

SUMMARY
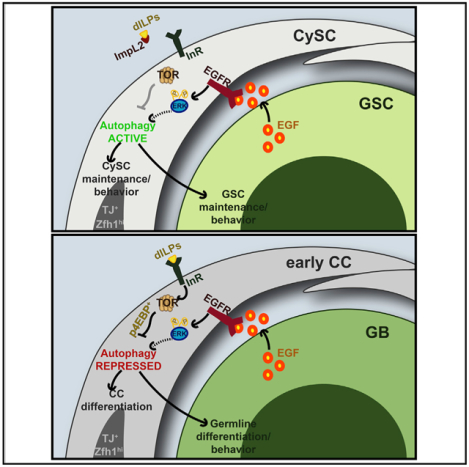
Although typically upregulated upon cellular stress, autophagy can also be utilized under homeostatic conditions as a quality control mechanism or in response to developmental cues. Here, we report that autophagy is required for the maintenance of somatic cyst stem cells (CySCs) in the Drosophila testis. Disruption of autophagy in CySCs and early cyst cells (CCs) by the depletion of autophagy-related (Atg) genes reduced early CC numbers and affected CC function, resembling decreased epidermal growth factor receptor (EGFR) signaling. Indeed, our data indicate that EGFR acts to stimulate autophagy to preserve early CC function, whereas target of rapamycin (TOR) negatively regulates autophagy in the differentiating CCs. Finally, we show that the EGFR-mediated stimulation of autophagy regulates lipid levels in CySCs and CCs. These results demonstrate a key role for autophagy in regulating somatic stem cell behavior and tissue homeostasis by integrating cues from both the EGFR and TOR signaling pathways to control lipid metabolism.
Graphical Abstract
In Brief
Sênos Demarco et al. demonstrate that Drosophila somatic cyst stem cells (CySCs) rely on basal levels of autophagy, which is regulated by EGFR signaling, for maintenance under homeostatic conditions. Blocking autophagy results in the accumulation of lipid droplets; therefore, one role of autophagy is to regulate lipid homeostasis in the soma.
INTRODUCTION
Adult stem cells are characterized by the ability to self-renew and generate highly specialized cells throughout life, which is crucial for tissue maintenance and regeneration and, in some cases, survival (Drummond-Barbosa, 2008). Hence, cellular control mechanisms are necessary to maintain pristine pools of adult stem cells. Autophagy is a highly conserved process by which cytosolic components are degraded and recycled (Levine and Klionsky, 2004). Macroautophagy (referred to as “autophagy” hereafter) involves the entrapment of cytosolic components, such as lipids, proteins, and organelles, within a specialized, double-membrane organelle called the autophagosome (AP). Autophagasomes then fuse with lysosomes to form autophagolysosomes (ALs), inside which lysosomal-derived enzymes break down the autophagic cargo, allowing small molecular byproducts to be recycled by the cell (Guan et al., 2013). A family of conserved Autophagy-related (Atg) proteins tightly regulates the formation of APs (Klionsky et al., 2003), with Atg1, the Drosophila homolog of mammalian Ulk1, being the most upstream (Suzuki et al., 2007; Figure 1A).
Figure 1. Basal Levels of Autophagy Are Required for CySC Maintenance and Function.
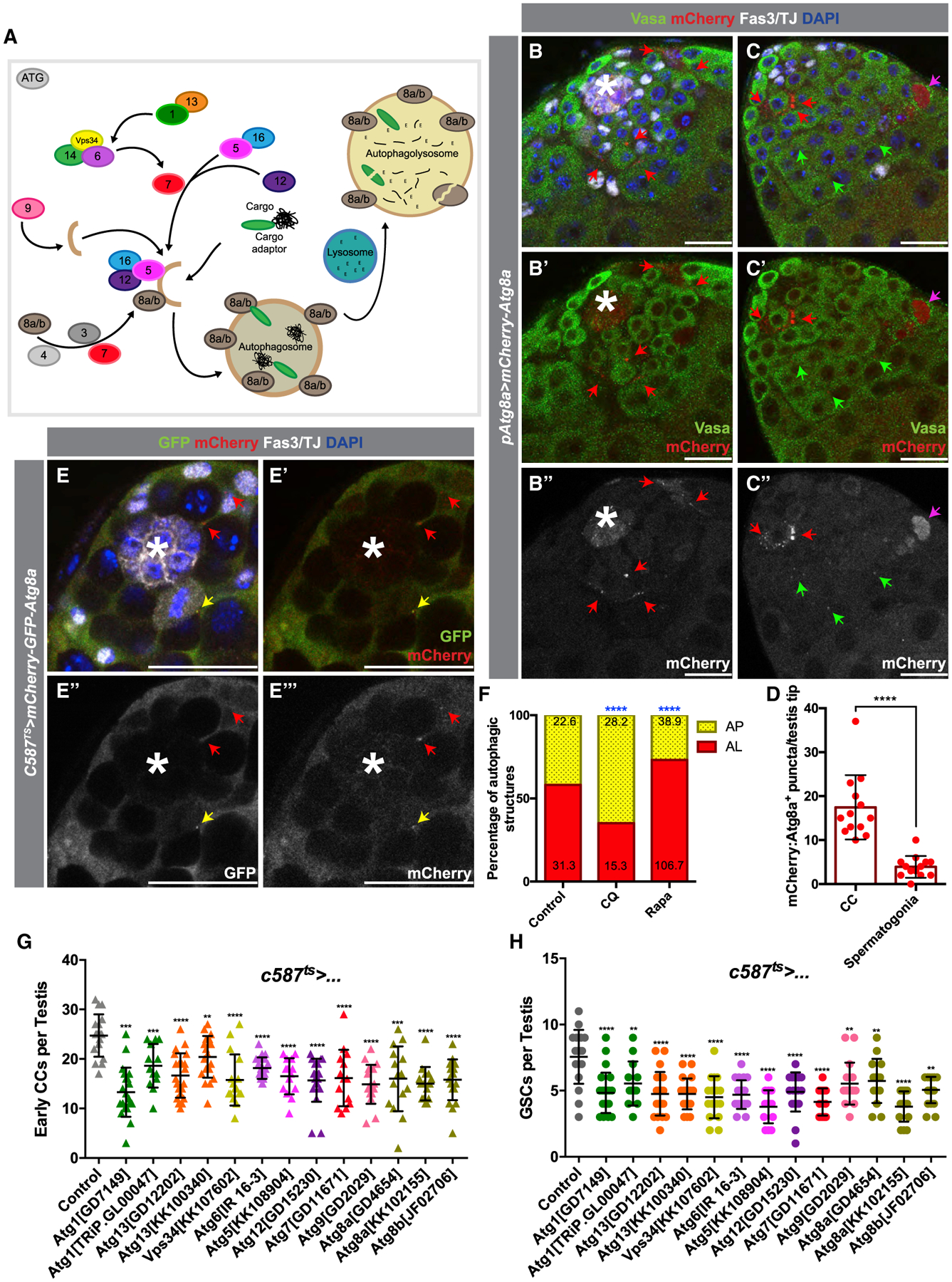
(A) Diagram of key proteins involved in autophagy, including many conserved Atg-family proteins required for AP formation.
(B–C’’) Two examples of testis tips in which APs (visualized by mCherry:Atg8a+ puncta) are shown to be enriched in early CCs (red arrows), with very few present in the germline (green arrows). The antibody against Vasa stains the germline, and the antibody against TJ marks the nuclei of early CCs, while Fas3 marks the hub (see STAR Methods). Occasionally, cytoplasmic mCherry:Atg8a is observed in dying germline cysts (pink arrow).
(D) Quantification of the number of APs per testis tips in the somatic CC tissue versus in early spermatogonia.
(E–E’’’) Example of the expression in CCs of a tandem-tagged GFP-mCherry-Atg8a probe. Autophagolysosomes (ALs) that can successfully acidify have quenched GFP signal (as seen by GFP−/mCherry+ puncta, red arrow), as opposed to APs that have not matured and acidified (double GFP+/mCherry+ puncta, yellow arrow).
(F) Quantification of the types of autophagic structures (see STAR Methods) in regular diet (RD), chloroquine (CQ) feeding, or rapamycin (RAPA) feeding. Numbers on each bar color represent the mean number of autophagic structures per testis tip (n = 10 testis per condition). Blue asterisks represent statistical information when compared to control.
(G and H) Quantification of very early CCs (including CySCs) (G) and GSCs (H) in testes from 10-day-old animals (see STAR Methods) in which autophagy-related genes have been knocked down by RNAi in early CCs with c587-Gal4ts.
In all images, asterisks (*) denote the hub; scale bars, 20 μm. For (D), (G), and (H), two-tailed t tests were used; error bars represent standard deviation. For (F), two-sided Fisher’s exact was used. *p < 0.05, **p < 0.01, ***p > 0.001, ****p < 0.0001; n.s., not significant.
Autophagy is typically considered a stress response. Upon starvation, levels of autophagy increase to satisfy cellular energetic demands and promote survival by recycling non-essential proteins and organelles (Chang and Neufeld, 2010). In fact, in tumors in which epidermal growth factor receptor (EGFR) signaling is upregulated, autophagy is often positively regulated by the Ras/mitogen-activated protein kinase (MAPK) signaling cascade (Sooro et al., 2018). However, recent evidence has revealed an underappreciated role for basal levels of autophagy in the maintenance of cellular and tissue homeostasis. Dysregulation of autophagy is associated with age-related pathologies, such as neurological disorders (Lynch-Day et al., 2012; Zare-Shahabadi et al., 2015; Rubinsztein et al., 2005) and cancer (Saito et al., 1993; Gao et al., 1995), and loss of autophagic activity has been shown to impact male fertility (Yang et al., 2017; Liu et al., 2016). Given the role of stem cells in maintaining tissue homeostasis throughout life, quality-control mechanisms are also important to ensure proper stem cell function (Guan et al., 2013). Indeed, it has been demonstrated that autophagy is required for hematopoietic stem cell (HSC) maintenance and proliferative activity (Liu et al., 2010; Mortensen et al., 2011; Ho et al., 2017), for DNA integrity and prevention of exhaustion of intestinal stem cells (ISCs) (Nagy et al., 2018), and for muscle stem cell activation (Tang and Rando, 2014). However, the precise mechanisms through which autophagy is regulated under homeostatic conditions is still not well understood.
Due to the characterization and validation of well-defined markers and genetic tools (reviewed in Demarco et al., 2014), as well as the conservation of developmental and metabolic signaling pathways (reviewed in Greenspan et al., 2015), the Drosophila testis presents an excellent model to investigate mechanisms regulating stem cell maintenance and function. Two stem cell populations are located at the apical tip, namely, the germline stem cells (GSCs) and somatic cyst stem cells (CySCs), which surround somatic support cells, referred to as the hub (Figure S1A). GSCs can divide asymmetrically to self-renew and give rise to a daughter gonialblast (GB). Each GB undergoes four rounds of mitotic, transit-amplification (TA) divisions with incomplete cytokinesis to generate a cyst of 16 interconnected spermatogonia. These spermatogonia will then mature into spermatocytes prior to meiosis, which produces 64 haploid spermatids and, eventually, mature sperm (Figure S1A; Hardy et al., 1979; Fuller, 1993). CySC division maintains the somatic stem cell population and gives rise to somatic cyst cells (CCs) that differentiate in close contact with the germline. During spermatogenesis, CCs encapsulate germ cells and provide signals for both self-renewal and differentiation, making them functionally analogous to mammalian Sertoli cells (Figures S1A and S1A’; Zoller and Schulz, 2012).
Two populations of stem cells residing within the same niche at the tip of the testis provide a means to compare and contrast how different stem cells respond to the local signals and how one population can regulate the behavior of the other. Numerous conserved signaling pathways, including the Janus kinase/signal transducer and activator of transcription (JAK/STAT) and EGFR pathway, are important regulators of stem cell behavior in the testis (Kiger et al., 2000, 2001; Tulina and Matunis, 2001; Leatherman and Dinardo, 2008, 2010; Lim and Fuller, 2012; Amoyel et al., 2016a; Greenspan et al., 2015). Importantly, EGFR signaling has been implicated in regulating CySC behavior, primarily by promoting CySC competitiveness and niche occupancy through MAPK/extracellularly regulated kinase (Erk) activity, in opposition to the JAK/STAT pathway (Amoyel et al., 2016a). In addition, a severe loss of EGFR signaling in CCs leads to the defective encapsulation of the GB and accumulation of early germ cells, due to the disruption of the specialized microenvironment that the CCs provide (Schulz et al., 2002; Kiger et al., 2000; Tran et al., 2000). Meanwhile, weaker reduction of EGFR signaling in CCs allows for germline encapsulation but disrupts the ability of the germline to proceed through synchronous TA divisions (Hudson et al., 2013). Additional studies have shown that EGFR signaling also contributes to CySC maintenance (Amoyel et al., 2016a).
Here, we show that basal levels of autophagy are required for stem cell maintenance in the Drosophila testis under homeostatic conditions. Disruption of the autophagic machinery in somatic CySCs, but not in GSCs, resulted in the loss of stem cells to differentiation. Moreover, loss of autophagy in CCs disrupted the synchronous mitotic divisions of germ cells, reminiscent of decreased EGFR signaling. Our data indicate that activation of basal autophagy is tightly controlled through the combined actions of the EGFR and TOR signaling pathways to influence lipid levels, which ultimately regulates the behavior of the somatic cyst lineage in the Drosophila testis.
RESULTS
Basal Levels of Autophagy Are Detected in Somatic Tissues
To determine whether basal levels of autophagy are important in the Drosophila testis, two assays were used to visualize autophagic structures. Atg8a is the Drosophila homolog of mammalian MAP-LC3, which specifically marks autophagic vesicles (Scott et al., 2007; Figure 1A). An mCherry-tagged Atg8a coding sequence, expressed under the transcriptional regulation of the Atg8a promoter (Denton et al., 2012), was used to visualize AP formation and localization. In testes from young flies raised under normal conditions and stained with antibodies for markers for germ cells (Vasa), early CCs (TJ), and hub cells (Fasciclin 3), mCherry-Atg8a+ puncta accumulated primarily in hub cells and CCs, with fewer puncta in the germline (Figures 1B–1D and S1B). Depletion of autophagy components in early CCs reduced the number of these structures in the somatic tissue, indicating that mCherry-Atg8a+ puncta truly represent APs (Figure S1C). Therefore, we conclude that basal autophagy occurs at low levels primarily in somatic tissues of the testis.
