed by Folkesson.80 TiCl4 was dissolved in 10% HCl with 2% Pt/Al2O3. The reduction with H2 exhibited a first-order dependence on [TiO2+]. A full fuel cell constructed using a vanadium/NOx/O2 cathode (H2|Pt/Al2O3|TiO2+||VO2+|NOx|O2) achieved a peak power density of 15–20 mW/cm2. The fuel cell was discharged through a 1-ohm resistor, and exhibited sustained performance for over 7 h (Figure 32).
Figure 32.
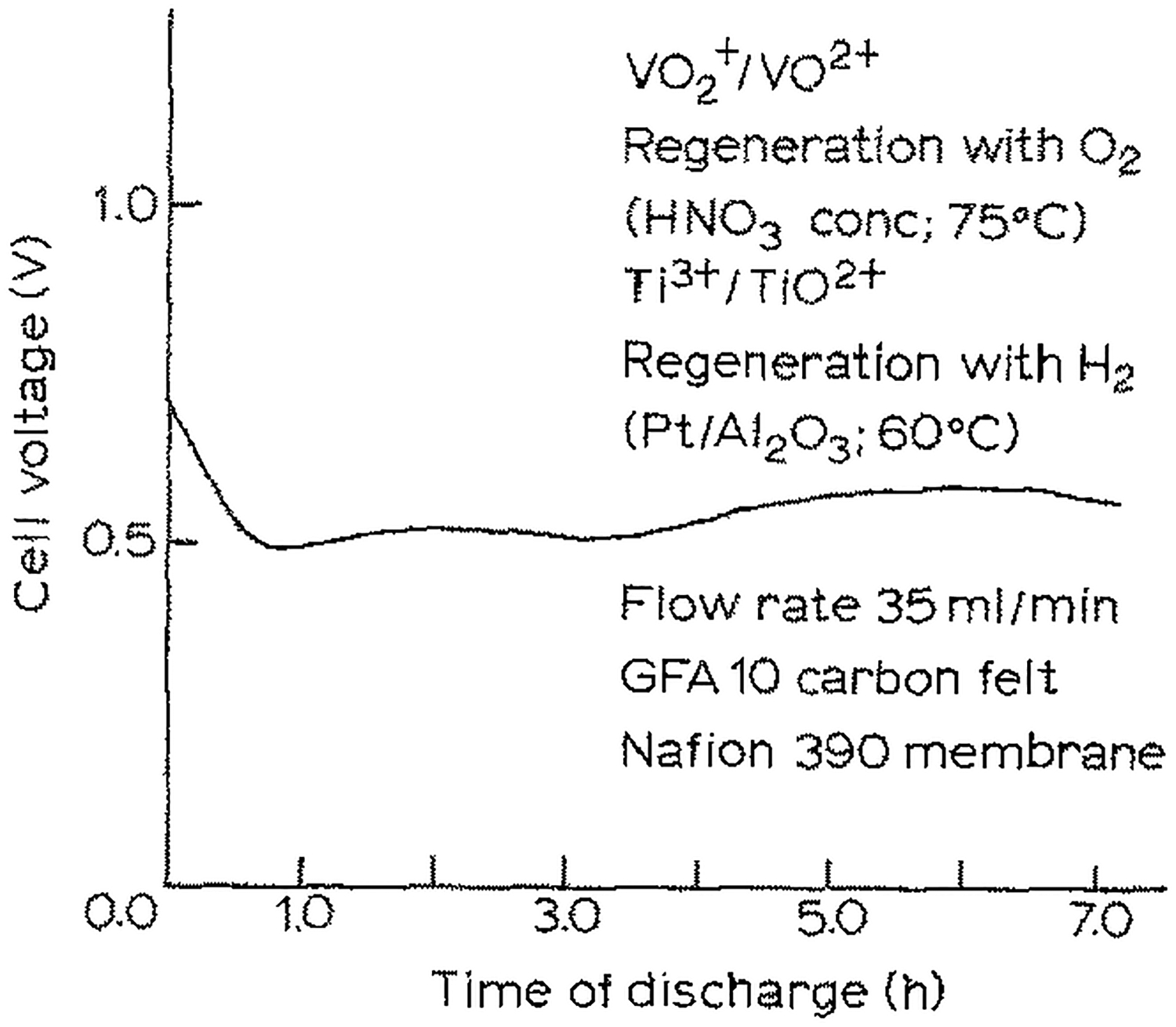
Discharge cell voltage vs. time for double MedFC. Cathode solution is 0.2 M VO2+ in 1 M H2SO4 regenerated with HNO3 and O2 at 75 °C. Anode solution is 1 M TiCl3 in 10% HCl regenerated with Pt/Al2O3 catalyst with H2 at 60 °C (H2|Pt/Al2O3|TiO2+||VO2+|NOx|O2). Adapted from ref. 80. Copyright 1990, Chapman and Hall Ltd.
3.1.3. Fe2+/Fe3+ mediators
The Fe2+/Fe3+ redox couple has been used as an anodic mediator for a variety of fuel oxidations. Complementary applications of the Fe2+/Fe3+ couple to mediate O2 reduction were presented in Section 2.3.
Coordination of EDTA to Fe3+ decreases the reduction potential from ~0.7 V vs. NHE to 0.117 V vs. NHE. Folkesson took advantage of this decrease in potential to use Fe(EDTA)2− as a mediator for H2 oxidation, catalyzed by Pd/Al2O3, at pH 5 (Figure 33).80 A fuel cell, incorporating VO2+/VO2+ and NOx in a mediated cathode for O2 reduction, exhibited rather low power densities of 3 mW/cm2 (H2|Pd/Al2O3|Fe(EDTA)2−||VO2+|NOx|O2). Some of the drawbacks of this system include poor solubility of Fe(EDTA)2− (max. solubility 0.075 M) and high resistance due to the raised anode solution pH. While the regeneration was shown to be effective, these issues have prevented further research into using Fe(EDTA)2−as a mediator for H2 oxidation.
Figure 33.

Redox scheme of Fe(EDTA)2−-mediated anode for H2 oxidation with Pd/Al2O3 catalyst. See ref. 80.
Whitesides and coworkers reported the use of the Fe2+/Fe3+ redox couple in a variety of fuel cell applications (cf. Section 2.3 for cathode examples). In the first report, the Fe2+/Fe3+ redox couple was used to mediate methane oxidation with a Pt black catalyst (c.f. Figure 7).82 An analogous MedFC approach was then demonstrated with methanol, ethylene glycol (and oxidized derivatives), and glycerol oxidation to CO2 as the anodic process.83 Other fuels, such as ethanol, acetic acid, or glucose, were oxidized, but CO2 was not detected. Using a gas buret, the rate of CO2 formation was measured. Potassium oxalate was oxidized to CO2 the fastest, followed by ethylene glycol and glyoxal (Figure 34). Using a cell similar to that used with methane (cf. Figure 7), a fuel cell utilizing ethylene glycol as the fuel produced a maximum power density of 9.9 mW/cm3 at a potential of 197 mV with a graphite felt electrode.
Figure 34.

Rate of CO2 formation vs. time for the oxidation for some organic compounds catalyzed by Pt black with Fe3+ as electron donor. Reactions were conducted at 80 °C in 3 M H2SO4. Adapted from ref. 83. Copyright 1996, Academic Press.
The Fe2+/Fe3+ mediator system was also extended to the use of sub-bituminous coal, Kraft lignin, and peat as fuels, without any added catalyst.84 These efforts demonstrated the ability of mediators to enable the use of fuels that would be highly challenging in conventional fuel cell configurations. While biomass-derived sources were capable to be used as fuels, primary efforts focused on the use of sub-bituminous coal. When oxidizing the fuel in 5 M H2SO4 saturated with Fe3+ at 100 °C, approximately 40% of the available electrons in the coal were liberated. Polarization curves for this system were collected, and a maximum power of 0.6 W per liter of slurry solution was obtained when using a solution containing 4 g of coal in 15 mL of 5 M H2SO4 saturated with Fe2(SO4)3. Simulations of the data suggested that the cell resistance limited the performance, and it was suggested that volumetric power densities of over 7 W/L should be achievable with improved cell engineering. The fuel cell was run continuously for 1000 h (with necessary additions of coal every ~100 h) with no significant degradation in cell performance.
Deng and coworkers used the Fe2+/Fe3+ mediator couple to oxidize wheat straw in a fuel cell using VO2+/VO2+ as the cathodic mediator.85 Using suspensions of wheat straw in aqueous solutions of FeCl3 and HCl, Fe3+ was reduced to Fe2+, eventually solubilizing the wheat straw. The fuel cell constructed using these redox couples in a two-stage process achieved power densities of over 100 mW/cm2 when 10 g of wheat straw was used in 100 mL of electrolyte (Figure 35A). The performance stability of the cell was examined in a one-stage process, and, with a discharge voltage of 0.3 V, a stable current density of 320 mA/cm2 was achieved for 12 h (Figure 35B). Based on the values given, each Fe2+ ion was oxidized approximately 1.4 times over the course of this test. The degradation products of wheat grass were hypothesized to contain vanillin and other aromatics, which over time were further oxidized to CO2. Analysis of the biomass after completing the fuel cell test suggested that hemicellulose was the primary reactive component, with cellulose and lignin remaining largely intact.
Figure 35.

A) Polarization curves and B) Continuous one-stage operation of a fuel cell using the Fe2+/Fe3+ redox couple as a mediator for wheat straw oxidation and a VO2+/VO2+-mediated, HNO3-catalyzed cathode for O2 reduction (wheat straw|Fe2+||VO2+|NOx|O2). Adapted from ref. 85. Copyright 2017, John Wiley & Sons, Inc.
In 2017, Li and coworkers reported a MedFC using various carbohydrates as the fuel. A combination of FeCl3 with low loadings of H3PW12O40 or H3PMo12O40 POM was the most effective mediator system for electricity generation.110 Their initial studies focused on testing high loadings (0.3 M) of H3PMo12O40 or H3PW12O40 as the sole catalyst/mediator for oxidation of glucose, but later studies found that using 1 M FeCl3 and 0.06 M H3PW12O40 or 1 M FeCl3 and 0.03 M H3PMo12O40 yielded similar results as 0.3 M H3PMo12O40 (Figure 36). A maximum power density of approximately 2.6 mW/cm2 was achieved. These experiments were conducted in a two-stage process, in which the glucose fuel was heated with the catalyst/mediator system at 85–95 °C for 2 h before introduction into the fuel cell at room temperature. Additionally, both 0.3 M H3PMo12O40 and 1 M FeCl3 + 0.06 M H3PW12O40 were competent catalyst systems for the oxidation of starch (max power densities of ~1.6 mW/cm2), while 1 M FeCl3 + 0.06 M H3PW12O40 was more efficient for the oxidation of cellulose, which enabled a maximum power density of 0.72 mW/cm2, compared to 0.46 mW/cm2 with 0.3 M H3PMo12O40. These results indicate that the addition of FeCl3 could provide a means to reduce the POM loading in biomass oxidation (see Section 3.1.4 for POM-only based methods).
Figure 36.
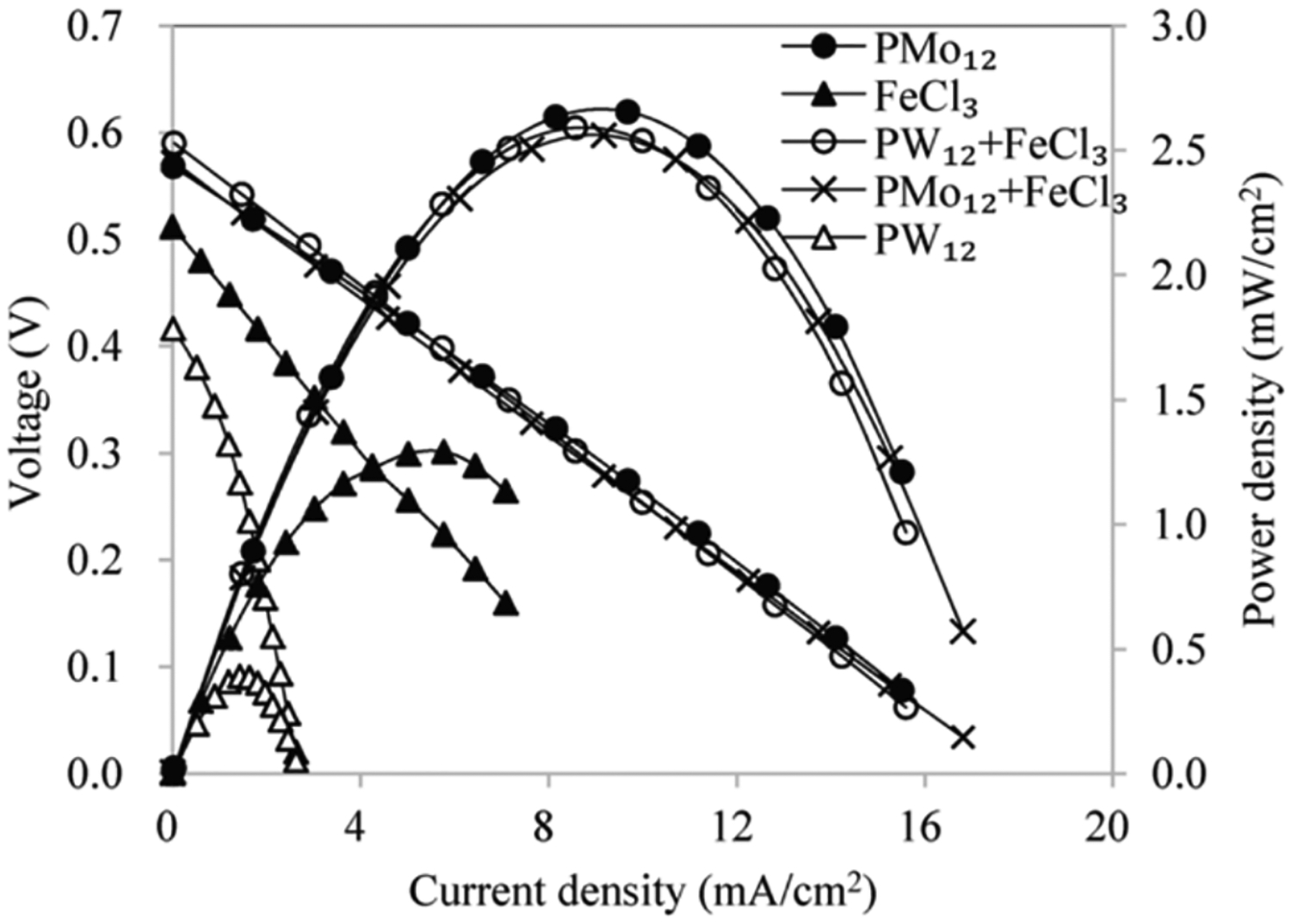
Polarization curves for mediated fuel cell using POM or POM + FeCl3 as mediator for glucose oxidation. H12P3Mo18V7O85 was used as a cathodic mediator. The anode contained 1.0 M glucose and 1.5 M phosphoric acid and was operated at 23–25 °C. Mediator concentrations in the various experiments were as follows: 0.3 M PMo12, 1.0 M FeCl3, 0.06 M PW12 + 1.0 M FeCl3, 0.03 M PMo12 + 1.0 M FeCl3, and 0.3 M PW12. Adapted from ref. 110.
FeCl3 was used as a mediator for the oxidation of glucose and other biomass sources by Deng and coworkers in 2018.86 The kinetics of this oxidation process were studied, and a nearly first-order dependence of the rate on [glucose] and [FeCl3] was observed under the acidic conditions used. The byproducts of glucose oxidation were identified as CO2, formic acid, acetic acid, and 4-hydroxybutyric acid. The best fuel cell performance, with a power density of approximately 125 mW/cm2, was achieved with a two-stage approach in which 0.33 M glucose and 2.0 M FeCl3 were heated at reflux for 4 h prior to electrochemical reoxidation of the Fe2+. The charged solution was capable of maintaining a current density of 250 mA/cm2 (stated 105 mW/cm2 power density) over the course of 1 h. In a closely related process, Fe3+-mediated oxidation of biomass (e.g., lignin and sugars) has been conducted in electrolysis cells to produce H2 via proton reduction at the cathode, rather than produce power via reduction of O2 at the cathode (Figure 37).156–158
Figure 37.

Schematic of H2 electrolysis system using Fe2+/Fe3+ mediator for the oxidation of biomass. Adapted from ref. 157. Copyright 2017, Elsevier Ltd.
3.1.4. Polyoxometalate anodic mediators
Polyoxometalates (POMs) have been described in several sections above, most extensively as catalysts/mediators in mediated O2 reduction (see Section 2.2). They have also been used as catalysts and/or mediators for the oxidation of hydrogen, alcohols and biomass in fuel cells, as briefly noted in the previous section. These soluble mediators interact effectively with suspensions of heterogeneous and/or polymeric biomass materials, and display higher activity for C–C bond cleavage and are less easily poisoned by biomass impurities relative to heterogeneous catalysts supported on electrodes.
In connection with studies of redox flow batteries, Oei and Kummer were investigating a series of mediators (e.g., Sn2+/Sn4+, E° = 0.15 V vs. NHE; Cu2+/Cu, E° = 0.153 V vs. NHE; Fe2+/Fe3+, E° = 0.77 V vs. NHE) and testing the ability of the oxidized mediator to be reduced by H2 following electrochemical oxidation.76 Silicotungstic acid (H4SiW12O40) exhibited especially favorable properties, with a reduction potential close to 0 V vs. NHE and an ability to be reduced by H2.63 While the uncatalyzed reduction at pH 0.5–1 is too slow to observe at room temperature, a variety of catalysts, including PtO2, PtCl4, or Pt/SiO2, in addition to tungsten carbide and Pd(4,4′,4″,4‴-tetrasulphophthalocyanine), enhanced the rate. At 75 °C, the silicotungstic acid was reduced to a lesser extent than at 25 °C due to a temperature-dependent shift in its E°, and H2 evolved when a solution reduced at 25 °C was heated to 75 °C. Polarization curves using a VO2+/VO2+ mediated cathode displayed a cell voltage of 568 mV at 80 mA/cm2 current density. In an extended test of the fully constituted system using a Pt/SiO2 catalyst for reduction of H4SiW12O40, good anode performance was maintained over 20 h, and the performance of the system was limited by the cathode performance. A follow-up study revealed improved cell performance using a VO2+/VO2+ mediated cathode with HNO3 as a catalyst, reflecting enhanced cathode system stability (Figure 38).79
Figure 38.

Sustained performance curve of a doubly mediated H2/O2 fuel cell. The cathode utilized a VO2+ mediator reoxidized with HNO3 in 1 M H2SO4 at 75–80 °C, and the anode contained H4SiW12O40 reduced by H2 with a Pt/silica catalyst at 25–30 °C (H2|Pt|H4SiW12O40||VO2+|NOx|O2). The cell was discharged through a 2.1-ohm resistor. Flow rate was 230 mL/min. I0, I4, and I12 correspond to the current at 0, 4, and 12 h. Adapted from ref. 79. Copyright 1985, Chapman and Hall Ltd.
In 2014, Deng and coworkers demonstrated photochemical and thermal oxidation of starch solutions and other biomass sources by POMs at the anode of a MedFC,159 using a conventional Pt/C O2 cathode. The biomass solution with POM was thermally or photochemically pre-treated and then flowed through the anode compartment in a two-stage process (c.f. Figure 30B). Using this approach with H3PMo12O40 as the POM, power densities of 0.28 mW/cm2 (photochemical pretreatment) and 0.44 mW/cm2 (thermal pretreatment) were measured. In a continuous experiment, in which the POM/starch solution was continuously exposed to light irradiation and heated to 95 °C, a steady current density of 2.5 mA/cm2 was observed for over 20 h. Polarization curves using poplar and switch grass as fuels and photochemical pretreatment displayed peak power densities of 0.65 and 0.62 mW/cm2, respectively.
A subsequent report by Deng and coworkers described a MedFC with POM mediators for both the anode and cathode (cf. Figure 12 in Section 2.2):107 H3PW11MoO40 (0.3 M) as the anodic mediator and H12P3Mo18V7O85 (0.3 M) as the cathodic mediator. A two-stage process was used for the anode, in which the H3PW11MoO40 solution was heated with the biomass at 100 °C for 4 h prior to electrochemical treatment and, following this pretreatment procedure, the two POM solutions were passed through their respective electrode compartments and polarization curves were recorded. A variety of biomass sources were tested using this two-stage protocol (Figure 39A). Fresh grass displayed the highest power densities (51 mW/cm2), followed by switchgrass (45 mW/cm2), starch solution (34 mW/cm2), and cellulose (22 mW/cm2). One-stage performance of this system (c.f. Figure 30A) using starch as the fuel source was also demonstrated, displaying a power density of 30 mW/cm2 at a constant discharge current of 160 mA/cm2 for greater than 10 h (Figure 39B). A large portion of the biomass was converted into CO2: 88% of the initial glucose, 82% of the cellulose, and 65% of the lignin was converted to CO2, as measured by total organic content remaining after 12 thermal treatment cycles. Photochemical treatment led to nearly complete conversion to CO2 after 200 hours of photolysis; however, the authors did not report electrochemical performance over these cycles. Overall, these studies highlight the ability of POMs to cleave C–C bonds in biomass while also being capable of regeneration at the anode.
Figure 39.
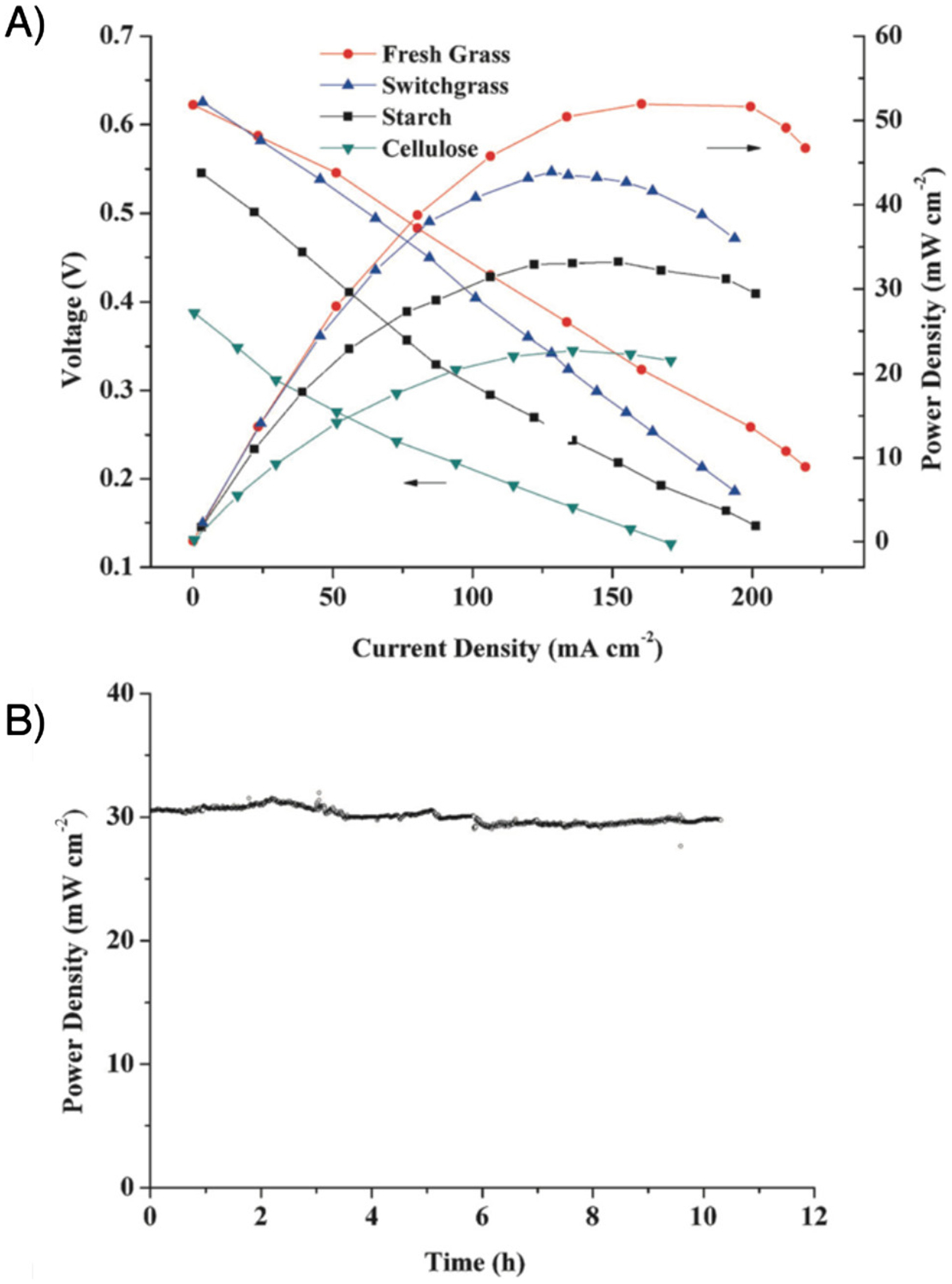
(A) Polarization curves for a variety of biomass sources at 80 °C using 0.3 M H3PW11MoO40 as the anodic mediator and 0.3 M H12P3Mo18V7O85 as the cathodic mediator. (B) Power density measured over 10 h of operation using starch as biomass source. Adapted from ref. 107. Copyright 2014 WILEY-VCH Verlag GmbH & Co.
A similar MedFC was reported by Zhu and coworkers using various lignins as the biomass source and H3PMo12O40 as the POM at the anode with a conventional Pt/C O2 cathode.113 Polarization curves were collected at room temperature following the reaction of a POM/lignin combinations under a variety of conditions (i.e., in a two-stage approach; cf. Figure 30B). Peak power densities approached 0.35 mW/cm2 for reaction mixtures derived from lignin loadings of 25 g/L and a POM concentration of 0.1 M. Replacement of the Pt/C O2 cathode with a POM-mediated cathode system generated power densities of up to 5 mW/cm2 at 90 °C. The POM source for the cathode was identical to that used at the anode (H3PMo12O40), and power generation was achieved from the different redox states of the POM in the two electrode compartments.
In 2016, Deng and coworkers reported a MedFC using polyols as the fuel in a two-stage anodic process.108 The anodic mediator/fuel solution, containing 2 M polyol and 0.2 M H3PMo12O40, was subjected to irradiation for 17.5 h with a 250 W metal halide lamp at 25 °C. This solution was then passed through the anode compartment of the MedFC, which employed a 0.3 M H12P3Mo18V7O85 as a cathodic POM mediator. Using this approach, polarization curves were collected using various alcohol-based fuels. In contrast to conventional fuel cells, in which methanol is typically the best alcohol fuel, the authors showed that glucose performed much better than methanol. The maximum power density attained with glucose was ~ 31.5 mW/cm2, while use of methanol led to a power density of ~8 mW/cm2. The authors also evaluated ethylene glycol, glycerol, erythritol, xylitol, and sorbitol (Figure 40A), and the fuels with more hydroxyl groups led to enhanced power densities at identical fuel concentration (2 M, Figure 40B) or at the same effective hydroxyl concentration of 6 M (i.e. 6 M methanol, 3 M ethylene glycol or 2 M glycerol). UV-visible experiments suggested that increased association between polyhydroxylated fuels and the POM increased the POM reduction by fuels containing more hydroxyl groups. This pre-association was also supported by NMR spectroscopic data. As observed previously,107 carbon dioxide is the major product arising from these POM-mediated photochemical oxidation reactions of polyols.
Figure 40.

(A) Structures of alcohols used as fuels. (B) Polarization curves obtained using anodic mediator solutions prepared by irradiation of solutions containing 2 M of the alcohol and 0.2 M H3PMo12O40 as the mediator. Adapted from reference 108. Copyright 2016, Elsevier B.V.
Chao and coworkers reported a biomass fuel cell with glucose and other sugars as the fuel, using H3PMo12O40 as the anodic mediator and H12P3Mo18V7O85 as the cathodic mediator.112 A two-stage anodic process was used, and a maximum MedFC performance of 40 mW/cm2 was obtained using an anodic solution prepared by photolysis of the POM and glucose solution at 80 °C for 8 hours. Thermal measurements showed that the temperature of the photolysis reaction increased substantially during irradiation, which in turn enhanced the rate of glucose oxidation. The overall efficiency, considering all energy inputs and outputs, of this fuel cell was calculated to be 36.7%. Other sugars, such as fructose and xylose, as well as polysaccharides, such as raffinose and maltose, were found to be nearly as effective of fuels as glucose.
Deng and coworkers reported a MedFC that used coal as a fuel and POM mediators in the anode.109 Lignite, the lowest grade of coal, was used as the coal source, and it was combined with H3PMo12O40 under a variety of conditions to examine reduction of the POM mediator in the first stage of a two-stage anodic process. The best fuel cell results were obtained by treatment of 10 g lignite in 0.1 L of a 0.3 M POM solution at 200 °C for 5 h. The resulting solution of reduced POM mediator was cooled, filtered to remove coal particulates, and then used at the anode of a MedFC operated at 80 °C with a H12P3Mo18V7O85-mediated cathode. The maximum power density observed was 120 mW/cm2. Continuous discharging of the fuel cell using a single batch of reduced anodic POM solution was conducted at 150 mA/cm2, and the maximum power density decreased from 60 to ~30 mW/cm2 over 10 h. The coal oxidation was repeated three times with the same batch of coal, consuming approximately 9% of the coal mass in each case. Diminished performance was observed on the third run. Characterization of the coal residue after the third reaction showed a decrease in the total organic carbon, indicating oxidation of the coal to CO2 or water-soluble low molecular weight species, and 13C NMR spectroscopic data suggested that aliphatic-like carbonaceous material was preferentially oxidized over aromatic content.
Deng and coworkers also reported a MedFC that utilized sewage sludge as the fuel.111 This system employed H3PMo12O40 as the anodic POM mediator and H12P3Mo18V7O85 as the cathodic POM mediator. The authors examined the role of temperature and treatment time on the oxidation of the sewage sludge by H3PMo12O40. Power densities of 13.56 mW/cm2, 16.07 mW/cm2, and 17.91 mW/cm2 were obtained from anodic solutions prepared by treatment of the sewage sludge at 80 °C for 6 h, 12 h, and 24 h, respectively. Further improvements in power densities were achieved by treatment of the sludge for 24 h at 100 °C and 150 °C (49.8 mW/cm2 and 61.4 mW/cm2, respectively; Figure 41). The first oxidation treatment removed over 60 wt% of the sludge sample, which increased to 85 wt% after a second treatment. The authors additionally showed that the POM could undergo repeated cycling with addition of fresh fuel after each cycle. Four cycles were conducted with each showing similar performance.
Figure 41.