Although the appearance of these structures indicates the presence of autophagic vesicles, they do not reflect the successful degradation of cargo by autophagy upon fusion to the lysosome (when the AP becomes the AL). To analyze the composition of the different autophagic structures, a genetic probe including the coding sequence of Atg8a fused with GFP and mCherry, in tandem, was used. In ALs, GFP fluorescence is more rapidly quenched than that of mCherry, due to the pH of the vesicle. As a result, Atg8a structures that are positive for mCherry, but not GFP, mark ALs that have successfully acidified and can degrade their internal cargo (Nagy et al., 2015). The GFP-mCherry-Atg8a transgene was expressed exclusively in CySCs and early CCs by using the Gal4-UAS system, which enables tissue-specific expression of target transgenes (Brand and Perrimon, 1993). GFP-negative and mCherry-positive Atg8a puncta were observed in CySCs and early CCs, indicating that autophagic structures are present in these cells and capable of fusion with lysosomes (Figures 1E and 1F). Accordingly, the relative proportion of ALs to APs in CCs was decreased when flies were fed chloroquine (CQ; Figure 1F; STAR Methods), a drug that blocks lysosomal acidification and further fusion with the AP, and increased when flies were fed with rapamycin (RAPA; Figure 1F; STAR Methods), which blocks the activity of the autophagy-inhibitor TOR (Blommaart et al., 1995). Together, these results indicate that autophagy is ongoing in the somatic lineage of the Drosophila testis.
Autophagy-Related Genes Are Required for CySC Maintenance and CC Function
To investigate the role of basal autophagy in CySCs and CCs, an RNAi-based screen targeting various components of AP formation was performed using the c587-GAL4 “driver” line (see STAR Methods; Figures S1D and S1D’). Disruption of autophagy exclusively during adulthood was achieved by using the ubiquitously expressed and temperature-sensitive (ts) tub-Gal80ts transgene. This mutant form of GAL80 represses the activity of GAL4 in a temperature-dependent manner, allowing for transient expression of upstream activating sequence (UAS) transgenes (Zeidler et al., 2004). For simplicity, this system will be referred to as GAL4ts.
The depletion of genes involved in AP formation with c587-GAL4ts for 10 days resulted in the reduction of CySCs and very early CCs, scored as TJ+ cells within the first two rows of cells from the hub (see STAR Methods). For example, depletion of the most upstream kinase required for AP formation Atg1 resulted in an average of 13.29 ± 5.0 of early CCs versus 24.7 ± 4.3 in controls (mean ± standard deviation, p < 0.0001), whereas depletion of the E3 ubiquitin ligase involved in autophagosomal membrane elongation Atg5 resulted in 16.50 ± 3.7 of very early CCs (p < 0.0001). Depletion of the ubiquitin-like autophagosomal membrane protein Atg8a/LC3 resulted in 16 ± 6.5 early CCs (p < 0.0001), and similar trends were observed for the majority of genes tested (Figure 1G; see STAR Methods). Disrupting autophagy in early CCs also resulted in a non-autonomous reduction in GSC number (7.6 ± 2.0 GSCs in controls versus 4.8 ± 1.5 GSCs in Atg1RNAi, p < 0.0001; 3.8 ± 1.3 GSCs in Atg5RNAi, p < 0.0001; 5.7 ± 1.7 GSCs in Atg8aRNAi, p = 0.0063) (Figure 1H). Importantly, however, autophagy does not appear to be required autonomously in the early germline under homeostatic conditions, as depletion of autophagy-related genes using nos-GAL4:VP16 (Van Doren et al., 1998) did not lead to the loss of GSCs (8.27 ± 1.16 GSCs in controls versus 7.93 ± 1.72 GSCs in Atg1RNAi, not significant [n.s.]); similar trends were observed for other autophagy-related genes tested (Figure S1E). Interestingly, disrupting autophagy in CySCs and early CCs for 10 days occasionally led to testes with no detectable hub cells (Figure S1F; note Figures 1G and 1H show CySCs and GSCs counted only in testes with hubs). As expression of c587-Gal4 under the temperature-shift paradigm is not detected in hub cells in adults (Figures S1D and S1D’), this would suggest that the effect on hub cells is likely due to altered CySC behavior (Voog et al., 2014; Hétié et al., 2014).
As another strategy to test the role of autophagy in CySCs, flippase/flippase recognition target (FLP/FRT)-mediated recombination was used to generate a CySC homozygous mutant for a null allele of Atg1, Atg1Δ3D (Scott et al., 2004), or of Atg13, Atg13Δ81 (Chang and Neufeld, 2009). In this paradigm, heat-shock-induced recombination generates Atg1 or Atg13 mutant CySCs that become permanently labeled by the expression of GFP. Progeny derived from marked CySCs are similarly marked, permitting characterization of cells derived from Atg1 and Atg13 mutant CySCs or wild-type CySCs in control animals (Figures 2A–2E and S2A–S2B’’; see STAR Methods). At 2 days post-heat shock (phs), 75% (n = 26/43) of testes from FRT80B (control for Atg1) animals and 83% (n = 41/49) of testes from mutant animals had at least one GFP+ CySC clone adjacent to the hub; GFP+ CySCs also stained positive for the very early CC marker Zfh1(Figures S2A–S2B’’). Although FRT80B control CySCs were maintained 9 days phs, as observed in 61.11% of testes examined (n = 22/36), only 18.18% (n = 8/44) of testes maintained Atg1Δ3D mutant CySC clones (Figures 2A–2E). A similar trend was observed for Atg13Δ81 mutant CySCs (Figure 2E). Therefore, both RNAi-mediated depletion of autophagy-related genes and the generation of Atg1 or Atg13 mutant CySCs indicate that autophagy is required for CySC maintenance. Taken together, these results indicate that autophagy in CySCs and very early CCs is required for maintenance and proper function in maintaining GSCs in the stem cell niche.
Figure 2. Depletion of Autophagy-Related Gene Phenocopies Reduced EGFR Signaling.
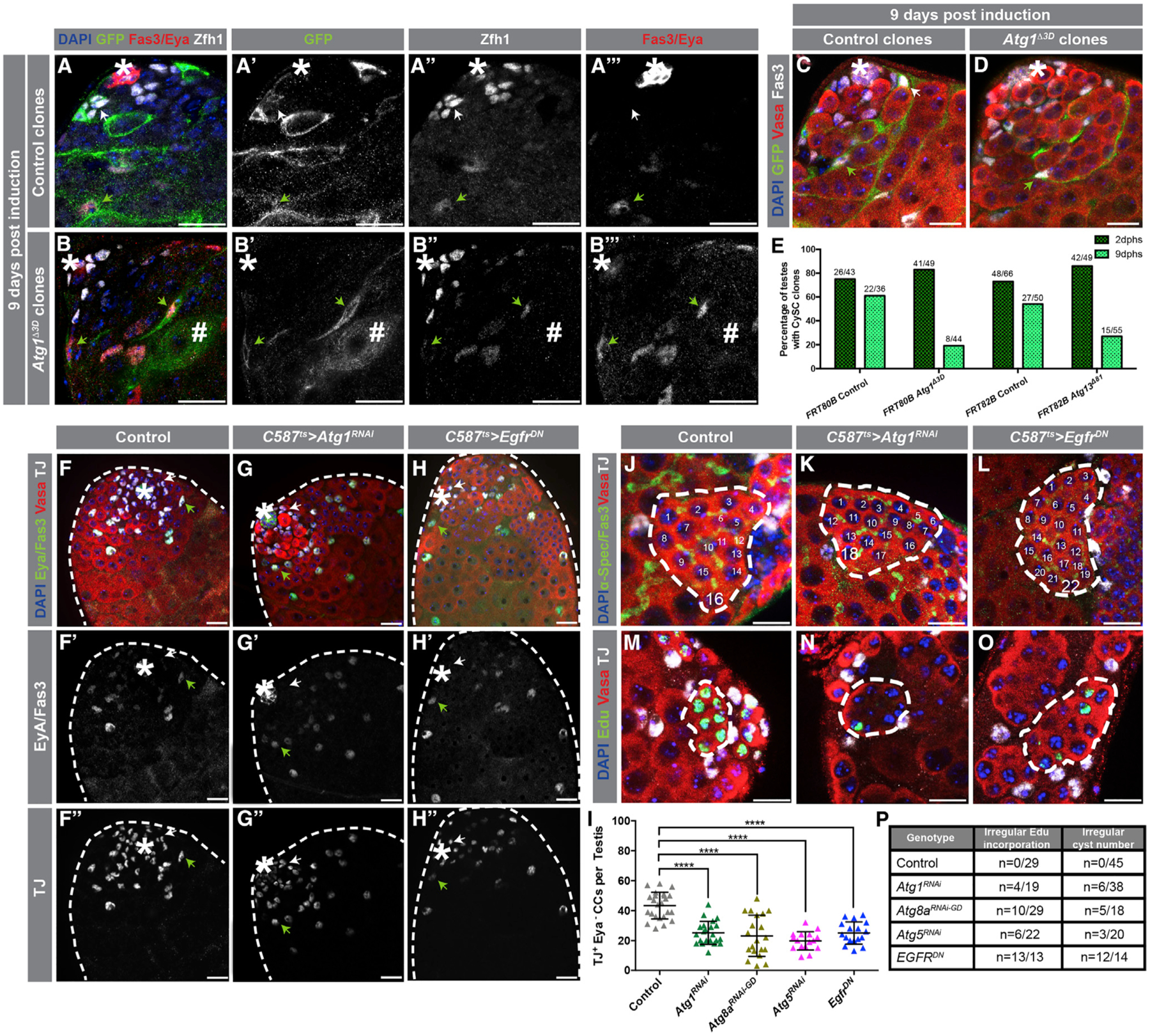
(A–B’’’’) Images of testes in which either control (A–A’’’) or Atg1−/− clones (B–B’’’) were generated 9 days prior using the mosaic analysis with a repressible cell marker (MARCM) technique (see STAR Methods). Clones are positively marked by GFP, and CySCs and very early CCs were stained with an antibody against Zfh1. Late CCs were stained with anti-Eya. White arrows denote CySC clones, and green arrows denote late CC clones. Hashmark denotes a germline cyst marked by GFP.
(C and D) Immunofluorescence (IF) of testes in which either control (C) or Atg1−/− clones (D) were generated and stained with the germline marker Vasa. Note that Atg1−/− somatic cell clones can still encapsulate developing germline cysts.
(E) Quantification of the percentage of CySC clones per testes. IF examples of 2 days post-heat shock (phs) clones can be found in Figure S2.
(F–H’’) Examples of testes from 10-day-old control (c587ts > +) (F–F’’), c587ts > Atg1RNAi (G–G’’), and c587ts > EgfrDN (H–H’’) animals stained with antibodies against TJ (early CCs, including CySCs) and Eya (late CCs). White arrows point to TJ+/Eya− CCs, and green arrows point to double TJ+/Eya+ CCs.
(I) Quantification of the number of early CCs (TJ+/Eya−) in 10-day-old animals driving somatic expression of transgenes with c587ts.
(J–O) Both knock down of Atg1 and overexpression of EgfrDN result in irregular spermatogonial cysts with more than 16 cells (stained with the fusome marker α-Spectrin) (J–L) and asynchronous mitotic divisions within a single germline cyst (stained for EdU incorporation) (M–O).
(P) Number of testes that contain at least one aberrant spermatogonial cyst is shown over the total amount of testes analyzed for different conditions. In all images, asterisks (*) denote the hub; scale bars, 20 μm. For (I), two-tailed t tests were used; error bars represent standard deviation. *p < 0.05, **p < 0.01, ***p > 0.001, ****p < 0.0001; n.s., not significant.
Autophagy Is Not Required for Mid-to-Late CC Differentiation
CC differentiation can be easily monitored by staining for a series of transcription factors that are expressed in a gradient from the apical tip to the base. The transcription factor Zfh-1 is highly expressed in CySCs and approximately the first two rows of CCs surrounding the hub (Leatherman and Dinardo, 2008). These cells also express Traffic Jam (TJ) (Li et al., 2003), which continues to be expressed in CCs surrounding spermatogonia throughout the TA zone. Late-stage CCs in contact with differentiating spermatocytes express high levels of Eyes absent (Eya) (Fabrizio et al., 2003; Figure S1A’). Thus, the number of early CCs, which have not yet begun differentiating into mid-to-late CCs, can be determined by the expression of TJ and absence of Eya (TJ+/Eya−). This approach allows for a more precise quantification of early progenitor cells without artifacts that could arise due to tissue mounting or morphological tissue variances when determining the location of Eya+ CCs (Amoyel et al., 2016a).
Fewer TJ+/Eya− CCs were observed when Atg1 was depleted from early CCs for 10 days, in comparison to controls (43.4 ± 8.9 TJ+/Eya− cells in controls versus 25.1 ± 7.7 TJ+/Eya− cells in Atg1RNAi, p < 0.0001) (Figures 2F–2G’’, and 2I). Furthermore, Eya+ CCs were observed close to the hub when Atg1RNAi was expressed (Figures 2F–2G’’, arrowheads), indicating that CCs lacking Atg1 are capable of differentiating into mid-to-late CCs. Similar results were obtained when either Atg5 or Atg8a were depleted for 10 days in early CCs (19.6 ± 6.1 TJ+/Eya− cells in Atg5RNAi, p < 0.0001; 23.2 ± 13.8 TJ+/Eya− cells in Atg8aRNAi; p < 0.0001) (Figure 2I). To test more directly whether Atg1 is required for CC differentiation, GFP+ Atg1Δ3D mutant CC clones were co-stained with antibodies to Eya (Figures 2A, 2A’’’, 2B, and 2B’’’), which revealed the presence of GFP+/Eya+ cells encapsulating germ cell cysts 9 days after heat -shock induction. Importantly, there was no evidence that the depletion of autophagy-related genes in CySCs and early CCs led to cell death (Figures S2C–S2E). Therefore, these data suggest that autophagy-deficient CySCs do not self-renew and are capable of differentiation.
Block in Autophagy Resembles Loss of EGFR Signaling
Although a number of pathways regulate communication between the germline and soma, EGFR signaling is one of the primary mechanisms used to orchestrate synchronous rounds of spermatogonial TA divisions (Kiger et al., 2000; Hudson et al., 2013). After 10 days of Atg1RNAi expression, testes that contained spermatogonial cysts with more than 16 cells were observed (16%, n = 6/38 testes) (Figures 2J, 2K, and 2P), as were cysts containing more than 16 spermatogonia that appeared to be dividing asynchronously (21%, n = 4/19 testes) (Figures 2M, 2N, and 2P), resembling phenotypes that occur when EGFR signaling is blocked through the expression of a dominant-negative (DN) construct (Perkins et al., 1996) (86%, n = 12/14 testes where spermatogonial cysts with more than 16 cells were present and 100%, n = 13/13 testes where germline cysts have uneven incorporation of the nucleotide analog 5-Ethynyl-2-deoxyuridine, EdU) (Figures 2L, 2O, and 2P). Similar observations were made upon depletion of Atg5 or Atg8a (Figure 2P). Furthermore, testes from animals in which Atg1 was depleted in CCs throughout development (Atg1RNAi(dev)) displayed a striking resemblance to testes mutant for the germline-derived EGF-ligand processing enzyme stet (Schulz et al., 2002; Figures S2F–S2H).