Polarization curves for sewage sludge-powered mediated fuel cell using H3PMo12O40 as anodic mediator and H12P3Mo18V7O85 as cathodic mediator. Sewage sludge was treated with H3PMo12O40 for 6, 12, 24, or 36 h at (a) 80 °C, (b) 100 °C, or (c) 150 °C prior to the fuel cell experiment. Adapted from reference 111. Copyright 2017 Elsevier Ltd.
In 2017, Zhao and coworkers demonstrated the oxidation of lignin in wheat straw to produce electricity using a two-stage process incorporating H3PMo12O40 as an anodic mediator.160,161 Enzyme-digestible carbohydrates were obtained as a by-product of the biomass oxidative pretreatment. The extent of deliginification, the level of xylan and glucan dissolution, the ability of the remaining glucans to be enzymatically hydrolyzed, and the degree of reduction of the POM were assessed under a variety of conditions. Solutions containing reduced POM were introduced into the anode compartment of a MedFC, in which the cathode solution contained a solution of FeCl3. After testing a variety of conditions, the maximum power density obtained was approximately 11 mW/cm2.
POMs have also been utilized as mediators for biomass oxidation in combination with electrochemical production of H2 and CO2 reduction at the cathode. In these systems, instead of reducing O2 at the cathode to generate power, power is applied to the system and either protons are oxidized to produce H2 (Figure 42)156,162–163 or CO2 is reduced to CO and hydrocarbons.164 The anodic performance of these systems is quite similar to the fuel cell systems.
Figure 42.

Schematic of electrochemical H2 evolution using biomass as H2 source and POM-based mediators. Adapted from reference 162 with permission from The Royal Society of Chemistry. Copyright 2016 The Royal Society of Chemistry.
3.1.5. Other transition metal-based mediators
Two additional examples of transition metal-based mediators have been only briefly studied. The first case was reported by Kummer and Oei, who were searching for a mediator solution for H2 oxidation with higher volumetric charge capacity than silicotungstic acid (c.f. Section 3.1.4).79 Their work was inspired by a precedent for 2 e− oxidation of a trinuclear molybdenum ion, [(Mo3+)2Mo4+], to [Mo4+]3 with an E° very close to 0 V vs. NHE. Using Pt catalysts, a solution containing Mo6+ was cleanly reduced to Mo4+ by H2, but only half of the species were further reduced to the Mo3+ state, compared to the expected value of two-thirds. This outcome fell short of expectations, but it still led to more reduced metal sites than the one-twelfth observed in silicotungstic acid. When used in a fuel cell along with a VO2+/VO2+-mediated, NOx-catalyzed O2 cathode (H2|Pt|Mo3+||VO2+|NOx|O2), a current density of 85 mA/cm2 at approximately 550 mV (iR-free voltage of 725 mV) was obtained. No long-term stability experiments were reported using this mediated anode.
In 2018, Bullock and coworkers reported a fundamental study of electrochemical H2 oxidation under non-aqueous conditions using a mediator/co-catalyst system comprising Cp*Cr(CO)3 and an Fe(P2N) complex (Figure 43).165 In this report, H2 underwent homolytic cleavage by two equivalent of Cp*Cr(CO)3 to form two Cp*Cr(CO)3H species. This complex can transfer an H-atom to [Fe(PEtNPhPEt)(CO)3]+, and the resulting species undergoes deprotonation and electrochemical oxidation to regenerate the five-coordinate Fe species. A catalytic wave was observed by cyclic voltammetry at the Fe+/0 redox wave, and it increased with higher 2-methylpyridine concentration. Each complex was required for competent H2 oxidation, and controlled potential electrolysis with 2,6-di-tert-butylpyridine as a base gave a lower limit of 5 turnovers for Co and 21 turnovers for Fe.
Figure 43.
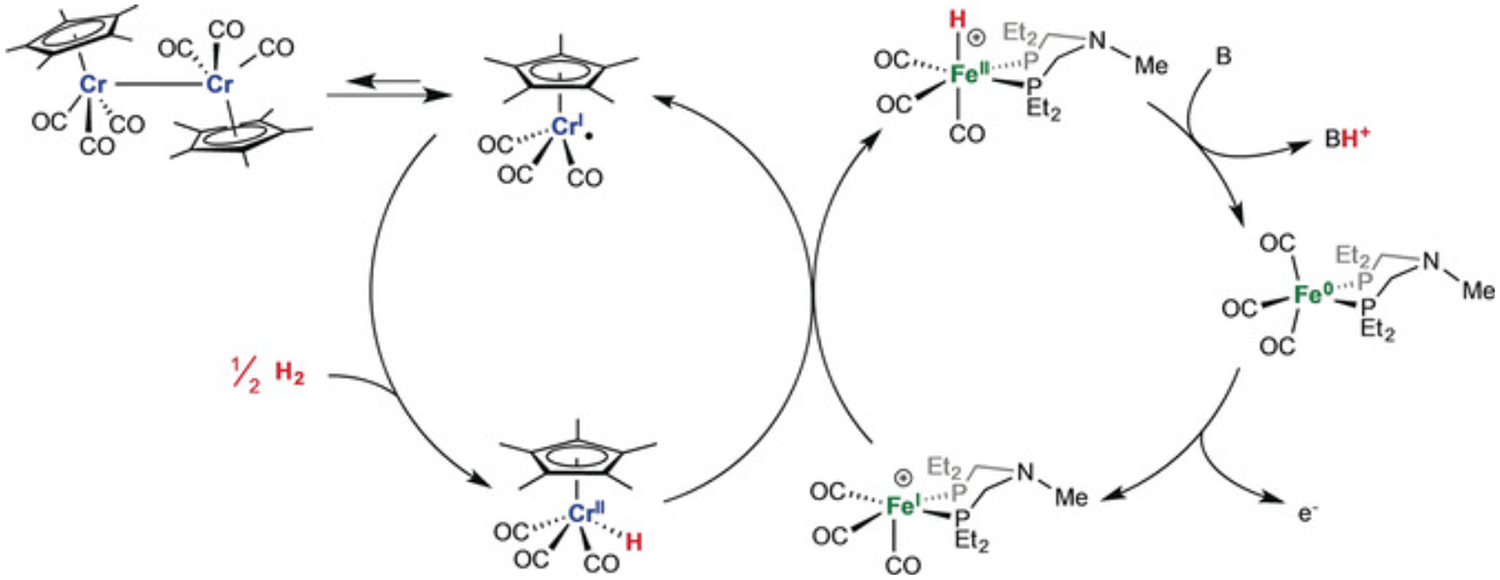
Proposed mechanism for electrochemical H2 oxidation using Cp*Cr(CO)3/Fe(P2N) system. Adapted from reference 165. Copyright 2018 Wiley-VCH Verlag GmbH & Co.
3.1.6. Quinone/Hydroquinone mediators
In the mid-1980s, Weetall and coworkers at Corning Glass Works investigated the use of various biomass sources as fuels for a MedFC, using sulfonated anthraquinone (AQ) derivatives as anodic mediators. They examined simple alcohols,166 carbohydrates,167 and sulfonated lignin or Kraft Black liquor168 (a byproduct of wood-pulping) as fuels. To facilitate the oxidation of the alcohol or carbohydrate fuels, visible light irradiation was necessary to excite the anthraquinone mediator. Basic (pH > 12) conditions were used. With ethylene glycol as the fuel (10 volume %), currents of approximately 1 mA were observed, corresponding to a current density of about 0.1 mA/cm2. No potentials, power densities, or sustained performance data for simple alcohol fuels were given. More thorough analysis was performed when carbohydrates, such as glucose, sucrose, or unpurified molasses were used. The highest power output was obtained with glucose as a fuel (0.57 mW/cm2), followed by invertase-treated sucrose (0.43 mW/cm2) and fructose (0.41 mW/cm2). In each of these experiments, 6 mM anthraquinone sulfonate was used as mediator. The authors calculated that 6.4 pounds of glucose would be required to generate one kWh of energy using their cell setup. Under similar conditions using lignin-based fuels, peak power densities of approximately 0.34 mW/cm2 were obtained. Further experiments indicated that the initial polymeric lignin material was converted into low-molecular weight fragments during fuel cell operation, though the identities of these fragments was not further investigated. It is likely that the performances of the cells could have been substantially increased through further engineering of the cell design. For example, the authors observed a 50% increase in currents merely by mixing the system.
Stahl and coworkers reported a MedFC utilizing a water-soluble anthraquinone mediator, anthraquinone-2,6-disulfonic acid (AQDS) in 1 M H2SO4 paired with an off-electrode Pt/C catalyst for mediated electrochemical H2 oxidation.105 The fuel cell was initially constructed using a conventional Pt/C O2 cathode; however, substantial mass-transport losses were observed in the polarization curves due to cathode flooding. To address these concerns, a POM-mediated cathode utilizing 0.3 M Na4H3PMo8V4O40 was used (Figure 44). Polarization curves were obtained, and a peak iR-free power density of 528 mW/cm2 was reported (228 mW/cm2 without iR-correction). The system also displayed sustained performance at a current density of 50 mA/cm2 for over 8 h. These MedFC performance metrics are substantially lower than those of conventional PEMFCs, but considerable better than metrics achieved with previous applications of MedFCs using H2 as the fuel (cf. sections 3.1.3, 3.1.4, and 3.1.5) Moreover, formic acid and methanol were shown to reduce the AQDS mediator using the Pt/C catalyst, suggesting this approach could potentially be adapted to achieve improved fuel cell performance with fuels more complex than H2.
Figure 44.

Schematic of MedFC utilizing AQDS as an anodic mediator for H2 oxidation with a Pt/C catalyst and a POM-mediated cathode. Adapted from reference 105.
3.1.7. V3+/VO2+ mediator
A patent published in 1997 by Larsson and Folkesson reported a MedFC using vanadium salts and a Pt catalyst to oxidize glucose to CO2 as the anodic reaction (Figure 45) and VO2+/VO2+ as the cathodic redox couple (glucose|Pt|VO3+||VO2+|catalyst|O2).169 In this cell, VO2+ was reduced to V3+ (with some further reduction to V2+). Using vanadium species in both the cathode and anode eliminated concerns with mediator crossover, resembling advantages evident in all-vanadium redox flow batteries.74 A cell capable of producing 10 kW with a power density of 10.5 mW/cm2 (cell voltage of 0.7 V with current density of 15 mA/cm2) was developed, using an anode solution of 180 liters containing 0.3 M VOSO4, 5 M H3PO4, 0.003 M sugar, and 229 g Pt. Overall, 4 stacks were connected in parallel, with each stack consisting of 130 cells in series, in order to deliver the desired power output. A further kinetic study of the oxidation of sugars by VO2+ was reported by the same authors in 2005.170 They found that low concentrations of sugar were necessary for complete oxidation to CO2. A variety of sugars (sucrose, fructose, glucose, arabinose, xylose) all behaved similarly. Continued optimization of this cell led to power conversion efficiencies of 41–45%, which were suggested to exceed efficiencies accessible by thermal combustion of the sugar.81
Figure 45.

Redox scheme for V3+-mediated anode using glucose fuel and Pt catalyst. See refs. 169–170.
3.1.8. Viologen-based mediators
Viologens (1,1’-dialkyl-4,4’-bipyridyls) are cationic, low-potential, redox-active organic species that have been used as mediators for the oxidation of glucose and other carbohydrate derivatives under basic conditions. In 2009, a pair of publications, one from Wheeler and coworkers171 and one from Scott and Liaw,172 reported fuel cells incorporating viologens as mediators for electrochemical glucose oxidation.
Wheeler and coworkers focused on the oxidation of glucose by methyl viologen (MV2+, Figure 46A) under mildly basic conditions (pH 9–12).171 Anaerobic experiments indicated ~8 equiv of MV2+ were reduced in the oxidation of glucose when high MV2+:glucose ratios were used, compared to the 24 equiv theoretically necessary for full oxidation. However, exposure to O2 under these conditions led to enhanced consumption of glucose, and, at a 12:1 MV2+/glucose ratio, significant amounts of carbonate and formate were observed. Preliminary experiments with a simpler substrate (dihydroxyacetone) indicated that a fuel cell could operate using MV2+ as a mediator and a cathode containing a cobalt oxide-based catalyst originally developed for zinc-air batteries. Liaw and coworkers demonstrated improved performance in a related fuel cell,172 showing that with 3 M KOH, 2 M glucose, and 28 mM MV2+, a power density of 2.5 mW/cm2 could be obtained with an air-breathing cathode (Figure 46B–C). Liaw proposed that glucose oxidation proceeded only to d-gluconolactone. However, no characterization data for this proposed species was reported. In a later study, Liaw and coworkers provided spectroscopic evidence that the major product of glucose oxidation was gluconic acid when indigo carmine was used as a mediator.173 One explanation for the discrepancy between the fate of glucose between the Liaw and Wheeler reports is the difference in solution basicity used in these two studies, which may influence the reaction pathways during glucose oxidation.
Figure 46.

A) Structure of methyl viologen. B) Schematic of MV2+-mediated (“Dye2+”) fuel cell using glucose fuel and air breathing cathode. C) Polarization curve for glucose-O2 fuel cells with MV2+ as anodic mediator. Cell contained 2 M glucose and 28 mM MV2+ in 3 M KOH solution. Adapted from ref. 172. Copyright 2009, The Royal Society of Chemistry.
A later paper, published in 2013 by Liu and coworkers, reported activities similar to those of Liaw and coworkers for their fuel cell using MV2+ as a mediator for glucose oxidation.174 Further work was performed by Liu, Zhang, and coworkers, who reported a power density of 0.52 mW/cm2 at room temperature with 1 M glucose, 3 M KOH, and 15 mM MV2+.175 They detected the formation of both acetate and formate products in their cell.
Wang, Liu, and coworkers reported the use of cellulose as a fuel with a MV2+ mediator using a solution containing 5 M NaOH, 1 wt% (w/v) cellulose, and 15 mM MV2+.176 This system achieved a peak power density of 45 μW/cm2. The degradation products of cellulose oxidation were identified by HPLC as oxalic acid, glucaric acid, glyceric acid, glycolic acid, formic acid, and acetic acid.
To increase the performance of a glucose-powered fuel cell using viologen mediators, Watt and coworkers investigated the mechanisms of glucose oxidation and mediator degradation.177 Under mildly basic conditions, they identified the reaction of an enediol formed from glucose under basic conditions with MV2+ as the important step for selectivity. High selectivity required high MV2+ loadings to prevent the formation of inactive intermediates. Additionally, it was noticed that dialkyl viologens decomposed under basic conditions (pH > 13) to monoalkyl viologens, so the activity of monoalkyl viologens for glucose oxidation was examined.178 While glucose oxidation proceeded approximately 50% slower using monoalkyl viologens, the monoalkyl viologens displayed substantially higher stability than the dialkyl variants. In this initial report, higher OCP values also were reported for the monomethyl viologen (0.9 V) than for the dimethyl viologen (0.72 V).179 Further kinetic studies were undertaken, and increased temperatures were found to substantially increase the rate of alcohol oxidations by methyl viologen and mono-methyl viologen.180 To assist in the recovery of the viologen mediators, Watt and coworkers also examined the ability of soluble, polymeric viologens to act as mediators in glucose oxidation. Less basic conditions (pH = 11) were required to prevent decomposition of the viologen polymers.181 While reoxidation with O2 allowed for effective glucose oxidation, attempts to perform the reoxidation electrochemically led to precipitation of the mediators from solution.
3.2. Fuel oxidations using enzymatic catalysts
3.2.1. Introduction
Enzymatic fuel cells are a broad class of devices, in which enzymes are used as catalysts for oxidant reduction or fuel oxidation.55–57 Enzymatic fuel cells are different from microbial fuel cells, which use entire organisms to catalyze the desired reactions.59 While the enzymes are occasionally used as soluble species in solution, they are typically tethered or otherwise immobilized onto the electrode. Some enzymes can undergo direct electron transfer at the electrode, but the buried active sites of many enzymes often leads to slow direct electron transfer. This problem is often addressed through the use of mediators, which can be soluble or immobilized on the electrode. Immobilization of both the enzyme and mediator is most commonly employed, as it has the benefit of removing the need for a membrane, thereby facilitating the miniaturization of enzymatic fuel cells, and minimizes the chance of releasing toxic mediators in implantable or wearable devices. Historical examples of common immobilized mediators include osmium- or ruthenium-containing redox polymers,182 and in recent years redox polymers containing organic mediators, such as quinones, viologens, or phenothiazines have been developed.183 The discussion here emphasizes dissolved mediators, analogous to the chemical systems described above. Most enzymatic fuel cells operate at or near ambient conditions (room temperature or slightly above) at moderate or neutral pH values and generate low amounts of power. Power densities are typically ≤1 mW/cm2, commensurate with that appropriate for implantable devices or sensors. A summary of enzymatic fuel cells using at least one soluble mediator is given in Table 8.
Table 8.
Enzymatic fuel cells using at least one soluble mediator in the anode or cathode.
| fuel | catalyst | mediator | mediator | catalyst | oxidant | ref |
|---|---|---|---|---|---|---|
| H2 | Pt | – | ABTS2− | laccase | O2 | 146 |
| Desulfovibrio vulgaris | viologen | bilirubin oxidase | 149 | |||
| glucose | glucose oxidase | Fc(CH2OH) | bilirubin oxidase or laccase | 147–151 | ||
| glucose | glucose oxidase/AuNP-graphene | – | laccase/AuNP-graphene | 152 | ||
| BPA | laccase | – | laccase | 153 | ||
| glucose | glucose dehydrogenase | PS-βCD-PQ | PS-βCD-P2ABTS | bilirubin oxidase | 154 | |
| MeOH | methanol dehydrogenase | PMS or PES | – | Pt | 184–185 | |
| MeOH | quinoprotein alcohol dehydrogenase | TMPD | 186 | |||
| glucose | glucose oxidase | Os polymer | laccase | 188 | ||
| MeOH | multi-enzyme cascade | BV2+ +NADH | – | Pt | 190 | |
| glucose | glucose oxidase | DCIP | 189 | |||
| quinone | 195 | |||||
| glucose-6-phosphate | G6PDH and diaphorase | quinone + NADH | 196 | |||
| MeOH | alcohol dehydrogenase | TMPD | – | KMnO4 | 187 | |
| H2 | NiFe hydrogenase | MV2+ | MV2+ | MoFe Nitrogenase | N2 | 193 |
3.2.2. Phenazine-based mediators
Hill and coworkers reported an enzymatic fuel cell in which methanol dehydrogenase was used to oxidize methanol to formate with phenazine methosulfate (PMS) or phenazine ethylsulfate (PES) as a mediator (Figure 47A)184 in combination with a conventional Pt/C O2 cathode. PES exhibits greater stability and was shown to give better performance. The fuel cell was operated at pH 9.5 and temperatures from 25–30 °C. The current reached a saturation limit at higher concentrations of mediator and enzyme, and sustained operation was examined with two loadings of the enzyme (Figure 47B). The open circuit potential was ~0.3 V, and a maximum current of ~3 mA (corresponding to a current density of 0.4 mA/cm2 based on the reported Pt electrode dimensions) was obtained across a 10 W resister with 4.6 mg enzyme, 3.5 mM PES, and 62 mM MeOH in 4 mL anode solution. Over the course of 80 min of fuel cell operation, the current dropped to ~1.5 mA (~0.2 mA/cm2). The authors suggested that this decrease could be due to PES blocking of proton flow through the membrane. A pH difference was observed between the anode and cathode compartments at the end of cell operation, supporting this conclusion.
Figure 47.