In addition to regulating germline differentiation in a non-autonomous manner, EGFR/MAPK signaling is also required for regulating CySC maintenance and behavior (Amoyel et al., 2016a). Indeed, when EGFR signaling was reduced for 10 days, the number of early CCs decreased (25.1 ± 7.4 TJ+/Eya−, p < 0.001) (Figures 2H and 2I) (Amoyel et al., 2016a), resembling what was observed as a consequence of depleting autophagy-related genes (Figure 2I). Altogether, these results suggest that the reduction in autophagy, which results in a loss of CySCs and aberrant spermatogonial TA divisions, mimics reduced EGFR signaling in CySCs and CCs.
EGFR Acts Upstream of Autophagy-Related Genes in Somatic Cells
To investigate a potential interaction between Egfr and autophagy-related genes, both EGFR/MAPK and autophagic activities were assessed. EGFR activity can be monitored using a specific antibody against the diphosphorylated form of Erk (dpErk) (Yung et al., 1997). Staining for dpERK was detected in CySCs and CCs, which correlated with the expression of EGFR (Figures S3A–S3A’’ and S3B–S3B’’). As expected, the dpERK signal intensity increased when EGFR was activated using a transgene expressing a constitutively active (CA) EGFR (Queenan et al., 1997; Figures S3C–S3C’’ and S3H) and decreased when EGFR signaling was reduced (Figures S3D–S3D’’ and S3H). However, no significant changes were observed in dpErk intensity when Atg1, Atg5, or Atg8a were depleted (Figures S3E–S3H), suggesting that autophagy does not act upstream of the EGFR pathway. However, significant changes to the composition of autophagic structures, as assayed by the GFP-mCherry-Atg8a probe, was observed when EGFR/MAPK signaling was activated in early somatic cells (Figures 3A and 3B), indicating that EGFR signaling may stimulate autophagy (i.e., a relative increase in ALs compared to APs). Conversely, the downregulation of EGFR signaling resulted in no detectable puncta for the GFP-mCherry-Atg8a analysis, as well as a severe decrease in the number of mCherry-Atg8a+ APs present in CCs (Figure S1C). These findings are consistent with a role for EGFR in promoting basal autophagy.
Figure 3. EGFR Acts Upstream of Autophagy-Related Genes in CCs to Regulate Autophagy.
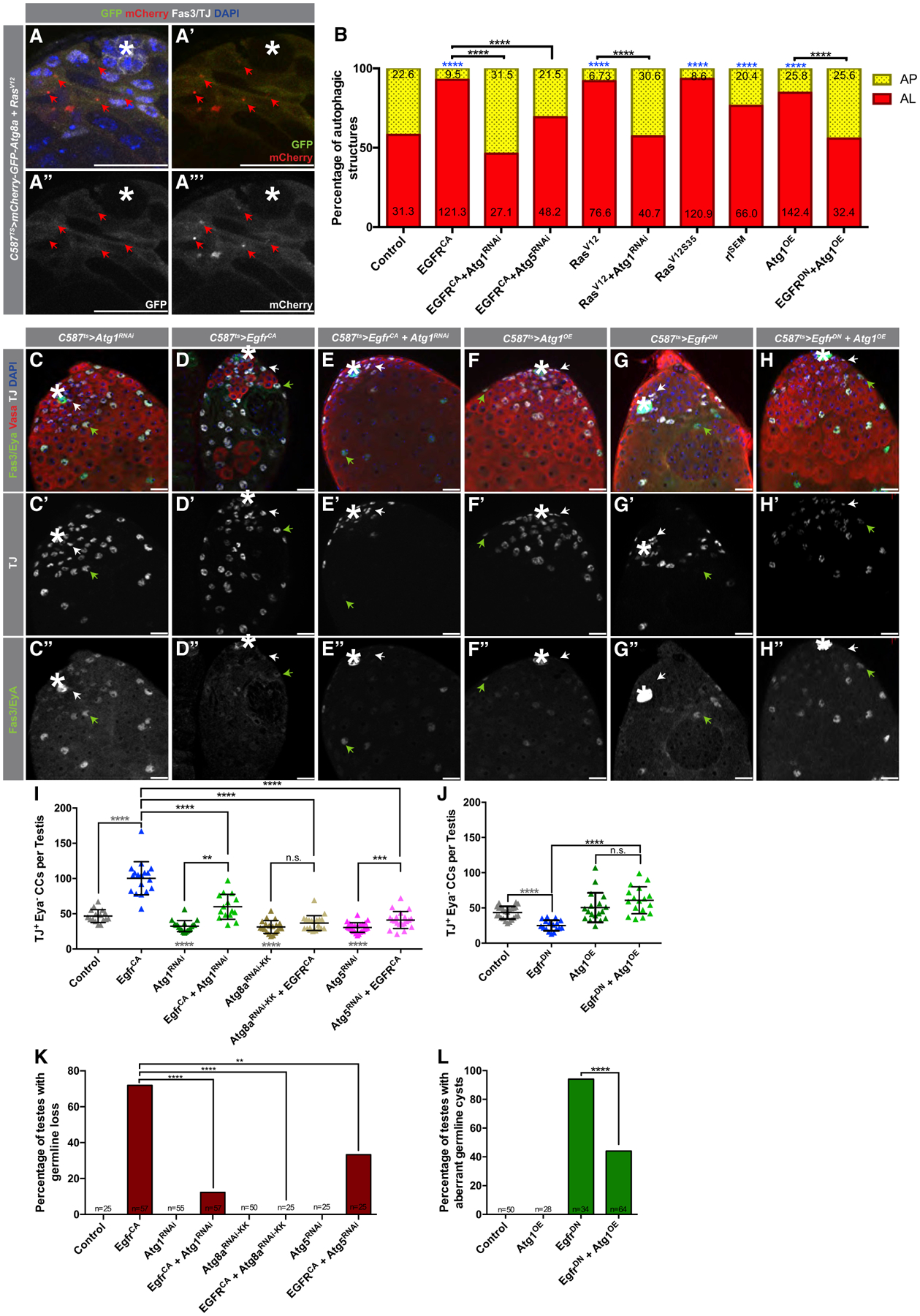
(A–A’’’) Representative image of a testis tip from 5-day-old animals expressing the autophagic flux probe GFP:mCherry:Atg8a in CCs together with the overexpression of an activated Ras mutant (compare to Figure 1E–E’’’).
(B) Quantification of autophagic structures in the noted genotypes. Numbers on each bar color represent the mean number of autophagic structures per testis tip (n = 10 testes per condition). Blue asterisks represent statistical information compared to control. Control data reproduced from Figure 1F (RD) for comparison.
(C–E’’) Representative images of testes from 5-day-old animals expressing c587ts > Atg1RNAi+TdTomato (C–C’’), c587ts > EgfrCA+TdTomato (D–D’’), and c587ts > Atg1RNAi+EgfrCA (E–E’’) stained with antibodies against Fas3/Eya, Vasa, and TJ. Note the accumulation in (D–D’’) of early CCs, marked by TJ expression and Eya absence (white arrows). Green arrows point to double TJ+/Eya+-stained CCs.
(F–H’’) Representative images of testes from 10-day-old animals expressing c587ts > Atg1OE+TdTomato (F–F’’), c587ts > EgfrDN +TdTomato (G–G’’), and c587ts > Atg1OE+EgfrDN (H–H’’).
(I) Quantification of the number of early CCs (as marked by TJ+/Eya−) in 5-day-old animals with the c587TS driver.
(J) Quantification of the number of early CCs (as marked by TJ+/Eya−) in 10-day-old animals with the c587TS driver. Note that for (I) and (J), the control is (c587TS > TdTomato), and all single UAS-based transgenes were paired with UAS-TdTomato to take into account Gal4/UAS levels compared to double UAS combinations (see STAR Methods). Gray statistical information compared to control.
(K) Quantification of the percentage of testes in which germline loss was observed (Fisher’s exact was used).
(L) Quantification of the percentage of testes in which spermatogonial cysts with aberrant numbers were observed (Fisher’s exact was used) (Figures S3M–S3R show α-Spectrin stains used to aid in categorizing aberrant cysts).
In all images, asterisks (*) denote the hub; scale bars, 20 μm. For (I) and (J), two-tailed t tests were used; error bars represent standard deviation. For (B), (K), and (L), two-sided Fisher’s exact was used. *p < 0.05, **p < 0.01, ***p > 0.001, ****p < 0.0001; n.s., not significant.
To further elucidate the relationship between the EGFR pathway and autophagy, a possible genetic interaction was tested. Similar to previous reports using a broadly expressed CC driver (TJ-Gal4), overexpression of EgfrCA or a CA form of Ras (RasV12) with c587-Gal4ts resulted in the expansion of TJ+/Eya− early CCs and an increase in CySC competitiveness and niche occupancy, as reflected by the loss of germ cells adjacent to the hub (Figures 3D–3D’’, 3I, and 3K; Figures S3I–S3I’’, S3K, and S3N; Amoyel et al., 2016a). Because the activation of Ras by EGFR signaling can lead to the activation of the RAF/MEK/ERK and/or PI3K/Akt/TOR cascades in other systems (Bergmann et al., 1998; Karim and Rubin, 1998; O’Keefe et al., 2007), the specificity of Ras activation in early CCs was tested genetically. Only the expression of an activated Ras mutant that is capable of activating the MAPK cascade (RasV12S35), but not one capable of activating the PI3K pathway (RasV12G37), resulted in the expansion of early CCs (Figure S3L), suggesting that Ras targets the MAPK but not the PI3K pathway in early CCs. Expression of RasV12S35 in early CCs also increased the proportion of ALs in these cells (Figure 3B).
Strikingly, co-expression of Atg1RNAi with EgfrCA (Figures 3C–3E’’, 3I, and 3K; Figures S3M–S3O) or RasV12 (Figure S3I–K) for 5 days suppressed the accumulation of early CCs and germline loss, indicating that Atg1 is required for the effects of constitutive EGFR signaling on CC behavior. Co-expression of either Atg5RNAi or Atg8aRNAi with EgfrCA also suppressed the accumulation of early CCs and germline loss caused by excessive EGFR signaling (Figures 3I and 3K; Figures S3S–S3V’’). Importantly, the knockdown of autophagy-related genes suppressed the increase in AL frequency caused by the overactivation of the EGFR pathway (Figure 3B), further indicating that changes in autophagic structures are correlated with the disruption in early CC behavior.
As noted above, overexpression of EgfrDN results in the reduction of early CC number and disruption of CC function, as reflected by excess spermatogonial divisions (Figures 3G–3G’’, 3J, and 3L; Figure S3Q). However, co-expression of Atg1 (with UAS-Atg1OE) (Scott et al., 2007) with EgfrDN partially suppressed these phenotypes (Figures 3F–3H’’, 3J, and 3L; Figures S3P–S3R) and decreased the AL to AP proportion (Figure 3B). Together, these results indicate that the EGFR pathway acts upstream of autophagy-related genes to stimulate autophagy in early CCs to regulate their behavior.
EGFR/MAPK Signals through AP-1/Fos to Stimulate Autophagy
Although a positive relationship between EGFR signaling and autophagy has been observed in other instances (Sooro et al., 2018), the precise mechanism(s) by which EGFR/MAPK activates autophagy is still unknown. One positive regulator of autophagy, the c-Jun N-terminal Kinase (JNK) pathway controls both the expression and activity of autophagy-related genes (Füllgrabe et al., 2016; Zhou et al., 2015). Under stressful conditions in the Drosophila testis, JNK activation correlated with the activation of autophagy in CCs (Yang and Yamashita, 2015; Tang et al., 2017). One mechanism by which JNK activates autophagy is through AP-1-mediated transcription and upregulation of Atg6/Beclin1, a heavily regulated component of the Vps34 complex required for autophagy progression (Figures 1A, 1G, and 1H; Füllgrabe et al., 2016; Zhou et al., 2015; Cao and Klionsky, 2007). In Drosophila, one of the two AP-1 heterodimer components Fos has been shown to be a substrate for phosphorylation by both ERK and JNK (Biteau and Jasper, 2011; Ciapponi et al., 2001). Hence, ERK-mediated phosphorylation of Fos is a possible mechanism for the modulation of autophagy by the EGFR/MAPK pathway.
Consistent with this hypothesis, blocking EGFR signaling in early CCs led to a significant decrease in mRNA levels for Atg6/BCL1 (Figure 4A). In addition, expression of dominant-negative Fos mutants, in which all JNK and ERK phosphorylation sites (FosPan-Ala) or only specific ERK phosphorylation sites (FosC-Ala) have been abolished, significantly decreased the amount of APs in CCs (Figure 4B). Furthermore, expression of these DN Fos mutants, as well as RNAi-mediated depletion of Fos, for 10 days significantly reduced the number of TJ+/Eya− CCs (Figures 4C–4E’’ and 4I), phenocopying the block in EGFR and autophagy. Finally, overexpressing Fos rescued the loss of early CCs, restored the balance of ALs to APs that was absent due to the block in EGFR signaling, and increased the mRNA levels for Atg6/BLC1, which was reduced by the inhibition of EGFR (Figures 4A, 4I, and 4J). In contrast, overexpression of Fos did not rescue the decrease in early CC number caused by the depletion of Atg8a (Figure 4I), which acts downstream of Atg6/BLC1. These results strongly suggest that Fos acts downstream of Egfr to stimulate autophagy.
Figure 4. Fos/AP-1 Acts Downstream of EGFR to Stimulate Autophagy.
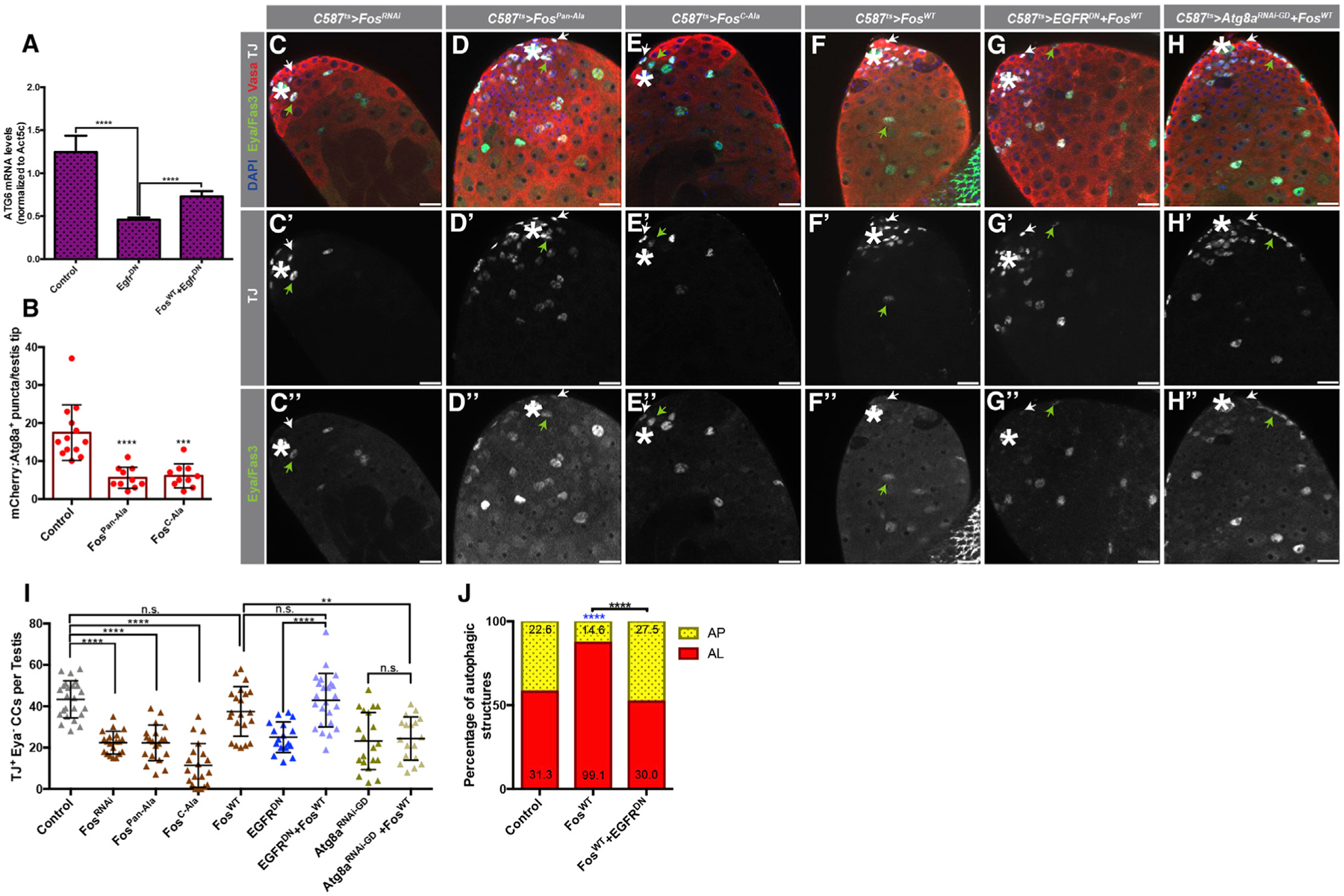
(A) qRT-PCR quantification of quintuplicates displaying the ratio between Atg6 and Atc5c mRNA in testes from 3-day-old animals of the noted genotypes.
(B) Quantification of the number of APs per testis tips in the somatic CC tissue of the noted genotypes.
(C–H’’) Representative images showing testes tips from 10-day-old animals expressing the early CC ‘driver’ c587-GAL4TS with: UAS-FosRNAi(C–C’’), UAS-FosPan-Ala(D–D’’), UAS-FosC-Ala(E–E’’), UAS-FosWT(F–F’’), UAS-EGFRDN, UAS-FosWT(G–G’’), and UAS-Atg8aRNAi-GD, UAS-FosWT(H–H’’). White arrows point to TJ+/Eya− CCs, and green arrows point to TJ+/Eya+ CCs. Note that all single UAS-based transgenes were paired with UAS-TdTomato to consider Gal4/UAS levels compared to double UAS combinations.
(I) Quantification of TJ+/Eya− CCs in 10-day-old animals of the noted genotypes. Control data reproduced from Figure 3J for comparison.
(J) Quantification of autophagic structures in the noted genotypes. Blue asterisks represent statistical information compared to control. Numbers on each barcolor represent the mean number of autophagic structures per testis tip (n = 10 testes per condition). Control data reproduced from Figure 1F for comparison. In all images, asterisks (*) denote the hub; scale bars, 20 μm. For (A), (B), and (I), two-tailed t tests were used; error bars represent standard deviation. For (J), two-sided Fisher’s exact was used. *p < 0.05, **p < 0.01, ***p > 0.001, ****p < 0.0001; n.s., not significant.
TOR Acts in Differentiating CCs to Suppress Autophagy
A major upstream regulator of autophagy is the target of rapamycin (TOR) pathway (Noda and Ohsumi, 1998). In response to elevated nutrient levels and/or extracellular signals (such as insulin/insulin-like peptides and growth factors), TOR is activated, together with regulator and effector proteins, to control multiple aspects of cell behavior and metabolism. When bound to insulin/insulin-like peptides, the insulin receptor pathway (InR) activates phosphoinositol 3 kinase (PI3K), which triggers the downstream phosphorylation of Akt by PDK1 (reviewed in Wullschleger et al., 2006). Activated Akt then inhibits the tuberous sclerosis complex (TSC)1/2 repressor complex, releasing the small GTPase Rheb from inhibition (Garami et al., 2003). Once uninhibited, Rheb can activate the TOR pathway. In response to active TOR, Atg1/ULK1 is phosphorylated and inactivated (Kamada et al., 2000), thereby suppressing autophagy.
Recently, activation of the TOR pathway has been implicated in regulating stem cell maintenance and differentiation in a range of systems (Meng et al., 2018). In the Drosophila testis, TOR signaling regulates CC differentiation downstream of the InR pathway (Figure S1A). Depleting components of the InR/TOR pathway in CCs or inhibiting TOR pharmacologically with rapamycin blocks CySC daughter cells from differentiating, leading to an accumulation of early progenitor cells (Amoyel et al., 2016b).
A significant accumulation of cytoplasmic GFP-mCherry-Atg8a was observed in differentiating CCs, along with the presence of APs that did not merge with the lysosome to acidify and complete autophagy (Figures 5A–5A’’’). This apparent block in autophagy correlates with the onset of CySC daughter cell differentiation, which is reflected by high InR/TOR activity (Figures S4A–S4B’), suggesting that the InR/TOR-mediated suppression of autophagy may be important for CC differentiation. Accordingly, pharmacological inhibition of TOR (Figure 1F) or RNAi-mediated depletion of TOR in CCs (Figure 5B) led to an increase in the proportion of ALs and reduction in APs in early CCs, providing further evidence that autophagy is subject to TOR-mediated regulation in these cells.
Figure 5. TOR Acts in Differentiating CCs to Suppress Autophagy.
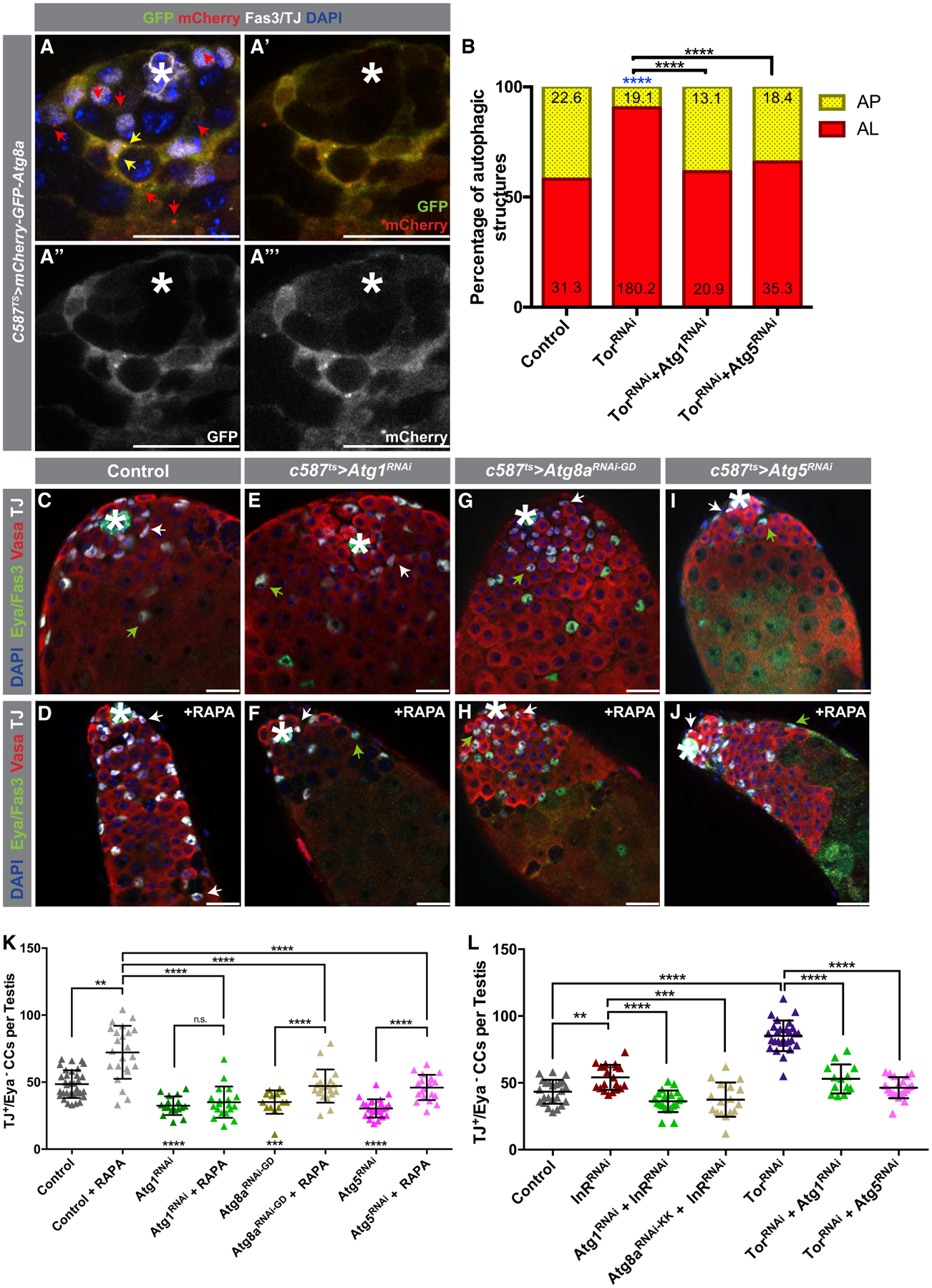
(A–A’’’) Representative IF of a testis tip from flies expressing the GFP:mCherry:Atg8a probe. Red arrows point to mCherry+-only APs, and yellow arrows point to GFP+/mCherry+ APs.
(B) Quantification of autophagic structures in the noted genotypes (all with c587TS). Blue asterisks represent statistical information compared to control. Numbers on each bar color represent the mean number of autophagic structures per testis tip (n = 10 testis per condition). Control data reproduced from Figure 1F for comparison.
C–J) Examples of testes from 5-day-old control animals (C and D) and from animals expressing c587ts > Atg1RNAi (E and F), c587ts > Atg8aRNAi (G and H), or c587ts > Atg5RNAi (I and J) that were either fed a RD (C, E, G, and I) or food containing RAPA for 5 days (D, F, H, and J). White arrows point to TJ+/Eya− early CCs, and green arrows point to TJ+/Eya+ CCs.
(K) Quantification of numbers of early CCs (TJ+/Eya−) in the genotypes shown in (C)–(J).
(L) Quantification of the numbers of early CCs in animals of the noted genotypes. Single UAS-based transgenes were paired with UAS-TdTomato for consideration of Gal4/UAS-levels.
In all images, asterisks (*) denote the hub; scale bars, 20 mm. For (B), Fisher’s exact was used. For (K) and (L), two-tailed t tests were used; error bars represent standard deviation. *p < 0.05, **p < 0.01, ***p > 0.001, ****p < 0.0001; n.s., not significant.
To directly test whether the block in CC differentiation caused by the inhibition of the InR/TOR pathway is due to an upregulation of autophagy, control and flies depleted of autophagy-related genes were fed rapamycin to inhibit TOR activity for 5 days. Although rapamycin treatment led to the accumulation of TJ+/Eya− early CCs in testes from control flies (Figures 5C, 5D, 5K, and S4C–S4D’), depleting Atg1, Atg5, or Atg8a in early CCs blocked this accumulation (Figures 5E–5K and S4E–S4J’). A similar trend was observed when InR or Tor were depleted simultaneously with the depletion of Atg genes (Figure 5L). Accordingly, depletion of either Atg1 or Atg5 also suppressed the increase in the AL to AP proportion in CCs caused by the depletion of Tor (Figure 5B). In summary, suppression of autophagy, induced by high InR/TOR signaling, is required in the CySC daughter cells to promote differentiation.
Lipid Levels Are Maintained by EGFR-Stimulated Autophagy in CCs
Our data indicate that proper regulation of basal autophagy by both EGFR (positive) and TOR (negative) is important for maintaining tissue homeostasis in the fly testis. However, the specific role that autophagy plays in influencing somatic CC behavior is unknown. Given that both the EGFR and InR/TOR pathways have been shown to influence metabolism (Choudhary et al., 2016; Oldham, 2011), we hypothesized that a change in metabolism, driven by the regulation of autophagy, would correlate with the differentiation of CySC daughter cells. Recent data from our lab indicated that lipid accumulation in Drosophila male GSCs correlated with stem cell loss, at least in part, due to ectopic TOR signaling (Sênos Demarco et al., 2019). Hence, we decided to investigate whether lipid levels would change upon the disruption of autophagy and EGFR signaling in the soma.