(A) Redox scheme for PES-mediated anode using methanol as a fuel and methanol dehydrogenase enzyme as catalyst and structures of PMS and PES. For systems using this mediated anode, see refs. 184–185. (B) Current output of mediated anode fuel cell using PES as anodic mediator. Anode compartment contained 2.3 mg (dashed line) or 4.6 mg (solid line) methanol dehydrogenase, 0.35 mL of 40 mM PES in water, 3.6 mL of buffer, and 10 μL methanol. Adapted from reference 184. Copyright 1981 Science and Technology Letters.
Later, Yue and Lowther revisited this system and attempted to improve on the results by including the enzyme formate dehydrogenase to oxidize the formate product obtained from methanol dehydrogenase.185 The authors used conditions similar to those of Hill and coworkers, but they incorporated a carbon anode instead of a Pt mesh anode. The authors selected PMS as the mediator since they observed approximately 10% higher activities for MeOH and formaldehyde oxidation with PMS than with PES in control experiments. Over the course of a 60 min experiment using immobilized enzymes and soluble PMS, the current decreased from 2.14 mA (0.054 mA/cm2) to 1.57 mA (0.039 mA/cm2). It is unclear why the current densities of this system were substantially lower than those of Hill and coworkers. Further testing indicated that the formate dehydrogenase enzyme used in these experiments led to negligible current (only 0.03 mA) when formic acid was directly used as a fuel. This result contrasted the enzyme activity tests, which showed similar activity for formic acid oxidation catalyzed by formate dehydrogenase using PMS and for methanol oxidation catalyzed by methanol dehydrogenase with PMS.
3.2.3. Tetramethyl-4-phenylenediamine mediator
In an effort to incorporate mediators more stable than PES or PMS (see section 3.2.2), Hill and coworkers examined a variety of organic species as oxidants for methanol oxidation catalyzed by the quinoprotein alcohol dehydrogenase.186 Through these efforts, N,N,N’,N’-tetramethyl-4-phenylenediamine (TMPD, Figure 48A) was identified as the best mediator. Cyclic voltammetry studies at pH 10.5 showed substantial catalytic activity at the TMPD reduction potential in the presence of methanol and enzyme. In the absence of substrate or enzyme, a reversible 1 e− wave was observed at 0.01 V vs. SCE (+ 0.25 V vs. NHE). The stability of the TMPD mediator was then compared to PES in a fuel cell. Over a 6 h electrolysis at pH 10.5, the current decayed nearly to zero when PES was used as the mediator (Figure 48B, trace B). When TMPD was used at the mediator under the same conditions, the current remained stable over the entire 6 h electrolysis (Figure 48B, trace A), and decreased by less than 10% over 24 h. Polarization curves were collected, and a peak power of 12 μW was obtained, corresponding to a power density of 2 μW/cm2. The low power density likely reflects, at least in part, the high mediator potential. While no open-circuit potential was reported, the potential at the lowest current indicated was only ~160 mV. The authors noted that this cell was not limited by the cathode chemistry, since no fuel cell improvement was observed when pure O2 was purged into the cathode compartment rather than air. Formate dehydrogenase was included to oxidize the formate since formate inhibits the alcohol dehydrogenase, but poor enzyme performance at the relevant pH led to no improvement in the fuel cell performance.
Figure 48.

(A) Structure of N,N,N’,N’-tetramethyl-4-phenylenediamine (TMPD). (B) Current output of a fuel cell using A) 4.0 mM TMPD or B) 4.0 mM PES with alcohol dehydrogenase as the enzyme and methanol as fuel. Adapted from reference 186. Copyright 1983 Elsevier Inc.
Ranta and coworkers also studied the stability of TMPD in fuel cells utilizing MeOH/methanol dehydrogenase at pH 10.187 A KMnO4 solution was used as the oxidant at the cathode. Polarization curves revealed a peak power density of 0.25 mW/cm2 at a current density of 0.38 mA/cm2 and cell voltage of 0.67 V. Most of the polarization losses arose from the anode. The product of methanol oxidation with this enzyme is formate, which led to a decrease in pH that led to a decrease in the system performance during extended operation. TMPD decay (via dimerization or oligomerization) was observed, with an estimated half-life of approximately 5 days during fuel cell operation.
In 2009, Leech and coworkers reported an enzymatic fuel cell using a glucose oxidase enzyme as a catalyst for glucose oxidation with TMPD as a mediator.188 The cathode consisted of a laccase enzyme co-immobilized with an osmium redox polymer acting as a redox mediator. This study primarily focused on the cathode reaction, but the authors required a highly active anode catalyst and mediator combination. A maximum power density of 52 μW/cm2 was observed, with the performance of the system being limited by the cathodic reaction.
3.2.4. Dichlorophenolindophenol mediator
In 1984, Laane and coworkers reported a biofuel cell using glucose as a fuel and D–glucose oxidase as the catalyst with a dichlorophenolindophenol (DCIP, Figure 49A) mediator.189 Glucose was oxidized to gluconic acid, which the authors identified as a valuable industrial chemical. In their initial studies, O2 reduction to H2O on a Pt electrode was the cathodic reaction, and sustained currents of 0.3 mA were obtained for over 200 min of cell operation. In a later experiment, a gold cathode was used to reduce O2 to H2O2, which the authors used to oxidize barbituric acid to 5-chlorbarbituric acid with chloroperoxidase as a catalyst (Figure 49B). Limited information was given about the power generation capabilities of this system. Over 3 days of operation, 10 mg gluconic acid and 8 mg 5-chlorobarbituric acid were generated, corresponding to 1.8 × 104 turnovers for D-glucose oxidase.
Figure 49.

(A) Structure of dichlorophenolindophenol (DCIP). (B) Schematic of biofuel cell used for the synthesis of 5-chlorobarbituric acid. The anode was mediated by DCIP. Adapted from reference 189. Copyright 1984 Elsevier Inc.
3.2.5. Viologen-based mediators
In efforts to increase the power densities of an enzymatic fuel cell using MeOH as a fuel, Palmore and coworkers were interested in decreasing the overpotential between the anodic mediator and MeOH.190 Previously reported mediators typically have overpotentials of at least 700 mV (cf. sections 3.2.2. and 3.2.3.), presumably because lower overpotential mediators were not sufficiently active with the chosen enzymes. However, the high potentials greatly reduce the potential power output of the fuel cell. To enable the use of lower-potential mediators, three NAD+-dependent enzymes were used to catalyze the oxidation of methanol to CO2: alcohol dehydrogenase (ADH), aldehyde dehydrogenase (AldDH), and formate dehydrogenase (FDH). Three equivalents of NAD+ were required to complete the 6 e− oxidation of MeOH. The NAD+/NADH redox couple has slow electrode kinetics, however, that often require overpotentials of >1 V to achieve suitable electron-transfer rates.191 To facilitate the oxidation of NADH, the enzyme diaphorase was used with another soluble mediator, benzyl viologen (BV2+), and a fuel cell was constructed using these mediators and enzymes in the anodic compartment and a Pt-black O2 cathode (Figure 50). The cell exhibited an OCP of approximately 0.8 V, and polarization measurements exhibited a peak power density of 0.68 mW/cm2. The rate-determining step in the fuel cell was identified as the oxidation of NADH to NAD+, catalyzed by diaphorase with BV2+ as the oxidant.
Figure 50.

Schematic of a methanol/oxygen biofuel cell, in which methanol is oxidized to CO2 by NAD+-dependent dehydrogenases ADH, AldDH, and FDH. Diaphorase (D) catalyzes the oxidation of NADH to NAD+ with benzylviologen, BV2+, as an electrochemical mediator. Adapted from reference 190. Copyright 1998 Elsevier S.A.
Enzymes have been extensively studied for electrocatalytic H2 oxidation,192 but most of these studies utilize heterogenized hydrogenase enzymes. Recently, Minteer and coworkers reported a fuel cell using a solubilized hydrogenase enzyme and methyl viologen (MV2+) as a mediator.193 N2 reduction, catalyzed by nitrogenase and mediated by MV2+, was performed at the cathode (Figure 51). With the same mediator used in both anode and cathode, this cell would not supply power at equilibrium, but the shift in equilibrium concentrations caused by substrate oxidation/reduction yielded a potential difference. This cell demonstrated an OCP of 228 mV and achieved a maximum power density of 1.50 μW/cm2. Dinitrogen was reduced to ammonia in this fuel cell, with an overall yield of 286 nmol NH3/mg MoFe protein and a cathodic Faradaic efficiency of 26.4%. This enzymatic fuel cell is noteworthy for its ability to produce NH3 from N2 and H2 while also generating electricity. The authors also demonstrated that an anode consisting of glucose directly oxidized by MV2+ under carbonate buffer (pH=11) conditions was also feasible (see section 3.1.7). The OCP under these conditions was 238 mV and the maximum power density was 0.76 μW/cm2.
Figure 51.

Schematic of biofuel cell using methyl viologen as a mediator in both anode and cathode. The cell produces NH3 from N2 reduction in the cathode compartment. Adapted from ref. 193. Copyright 2017, John Wiley & Sons, Inc.
3.2.6. Ferrocene-based mediators
Ferrocene derivatives, such as ferrocene methanol or ferrocene carboxylic acid, have found widespread use as mediators for enzymatic reactions, though many of these examples employed in sensing applications rather than power generation.194 Dong and coworkers employed a porous carbon electrode with an immobilized glucose oxidase enzyme was paired with ferrocene monocarboxylic acid as a mediator in the anodic compartment of an enzymatic fuel cell.147 The cathode compartment contained a laccase enzyme and ABTS2− as the mediator. A peak power density of 99.8 μW/cm2 at pH 4 decreased to 2.0 μW/cm2 at pH 7 due to the lower activity of the laccase enzyme in the cathode compartment at higher pH (see section 2.7 for further discussion).
Dunn and coworkers reported an enzymatic fuel cell with glucose oxidase immobilized in a carbon nanotube-containing sol-gel matrix, using ferrocene methanol as a mediator and glucose as the fuel.150 The cathode compartment contained immobilized bilirubin oxidase and ABTS2− as a soluble mediator for O2 reduction (see Section 2.7). An open circuit potential of 480 mV was obtained with this cell. This value is substantially larger than the difference in redox potentials between ABTS2− (0.480 V vs. Ag/AgCl) and (Fc)CH2OH (0.21 V vs. Ag/AgCl). The authors suggested that glucose oxidase had sufficient electron transport to the electrode to influence the open circuit potential (the potential of the enzyme was reported to be −0.340 V vs. Ag/AgCl). The system displayed minimal activation polarization, suggesting that this electron transfer continues to be reasonably facile. Similar work was published by Xu and coworkers, using ABTS2− as a mediator with laccase at the cathode and glucose oxidase with ferrocene carboxylic acid as the anodic mediators.151
3.2.7. Quinone/Hydroquinone mediators
Wang, Kim, and coworkers reported a biofuel cell using glucose oxidase attached to carbon nanotubes with 1,4-benzoquinone as a soluble mediator.195 Polarization tests using a conventional Pt/C O2 cathode indicated minimal cell polarization up to current densities approaching 0.1 mA/cm2, until mass-transport limitations led to a significant decrease in performance (Figure 52). A maximum power density of 77 μW/cm2 was observed. The enzyme activity was suggested to be capable to supporting power densities approaching 270 μW/cm2.
Figure 52.

Polarization curve of glucose/O2 biofuel cells using benzoquinone as an anodic mediator with a CNT-GOx bioanode. Adapted from reference 195. Copyright 2009 Wiley Periodicals, Inc.
Zhu and Zhang reported the use of a water-soluble anthraquinone derivative, 2,7-anthraquinone-disulfonic acid (AQDS), as a mediator for oxidation of glucose 6-phosphate using glucose 6-phosphate dehydrogenase (G6PDH) and diaphorase as enzymatic catalysts.196 NADH was used to mediate electron transfer between glucose 6-phosphate dehydrogenase and diaphorase. The authors compared the use of soluble mediators and enzymes with immobilized mediators and enzymes (Figure 53A), including (a) immobilized enzymes and an immobilized naphthoquinone mediator, menadione (VK3), (b) soluble enzymes with immobilized VK3, and (c and d) soluble enzymes with either soluble benzyl viologen or soluble AQDS. The results with AQDS as a soluble mediator and non-immobilized enzymes gave the best performance, as determined from polarization curves, with a maximum power density of 1.1 mW/cm2 at room temperature. This value could be increased to 2.4 mW/cm2 at 37 °C. Long-term performance tests showed that the cell with AQDS and immobilized mediators continued to display optimal activity, though overall the performance decreased by 65%, while a cell with immobilized mediator and enzymes only decreased by 35% (Figure 53B).
Figure 53.

A) Schematic diagrams of enzymatic fuel cell anodes with glucose 6-phosphate as the fuel. The four formulations included (a) immobilized enzymes with immobilized vitamin K3 mediator, (b) soluble enzymes with immobilized vitamin K3 mediator, and (c and d) soluble enzymes with free benzyl viologen (BV) or free AQDS mediators. G6P = glucose 6-phosphate, G6PDH = glucose 6-phosphate dehydrogenase, DI = diaphorase. B) Current vs. time plots for the four O2/glucose 6-phosphate fuel cell strategies. Cell was discharged through a 150-ohm resistor. Fresh G6P substrate was added every 3 days. The electrolyte contained 5 mM mediator, 20mM G6P, 8mM NAD+, 0.1 M pH 7.3 HEPES buffer, 10mM MgCl2, 0.5mM MnCl2, 0.1 M NaCl, 2 U/mL G6PDH, and 2.7 U/mL diaphorase. Adapted from reference 196.
Cosnier and coworkers encapsulated 9,10-phenanthrenequinone in a glyconanoparticle (PS-bCD-PQ) and used fungal FAD-dependent glucose dehydrogenase for the oxidation of glucose in a “solubilized enzymatic fuel cell”.154 This encapsulation increases the concentration of the hydrophobic quinone mediator from ~ 5 μM to 68.4 μM (for a 2:1 quinone/glyconanoparticle ratio). Discussion of the performance of the assembled fuel cell is provided in Section 2.7.
3.2.8. Nitroxyl/Oxoammonium mediators
Minteer, Sigman, and coworkers use a nitroxyl-based mediator in combination with an enzyme for the complete oxidation of glycerol to CO2. The proposed pathway features non-enzymatic nitroxyl-mediated oxidation of glycerol to mesoxalic acid, followed by decarboxylation of mesoxalic acid by oxalate oxidase.197 Further nitroxyl-mediated (non-enzymatic) oxidation of glyoxylic acid forms oxalic acid, which undergoes enzymatic oxidation to CO2. Most nitroxyl mediators were found to be inactive as oxidation catalysts under the acidic conditions required for oxalate oxidase (i.e., pH 4–5), but 4-amino-TEMPO exhibited suitable activity. Bulk electrolysis studies with glycerol, 4-amino-TEMPO and oxalate oxidase at pH 5.2 generated current densities up to 1.2 mA/cm2. Intermediates such as glyceric acid, tartronic acid, mesoxalic acid, and glyoxylic acid were detected by HPLC. Using 13C-labeled glycerol, 13CO2 was also detected by NMR. This mediated electrochemical system is different from the other enzymatic processes discussed in this article in that the organic nitroxyl mediator does not shuttle electrons between the enzyme and the electrode, but rather directly mediates electrochemical oxidation of the fuel (glycerol). The enzyme processes intermediates derived from glycerol oxidation that are not amenable to nitroxyl-mediated oxidation, and thereby enables complete oxidation of glycerol to CO2.
Oxalate oxidase requires O2 to support decarboxylative production of CO2 from mesoxalic acid and oxalic acid. Consequently, the four electrons transferred in these steps are not captured electrochemically. To address this issue, 4-amino-TEMPO was paired with other enzymes capable of promoting non-oxidative decarboxylation. Such performance was achieved with oxalate decarboxylase, a redox-neutral enzyme that enabled collection of all possible electrons in the electrochemical oxidation of glycerol (Figure 54).198 Another study employed the NAD-dependent formate hydrogenase with 4-amino-TEMPO to oxidize glycerol to CO2.199 Unlike oxalate decarboxylase and oxalate oxidase, which require acidic pH (e.g., pH 4–5), formate hydrogenase is effective at higher pH (e.g., pH 8) where the 4-aminoTEMPO exhibits higher activity. The catalytic activity of the combined system, with NAD+, formate hydrogenase, and 4-amino-TEMPO, for the oxidation of mesoxalate was evident by cyclic voltammetry. Removal of any of the components led to significant or complete loss of catalytic activity. Complete oxidation of glycerol to CO2 from bulk electrolysis was confirmed by NMR.
Figure 54.

Electro-oxidation cascade pathway for the complete oxidation of glycerol to CO2 catalyzed by TEMPO-NH2 and oxalate decarboxylase. The red arrows indicate the oxidations by TEMPO-NH2. Adapted from reference 198. Copyright 2015 The Royal Society of Chemistry.
One problem in using nitroxyl species as mediators for fuel oxidation at the anode of a fuel cell is the high redox potential of these species, which limits the cell voltage that can be achieved in a fuel cell. In an effort to identify nitroxyls with a more suitable redox potential while retaining high rates of electrocatalytic alcohol oxidation, Sigman, Minteer, and coworkers developed a structure-activity relationship for nitroxyl catalysts.200 The catalytic activity of the nitroxyl radical was found to be empirically correlated with the potential difference between proton-coupled oxidation of the (protonated) hydroxylamine and oxidation of the nitroxyl. By analyzing this relationship, the authors were able to assign different nitroxyl species to one of four groups, associated with their catalytic activity (y-axis) and their nitroxyl/oxoammonium redox potential (x-axis) (Figure 55A). The four groups correspond to (a) good anodic catalysts (low redox potential and high catalytic activity), (b) good synthetic catalysts (high potentials and high catalytic activity), (c) anti-oxidants (low catalytic activity and low potential), and (d) EPR labels (high potential and low catalytic activity). From this analysis, two species were highlighted as optimal targets for fuel cell applications (numbered 14 and 19 in the original study) (Figure 55B).
Figure 55.