Intracellular lipids can be stored in lipid droplets (LDs) that act as reservoirs of lipids to satisfy energy demands and to participate in important metabolic and signaling events (Fujimoto and Parton, 2011). To visualize whether excess lipids could be catabolized through autophagy in CCs, testes were cultured ex vivo for 1 h in the presence of BODIPY-C12, which can be incorporated into LDs (Rambold et al., 2015), with or without the addition of chloroquine to inhibit autophagy. Subsequent imaging revealed that the number of LDs increased significantly when autophagy was blocked, suggesting that lipid levels are regulated by autophagy (Figures S5A–S5D). To visualize whether LDs can be found in association with APs in CCs, testes from animals expressing GFP:Atg8a in CCs were cultured ex vivo in the presence of the fluorescently labeled fatty acid (FA) analog BODIPY-C12. Under these conditions, the majority of Atg8a+ APs co-localized with LDs (Figures S5E–S5G). To determine if autophagy is utilized in early CCs to regulate lipid levels in vivo, the lipophilic dye BODIPY 493/403 was used to stain neutral lipids in LDs. Depletion of Atg1 or Atg8a in early CCs led to a significant increase in the number of LDs (Figures 6A–6C’ and 6F) and an overall increase in the levels of triacylglycerides (TAGs), the main lipid species in LDs, in testes, as determined by thin-layer chromatography (Figure 6E). Altogether, these results suggest that autophagy is required for regulating lipid levels in early CCs.
Figure 6. EGFR-Stimulated Autophagy Controls Lipid Metabolism in CCs.
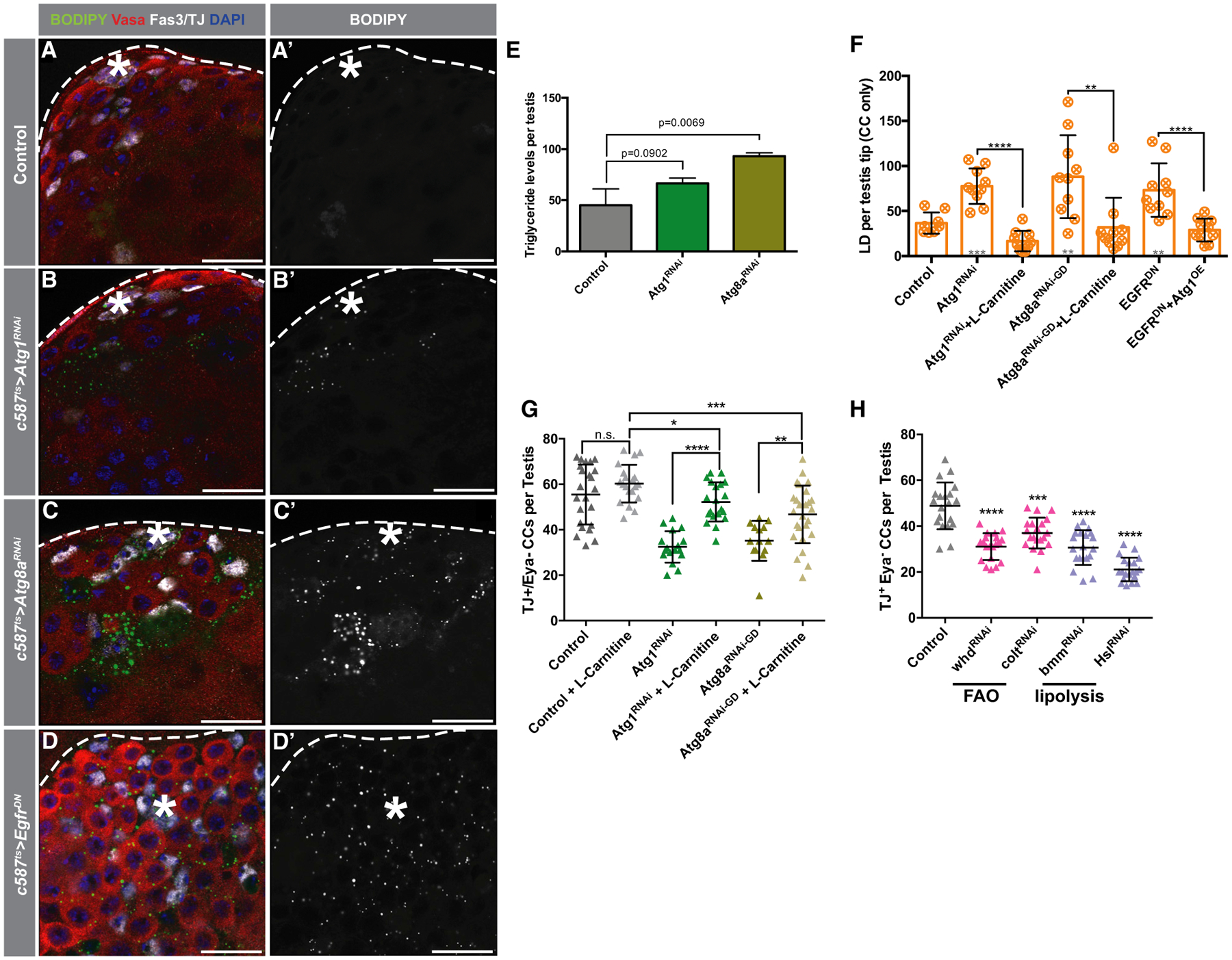
(A–D’) Representative images of testes from 5-day-old animals in control (A–A’), c587TS>Atg1RNAi(B–B’), c587TS>Atg8aRNAi(C–C’) and c587TS>EgfrDN(D–D’) stained with BODIPY. Note the dramatic accumulation of LDs when autophagy or EGFR is hindered.
(E) Quantification of the levels of triglycerides per testis of the noted genotypes. Pixel intensity quantified using ImageJ as described (STAR Methods). Average of 3 biological replicates (50 testes each replicate) represented in bars.
(F) Quantification of LDs in somatic tissue per testis tips (see STAR Methods) in 5-day-old flies of the noted genotypes/conditions.
(G) Quantification of early CCs per testis of 5-day-old flies of the noted genotypes/conditions. For (F) and (G), single UAS-based transgenes were paired with UAS-TdTomato for consideration of Gal4/UAS-levels.
(H) Quantification of early CCs per testis of 10-day-old c587-GAL4TS flies crossed to control (w1118) or UAS-based transgenes triggering RNAi-mediated depletion of genes involved in fatty acid oxidation (FAO; colt/CACT and whd/CPT1) and lipolysis (bmm/ATGL and Hsl).
In all images, asterisks (*) denote the hub; scale bars, 20 μm. For (E)–(H), two-tailed t tests were used; error bars represent standard deviation. *p < 0.05, **p < 0.01, ***p > 0.001, ****p < 0.0001; n.s., not significant.
To assess whether the increase in lipid accumulation contributes to the loss of early CCs caused by the disruption of autophagy, control and autophagy-depleted animals were fed a diet with L-carnitine to stimulate lipid utilization by the mitochondria (Sênos Demarco et al., 2019; see STAR Methods). Strikingly, the enhancement of mitochondrial lipid catabolism was sufficient to suppress the loss of early CCs triggered by the depletion of either Atg1 or Atg8a, as well as LD accumulation (Figures 6F and 6G), providing evidence that lipid accumulation contributes to the loss of CySCs and early CCs caused by the block in auto/lipophagy. As our data indicate that EGFR signaling acts upstream to stimulate autophagy, we hypothesized that reducing EGFR signaling would also lead to the accumulation of lipids in CCs. Indeed, expression of EGFRDN resulted in LD accumulation in early CCs (Figures 6D, 6D’, and 6F), which was suppressed by the enhancement of autophagy achieved by Atg1 overexpression (Figure 6F). In summary, these results reveal a role for autophagy in controlling lipid metabolism in early CCs.
Importantly, CySCs and early CCs appear to be sensitive to intracellular lipid levels, as reducing FA utilization by knocking down components of the FA/carnitine shuttle, which allows FA entry into the mitochondria for FA oxidation (Hartenstein et al., 1997; Palanker et al., 2009; Strub et al., 2008), resulted in significant decreases in the number of TJ+/Eya− CCs (Figure 6H). Similar observations were made when the cytoplasmic lipase Brummer (Bmm) was depleted (Grönke et al., 2005; Figure 6H). Together, these data provide evidence for a role for lipid metabolism in regulating CC behavior.
DISCUSSION
Our findings establish a role for EGFR in promoting autophagy, in opposition to InR/TOR signaling, to support stem cell maintenance and regulate progenitor cell differentiation in the Drosophila testis. An RNAi-based screen of components required for AP formation revealed that basal autophagy is required in CySCs and CCs for stem cell maintenance and function (Figures 1G, 2H, and 2A–2P). Both the analysis of autophagic markers in response to changes in EGFR activity (Figures 3A and 3B), as well as genetic epistasis experiments (Figures 3C–3L; Figures S3M–S3V’’), revealed that autophagy can be stimulated by EGFR activity to regulate tissue homeostasis. The stimulation of autophagy by EGFR signaling is counteracted by InR/TOR in differentiating CCs (Figures 1F, 5A, and 5B), due to the well-characterized role TOR plays in regulating autophagic activity(Noda and Ohsumi, 1998). Accordingly, the inhibition of autophagy in CySCs and early CCs blocks the accumulation of early progenitor cells when InR/TOR activity is inhibited (Figures 5C–5L). Altogether, these data demonstrate that autophagy is an important process involved in regulating somatic cell fate decisions in the fly testis.
Somatic CCs must grow continuously to encapsulate the differentiating germline cyst; therefore, we speculated that CySCs, the daughter cells that initiate differentiation, and later stage CCs likely have distinct metabolic needs. Indeed, our data indicate that metabolic “remodeling” or “reprogramming” may be required during the transition from CySCs to differentiating CCs. Upon repression of either autophagy or EGFR signaling, an accumulation of LDs was observed, which accompanied a loss of early CCs (Figures 6A–6F). Moreover, directly increasing lipid accumulation in early CCs also led to stem cell loss, suggesting that these cells are sensitive to intracellular lipid levels (Figure 6H). Pharmacological enhancement of mitochondrial FA oxidation in autophagy-depleted animals was sufficient to suppress early CC loss (Figures 6F and 6G), similar to the manner in which the genetic enhancement of autophagy suppressed LD accumulation in EGFRDN animals (Figure 6F). Hence, the dynamic regulation of autophagy by EGFR and TOR regulates differentiation of the CCs, as well as the germ cells that it supports, in part, by controlling lipid metabolism.
Multiple scenarios could lead to LD accumulation as a result of the disruption of autophagy. Lipophagy has emerged as an important branch of the autophagic pathway, which is required for regulating lipid catabolism (Liu and Czaja, 2013; Schulze et al., 2017). When needed, lipids can be released from LDs by lipophagy or lipolysis and transferred into the mitochondria for energy production or used for other catabolic processes (Liu and Czaja, 2013). Conversely, cells with sufficient nutrients signal through TOR to inhibit lipophagy and instead promote lipogenesis and/or lipid storage (Schulze et al., 2017). However, the accumulation of LDs could also be due to an acceleration of organelle turnover or as a consequence of mitochondrial damage, resulting in faulty mitochondria that cannot oxidize FAs, which then accumulate into LDs (Nguyen et al., 2017; Zhang et al., 2018). We provide evidence that defective autophagy, rather than hyperactive autophagy, leads to the accumulation of LDs (Figures 6A–6D’, 6F, and S5A–S5D). In addition, we show that feeding flies L-carnitine suppresses the increase in LDs (Figure 6F) and the loss of early CCs (Figure 6G) observed when autophagy is disrupted, indicating that the mitochondria in these cells are still active and capable of oxidizing FAs. As independent evidence that these cells are sensitive to intracellular lipid levels, we demonstrate that the depletion of FA catabolism enzymes also leads to a loss in early CCs (Figure 6H). These data strongly support a model in which autophagy is an important mechanism used to regulate lipid levels under homeostatic conditions; however, autophagy is likely important for the turnover of organelles, in addition to LDs, and prevention of cellular damage in the context of aging (Nagy et al., 2018). In addition, lysosomal lipases (Folick et al., 2015; Lapierre et al., 2011; Wang et al., 2008) may also be important for controlling lipid levels in early CCs. Future experiments will be necessary to identify additional mechanisms involved in maintaining lipid homeostasis in the soma of the Drosophila testis and for determining other processes regulated by autophagy to control aspects of CC maintenance and metabolism.
Although EGFR signaling has been reported to suppress autophagy in some mammalian cancer cell lines (Fan et al., 2009; Tan et al., 2015), it has also been shown to positively regulate autophagy in others (Sooro et al., 2018). The discrepancy between activation and inhibition may lie in secondary mutations and/or the ability of cancer cells to modulate both MAPK and PI3K through EGFR. The data presented here, however, illustrate a positive relationship between EGFR signaling and autophagy under homeostatic conditions. In agreement with our work, a recent study has shown that the GTPase Ras, which is activated by EGFR, can induce autophagy in mammalian tissues (Alves et al., 2015). A positive relationship between activated Ras and autophagy has also been reported in a Drosophila model of tumorigenesis (Manent et al., 2017). Importantly, our work demonstrates that Ras activation selectively stimulates the MAPK cascade in CCs, which has been shown to activate autophagy in tumors (Sooro et al., 2018). EGFR can also contribute to the activation of the JNK pathway, which is a well-established, positive regulator of autophagy (Oda et al., 2005; Pearson et al., 2001; Sui et al., 2014; Xu et al., 2011; Zhou et al., 2015; Ciapponi et al., 2001; Kockel et al., 2001). Indeed, our work also reveals a putative path, through Fos, by which EGFR and JNK may cross talk during stressful conditions to control early CC behavior. The many roles that EGFR plays in regulating homeostasis in the testis suggests that EGFR does not act only to stimulate autophagy. Notably, signaling through Vav/Rac1/Rho1 has been described to act in the EGFR-mediated encapsulation of germ cells (Sarkar et al., 2007). In addition, because the overexpression of Atg1 is not sufficient to drive early CC accumulation (as observed upon strong activation of the EGFR pathway; Figure 3J), it suggests that the MAPK cascade likely acts not only through autophagy to control early CC behavior. Nevertheless, our data indicate that stimulation of autophagy is an important and required consequence of EGFR signaling in early CCs.
Stem cells often have distinct metabolic profiles compared to differentiated cells (Chandel et al., 2016). Indeed, autophagy is required for the increase in energy demands required during muscle stem cell (satellite cell) activation and differentiation (Tang and Rando, 2014). In addition, a recent report demonstrated that autophagy maintains HSC self-renewal and regenerative capacity by clearing damaged mitochondria (Ho et al., 2017). Our work sheds light on another role of autophagy in stem cells by demonstrating that autophagy influences stem cell fate decisions and tissue homeostasis by controlling lipid catabolism, a role that may be conserved in more complex mammalian stem cell systems.
STAR★METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, D. Leanne Jones (leannejones@ucla.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Flies were raised on a standard cornmeal and molasses diet with no more than 25 flies per vial. For pharmacological regimens, Rapamycin (final concentration - 400 mM; TSZ Chem, Cat# R1017), Chloroquine Diphosphate (final concentration - 50mM; Sigma, Cat#C6628) or L-Carnitine inner salt (final concentration - 5mg/mL; BeanTown Chemical, Cat# 215415) was mixed with regular molten Drosophila media and poured into vials. Eclosed flies of the specific genotypes were transferred to Rapamycin, Chloroquine or L-Carnitine-containing food vials and transferred every 2–3 days.
METHOD DETAILS
Tissue-specific genetic manipulation
To knock down genes in CySCs and early CCs using c587TS (c587-Gal4/Y;tub-Gal80ts/+;+/+), crosses were performed and maintained at 18°C until eclosion [for UAS-RasV12G37 (transgene on the X chromosome), TJTS was used (+/Y;TJ-Gal4/+;tub-Gal80TS/+)]. Males were then shifted to 29°C after eclosion to induce the expression of UAS-transgenes. Flies maintained at 29°C were transferred onto new food every 2–3 days and were dissected after 5 or 10 days as stated. Control flies were the progeny of outcrosses from the Gal4 driver line to w1118 flies. When determining the genetic interaction between two UAS-based constructs, UAS-levels were taken into consideration – UAS-TdTomato was incorporated into every cross using a single UAS-element (i.e., UAS-Atg1RNAi or UAS-EGFRCA) but not incorporated in crosses using 2 UAS-elements (i.e., UAS-Atg1RNAi + UAS-EGFRCA). The control used in such cases was the progeny of the cross between c587TS and UAS-TdTomato. To knock down genes in GSCs and early germ cells using nos-Gal4:VP16, crosses were performed and maintained at 25°C, and males were aged to 10 days. Precise genotypes for all representative figures and experiments described on this manuscript can be found in Table S1.
RNAi screen targeting autophagy-related genes
Disruption of autophagy in the testis was restricted to adulthood by using the ubiquitously expressed and temperature-sensitive (ts) tub-Gal80ts transgene in combination with the CC driver c587-Gal4. GAL80ts represses the activity of GAL4 in a temperature-dependent manner, allowing for transient expression of UAS-transgenes (Zeidler et al., 2004). For simplicity, this system was referred to as Gal4ts in the manuscript.
Immunostaining
With the exception of dpErk stains, testes from adult flies were dissected and fixed in a 2% paraformaldehyde solution for 1 hour. Samples were washed 15 minutes twice with 0.1% Triton X-100 in PBS (PBS-T) with 0.3% sodium deoxycholate, then washed once for 10 minutes with PBS-T. Testes were blocked with a 3% bovine serum albumin (BSA) solution in PBS-T and incubated overnight at 4°C in primary antibodies diluted in the block solution. Samples were then washed for 10 minutes three times with PBS-T, incubated in Alexa-conjugated secondary antibody (1:500, Invitrogen) with 3% BSA in PBS-T, washed 10 minutes three times, and mounted in vectashield with DAPI (Vector Labs). Primary antibodies used were mouse anti-Fasciclin 3 (1:50; DSHB), mouse anti-α-Spectrin (1:10; DSHB), mouse anti-Eyes absent (1:10, DSHB), rat anti-Vasa (1:50, DSHB), rabbit anti-Vasa (1:100; Santa Cruz Biotechnology), mouse anti-Phospho-Histone H3 (Ser10) (6G3) (1:100; Cell Signaling), chicken anti-Green Fluorescent Protein (1:1000; Aves Labs), rabbit anti-P-4E-BP1 (1:500, Cell Signaling), mouse anti-dEGF Receptor (clone C-273) (1:1000; Sigma), rabbit anti-phospho-p44/42 MAPK (1:100), rabbit anti-Zfh1 (1:500; a gift from R. Lehmann, New York University), and guinea pig anti-Traffic Jam (1:100; a gift from D. Godt, University of Toronto, Canada). For LD stains – samples were incubated for 20min in BODIPY 493/503 (1:1000, Thermo Fisher) after secondary antibody stain and washes, followed by two more washes in PBS-T and mounting in vectashield with DAPI. Samples were imaged with a Carl Zeiss LSM 780 or 880 Confocal microscope, or Axio Vert.A1 inverted light microscope with a 40x, 40x water-immersion, or 63x oil-immersion objective. Digital images were processed using ZEN digital imaging (version 4.1, Zeiss), ImageJ (v2.0.0, Wayne Rasband, National Institute of Health, https://imagej.nih.gov/ij), Adobe Photoshop and Adobe Illustrator softwares.
dpErk staining protocol
Flies were fed yeast paste placed on top of regular food overnight prior to dissection into Schneider’s media supplemented with phosphatase inhibitor cocktail 2 (1:100, Sigma, cat#P5726). Dissected testes were then fixed for 30 min in 4% paraformaldehyde supplemented with phosphatase inhibitor, washed 4x in testis buffer (10mM Tris-HCl pH6.8, 180mM KCl; as seen in Schulz et al. (2002) supplemented with phosphatase inhibitor, 0.2% BSA and 0.3% Triton TX100, prior to incubation in 1° antibody overnight supplemented with phosphatase inhibitor.
Generation of Atg1−/− and Atg13−/− clones
Clones positively marked with GFP were generated using the MARCM technique (Lee and Luo, 1999). Two 2h-heat shock treatments at 37°C were performed on the same day to induce the expression of the flippase enzyme and consequent FRT-mediated recombination. The genotypes utilized were: control for Atg1−/− – hs-flp, UAS-GFP/Y; tub-GAL4/+;FRT80B tub-Gal80/FRT80B +, Atg1−/− – hs-flp, UAS-GFP/Y; tub-GAL4/+; FRT80B tub-Gal80/FRT80B Atg1Δ3D, control for Atg13−/− – hs-flp, UAS-GFP/Y; tub-GAL4/+; FRT82B tub-Gal80/FRT82B + and Atg13−/− – hs-flp, UAS-GFP/Y; tub-GAL4/+;FRT80B tub-Gal80/FRT80B Atg1Δ81.
Thin layer chromatography for triacylglycerides (TLC-TAG)
50 testes were quickly dissected into PBS and snap frozen in an Eppendorf tube in liquid nitrogen. Samples were dissolved in 50 μL solvent (2pt chloroform:1pt methanol) on ice, then homogenized with a pestle and spun for 10 min at 4°C 20,000 g. Samples were then immediately loaded into silica gel plates (Analtech cat# P11521) and placed into a chamber containing 80mL hexane, 20mL diethyl ether and 1mL acetic acid for approximately 7min. After drying, plates were stained for 20min with 0.2% naphthol blue black in 1M NaCl, and washed several times with 1M NaCl until little to no blue stain was observed in solution. Plates were then left to dry, prior to being scanned and analyzed on ImageJ (v2.0.0) software. Three biological replicates for each genotype were assayed.
TUNEL assay
TUNEL assay was performed using ApopTag® Red In Situ Apoptosis Detection Kit (Millipore Sigma) according to manufacturer instructions. Testes were dissected in 1x PBS and fixed in 2% PFA for 30 minutes. Samples were washed twice for 10 minutes each in 0.3% Sodium Deoxycholate 0.3% PBS-T and were then incubated in Equilibrium Buffer for 10 minutes. Samples were incubated at 37°C for 1 hour in Reaction Buffer with Tdt enzyme in a 7:3 (buffer:enzyme) ratio. Samples were incubated for 10 minutes in Stop/Wash Solution and then washed in 0.1% PBS-T for 10 minutes. Samples were then transferred to diluted anti-DIG Rhodamine in Blocking Solution at a 47:53 ratio for 30 minutes, after which they were washed in 0.1% PBS-T twice for 10 minutes each and blocked in 3% BSA in 0.1% PBS-T. The aforementioned protocol for immunofluorescence was then resumed.
RNA extraction and quantitative RT-PCR
Two hundred testes per condition after dissection were frozen at −80°C in fresh Trizol buffer (Trizol Life Technologies, 15596026; 5 μg Linear Poly-Acrylamide Sigma 56575, 100ηg of tRNA). Total RNA was extracted pooling testes samples, followed by 5 rounds of freezing (liquid nitrogen)/thawing at 37°C in a water bath. Then 5 Vortex rounds at RT for 30”, letting stand at RT for 5 min to disrupt all RNA-protein complexes. Finally, RNA was isolated by phenol/chloroform extraction.
Purified RNA was treated with DNase Q1 (Promega, M610A). RNA (1 mg) from testes dissected from 3 do control or EGFRDN or FosWT+EGFRDN flies (genotypes: c587-Gal4/Y; tub-Gal80TS/+; +/+, c587-Gal4/Y; tub-Gal80TS/+; UAS-EGFRDN/UAS-TdTomato or c587-Gal4/Y; tub-Gal80TS/UAS-FosWT; UAS-EGFRDN/+) was reverse-transcribed using the iScriptkit (Bio-Rad, 170–8841). Standard qPCRs were carried out on a Bio-Rad CFX96/C1000 Touch system (Bio-Rad), using Sso Advanced SYBR Green (Bio-Rad, 1725–264). The following primer sequences were used: Act5c Fwd: TTGTCTGGGCAAGAGGATCAG; Act5cRev: ACC ACTCGCACTTGCACTTTC; Atg6 Fwd: CGACAATGAGTGAGGCGGAA; Atg6 Rev: TCTCCGTAGATGGGCAAAGA. Cycling conditions were as follows: 95°C for 30 s; 95°C for 5 s then 55°C for 30 s, cycled 40 times. All calculated gene expression values were normalized to the value of the loading control gene, Actin5c.
QUANTIFICATION AND STATISTICAL ANALYSIS
dpErk quantification
For acquisition - all samples were scanned at the same time with identical laser and microscopic settings. For quantification, at least 10 testes from each condition were used. ImageJ software was used to draw a 25×25pi circle inside cells in the CySC position (i.e., in very close proximity to the hub) and the mean of fluorescence of the dpErk signal was obtained, followed by a second measure of same area of the most nearby hub cell. The ratio was then determined (i.e., CySC/hub dpErk) for that particular cell. No more than one ratio per same cell was obtained. Several cells in the CySC position from each testis were measured and used to determine the average intensity for each condition.
Autophagosome number and autophagic structure quantification
To determine the overall number of autophagosomes per testis tips, flies carrying the pAtg8a>mCherry-Atg8a transgene were used in combination with antibody stains against Vasa, Fas3, and TJ. Samples were acquired with a 63x objective, 1.5 optical zoom in a Zeiss LSM780 microscope. The number of autophagosomes was then determined in the germline (all mCherry-Atg8a+ puncta within the Vasa+ area) and somatic (all area outside Vasa+, Fas3+) components. For the 3D-reconstruction of Figure S1B, Imaris software (v8.4, Bitplane) was used.
To determine the number of autophagic structures, flies carrying the UAS-GFP-mCherry-Atg8a transgene were crossed to the c587-Gal4; Gal80TS driver line, and the endogenous fluorescences of GFP and mCherry were obtained along with IF stains using antibodies against TJ and Fas3. Samples were acquired as described above. The number of autophagic structures (ALs and APs) was determined by counting the number of red-only ALs (GFP fluorescence quenched in low pH) and yellow APs (GFP fluorescence not yet quenched) per testis tips, and their mean numbers and percentages (ALs/total autophagic structures and APs/total autophagic structures) were determined and displayed on Figures 1F, 3B, 4J, and 5B.
Lipid droplet number quantification and incorporation assays
To determine the overall number of LDs per testis tips, IF was performed with antibodies against Vasa, Fas3, and TJ, followed by a 20 min incubation with BODIPY 493/503 (ThermoFisher), a final wash and mouting in Vectashield. Samples were acquired with a 63x objective, 1.5 optical zoom in a Zeiss LSM780 microscope. The number of LDs was then determined in CCs as all the area outside Vasa+, Fas3+ domains.
For the LD incorporation assays, dissected testes were cultured ex vivo in Schneider’s medium supplemented with 1 μM BODIPY 558/568 C12 (ThermoFisher) and presence or absence of 50mM Chloroquine for 1h, prior to regular IF procedure. When noted, a 20min final stain for LDs with LipidTox Red (ThermoFisher) was used to stain all LDs. Quantifications were performed as described above.
CySC and TJ+/Eya− counts
CySCs were counted (for Figure 1G) as TJ+ CCs located within two rows of CC nuclei away from the hub. CySC division leads to two daughter cells that could potentially become the new CySC (Amoyel et al., 2014, 2016a). In addition, we have noticed mitotically active cells within this region. Since CySCs are the only dividing cells in the CC lineage, all cells within this region were counted. For a more precise definition of early progenitor CCs (including CySCs), a similar approach as to the one described in Amoyel et al. (2016a) was used – CCs were co-stained with TJ (which marks early CCs including CySCs) and EyA (which marks late CCs), and counted only CCs that expressed TJ but not Eya.
Statistics
All quantitative experiments were evaluated for statistical significance using the software Graphpad Prism v6.0, after verifying the normality of values and equivalence of variances. For stem cell counts, means with standard deviations are displayed, and the statistical differences between mutant or RNAi-treated samples and controls were addressed using a Student’s two-tailed t test. For the frequency of cells positive for cell cycle markers, or frequency of testes with noted phenotypes, results were translated into individual contingency tables for each condition, where each row defines a genetic background (for example Atg1RNAi versus control), each column defines an outcome (for example, germ cysts with > 16 cells versus normal germ cyst numbers) and each value is an exact count. Statistical significance was assayed using a two-sided Fisher’s exact test. Statistical significance was denoted as *p < 0.05, ** p < 0.01, *** p < 0.001 and **** p < 0.0001; n.s. – not significant (p > 0.05). In all cases, the specific statistical test used, along with numbers and further statistical information, can be found in figure legends and Table S1 with statistical details and listed genotypes.