A) Plot of catalytic activity (ipa/ipc)cat vs. Ea1 (nitroxyl/oxoammonium redox potential) separated into four quadrants based on catalyst characteristics. Adapted from reference 200. Copyright 2015, American Chemical Society. B) Structure of two optimal nitroxyl derivatives in the “Anodic Catalyst” quadrant.
4. Conclusions and Outlook
This content above highlights historical and recent advances in the applications of soluble redox mediators to electrochemical power generation. The development and testing of mediated fuel cells (MedFCs) have greatly expanded in recent years, with advances leading to new approaches to the catalytic reduction of O2 at the cathode and to the oxidation of H2, alcohols, biomass, and other fuels at the anode. Replacement of Pt with non-PGM cathodes in fuel cell cathodes remains a high priority. Whereas historical efforts commonly featured Br2- or NOx-based mediators, these materials seem unlikely to provide long-term solutions due to challenges related to the volatility, toxicity and/or corrosivity of these species. Polyoxometalates continue to be the focus of attention because their excellent stability and good reactivity with O2, and their ability to access reasonable overpotentials makes them promising mediators and/or catalysts for the ORR and mediated cathode systems. POM mediators have some disadvantages related to their high molecular weight, which contributes to a decreased specific energy density. The recent demonstration of organic cathode mediators in combination with off-electrode ORR catalysts provides a novel approach to implement Pt-free ORR catalysts (e.g., Fe- or Co-N/C materials) and may allow for improved utilization of these catalytic materials. The design of highly stable, high-potential mediators represents a key challenge that will need to be addressed to expand on recent demonstrations. In addition, exploration of other conditions, such as the use alkaline conditions and anion exchange membranes, may open new avenues for catalyst and mediator compositions. Mediated electrochemistry introduces significant opportunities for fuel cell power generation with complex fuels, such as polyols and other biomass-derived materials, hydrocarbons, and even low-grade coal and sewage. Low-potential POMs have emerged as some of the most effective anodic mediators, reflecting their low cost, good stability, effective redox behavior, and compatibility with chemicals present in complex fuels that inhibit or poison many other types of catalysts (e.g., sulfur or other heteroatom-containing compounds). Thus far, many implementations of mediated fuels with complex chemicals employ a two-stage process in which the first stage features oxidation of the fuel by the mediator in a (non-electrochemical) batch process, and the mediator is regenerated electrochemically in a second stage. Assessment of the merits of this approach relative to a one-stage continuous electrolysis process represents an important issue for future efforts in the field. Organic mediators have been commonly used to support fuel oxidation, especially in enzymatic fuel cells. Organic mediators are appealing because of the ability to tune their redox kinetics and electrochemical potentials, as revealed by studies of viologens and organic nitroxyls. For enzymatic fuel cell systems, efforts will likely continue to focus on immobilized enzymes and mediators, but recent reports highlight potential performance enhancements possible when the mediator and/or enzyme are dissolved in solution. The latter strategy may find utility for systems in which refreshing the solutions to restore the enzyme and mediator activity is feasible.
Thus far, mediated fuel cells, especially those using alcohols, biomass, or other complex fuels achieve lower power output than conventional fuel cells. Nonetheless, the ability to eliminate the requirement for Pt and other PGM catalysts and derive power from non-conventional fuel sources provides strong motivation to continue exploration of this technology, including the development of new mediators that achieve improved stability, lower overpotentials for the processes of interest, better kinetics; and the development of new catalysts that serve as effective partners with mediators to allow electrochemically driven off-electrode redox reactions or that exhibit cooperative redox behavior with the mediator, allowing for faster rates or lower overpotentials.
Acknowledgements
Our research on in this field has been exploring a number of different topics and has been supported different funding sources: the Center for Molecular Electrocatalysis, an Energy Frontier Research Center funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences (molecular mediators for catalytic oxygen reduction); the Great Lakes Bioenergy Research Center, DOE Office of Science DE-SC0018409 (research on biomass-based fuels; U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences, Catalysis Program, DE-FG02-05ER15690 (copper-catalyzed oxidation reactions); and the NIH NIGMS, R01 GM100143 and R35 GM134929 (use of molecular mediators for organic chemical synthesis).
Biographies
Colin W. Anson obtained his B.A. in Chemistry in 2011 from Colby College, located in Waterville, Maine. Later that year, he joined the research group of Professor Shannon Stahl at the University of Wisconsin-Madison, where he obtained his Ph.D. in Chemistry in 2017. His doctoral work focused on the use of organic quinones as mediators for electrocatalytic O2 reduction using molecular cobalt catalysts. He continued this research during his postdoctoral work, targeting fuel cell-relevant applications of this concept. Currently, he is an Associate Scientist in the Stahl group, where he works on electrochemical energy conversion and biomass valorization projects.
Shannon S. Stahl obtained his B.S. in Chemistry in 1992 from the University of Illinois at Urbana–Champaign and earned his Ph.D from Caltech in 1997, where he worked with Professor John Bercaw. He was an NSF postdoctoral fellow with Professor Stephen Lippard at Massachusetts Institute of Technology from 1997 to 1999. He is currently the Steenbock Professor of Chemical Sciences at the University of Wisconsin–Madison, where he began his independent career in 1999. His research group specializes in catalysis and electrochemistry, with an emphasis on catalytic aerobic oxidation reactions and electrocatalytic reactions related to chemical synthesis and energy conversion.
Appendix I. Summary of Mediated Fuel Cells Using Chemical Catalysts. (See Table 8 for a Summary of Mediated Fuel Cells Using Enzymatic Catalysts.)
| fuel | catalyst | mediator | mediator | catalyst | oxidant | ref. |
|---|---|---|---|---|---|---|
| H2 | Pd black | TiO2+/Ti3+ | Br−/Br2 | NOx | O2 | 40 |
| Pt | – | 73 | ||||
| VO2+/VO2+ | HCl | 75 | ||||
| Mo3+/Mo4+ | NOx | 79 | ||||
| H4SiW12O40 | 79 | |||||
| Ti3+/TiO2+ | 80 | |||||
| Pd/Al2O3 | Fe(EDTA)2−/Fe(EDTA)− | 80 | ||||
| Pt | H4SiW12O40 | POM | 76–78 | |||
| Pt/C | AQ | V-containing POM | 105 | |||
| Pt/C | – | 63, 95–102, 104 | ||||
| Fe2+/Fe3+ | Fe(pc) | 61 | ||||
| K3Fe(CN)6 | Mn complex | 118 | ||||
| Fc species | Fe complex | 119–123 | ||||
| HNO3 | 128–129 | |||||
| nitroxyl | – | 137 | ||||
| quinone | Co–N/C | 143 | ||||
| alcohols | Pt | – | HNO3 | 124–127 | ||
| coal | – | Sn4+/Sn2+ | Br−/Br2 | NOx | 70 | |
| CH4, alcohols, coal | Pt | Fe2+/Fe3+ | VO2+/VO2+ | NOx | 82–84 | |
| biomass | – | 85, 86 | ||||
| sugars | Pt | V3+/VO2+ | Pt | 81, 169 | ||
| sugars | POM | Fe2+/Fe3+ | V-containing POM | 110 | ||
| biomass | POM | 107, 111, 112 | ||||
| alcohols | 108 | |||||
| lignin | 113 | |||||
| coal | 109 | |||||
| biomass | – | Pt/C | 113, 159 | |||
| alcohols, sugars or lignin | – | AQ | – | – | 166–168 | |
| sugars | – | viologen | – | Co | 171–175, 177 | |
| – | – | V2+/V3+ | VO2+/VO2+ | NOx | 79 | |
| Zn | – | – | Fe2+/Fe3+ | charcoal | 114 | |
References
- (1).Gyuk I; Johnson M; Vetrano J; Lynn K; Parks W; Handa R; Kannberg L; Hearne S; Waldrip K; Braccio R Grid Energy Storage; U.S. Department of Energy, Office of Electricity Delivery & Energy Reliability: Washington, DC, 2013. [Google Scholar]
- (2).Annual Energy Outlook 2019 with Projections to 2050; U.S. Department of Energy, Office of Energy Analysis, U.S. Energy Information Administration: Washington, DC, 2019. [Google Scholar]
- (3).Jamison L Monthly Energy Review May 2019, Section 10. Renewable Energy; pgs 171–191; U.S. Department of Energy, Office of Energy Analysis, U.S. Energy Information Administration: Washington, DC, 2019. [Google Scholar]
- (4).Goodenough JB; Park K-S The Li-Ion Rechargable Battery: A Perspective. J. Am. Chem. Soc 2013, 135, 1167–1176. [DOI] [PubMed] [Google Scholar]
- (5).Yabuuchi N; Kubota K; Dahbi M; Komaba S Research Development on Sodium-Ion Batteries. Chem. Rev 2014, 114, 11636–11682. [DOI] [PubMed] [Google Scholar]
- (6).Winter M; Barnett B; Xu K Before Li Ion Batteries. Chem. Rev 2018, 118, 11433–11456. [DOI] [PubMed] [Google Scholar]
- (7).Zhao C; Liu L; Qi X; Lu Y; Wu F; Zhao J; Yu Y; Hu Y-S; Chen L Solid-State Sodium Batteries. Adv. Energy Mater 2018, 8, 1703012. [Google Scholar]
- (8).Soloveichik GL Flow Batteries: Current Status and Trends. Chem. Rev 2015, 115, 11533–11558. [DOI] [PubMed] [Google Scholar]
- (9).Winsberg J; Hagemann T; Janoschka T; Hager MD; Schubert US Redox-Flow Batteries: From Metal to Organic Redox-Active Materials. Angew. Chem. Int. Ed 2017, 56, 686–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Yang Z; Zhang J; Kintner-Meyer MCW; Lu X; Choi D; Lemmon JP; Liu J Electrochemical Energy Storage for Green Grid. Chem. Rev 2011, 111, 3577–3613. [DOI] [PubMed] [Google Scholar]
- (11).Winter M; Brodd RJ What Are Batteries, Fuel Cells, and Supercapacitors? Chem. Rev 2004, 104, 4245–4270. [DOI] [PubMed] [Google Scholar]
- (12).Nocera DG Solar Fuels and Solar Chemicals Industry. Acc. Chem. Res 2017, 50, 616–619. [DOI] [PubMed] [Google Scholar]
- (13).Detz RJ; Reek JNH; van der Zwaan BCC The Future of Solar Fuels: When Could They Become Competitive? Energy Environ. Sci 2018, 11, 1653–1669. [Google Scholar]
- (14).Fuel Cells; U.S. Department of Energy, Energy Efficiency & Renewable Energy, Fuel Cell Technologies Office, November 2015. https://www.energy.gov/sites/prod/files/2015/11/f27/fcto_fuel_cells_fact_sheet.pdf, accessed 08/26/2019. [Google Scholar]
- (15).Mehta V; Cooper JS Review and Analysis of PEM Fuel Cell Design and Manufacturing. J. Power Sources 2003, 114, 32–53. [Google Scholar]
- (16).The Future of Hydrogen, International Energy Agency: Paris, 2019. [Google Scholar]
- (17).Gottesfeld S; Dekel DR; Page M; Bae C; Yan Y; Zelenay P; Kim YS Anion Exchange Membrane Fuel Cells: Current Status and Remaining Challenges. J. Power Sources 2018, 375, 170–184. [Google Scholar]
- (18).Jiao K; Li X Water Transport in Polymer Electrolyte Membrane Fuel Cells. Prog. Energy Combust. Sci 2011, 37, 221–291. [Google Scholar]
- (19).Kreuer K-D; Paddison SJ; Spohr E; Schuster M Transport in Proton Conductors for Fuel-Cell Applications: Simulations, Elementary Reactions, and Phenomenology. Chem. Rev 2004, 104, 4637–4678. [DOI] [PubMed] [Google Scholar]
- (20).De las Heras A; Vivas FJ; Segura F; Andújar JM From the Cell to the Stack. A Chronological Walk through the Techniques to Manufacture the PEFCs Core. Renewable Sustainable Energy Rev 2018, 96, 29–45. [Google Scholar]
- (21).Hermann A; Chaudhuri T; Spagnol P Bipolar Plates for PEM Fuel Cells: A Review. Int. J. Hydrogen Energy 2005, 30, 1297–1302. [Google Scholar]
- (22).Garsany Y; Atkinson RW III; Gould BD; Swider-Lyons KE High Power, Low-Pt Membrane Electrode Assemblies for Proton Exchange Membrane Fuel Cells. J. Power Sources 2018, 408, 38–45. [Google Scholar]
- (23).Gasteiger HA; Kocha SS; Sompalli B; Wagner FT Activity Benchmarks and Requirements for Pt, Pt-alloy, and Non-Pt Oxygen Reduction Catalysts for PEMFCs. Appl. Catal. B 2005, 56, 9–35. [Google Scholar]
- (24).Martinez U; Babu SK; Holby EF; Zelenay P Durability Challenges and Perspective in the Development of PGM-Free Electrocatalysts for the Oxygen Reduction Reaction. Curr. Opin. Electrochem 2018, 9, 224–232. [Google Scholar]
- (25).Thompson ST; Wilson AR; Zelenay P; Myers DJ; More KL; Neyerlin KC; Papageorgopoulos D ElectroCat: DOE’s Approach to PGM-Free Catalyst and Electrode R&D. Solid State Ionics 2018, 319, 68–76. [Google Scholar]
- (26).Gewirth AA; Varnell JA; DiAscro AM Nonprecious Metal Catalysts for Oxygen Reduction in Heterogeneous Aqueous Systems. Chem. Rev 2018, 118, 2313–2339. [DOI] [PubMed] [Google Scholar]
- (27).Ballard Plans World’s First PEMFC Product with Low-Cost Nisshinbo Non-Precious Metal Catalyst. Fuel Cell Bull 2017, 9, 1. [Google Scholar]
- (28).Banham D; Choi J-Y; Kishimoto T; Ye S Integrating PGM-Free Catalysts into Catalyst Layers and Proton Exchange Membrane Fuel Cell Devices. Adv. Mater 2019, 31, 1804846. [DOI] [PubMed] [Google Scholar]
- (29).Liu H; Song C; Zhang L; Zhang J; Wang H; Wilkinson DP A Review of Anode Catalysis in the Direct Methanol Fuel Cell. J. Power Sources 2006, 155, 95–110. [Google Scholar]
- (30).Li X; Faghri A Review and Advances of Direct Methanol Fuel Cells (DMFCs) Part I: Design, Fabrication, and Testing with High Concentration Methanol Solutions. J. Power Sources 2013, 226, 223–240. [Google Scholar]
- (31).Soloveichik GL Liquid Fuel Cells. Beilstein J. Nanotechnol 2014, 5, 1399–1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Kurzweil P History: Secondary Batteries In Encyclopedia of Electrochemical Power Systems; Garche J, Ed.; Elsevier: Amsterdam, 2009, pp 565–578. [Google Scholar]
- (33).Bartolozzi M Development of Redox Flow Batteries. A Historical Bibliography. J. Power Sources 1989, 27, 219–234. [Google Scholar]
- (34).Noack J; Roznyatovskaya N; Herr T; Fischer P The Chemistry of Redox-Flow Batteries. Angew. Chem. Int. Ed 2015, 54, 9776–9809. [DOI] [PubMed] [Google Scholar]
- (35).Luo J; Hu B; Hu M; Zhao Y; Liu TL Status and Prospects of Organic Redox Flow Batteries toward Sustainable Energy Storage. ACS Energy Lett 2019, 4, 2220–2240. [Google Scholar]
- (36).Huskinson B; Marshak MP; Suh C; Er S; Gerhardt MR; Galvin CJ; Chen X; Aspuru-Guzik A; Gordon RG; Aziz MJ A Metal-Free Organic-Inorganic Aqueous Flow Battery. Nature 2014, 505, 195–198. [DOI] [PubMed] [Google Scholar]
- (37).Lin K; Chen Q; Gerhardt MR; Tong L; Kim SB; Eisenach L; Valle AW; Hardee D; Gordon RG; Aziz MJ et al. Alkaline Quinone Flow Battery. Science 2015, 349, 1529–1532. [DOI] [PubMed] [Google Scholar]
- (38).Janoschka T; Martin N; Hager MD; Schubert US An Aqueous Redox-Flow Battery with High Capacity and Power: The TEMPTMA/MV System. Angew. Chem. Int. Ed 2016, 55, 14427–14430. [DOI] [PubMed] [Google Scholar]
- (39).Gong K; Fang Q; Gu S; Li SFY; Yan Y Nonaqueous Redox-Flow Batteries: Organic Solvents, Supporting Electrolytes, and Redox Pairs. Energy Environ. Sci 2015, 8, 3515–3530. [Google Scholar]
- (40).Carson WN Jr.; Feldman ML A Redox Type. Proc. Ann. Power Sources Conf 1959, 13, 111–113. [Google Scholar]
- (41).Matsen JM Chemically Regenerative, Fuel Cell Systems. Adv. Chem. Ser 1967, 64, 277–291. [Google Scholar]
- (42).Tolmachev YV; Vorotyntsev MA Fuel Cells with Chemically Regenerative Redox Cathodes (Review). Russ. J. Electrochem 2014, 50, 403–411. [Google Scholar]