DATA AND CODE AVAILABILITY
This study did not generate new unique reagents.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-Fasciclin 3 (7G10) | Developmental studies hybridoma bank (DSHB) | RRID: AB_528238 |
| Mouse monoclonal anti-alfa-Spectrin (3A9) | DSHB | RRID: AB_528473 |
| Mouse monoclonal anti-Eyes absent (10H6) | DSHB | RRID: AB_528232 |
| Rat monoclonal anti-Vasa | DSHB | RRID: AB_760351 |
| Mouse monoclonal anti-phospho-Histone H3 (Ser10) (6G3) | Cell Signaling Technology | Cat#9706; RRID: AB_331748 |
| Chicken polyclonal anti-GFP | Aves Labs | Cat#GFP-1020; RRID: AB_10000240 |
| Rabbit monoclonal anti-P-4E-BP1 (T37/46) (236B4) | Cell Signaling Technology | Cat#2855; RRID: AB_560835 |
| Mouse monoclonal anti-dEGF Receptor (clone C-273) | Sigma-Aldrich | Cat#E2906; RRID: AB_609900 |
| Rabbit polyclonal anti-Zfh1 | Laboratory of Ruth Lehmann (NYU) | N/A |
| Guinea pig polyclonal anti-Traffic Jam | Li et al., 2003 | RRID: AB_2568583 |
| Rabbit monoclonal anti-Phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (D13.14.4E) XP | Cell Signaling Technology | Cat#4370; RRID: AB_2315112 |
| Rabbit polyclonal anti-Vasa (d-260) | Santa Cruz Biotechnology | Cat#sc-30210; RRID: AB_793874 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Rapamycin | TSZCHEM | Cat#R1017; CAS#53123-88-9 |
| Chloroquine diphosphate salt | Sigma-Aldrich | Cat#C6628; CAS#50-63-5 |
| L-Carnitine inner salt | BeanTown Chemical | Cat#215415; CAS#541-12-1 |
| BODIPY™ 493/503 | ThermoFisher | Cat#D3922 |
| BODIPY™ 558/568 C12 | ThermoFisher | Cat#D3835 |
| HCS LipidTox™ Red Neutral Lipid Stain | ThermoFisher | Cat#H34476 |
| Critical Commercial Assays | ||
| ApopTag® Red In Situ Apoptosis Detection Kit | Millipore/Sigma- Aldrich | Cat#S7165 |
| Experimental Models: Organisms/Strains | ||
| D. melanogaster: tub-Gal80TS | Bloomington Drosophila Stock Center | BDSC:7108 |
| D. melanogaster: UAS-EGFRDN | Bloomington Drosophila Stock Center | BDSC:5364 |
| D. melanogaster: UAS-EGFRCA | Bloomington Drosophila Stock Center | BDSC:9533 |
| D. melanogaster: UAS-TorRNAi [HMS0114] | Bloomington Drosophila Stock Center | BDSC:34639 |
| D. melanogaster: UAS-Atg1RNAi [GL00047] | Bloomington Drosophila Stock Center | BDSC:35177 |
| D. melanogaster: UAS-Atg1RNAi [HMS02750] | Bloomington Drosophila Stock Center | BDSC:44034 |
| D. melanogaster: UAS-Atg8bRNAi [HMS01245] | Bloomington Drosophila Stock Center | BDSC:34900 |
| D. melanogaster: UAS-Atg7RNAi [HMS01358] | Bloomington Drosophila Stock Center | BDSC:34369 |
| D. melanogaster: UAS-RasV12 | Bloomington Drosophila Stock Center | BDSC:4847 |
| D. melanogaster: UAS-InRRNAi [JF01482] | Bloomington Drosophila Stock Center | BDSC:31037 |
| D. melanogaster: UAS-SREBP[1–452] (‘SREBPCA’) | Bloomington Drosophila Stock Center | BDSC:41018 |
| D. melanogaster: Grb1(PH)-GFP [tGPH2] | Bloomington Drosophila Stock Center | BDSC:8163 |
| D. melanogaster: TJ-Gal4 | Kyoto Stock Center | BDSC:104055 |
| D. melanogaster: UAS-Atg1 [GD7149] | Vienna Drosophila RNAi Center | v16133 |
| D. melanogaster: UAS-Atg5RNAi [KK108904] | Vienna Drosophila RNAi Center | v104461 |
| D. melanogaster: UAS-Atg7RNAi [GD11671] | Vienna Drosophila RNAi Center | v27432 |
| D. melanogaster: UAS-Atg8aRNAi [KK102155] | Vienna Drosophila RNAi Center | v109654 |
| D. melanogaster: UAS-Atg8aRNAi [GD4654] | Vienna Drosophila RNAi Center | v43097 |
| D. melanogaster: UAS-Atg9RNAi [GD2029] | Vienna Drosophila RNAi Center | v10045 |
| D. melanogaster: UAS-Atg12RNAi [GD15230] | Vienna Drosophila RNAi Center | v29781 |
| D. melanogaster: UAS-Atg13RNAi [GD12202] | Vienna Drosophila RNAi Center | v27956 |
| D. melanogaster: UAS-Atg13RNAi [KK100340] | Vienna Drosophila RNAi Center | v103381 |
| D. melanogaster: UAS-Vps34RNAi [KK107602] | Vienna Drosophila RNAi Center | v100296 |
| D. melanogaster: UAS-bmmRNAi [GD5139] | Vienna Drosophila RNAi Center | v37877 |
| D. melanogaster: UAS-coltRNAi [KK107449] | Vienna Drosophila RNAi Center | v106089 |
| D. melanogaster: UAS-whdRNAi [GD1917] | Vienna Drosophila RNAi Center | v4046 |
| D. melanogaster: pAtg8a > mCherry-Atg8a | Laboratory of T. Neufeld (U. Minnesota) | N/A |
| D. melanogaster: UAS-GFP-mCherry-Atg8a | Laboratory of T. Neufeld (U. Minnesota) | N/A |
| D. melanogaster: UAS-Atg16A (‘Atg1OE’) | Laboratory of T. Neufeld (U. Minnesota) | N/A |
| D. melanogaster: UAS-Atg6RNAi [IR 16–3] | Laboratory of T. Neufeld (U. Minnesota) | N/A |
| D. melanogaster: UAS-Fos | Laboratory of H. Jasper (The Buck Institute for Aging Research) | N/A |
| D. melanogaster: UAS-FosRNAi-strong | Laboratory of H. Jasper (The Buck Institute for Aging Research) | N/A |
| D. melanogaster: UAS-FosC-Ala | Laboratory of F. Serras (Universitat de Barcelona, Spain) | N/A |
| D. melanogaster: UAS-FosPan-Ala | Laboratory of F. Serras (Universitat de Barcelona, Spain) | N/A |
| D. melanogaster: UAS-RasV12.S35 | Laboratory of H. Steller (The Rockefeller University) | N/A |
| D. melanogaster: UAS-RasV12.G37 | Laboratory of H. Steller (The Rockefeller University) | N/A |
| D. melanogaster: UAS-FosPan-Ala | Laboratory of F. Serras (Universitat de Barcelona, Spain) | N/A |
| D. melanogaster: nos-Gal4:VP16 | Laboratory of M. van Doren (Johns Hopkins) | N/A |
| D. melanogaster: c587-Gal4 | Laboratory of T. Xie (Stowers Institute of Biomedical Research) | N/A |
| D. melanogaster: stet871 | Laboratory of M. Fuller (Stanford University) | N/A |
| D. melanogaster: Df(3L)PX62 | Laboratory of M. Fuller (Stanford University) | N/A |
| Oligonucleotides | ||
| Primer Act5C For: TTGTCTGGGCAAGAGGATCAG | This paper | N/A |
| Primer Act5C Rev: ACCACTCGCACTTGCACTTTC | This paper | N/A |
| Primer Atg6 For: CGACAATGAGTGAGGCGGAA | This paper | N/A |
| Primer Atg6 Rev: TCTCCGTAGATGGGCAAAGA | This paper | N/A |
| Software and Algorithms | ||
| Prism v6.0 | Graphpad | N/A |
| Illustrator CC 2015 | Adobe | N/A |
| Photoshop CC 2015 | Adobe | N/A |
| ImageJ v2.0.0 | Wayne Rasband, NIH | https://imagej.nih.gov/ij |
| Imaris v8.4 | Bitplane | N/A |
| CFX ManagerTM | Biorad | N/A |
Highlights.
Autophagy is required for regulating CySC maintenance and cyst cell function
EGFR signaling stimulates autophagy to control early CC maintenance and behavior
Autophagy suppression by TOR in CCs is required for somatic/germline differentiation
Defective autophagy results in lipid accumulation and loss of early cyst cells
ACKNOWLEDGMENTS
The authors thank T. Neufeld (University of Minnesota, USA), M. Van Doren (Johns Hopkins University, USA), H. Jasper (Genentech and The Buck Institute for Research on Aging, USA), H. Steller (The Rockefeller University, USA), F. Serras (Univesitat de Barcelona, Spain), the Vienna Drosophila RNAi Center (VDRC), and Bloomington Stock Center for reagents; D. Walker (UCLA) and Todd Nystul (UCSF, USA) for helpful discussions, protocols, and reagents; the BSCRC/MCDB microscopy core at UCLA; and the Jones laboratory for comments on the manuscript. This work was supported by the Wasserman Foundation through the Undergraduate Research Center—Sciences at the University of California, Los Angeles (B.S.U.), the Eli & Edythe Broad Center of Regenerative Medicine & Stem Cell Research (R.S.D. and D.L.J.), and the NIH AG028092, AG040288, and AG052732 (D.L.J.)
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found online at https://doi.org/10.1016/j.celrep.2019.12.086.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Alves S, Castro L, Fernandes MS, Francisco R, Castro P, Priault M, Chaves SR, Moyer MP, Oliveira C, Seruca R, et al. (2015). Colorectal cancer-related mutant KRAS alleles function as positive regulators of autophagy. Oncotarget 6, 30787–30802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amoyel M, Simons BD, and Bach EA (2014). Neutral competition of stem cells is skewed by proliferative changes downstream of Hh and Hpo. EMBO J. 33, 2295–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amoyel M, Anderson J, Suisse A, Glasner J, and Bach EA (2016a). Socs36E Controls Niche Competition by Repressing MAPK Signaling in the Drosophila Testis. PLoS Genet. 12, e1005815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amoyel M, Hillion KH, Margolis SR, and Bach EA (2016b). Somatic stem cell differentiation is regulated by PI3K/Tor signaling in response to local cues. Development 143, 3914–3925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergmann A, Agapite J, McCall K, and Steller H (1998). The Drosophila gene hid is a direct molecular target of Ras-dependent survival signaling. Cell 95, 331–341. [DOI] [PubMed] [Google Scholar]
- Biteau B, and Jasper H (2011). EGF signaling regulates the proliferation of intestinal stem cells in Drosophila. Development 138, 1045–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blommaart EF, Luiken JJ, Blommaart PJ, van Woerkom GM, and Meijer AJ (1995). Phosphorylation of ribosomal protein S6 is inhibitory for autophagy in isolated rat hepatocytes. J. Biol. Chem 270, 2320–2326. [DOI] [PubMed] [Google Scholar]
- Brand AH, and Perrimon N (1993). Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development 118, 401–415. [DOI] [PubMed] [Google Scholar]
- Cao Y, and Klionsky DJ (2007). Physiological functions of Atg6/Beclin 1: a unique autophagy-related protein. Cell Res. 17, 839–849. [DOI] [PubMed] [Google Scholar]
- Chandel NS, Jasper H, Ho TT, and Passegué E (2016). Metabolic regulation of stem cell function in tissue homeostasis and organismal ageing. Nat. Cell Biol 18, 823–832. [DOI] [PubMed] [Google Scholar]
- Chang YY, and Neufeld TP (2009). An Atg1/Atg13 complex with multiple roles in TOR-mediated autophagy regulation. Mol. Biol. Cell 20, 2004–2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang YY, and Neufeld TP (2010). Autophagy takes flight in Drosophila. FEBS Lett. 584, 1342–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudhary KS, Rohatgi N, Halldorsson S, Briem E, Gudjonsson T, Gudmundsson S, and Rolfsson O (2016). EGFR Signal-Network Reconstruction Demonstrates Metabolic Crosstalk in EMT. PLoS Comput. Biol 12, e1004924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciapponi L, Jackson DB, Mlodzik M, and Bohmann D (2001). Drosophila Fos mediates ERK and JNK signals via distinct phosphorylation sites. Genes Dev. 15, 1540–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demarco RS, Eikenes AH, Haglund K, and Jones DL (2014). Investigating spermatogenesis in Drosophila melanogaster. Methods 68, 218–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denton D, Chang TK, Nicolson S, Shravage B, Simin R, Baehrecke EH, and Kumar S (2012). Relationship between growth arrest and autophagy in midgut programmed cell death in Drosophila. Cell Death Differ. 19, 1299–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drummond-Barbosa D (2008). Stem cells, their niches and the systemic environment: an aging network. Genetics 180, 1787–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fabrizio JJ, Boyle M, and DiNardo S (2003). A somatic role for eyes absent (eya) and sine oculis (so) in Drosophila spermatocyte development. Dev. Biol 258, 117–128. [DOI] [PubMed] [Google Scholar]
- Fan QW, Cheng C, Knight ZA, Haas-Kogan D, Stokoe D, James CD, McCormick F, Shokat KM, and Weiss WA (2009). EGFR signals to mTOR through PKC and independently of Akt in glioma. Sci. Signal 2, ra4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Folick A, Oakley HD, Yu Y, Armstrong EH, Kumari M, Sanor L, Moore DD, Ortlund EA, Zechner R, and Wang MC (2015). Aging. Lysosomal signaling molecules regulate longevity in Caenorhabditis elegans. Science 347, 83–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujimoto T, and Parton RG (2011). Not just fat: the structure and function of the lipid droplet. Cold Spring Harb. Perspect. Biol 3, a004838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller MT (1993). Spermatogenesis In The Development of Drosophila melanogaster, Bate M and Arias AM, eds. (Cold Spring Harbor Press; ). [Google Scholar]
- Füllgrabe J, Ghislat G, Cho DH, and Rubinsztein DC (2016). Transcriptional regulation of mammalian autophagy at a glance. J. Cell Sci 129, 3059–3066. [DOI] [PubMed] [Google Scholar]
- Gao X, Zacharek A, Salkowski A, Grignon DJ, Sakr W, Porter AT, and Honn KV (1995). Loss of heterozygosity of the BRCA1 and other loci on chromosome 17q in human prostate cancer. Cancer Res. 55, 1002–1005. [PubMed] [Google Scholar]
- Garami A, Zwartkruis FJ, Nobukuni T, Joaquin M, Roccio M, Stocker H, Kozma SC, Hafen E, Bos JL, and Thomas G (2003). Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol. Cell 11, 1457–1466. [DOI] [PubMed] [Google Scholar]
- Greenspan LJ, de Cuevas M, and Matunis E (2015). Genetics of gonadal stem cell renewal. Annu. Rev. Cell Dev. Biol 31, 291–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grönke S, Mildner A, Fellert S, Tennagels N, Petry S, Müller G, Jäckle H, and Kühnlein RP (2005). Brummer lipase is an evolutionary conserved fat storage regulator in Drosophila. Cell Metab. 1, 323–330. [DOI] [PubMed] [Google Scholar]
- Guan JL, Simon AK, Prescott M, Menendez JA, Liu F, Wang F, Wang C, Wolvetang E, Vazquez-Martin A, and Zhang J (2013). Autophagy in stem cells. Autophagy 9, 830–849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardy RW, Tokuyasu KT, Lindsley DL, and Garavito M (1979). The germinal proliferation center in the testis of Drosophila melanogaster. J. Ultrastruct. Res 69, 180–190. [DOI] [PubMed] [Google Scholar]
- Hartenstein K, Sinha P, Mishra A, Schenkel H, Török I, and Mechler BM (1997). The congested-like tracheae gene of Drosophila melanogaster encodes a member of the mitochondrial carrier family required for gas-filling of the tracheal system and expansion of the wings after eclosion. Genetics 147, 1755–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hétié P, de Cuevas M, and Matunis E (2014). Conversion of quiescent niche cells to somatic stem cells causes ectopic niche formation in the Drosophila testis. Cell Rep. 7, 715–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, Figueroa ME, and Passegué E (2017). Autophagy maintains the metabolism and function of young and old stem cells. Nature 543, 205–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson AG, Parrott BB, Qian Y, and Schulz C (2013). A temporal signature of epidermal growth factor signaling regulates the differentiation of germline cells in testes of Drosophila melanogaster. PLoS One 8, e70678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamada Y, Funakoshi T, Shintani T, Nagano K, Ohsumi M, and Ohsumi Y (2000). Tor-mediated induction of autophagy via an Apg1 protein kinase complex. J. Cell Biol 150, 1507–1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karim FD, and Rubin GM (1998). Ectopic expression of activated Ras1 induces hyperplastic growth and increased cell death in Drosophila imaginal tissues. Development 125, 1–9. [DOI] [PubMed] [Google Scholar]
- Kiger AA, White-Cooper H, and Fuller MT (2000). Somatic support cells restrict germline stem cell self-renewal and promote differentiation. Nature 407, 750–754. [DOI] [PubMed] [Google Scholar]
- Kiger AA, Jones DL, Schulz C, Rogers MB, and Fuller MT (2001). Stem cell self-renewal specified by JAK-STAT activation in response to a support cell cue. Science 294, 2542–2545. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Cregg JM, Dunn WA Jr., Emr SD, Sakai Y, Sandoval IV, Sibirny A, Subramani S, Thumm M, Veenhuis M, and Ohsumi Y (2003). A unified nomenclature for yeast autophagy-related genes. Dev. Cell 5, 539–545. [DOI] [PubMed] [Google Scholar]
- Kockel L, Homsy JG, and Bohmann D (2001). Drosophila AP-1: lessons from an invertebrate. Oncogene 20, 2347–2364. [DOI] [PubMed] [Google Scholar]
- Lapierre LR, Gelino S, Meléndez A, and Hansen M (2011). Autophagy and lipid metabolism coordinately modulate life span in germline-less C. elegans. Curr. Biol 21, 1507–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leatherman JL, and Dinardo S (2008). Zfh-1 controls somatic stem cell self-renewal in the Drosophila testis and non-autonomously influences germline stem cell self-renewal. Cell Stem Cell 3, 44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leatherman JL, and Dinardo S (2010). Germline self-renewal requires cyst stem cells and stat regulates niche adhesion in Drosophila testes. Nat. Cell Biol 12, 806–811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee T, and Luo L (1999). Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 22, 451–461. [DOI] [PubMed] [Google Scholar]
- Levine B, and Klionsky DJ (2004). Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell 6, 463–477. [DOI] [PubMed] [Google Scholar]
- Li MA, Alls JD, Avancini RM, Koo K, and Godt D (2003). The large Maf factor Traffic Jam controls gonad morphogenesis in Drosophila. Nat. Cell Biol 5, 994–1000. [DOI] [PubMed] [Google Scholar]
- Lim JG, and Fuller MT (2012). Somatic cell lineage is required for differentiation and not maintenance of germline stem cells in Drosophila testes. Proc. Natl. Acad. Sci. USA 109, 18477–18481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, and Czaja MJ (2013). Regulation of lipid stores and metabolism by lipophagy. Cell Death Differ. 20, 3–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu F, Lee JY, Wei H, Tanabe O, Engel JD, Morrison SJ, and Guan JL (2010). FIP200 is required for the cell-autonomous maintenance of fetal hematopoietic stem cells. Blood 116, 4806–4814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Wang H, Shang Y, Liu W, Song Z, Zhao H, Wang L, Jia P, Gao F, Xu Z, et al. (2016). Autophagy is required for ectoplasmic specialization assembly in sertoli cells. Autophagy 12, 814–832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch-Day MA, Mao K, Wang K, Zhao M, and Klionsky DJ (2012). The role of autophagy in Parkinson’s disease. Cold Spring Harb. Perspect. Med 2, a009357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manent J, Banerjee S, de Matos Simoes R, Zoranovic T, Mitsiades C, Penninger JM, Simpson KJ, Humbert PO, and Richardson HE (2017). Autophagy suppresses Ras-driven epithelial tumourigenesis by limiting the accumulation of reactive oxygen species. Oncogene 36, 5658–5660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng D, Frank AR, and Jewell JL (2018). mTOR signaling in stem and progenitor cells. Development 145, dev152595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortensen M, Soilleux EJ, Djordjevic G, Tripp R, Lutteropp M, SadighiAkha E, Stranks AJ, Glanville J, Knight S, Jacobsen SE, et al. (2011). The autophagy protein Atg7 is essential for hematopoietic stem cell maintenance. J. Exp. Med 208, 455–467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy P, Varga Á, Kovács AL, Takáts S, and Juhász G (2015). How and why to study autophagy in Drosophila: it’s more than just a garbage chute. Methods 75, 151–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy P, Sándor GO, and Juhász G (2018). Autophagy maintains stem cells and intestinal homeostasis in Drosophila. Sci. Rep 8, 4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TB, Louie SM, Daniele JR, Tran Q, Dillin A, Zoncu R, Nomura DK, and Olzmann JA (2017). DGAT1-Dependent Lipid Droplet Biogenesis Protects Mitochondrial Function during Starvation-Induced Autophagy. Dev. Cell 42, 9–21.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noda T, and Ohsumi Y (1998). Tor, a phosphatidylinositol kinase homologue, controls autophagy in yeast. J. Biol. Chem 273, 3963–3966. [DOI] [PubMed] [Google Scholar]
- O’Keefe DD, Prober DA, Moyle PS, Rickoll WL, and Edgar BA (2007). Egfr/Ras signaling regulates DE-cadherin/Shotgun localization to control vein morphogenesis in the Drosophila wing. Dev. Biol 311, 25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oda K, Matsuoka Y, Funahashi A, and Kitano H (2005). A comprehensive pathway map of epidermal growth factor receptor signaling. Mol. Syst. Biol 1, 2005.0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oldham S (2011). Obesity and nutrient sensing TOR pathway in flies and vertebrates: Functional conservation of genetic mechanisms. Trends Endocrinol. Metab 22, 45–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palanker L, Tennessen JM, Lam G, and Thummel CS (2009). Drosophila HNF4 regulates lipid mobilization and beta-oxidation. Cell Metab. 9, 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, and Cobb MH (2001). Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr. Rev 22, 153–183. [DOI] [PubMed] [Google Scholar]
- Perkins LA, Johnson MR, Melnick MB, and Perrimon N (1996). The non-receptor protein tyrosine phosphatase corkscrew functions in multiple receptor tyrosine kinase pathways in Drosophila. Dev. Biol 180, 63–81. [DOI] [PubMed] [Google Scholar]
- Queenan AM, Ghabrial A, and Schüpbach T (1997). Ectopic activation of torpedo/Egfr, a Drosophila receptor tyrosine kinase, dorsalizes both the eggshell and the embryo. Development 124, 3871–3880. [DOI] [PubMed] [Google Scholar]
- Rambold AS, Cohen S, and Lippincott-Schwartz J (2015). Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev. Cell 32, 678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC, DiFiglia M, Heintz N, Nixon RA, Qin ZH, Ravikumar B, Stefanis L, and Tolkovsky A (2005). Autophagy and its possible roles in nervous system diseases, damage and repair. Autophagy 1, 11–22. [DOI] [PubMed] [Google Scholar]
- Saito H, Inazawa J, Saito S, Kasumi F, Koi S, Sagae S, Kudo R, Saito J, Noda K, and Nakamura Y (1993). Detailed deletion mapping of chromosome 17q in ovarian and breast cancers: 2-cM region on 17q21.3 often and commonly deleted in tumors. Cancer Res. 53, 3382–3385. [PubMed] [Google Scholar]
- Sarkar A, Parikh N, Hearn SA, Fuller MT, Tazuke SI, and Schulz C (2007). Antagonistic roles of Rac and Rho in organizing the germ cell microenvironment. Curr. Biol 17, 1253–1258. [DOI] [PubMed] [Google Scholar]
- Schulz C, Wood CG, Jones DL, Tazuke SI, and Fuller MT (2002). Signaling from germ cells mediated by the rhomboid homolog stet organizes encapsulation by somatic support cells. Development 129, 4523–4534. [DOI] [PubMed] [Google Scholar]
- Schulze RJ, Sathyanarayan A, and Mashek DG (2017). Breaking fat: the regulation and mechanisms of lipophagy. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 1862, 1178–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott RC, Schuldiner O, and Neufeld TP (2004). Role and regulation of starvation-induced autophagy in the Drosophila fat body. Dev. Cell 7, 167–178. [DOI] [PubMed] [Google Scholar]
- Scott RC, Juhász G, and Neufeld TP (2007). Direct induction of autophagy by Atg1 inhibits cell growth and induces apoptotic cell death. Curr. Biol 17, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sênos Demarco R, Uyemura BS, D’Alterio C, and Jones DL (2019). Mitochondrial fusion regulates lipid homeostasis and stem cell maintenance in the Drosophila testis. Nat. Cell Biol 21, 710–720. [DOI] [PubMed] [Google Scholar]
- Sooro MA, Zhang N, and Zhang P (2018). Targeting EGFR-mediated autophagy as a potential strategy for cancer therapy. Int. J. Cancer 143, 2116–2125. [DOI] [PubMed] [Google Scholar]
- Strub BR, Parkes TL, Mukai ST, Bahadorani S, Coulthard AB, Hall N, Phillips JP, and Hilliker AJ (2008). Mutations of the withered (whd) gene in Drosophila melanogaster confer hypersensitivity to oxidative stress and are lesions of the carnitine palmitoyltransferase I (CPT I) gene. Genome 51, 409–420. [DOI] [PubMed] [Google Scholar]
- Sui X, Kong N, Ye L, Han W, Zhou J, Zhang Q, He C, and Pan H (2014). p38 and JNK MAPK pathways control the balance of apoptosis and autophagy in response to chemotherapeutic agents. Cancer Lett. 344, 174–179. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Kubota Y, Sekito T, and Ohsumi Y (2007). Hierarchy of Atg proteins in pre-autophagosomal structure organization. Genes Cells 12, 209–218. [DOI] [PubMed] [Google Scholar]
- Tan X, Thapa N, Sun Y, and Anderson RA (2015). A kinase-independent role for EGF receptor in autophagy initiation. Cell 160, 145–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang AH, and Rando TA (2014). Induction of autophagy supports the bio-energetic demands of quiescent muscle stem cell activation. EMBO J. 33, 2782–2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y, Geng Q, Chen D, Zhao S, Liu X, and Wang Z (2017). Germline Proliferation Is Regulated by Somatic Endocytic Genes via JNK and BMP Signaling in Drosophila. Genetics 206, 189–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran J, Brenner TJ, and DiNardo S (2000). Somatic control over the germline stem cell lineage during Drosophila spermatogenesis. Nature 407, 754–757. [DOI] [PubMed] [Google Scholar]
- Tulina N, and Matunis E (2001). Control of stem cell self-renewal in Drosophila spermatogenesis by JAK-STAT signaling. Science 294, 2546–2549. [DOI] [PubMed] [Google Scholar]
- Van Doren M, Williamson AL, and Lehmann R (1998). Regulation of zygotic gene expression in Drosophila primordial germ cells. Curr. Biol 8, 243–246. [DOI] [PubMed] [Google Scholar]
- Voog J, Sandall SL, Hime GR, Resende LP, Loza-Coll M, Aslanian A, Yates JR 3rd, Hunter T, Fuller MT, and Jones DL (2014). Escargot restricts niche cell to stem cell conversion in the Drosophila testis. Cell Rep. 7, 722–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang MC, O’Rourke EJ, and Ruvkun G (2008). Fat metabolism links germline stem cells and longevity in C. elegans. Science 322, 957–960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wullschleger S, Loewith R, and Hall MN (2006). TOR signaling in growth and metabolism. Cell 124, 471–484. [DOI] [PubMed] [Google Scholar]
- Xu P, Das M, Reilly J, and Davis RJ (2011). JNK regulates FoxO-dependent autophagy in neurons. Genes Dev. 25, 310–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, and Yamashita YM (2015). The regulated elimination of transit-amplifying cells preserves tissue homeostasis during protein starvation in Drosophila testis. Development 142, 1756–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang W, Li L, Huang X, Kan G, Lin L, Cheng J, Xu C, Sun W, Cong W, Zhao S, and Cui S (2017). Levels of Leydig cell autophagy regulate the fertility of male naked mole-rats. Oncotarget 8, 98677–98690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung Y, Dolginov Y, Yao Z, Rubinfeld H, Michael D, Hanoch T, Roubini E, Lando Z, Zharhary D, and Seger R (1997). Detection of ERK activation by a novel monoclonal antibody. FEBS Lett. 408, 292–296. [DOI] [PubMed] [Google Scholar]
- Zare-Shahabadi A, Masliah E, Johnson GV, and Rezaei N (2015). Autophagy in Alzheimer’s disease. Rev. Neurosci 26, 385–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeidler MP, Tan C, Bellaiche Y, Cherry S, Häder S, Gayko U, and Perrimon N (2004). Temperature-sensitive control of protein activity by conditionally splicing inteins. Nat. Biotechnol 22, 871–876. [DOI] [PubMed] [Google Scholar]
- Zhang X, Evans TD, Jeong SJ, and Razani B (2018). Classical and alternative roles for autophagy in lipid metabolism. Curr. Opin. Lipidol 29, 203–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou YY, Li Y, Jiang WQ, and Zhou LF (2015). MAPK/JNK signalling: a potential autophagy regulation pathway. Biosci. Rep 35, e00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoller R, and Schulz C (2012). The Drosophila cyst stem cell lineage: partners behind the scenes? Spermatogenesis 2, 145–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate new unique reagents.