- (43).Steckhan E Indirect Electroorganic Syntheses–A Modern Chapter of Organic Electrochemistry. Angew. Chem. Int. Ed 1986, 25, 683–701. [Google Scholar]
- (44).Ogibin YN; Elinson MN; Nikishin GI Mediator Oxidation Systems in Organic Electrosynthesis. Russ. Chem. Rev 2009, 78, 89–140. [Google Scholar]
- (45).Francke R; Little RD Redox Catalysis in Organic Electrosynthesis: Basic Principles and Recent Developments. Chem. Soc. Rev 2014, 43, 2492–2521. [DOI] [PubMed] [Google Scholar]
- (46).Nutting JE; Rafiee M; Stahl SS Tetramethylpiperidine N-Oxyl (TEMPO), Phthalimide N-Oxyl (PINO), and Related N-Oxyl Species: Electrochemical Properties and Their Use in Electrocatalytic Reactions. Chem. Rev 2018, 118, 4834–4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Hagfeldt A; Boschloo G; Sun L; Kloo L; Pettersson H Dye-Sensitized Solar Cells. Chem. Rev 2010, 110, 6595–6663. [DOI] [PubMed] [Google Scholar]
- (48).Bella F; Gerbaldi C; Barolo C; Grätzel M Aqueous Dye-Sensitized Solar Cells. Chem. Soc. Rev 2015, 44, 3431–3473. [DOI] [PubMed] [Google Scholar]
- (49).McCloskey BD; Addison D A Viewpoint on Heterogeneous Electrocatalysis and Redox Mediation in Nonaqueous Li-O2 Batteries. ACS Catal 2017, 7, 772–778. [Google Scholar]
- (50).Yan R; Wang Q Redox-Targeting-Based Flow Batteries for Large-Scale Energy Storage. Adv. Mater 2018, 30, 1802406. [DOI] [PubMed] [Google Scholar]
- (51).Wallace AG; Symes MD Decoupling Strategies in Electrochemical Water Splitting and Beyond. Joule 2018, 2, 1390–1395. [Google Scholar]
- (52).Hu L; Zhai T; Li H; Wang Y Redox-Mediator-Enhanced Electrochemical Capacitors: Recent Advances and Future Perspectives. ChemSusChem 2019, 12, 1118–1132. [DOI] [PubMed] [Google Scholar]
- (53).Cao L; Skyllas-Kazacos M; Wang D-W Solar Redox Flow Batteries: Mechanism, Design, and Measurement. Adv. Sustainable Syst 2018, 1800031. [Google Scholar]
- (54).Pattabiraman R; Venkatesan VK; Udupa HVK Applications of Redox Systems in Fuel Cells. J. Sci. Ind. Res 1981, 40, 432–447. [Google Scholar]
- (55).Cracknell JA; Vincent KA; Armstrong FA Enzymes as Working or Inspirational Electrocatalysts for Fuel Cells and Electrolysis. Chem. Rev 2008, 108, 2439–2461. [DOI] [PubMed] [Google Scholar]
- (56).Kavanagh P; Leech D Mediated Electron Transfer in Glucose Oxidising Enzyme Electrodes for Applications to Biofuel Cells: Recent Progress and Perspectives. Phys. Chem. Chem. Phys 2013, 15, 4859–4869. [DOI] [PubMed] [Google Scholar]
- (57).Rasmussen M; Abdellaoui S; Minteer SD Enzymatic Biofuel Cells: 30 Years of Critical Advancements. Biosens. Bioelectron 2016, 76, 91–102. [DOI] [PubMed] [Google Scholar]
- (58).Logan BE; Hamelers B; Rozendal R; Schröder U; Keller J; Freguia S; Aelterman P; Verstraete W; Rabaey K Microbial Fuel Cells: Methodology and Technology. Environ. Sci. Tech 2006, 40, 5181–5192. [DOI] [PubMed] [Google Scholar]
- (59).Santoro C; Arbizzani C; Erable B; Ieropoulos I Microbial Fuel Cells: From Fundamentals to Applications. A review. J. Power Sources 2017, 356, 225–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Mohan SV; Velvizhi G; Krishna KV; Babu ML Microbial Catalyzed Electrochemical Systems: A Bio-Factory with Multi-Facet Applications. Bioresour. Technol 2014, 165, 355–364. [DOI] [PubMed] [Google Scholar]
- (61).Han S-B; Kwak D-H; Park HS; Choi I-A; Park J-Y; Ma K-B; Won J-E; Kim D-H; Kim S-J; Kim M-C et al. Chemically Regenerative Redox Fuel Cells Using Iron Redox Couples as a Liquid Catalyst with Cocatalysts. ACS Catal 2016, 6, 5302–5306. [Google Scholar]
- (62).Sen K; Creeth A; Metz S A Combined Experimental/Theoretical Approach to Accelerated Fuel Cell Development by Quantitative Prediction of Redox Potentials. J. Power Sources 2018, 399, 443–447. [Google Scholar]
- (63).Gunn NLO; Ward DB; Menelaou C; Herbert MA; Davies TJ Investigation of a Chemically Regenerative Redox Cathode Polymer Electrolyte Fuel Cell using a Phosphomolybdovanadate Polyoxoanion Catholyte. J. Power Sources 2017, 348, 107–117. [Google Scholar]
- (64).Oei D-G. Chemically Regenerative Redox Fuel Cells. J. Appl. Electrochem 1982, 12, 41–51. [Google Scholar]
- (65).Kongkanand A; Mathias MF The Priority and Challenge of High-Power Performance of Low-Platinum Proton-Exchange Membrane Fuel Cells. J. Phys. Chem. Lett 2016, 7, 1127–1137. [DOI] [PubMed] [Google Scholar]
- (66).Banham D; Ye S Current Status and Future Development of Catalyst Materials and Catalyst Layers for Proton Exchange Membrane Fuel Cells: An Industrial Perspective. ACS Energy Lett 2017, 2, 629–638. [Google Scholar]
- (67).Wilson A; Marcinkoski J; Papageorgopoulos D Fuel Cell System Cost – 2016. DOE Hydrogen and Fuel Cells Program Record # 16020 Approved November 21, 2016.
- (68).Nie Y; Li L; Wei Z Recent Advancements in Pt and Pt-Free Catalysts for Oxygen Reduction Reaction Chem. Soc. Rev 2015, 44, 2168–2201. [DOI] [PubMed] [Google Scholar]
- (69).Shao M; Chang Q; Dodelet J-P; Chenitz R Recent Advances in Electrocatalysts for Oxygen Reduction Reaction. Chem. Rev 2016, 116, 3594–3657. [DOI] [PubMed] [Google Scholar]
- (70).Posner AM Redox Fuel Cell. Fuel 1955, 34, 330–338. [Google Scholar]
- (71).Merton-Bingham BE; Posner AM Nitrogen Peroxide as a Catalyst for the Autoxidation of Hydrobromic Acid. J. Am. Chem. Soc 1955, 77, 2634–2638. [Google Scholar]
- (72).Reneke WE Air Regeneration of Bromine-Bromide Fuel Cell Catholyte Ph.D. Dissertation, University of Florida, Gainesville, FL, 1961. [Google Scholar]
- (73).Juda W Fuel Cell U.S. Patent 3,152,013, October 6, 1964.
- (74).Skyllas-Kazacos M; Rychick M; Robins R All-Vanadium Redox Battery U.S. Patent 4,786,567, November 22, 1988.
- (75).Schaefer HF; Kordesch KV Fuel Cell Half-Cell Containing Vanadium Redox Couple U.S. Patent 3,279,949, October 18, 1966.
- (76).Kummer JT; Oei D-G. A Chemically Regenerative Redox Fuel Cell. J. Appl. Electrochem 1982, 12, 87–100. [Google Scholar]
- (77).Kummer JT; Oei D-G Chemically Regenerable Redox Fuel Cell and Method of Operating the Same U.S. Patent 4,396,687, August 2, 1983.
- (78).Kummer JT; Oei D-G Chemically Regenerable Redox Fuel Cell and Method of Operating the Same U.S. Patent 4,407,902, October 4, 1983.
- (79).Kummer JT; Oei D-G. A Chemically Regenerative Redox Fuel Cell. II. J. Appl. Electrochem 1985, 15, 619–629. [Google Scholar]
- (80).Folkesson B Chemically Regenerative Redox Fuel Cells II. Regeneration Reaction Studies. J. Appl. Electrochem 1990, 20, 907–911. [Google Scholar]
- (81).Larsson R; Folkesson B; Spaziante PM; Veerasai W; Exell RHB A High-Power Carbohydrate Fuel Cell. Renewable Energy 2006, 31, 549–552. [Google Scholar]
- (82).Bergens SH; Gorman CB; Palmore GTR; Whitesides GM A Redox Fuel Cell That Operates with Methane as Fuel at 120 °C. Science 1994, 265, 1418–1420. [DOI] [PubMed] [Google Scholar]
- (83).Gorman CB; Bergens SH; Whitesides GM Platinum-Catalyzed Oxidations of Organic Compounds by Ferric Sulfate: Use of a Redox Fuel Cell to Mediate Complete Oxidation of Ethylene Glycol by Dioxygen at 80 °C. J. Catal 1996, 158, 92–96. [Google Scholar]
- (84).Weibel DB; Boulatov R; Lee A; Ferrigno R; Whitesides GM Modeling the Anodic Half-Cell of a Low-Temperature Coal Fuel Cell. Angew. Chem. Int. Ed 2005, 44, 5682–5686. [DOI] [PubMed] [Google Scholar]
- (85).Gong J; Liu W; Du X; Liu C; Zhang Z; Sun F; Yang L; Xu D; Guo H; Deng Y Direct Conversion of Wheat Straw into Electricity with a Biomass Flow Fuel Cell Mediated by Two Redox Ion Pairs. ChemSusChem 2017, 10, 506–513. [DOI] [PubMed] [Google Scholar]
- (86).Zu X; Sun L; Gong J; Liu X; Liu Y; Du X; Liu W; Chen L; Yi G; Zhang W et al. Ferric Ion Pair Mediated Biomass Redox Flow Fuel Cell and Related Chemical Reaction Kinetics Study. Chem. Eng. J 2018, 348, 476–484. [Google Scholar]
- (87).Odyakov VF; Zhizhina EG; Maksimovskaya RI; Matveev KI New Methods for the Synthesis of Molybdenum-Vanadium-Phosphoric Heteropoly Acids. Kinet. Catal 1995, 36, 733–738. [Google Scholar]
- (88).Zhizhina EG; Odyakov VF; Matveev KI Thermochemical Study of Reduction and Oxidation Reactions of Molybdovanadophosphoric Heteropolyacids in Aqueous Solutions. Eur. J. Inorg. Chem 1999, 1009–1014. [Google Scholar]
- (89).Odyakov VF; Zhizhina EG; Matveev KI Redox Potentials of Molydovanadophosphoric Heteropoly Acids in Aqueous Solutions. J. Mol. Catal. A: Chem 2000, 158, 453–465. [Google Scholar]
- (90).Zhizhina EG; Odyakov VF; Simonova MV; Matveev KI Kinetics of Oxidation of Reduced Phosphorus-Molybdenum-Vanadium Heteropoly Acid Species with Dioxygen in Aqueous Solutions. Kinet. Catal 2005, 46, 354–363. [Google Scholar]
- (91).Odyakov VF; Zhizhina EG; Maksimovskaya RI Synthesis of Molybdovanadophosphoric Heteropoly Acid Solutions Having Modified Composition. Appl. Catal. A 2008, 342, 126–130. [Google Scholar]
- (92).Zhizhina EG; Odyakov VF Regeneration of the Oxidation Catalysts Based on the Aqueous Solutions of Non-Keggin Mo-V-P Heteropoly Acids by Molecular Oxygen. Int. J. Chem. Kinet 2014, 46, 567–576. [Google Scholar]
- (93).Odyakov VF; Zhizhina EG; Rodikova YA; Gogin LL Mo-V-Phosphoric Heteropoly Acids and Their Salts: Aqueous Solution Preparation – Challenges and Perspectives. Eur. J. Inorg. Chem 2015, 3618–3631. [Google Scholar]
- (94).Weinstock IA; Schreiber RE; Neumann R Dioxygen in Polyoxometalate Mediated Reactions. Chem. Rev 2018, 118, 2680–2717 [DOI] [PubMed] [Google Scholar]
- (95).Creeth A Pt-Free PEM Cathode Technology with Fundamental Durability Benefits: FlowCathâ. Fuel Cell Bull 2011, 2011, 12–15. [Google Scholar]
- (96).Singh R; Shah AA; Potter A; Clarkson B; Creeth A; Downs C; Walsh FC Performance and Analysis of a Novel Polymer Electrolyte Membrane Fuel Cell using a Solution Based Redox Mediator. J. Power Sources 2012, 201, 159–163. [Google Scholar]
- (97).Creeth AM; Potter AR Redox Fuel Cells with a Catholyte Solution Containing a Polyoxometallate U.S. Patent 9,005,828, April 14, 2015.
- (98).Knuckey K; Kangati B; Downs C; Potter A Regenerative Fuel Cell with Catholyte Comprising a Polyoxometalate and a Vanadium (IV)-Compound U.S. Patent 9,362,584, June 7, 2016.
- (99).Rochester DL; Herbert MA; Creeth AM Polyozometallates for Use at Elevated Temperatures and Pressures U.S. Patent App. 2016/0156059, June 2, 2016.
- (100).Knuckey KJ; Creeth AM; Baynes N. d. B.; Rochester D; Clarkson B Fuel Cells for use at Elevated Temperatures and Pressures U.S. Patent 10,326,156, June 18, 2019.
- (101).Matsui T; Morikawa E; Nakada S; Okanishi T; Muroyama H; Hirao Y; Takahashi T; Eguchi K Polymer Electrolyte Fuel Cells Employing Heteropolyacids as Redox Mediators for Oxygen Reduction Reactions: Pt-Free Cathode Systems. ACS Appl. Mater. Interfaces 2016, 8, 18119–18125. [DOI] [PubMed] [Google Scholar]
- (102).Ward DB; Gunn NLO; Uwigena N; Davies TJ Performance Comparison of Protonic and Sodium Phosphomolybdovanadate Polyoxoanion Catholytes within a Chemically Regenerative Redox Cathode Polymer Electrolyte Fuel Cell. J. Power Sources 2018, 375, 68–76. [Google Scholar]
- (103).Longman R; Clarkson B Fuel Cells U.S. Patent 9,385,391, July 5, 2016.
- (104).Ward DB; Davies TJ Effect of Temperature and Catholyte Concentration on the Performance of a Chemically Regenerative Fuel Cell. Johnson Matthey Technol. Rev 2018, 62, 189–203. [Google Scholar]
- (105).Preger Y; Johnson MR; Biswas S; Anson CW; Root T; Stahl SS Anthraquinone-Mediated Fuel Cell Anode Accessing High Power Density with an Off-Electrode Heterogeneous Catalyst. Manuscript under review, ACS Energy Lett [DOI] [PMC free article] [PubMed] [Google Scholar]
- (106).Zhao X; Liu W; Deng Y; Zhu JY Low-Temperature Microbial and Direct Conversion of Lignocellulosic Biomass to Electricity: Advances and Challenges. Renewable Sustainable Energy Rev 2017, 71, 268–282. [Google Scholar]
- (107).Liu W; Mu W; Deng Y High-Performance Liquid-Catalyst Fuel Cell for Direct Biomass-into-Electricity Conversion. Angew. Chem. Int. Ed 2014, 53, 13558–13562. [DOI] [PubMed] [Google Scholar]
- (108).Wu W; Liu W; Mu W; Deng Y Polyoxometalate Liquid-Catalyzed Polyol Fuel Cell and the Related Photoelectrochemical Reaction Mechanism Study. J. Power Sources 2016, 318, 86–92. [Google Scholar]
- (109).Zhang Z; Liu C; Liu W; Cui Y; Du X; Xu D; Guo H; Deng Y Innovative Design of Coal Utilization – A Green Pathway for Direct Conversion of Coal to Electricity through Flow Fuel Cell Technology. Appl. Energy 2017, 200, 226–236. [Google Scholar]
- (110).Xu F; Li H; Liu Y; Jing Q Advanced Redox Flow Fuel Cell using Ferric Chloride as Main Catalyst for Complete Conversion from Carbohydrates to Electricity. Sci. Rep 2017, 7, 5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Zhang Z; Liu C; Liu W; Du X; Cui Y; Gong J; Guo H; Deng Y Direct Conversion of Sewage Sludge to Electricity using Polyoxometalate Catalyzed Flow Fuel Cell. Energy 2017, 141, 1019–1026. [Google Scholar]
- (112).Liu W; Gong Y; Wu W; Yang W; Liu C; Deng Y; Chao Z.-s. Efficient Biomass Fuel Cell Powered by Sugar with Photo- and Thermal-Catalysis by Solar Irradiation. ChemSusChem 2018, 11, 2229–2238. [DOI] [PubMed] [Google Scholar]
- (113).Zhao X; Zhu JY Efficient Conversion of Lignin to Electricity Using a Novel Direct Biomass Fuel Cell Mediated by Polyoxometalates at Low Temperatures. ChemSusChem 2016, 9, 197–207. [DOI] [PubMed] [Google Scholar]
- (114).Lamb AB; Elder LW Jr. The Electromotive Activation of Oxygen. J. Am. Chem. Soc 1931, 53, 137–163. [Google Scholar]
- (115).Posner AM The Kinetics of Autoxidation of Ferrous Ions in Concentrated HCl Solutions. Trans. Faraday Soc 1953, 49, 382–388. [Google Scholar]
- (116).Posner AM The Kinetics of the Charcoal Catalyzed Autoxidation of Fe2+ Ion in Dilute HCl Solutions. Trans. Faraday Soc 1953, 49, 389–395. [Google Scholar]
- (117).Chen Y-WD; Santhanam KSV; Bard AJ Solution Redox Couples for Electrochemical Energy Storage I. Iron (III)-Iron(II) Complexes with O-Phenanthroline and Related Ligands. J. Electrochem. Soc 1981, 128, 1460–1467. [Google Scholar]
- (118).Creeth AM Fuel Cells U.S. Patent Appl. 2008/0274385, November 6, 2008.
- (119).Knuckey K; Creeth A Fuel Cells U.S. Patent 8,492,048, July 23, 2013.
- (120).Knuckey K; Creeth A Fuel Cells U.S. Patent 9,136,554, September 15, 2015.
- (121).Knuckey K; Rochester D Redox Fuel Cell with Catholyte Redox Mediator U.S. Patent 8,951,695, February 10, 2015.
- (122).Knuckey K; Rochester D; Creeth AM Redox Fuel Cells U.S. Patent 8,647,781, February 11, 2014.
- (123).Knuckey K; Rochester D Fuel Cells U.S. Patent Appl. 2011/0027671, February 3, 2011.
- (124).Heath CE; Tarmy BL; Holt EL; Levine DG; Moerikofer AW; Shropshire JA; Worsham CH Soluble Carbonaceous Fuel-Air Fuel Cell. Report No. 1, Contract No. DA 36–039 SC-89156, June 1962.
- (125).Shropshire JA; Tarmy BL The Nitric Acid-Oxygen Redox Electrode in Acid Electrolyte. Adv. Chem 1969, 47, 153–165. [Google Scholar]
- (126).Tarmy BL; Holt EL; Levine DG; Moerikofer AW; Shropshire JA; Worsham CH Soluble Carbonaceous Fuel-Air Fuel Cell. Report No. 2, Contract No. DA 36–039 SC-89156, December 1962.
- (127).Tarmy BL; Feng I-M; Holt EL; Levine DG; Lewis K; Moerikofer AW; Okrent EH; Shropshire JA; Wilson JA; Worsham CH Soluble Carbonaceous Fuel-Air Fuel Cell. Report No. 3, Contract No. DA 36–039 AMC-00134, June 1963.
- (128).Han S-B; Song Y-J; Lee Y-W; Ko A-R; Oh J-K; Park K-W High-Performance Hydrogen Fuel Cell using Nitrate Reduction Reaction on a Non-Precious Catalyst. Chem. Commun 2011, 47, 3496–3498. [DOI] [PubMed] [Google Scholar]
- (129).Han S-B; Kwak D-H; Park HS; Choi I-A; Park J-Y; Kim S-J; Kim M-C; Hong S; Park K-W High-Performance Chemically Regenerative Redox Fuel Cell Using a NO3–/NO Regeneration Reaction. Angew. Chem. Int. Ed 2017, 56, 2893–2897. [DOI] [PubMed] [Google Scholar]
- (130).Ma K-B; Han S-B; Kwon S-H; Kwak D-H; Park K-W. High-Performance Direct Ethanol Fuel Cell using Nitrate Reduction Reaction. Int. J. Hydrogen Energy 2018, 43, 17265–17270. [Google Scholar]
- (131).Rafiee M; Miles KC; Stahl SS Electrocatalytic Alcohol Oxidation with TEMPO and Bicyclic Nitroxyl Derivatives: Driving Force Trumps Steric Effects. J. Am. Chem. Soc 2015, 137, 14751–14757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Lauber MB; Stahl SS Efficient Aerobic Oxidation of Secondary Alcohols at Ambient Temperature with an ABNO/NOx Catalyst System. ACS Catal 2013, 3, 2612–2616. [Google Scholar]
- (133).Shibuya M; Osada Y; Sasano Y; Tomizawa M; Iwabuchi Y Highly Efficient, Organocatalytic Aerobic Alcohol Oxidation. J. Am. Chem. Soc 2011, 133, 6497–6500. [DOI] [PubMed] [Google Scholar]
- (134).Gerken JB; Stahl SS High-Potential Electrocatalytic O2 Reduction with Nitroxyl/NOx Mediators: Implications for Fuel Cells and Aerobic Oxidation Catalysis. ACS Cent. Sci 2015, 1, 234–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (135).Roberts JAS; Bullock RM Direct Determination of Equilibrium Potentials for Hydrogen Oxidation/Production by Open Circuit Potential Measurements in Acetonitrile. Inorg. Chem 2013, 52, 3823–3835. [DOI] [PubMed] [Google Scholar]
- (136).Pegis ML; Roberts JAS; Wasylenko DJ; Mader EA; Appel AM; Mayer JM Standard Reduction Potentials for Oxygen and Carbon Dioxide Couples in Acetonitrile and N,N-Dimethylformamide. Inorg. Chem 2015, 54, 11883–11888. [DOI] [PubMed] [Google Scholar]
- (137).Han S-B; Kwak D-H; Park HS; Park J-Y; Ma K-B; Won J-E; Kim D-H; Kim M-C; Park K-W A Chemically Regenerative Redox Fuel Cell using (2,2,6,6-tetramethylpiperidin-1-yl)oxyl Redox Reaction in Acid Medium. J. Power Sources 2018, 393, 32–36. [Google Scholar]
- (138).Gerken JB; Pang YQ; Lauber MB; Stahl SS Structural Effects on the pH-Dependent Redox Properties of Organic Nitroxyls: Pourbaix Diagrams for TEMPO, ABNO, and Three TEMPO Analogs. J. Org. Chem 2018, 83, 7323–7330. [DOI] [PubMed] [Google Scholar]
- (139).Piera J; Bäckvall J-E Catalytic Oxidation of Organic Substrates by Molecular Oxygen and Hydrogen Peroxide by Multistep Electron Transfer–A Biomimetic Approach. Angew. Chem. Int. Ed 2008, 47, 3506–3523. [DOI] [PubMed] [Google Scholar]
- (140).Wendlandt AE; Stahl SS Quinone-Catalyzed Selective Oxidation of Organic Molecules. Angew. Chem. Int. Ed 2015, 54, 14638–14658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (141).Anson CW; Ghosh S; Hammes-Schiffer S; Stahl SS Co(salophen)-Catalyzed Aerobic Oxidation of p-Hydroquinone: Mechanism and Implications for Aerobic Oxidation Catalysis. J. Am. Chem. Soc 2016, 138, 4186–4193. [DOI] [PubMed] [Google Scholar]
- (142).Anson CW; Stahl SS Cooperative Electrocatalytic O2 Reduction Involving Co(salophen) with p-Hydroquinone as an Electron–Proton Transfer Mediator. J. Am. Chem. Soc 2017, 139, 18472–18475. [DOI] [PubMed] [Google Scholar]
- (143).Preger Y; Gerken JB; Biswas S; Anson CW; Johnson MR; Root TW; Stahl SS Quinone-Mediated Electrochemical O2 Reduction Accessing High Power Density with an Off-Electrode Co-N/C Catalyst. Joule 2018, 2, 2722–2731. [Google Scholar]
- (144).Hoober-Burkhardt L; Krishnamoorthy S; Yang B; Murali A; Nirmalchandar A; Prakash GKS; Narayanan SR A New Michael-Reaction-Resistant Benzoquinone for Aqueous Organic Redox Flow Batteries. J. Electrochem. Soc 2017, 164, A600–A607. [Google Scholar]
- (145).Mano N; de Poulpiquet A O2 Reduction in Enzymatic Biofuel Cells. Chem. Rev 2018, 118, 2392–2468. [DOI] [PubMed] [Google Scholar]
- (146).Palmore GTR; Kim H-H Electro-Enzymatic Reduction of Dioxygen to Water in the Cathode Compartment of a Biofuel Cell. J. Electroanal. Chem 1999, 464, 110–117. [Google Scholar]
- (147).Liu Y; Wang M; Zhao F; Liu B; Dong S A Low-Cost Biofuel Cell with pH-Dependent Power Output Based on Porous Carbon as Matrix. Chem. Eur. J 2005, 11, 4970–4974. [DOI] [PubMed] [Google Scholar]
- (148).Tsujimura S; Tatsumi H; Ogawa J; Shimizu S; Kano K; Ikeda T Bioelectrocatalytic Reduction of Dioxygen to Water at Neutral pH using Bilirubin Oxidase as an Enzyme and 2,2’-azinobis(3-ethylbenzothiazolin-6-sulfonate) as an Electron Transfer Mediator. J. Electroanal. Chem 2001, 496, 69–75. [Google Scholar]
- (149).Tsujimura S; Fujita M; Tatsumi H; Kano K; Ikeda T Bioelectrocatalysis-Based Dihydrogen/Dioxygen Fuel Cell Operating at Physiological pH. Phys. Chem. Chem. Phys 2001, 3, 1331–1335. [Google Scholar]
- (150).Lim J; Malati P; Bonet F; Dunn B Nanostructured Sol-Gel Electrodes for Biofuel Cells. J. Electrochem. Soc 2007, 154, A140–A145. [Google Scholar]
- (151).Xu W; Danilov D; Gao L; Oudenhoven JFM; Pop V; Notten PHL Kinetic Study of Homogeneously Mediated Electrode Reactions in Glucose-Based BioFuel Cells. ECS Trans 2014, 48, 59–71. [Google Scholar]
- (152).Chen Y; Gai P; Zhang J; Zhu J-J Design of an Enzymatic Biofuel Cell with Large Power Output. J. Mater. Chem. A 2015, 3, 11511–11516. [Google Scholar]
- (153).Ji C; Hou J; Wang K; Ng YH; Chen V Single-Enzyme Biofuel Cells. Angew. Chem. Int. Ed 2017, 56, 9762–9766. [DOI] [PubMed] [Google Scholar]
- (154).Hammond JL; Gross AJ; Giroud F; Travelet C; Borsali R; Cosnier S Solubilized Enzymatic Fuel Cell (SEFC) for Quasi-Continuous Operation Exploiting Carbohydrate Block Copolymer Glyconanoparticle Mediators. ACS Energy Lett 2019, 4, 142–148. [Google Scholar]
- (155).Gross AJ; Chen X; Giroud F; Travelet C; Borsali R; Cosnier S Redox-Active Glyconanoparticles as Electron Shuttles for Mediated Electron Transfer with Bilirubin Oxidase in Solution. J. Am. Chem. Soc 2017, 139, 16076–16079. [DOI] [PubMed] [Google Scholar]
- (156).Du X; Liu W; Zhang Z; Mulyadi A; Brittain A; Gong J; Deng Y Low-Energy Catalytic Electrolysis for Simultaneous Hydrogen Evolution and Lignin Depolymerization. ChemSusChem 2017, 10, 847–854. [DOI] [PubMed] [Google Scholar]
- (157).Yang L; Liu W; Zhang Z; Du X; Gong J; Dong L; Deng Y Hydrogen Evolution from Native Biomass with Fe3+/Fe2+ Redox Couple Catalyzed Electrolysis. Electrochim. Acta 2017, 246, 1163–1173. [Google Scholar]
- (158).Ju H; Giddey S; Badwal SPS Role of Iron Species as Mediator in a PEM Based Carbon-Water Co-Electrolysis for Cost-Effective Hydrogen Production. Int. J. Hydrogen Energy 2018, 43, 9144–9152. [Google Scholar]
- (159).Liu W; Mu W; Liu M; Zhang X; Cai H; Deng Y Solar-Induced Direct Biomass-to-Electricity Hybrid Fuel Cell using Polyoxometalates as Photocatalyst and Charge Carrier. Nat. Commun 2014, 5, 3208. [DOI] [PubMed] [Google Scholar]
- (160).Zhao X; Ding Y; Du B; Zhu JY; Liu D Polyoxometalate-Mediated Lignin Oxidation for Efficient Enzymatic Production of Sugars and Generation of Electricity from Lignoellulosic Biomass. Energy Technol 2017, 5, 1179–1185. [Google Scholar]
- (161).Ding Y; Du B; Zhao X; Zhu JY; Liu D Phosphomolybdic Acid and Ferric Iron as Efficient Electron Mediators for Coupling Biomass Pretreatment to Produce Bioethanol and Electricity Generation from Wheat Straw. Bioresour. Technol 2017, 228, 279–289. [DOI] [PubMed] [Google Scholar]
- (162).Liu W; Cui Y; Du X; Zhang Z; Chao Z; Deng Y High Efficiency Hydrogen Evolution from Native Biomass Electrolysis. Energy Environ. Sci 2016, 9, 467–472. [Google Scholar]
- (163).Sarma BB; Neumann R Polyoxometalate-Mediated Electron Transfer-Oxygen Transfer Oxidation of Cellulose and Hemicellulose to Synthesis Gas. Nat. Commun 2014, 5, 4621. [DOI] [PubMed] [Google Scholar]
- (164).Yang L; Liu W; Zhang Z; Du X; Dong L; Deng Y Low Energy Electro-Reduction of Carbon Dioxide Coupling with Anodic Glycerol Oxidation Catalyzed by Chemical Regenerative Phosphomolybdic Acids. J. Power Sources 2019, 420, 99–107. [Google Scholar]
- (165).Chambers GM; Wiedner ES; Bullock RM H2 Oxidation Electrocatalysis Enabled by Metal-to-Metal Hydrogen Atom Transfer: A Homolytic Approach to a Heterolytic Reaction. Angew. Chem. Int. Ed 2018, 57, 13523–13527. [DOI] [PubMed] [Google Scholar]
- (166).Hertl W; Weetall HH A Photo-Chemical Electrical Fuel Cell Part I. Alcohol Fuels. Bioelectrochem. Bioenerg 1985, 14, 357–366. [Google Scholar]
- (167).Hertl W; Weetall HH A Photo-Chemical Electrical Fuel Cell Part II. Carbohydrate Fuels. Bioelectrochem. Bioenerg 1985, 14, 367–373. [Google Scholar]
- (168).Weetall HH; Forsyth BD; Hertl W A Direct Fuel Cell for the Production of Electricity from Lignin. Biotechnol. Bioeng 1985, 27, 927–979. [DOI] [PubMed] [Google Scholar]
- (169).Larsson R; Folkesson B Method for Producing Electric Energy in a Biofuel-Powered Fuel Cell U.S. Patent 5,660,940, August 26, 1997.
- (170).Larsson R; Folkesson B A Catalytic Oxidation of Sugar by Vanadium(IV). J. Mol. Catal. A: Chem 2005, 229, 183–190. [Google Scholar]
- (171).Wheeler DR; Nichols J; Hansen D; Andrus M; Choi S; Watt GD Viologen Catalysts for a Direct Carbohydrate Fuel Cell. J. Electrochem. Soc 2009, 156, B1201–B1207. [Google Scholar]
- (172).Scott D; Liaw BY Harnessing Electric Power from Monosaccharides – a Carbohydrate-Air Alkaline Fuel Cell Mediated by Redox Dyes. Energy Environ. Sci 2009, 2, 965–969. [Google Scholar]
- (173).Scott DM; Tsang TH; Chetty L; Aloi S; Liaw BY Mechanistic Understanding of Monosaccharide-Air Flow Battery Electrochemistry. J. Power Sources 2011, 196, 10556–10562. [Google Scholar]
- (174).Liu X; Hao M; Feng M; Zhang L; Zhao Y; Du X; Wang G A One-Compartment Direct Glucose Alkaline Fuel Cell with Methyl Viologen as Electron Mediator. Appl. Energy 2013, 106, 176–183. [Google Scholar]
- (175).Yang Y-L; Liu X-H; Hao M-Q; Zhang P-P Performance of a Low-Cost Direct Glucose Fuel Cell with an Anion-Exchange Membrane. Int. J. Hydrogen Energy 2015, 40, 10979–10984. [Google Scholar]
- (176).Hao M; Liu X; Feng M; Zhang P; Wang G Generating Power from Cellulose in an Alkaline Fuel Cell Enhanced by Methyl Viologen as an Electron-Transfer Catalyst. J. Power Sources 2014, 251, 222–228. [Google Scholar]
- (177).Watt GD; Hansen D; Dodson D; Andrus M; Wheeler D Electrical Energy from Carbohydrate Oxidation during Viologen-Catalyzed O2-Oxidation: Mechanistic Insights. Renewable Energy 2011, 36, 1523–1528. [Google Scholar]
- (178).Read A; Hansen D; Aloi S; Pitt WG; Wheeler DR; Watt GD Monoalkyl Viologens are Effective Carbohydrate O2-Oxidation Catalysts for Electrical Energy Generation by Fuel Cells. Renewable Energy 2012, 46, 218–223. [Google Scholar]
- (179).Hansen DC; Pan Y; Stockton J; Pitt WG; Wheeler DR Cyclic Voltammetry Investigation of Organic Species Considered for Use as Catalysts in Direct-Carbohydrate Fuel Cells. J. Electrochem. Soc 2012, 159, H834–H841. [Google Scholar]
- (180).Watt GD A New Future for Carbohydrate Fuel Cells. Renewable Energy 2014, 72, 99–104. [Google Scholar]
- (181).Rigby CR; Han H; Bhowmik PK; Bahari M; Chang A; Harb JN; Lewis RS; Watt GD Soluble Viologen Polymers as Carbohydrate Oxidation Catalysts for Alkaline Carbohydrate Fuel Cells. J. Electroanal. Chem 2018, 823, 416–421. [Google Scholar]
- (182).Barton SC; Gallaway J; Atanassov P Enzymatic Biofuel Cells for Implantable and Microscale Devices. Chem. Rev 2004, 104, 4867–4886. [DOI] [PubMed] [Google Scholar]
- (183).Ruff A Redox Polymers in Bioelectrochemistry: Common Playgrounds and Novel Concepts. Curr. Opin. Electrochem 2017, 5, 66–73. [Google Scholar]
- (184).Plotkin EV; Higgins IJ; Hill HAO Methanol Dehydrogenase Bioelectrochemical Cell and Alcohol Detector. Biotechnol. Lett 1981, 3, 187–192. [Google Scholar]
- (185).Yue PL; Lowther K Enzymatic Oxidation of C1 Compounds in a Biochemical Fuel Cell. Chem. Eng. J 1986, 33, B69–B77. [Google Scholar]
- (186).Davis G; Hill HAO; Aston WJ; Higgins IJ; Turner APF Bioelectrochemical Fuel Cell and Sensor Based on a Quinoprotein, Alcohol Dehydrogenase. Enzyme Microb. Technol 1983, 5, 383–388. [Google Scholar]
- (187).Zhang X-C; Ranta A; Halme A Direct Methanol Biocatalytic Fuel Cell–Considerations of Restraints on Electron Transfer. Biosens. Bioelectron 2006, 21, 2052–2057. [DOI] [PubMed] [Google Scholar]
- (188).Kavanagh P; Boland S; Jenkins P; Leech D Performance of a Glucose/O2 Enzymatic Biofuel Cell Containing a Mediated Melanocarpus albomyces Laccase Cathode in a Physiological Buffer. Fuel Cells 2009, 9, 79–84. [Google Scholar]
- (189).Laane C; Pronk W; Franssen M; Veeger C Use of a Bioelectrochemical Cell for the Synthesis of (Bio)chemicals. Enzyme Microb. Technol 1984, 6, 165–168. [Google Scholar]
- (190).Palmore GTR; Bertschy H; Bergens SH; Whitesides GM A Methanol/Dioxygen Biofuel Cell that Uses NAD+-Dependent Dehydrogases as Catalysts: Application of an Electro-Enzymatic Method to Regenerate Nicotinamide Adenine Dinucleotide at Low Overpotentials. J. Electroanal. Chem 1998, 443, 155–161. [Google Scholar]
- (191).Moiroux J; Elving PJ Effects of Absorption, Electrode Material, and Operational Variables on the Oxidation of Dihydronicotinamide Adenine Dincucleotide at Carbon Electrodes. Anal. Chem 1978, 50, 1056–1062. [Google Scholar]
- (192).Lojou E Hydrogenases as Catalysts for Fuel Cells: Strategies for Efficient Immobilization at Electrode Interfaces. Electrochim. Acta 2011, 56, 10385–10397. [Google Scholar]
- (193).Milton RD; Cai R; Abdellaoui S; Leech D; De Lacey AL; Pita M; Minteer SD Bioelectrochemical Haber-Bosch Process: An Ammonia-Producing H2/N2 Fuel Cell. Angew. Chem. Int. Ed 2017, 56, 2680–2683. [DOI] [PubMed] [Google Scholar]
- (194).Cass AEG; Davis G; Francis GD; Hill HAO; Aston WJ; Higgins IJ; Plotkin EV; Scott LDL; Turner APF Ferrocene-Mediated Enzyme Electrode for Amperometric Determination of Glucose. Anal. Chem 1984, 56, 667–671. [DOI] [PubMed] [Google Scholar]
- (195).Zhao X; Jia H; Kim J; Wang P Kinetic Limitations of a Bioelectrochemical Electrode Using Carbon Nanotube-Attached Glucose Oxidase for Biofuel Cells. Biotech. Bioengin 2009, 104, 1068–1074. [DOI] [PubMed] [Google Scholar]
- (196).Zhu Z; Zhang Y-HP Use of Nonimmobilized Enzymes and Mediators Achieved High Power Densities in Closed Biobatteries. Energy Sci. Eng 2015, 3, 490–497. [Google Scholar]
- (197).Hickey DP; McCammant MS; Giroud F; Sigman MS; Minteer SD Hybrid Enzymatic and Organic Electrocatalytic Cascade for the Complete Oxidation of Glycerol. J. Am. Chem. Soc 2014, 136, 15917–15920. [DOI] [PubMed] [Google Scholar]
- (198).Abdellaoui S; Hickey DP; Stephens AR; Minteer SD Recombinant Oxalate Decarboxylase: Enhancement of a Hybrid Catalytic Cascade for the Complete Electro-Oxidation of Glycerol. Chem. Commun 2015, 51, 14330–14333. [DOI] [PubMed] [Google Scholar]
- (199).Abdellaoui S; Chavez MS; Matanovic I; Stephens AR; Atanassov P; Minteer SD Hybrid Molecular/Enzymatic Catalytic Cascade for Complete Electro-Oxidation of Glycerol using a Promiscuous NAD-Dependent Formate Dehydrogenase from Candida boidinii. Chem. Commun 2017, 53, 5368–5371. [DOI] [PubMed] [Google Scholar]
- (200).Hickey DP; Schiedler DA; Matanovic I; Doan PV; Atanassov P; Minteer SD; Sigman MS Predicting Electrocatalytic Properties: Modeling Structure-Activity Relationships of Nitroxyl Radicals. J. Am. Chem. Soc 2015, 137, 16179–16186. [DOI] [PubMed] [Google Scholar]