

Abstract
N-[18F]fluoroacetylcrizotinib, a fluorine-18 labeled derivative of the first FDA approved tyrosine kinase inhibitor (TKI) for the treatment of Anaplastic lymphoma kinase (ALK)-rearranged non-small cell lung cancer (NSCLC), crizotinib, was successfully synthesized for use in positron emission tomography (PET). Sequential in vitro biological evaluation of fluoracetylcrizotinib and in vivo biodistribution studies of [18F]fluoroacetylcrizotinib demonstrated that the biological activity of the parent compound remained unchanged, with potent ALK kinase inhibition and effective tumor growth inhibition. These results show that [18F]fluoroacetylcrizotinib has the potential to be a promising PET ligand for use in NSCLC imaging. The utility of PET in this context provides a non-invasive, quantifiable method to inform on the pharmacokinetics of an ALK-inhibitor such as crizotinib prior to a clinical trial, as well as during a trial in the event of acquired drug resistance.
Keywords: Crizotinib, NSCLC, PET, Cancer, Therapy
Graphical Abstract
Anaplastic lymphoma kinase (ALK), first discovered in anaplastic large-cell lymphoma, is a receptor tyrosine kinase with an altered expression implicated in the pathogenesis of multiple human cancers, including non–small cell lung cancer (NSCLC)1. Of the estimated 2.1 million new cases of lung cancer diagnosed globally in 2018, 85% were NSCLC2,3, making it a leading cause of cancer death in industrialized countries2,4. Activating mutations in the ALK tyrosine kinase domain, aberrant chromosomal rearrangements, and fusion products tend to be transforming in vitro and in vivo1. Moreover, ALK-rearranged NSCLC comprises nearly 5–6% of all NSCLC, accounting for up to 125,000 new cases of lung cancer globally5. As such, ALK represents a rational therapeutic target, as preclinical and clinical studies have demonstrated promising efficacy of small-molecule ALK tyrosine kinase inhibitors (TKIs)3,4.
One ALK inhibitor of particular note is the 3- benzyloxy-2-aminopyridine crizotinib (Figure 1; Scheme 1, compound 1) (Xalkori®, Pfizer), an orally available, multi-targeted small-molecule ATP-mimetic active against both ALK and mesenchymal–epithelial transition factor kinase. Crizotinib was the first TKI approved for lung cancer by the FDA for the treatment of ALK-rearranged NSCLC and remains a viable treatment even with more recent options available. The National Comprehensive Cancer Network guidelines currently recommends crizotinib as a first-line therapy option in ALK-positive patients with locally advanced or metastatic NSCLC6.
Figure 1:
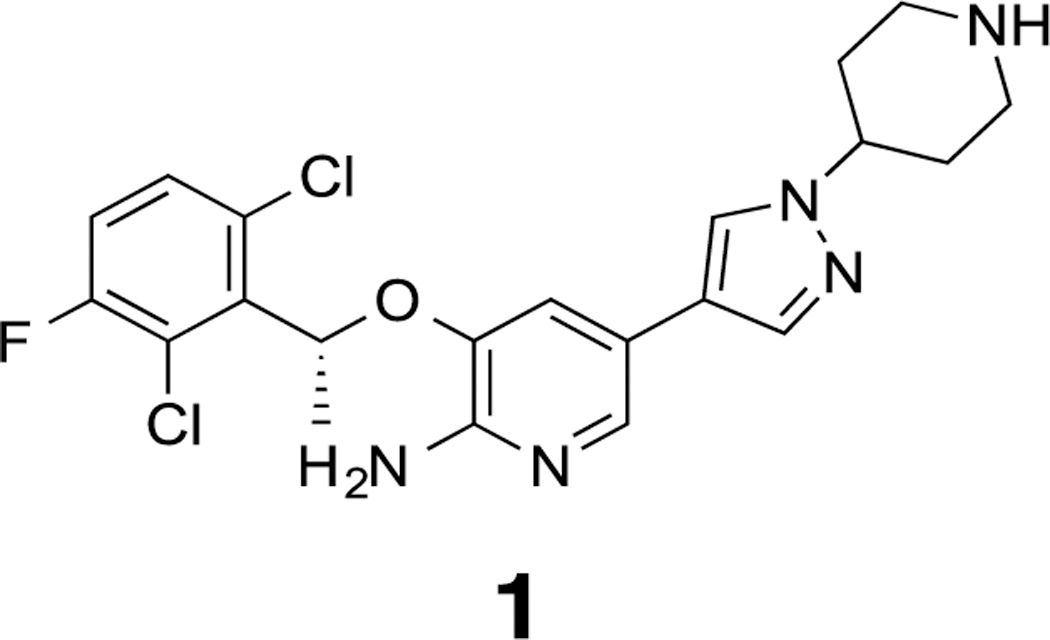
Structure of crizotinib, an ALK inhibitor.
Scheme 1:
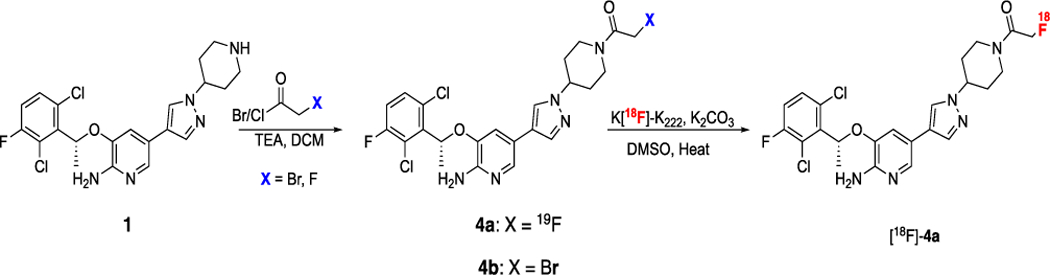
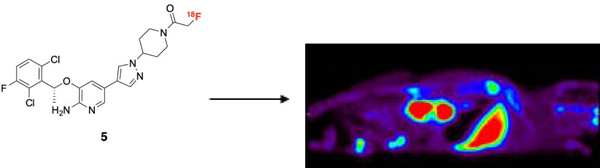
Synthesis of nonradioactive 4a and the radio-fluorination precursor 4b to yield [18F]-4a, a PET ligand for NSCLC imaging.
Application of positron emission tomography (PET) toward drug discovery and development is becoming prevalent as it can effectively guide and inform biological target selection, drug biodistribution, and therapeutic dosing. Moreover, as companion diagnostics, drugs labeled with radioisotopes (fluorine-18; 18F) could potentially aid in targeted therapy efficacy through improved patient selection. One example of this is the PRECEDENT study that used single photon emission computed tomography (SPECT) imaging with EC20 to identify a sub-population of ovarian cancer subjects more responsive to targeted therapy with EC1457. Since then, the EC20 SPECT tracer has been developed and used as an in vivo imaging companion diagnostic.
Our laboratory has previously explored the use of targeted PET imaging ligands in the context of various cancers to determine if they could potentially serve as useful cancer imaging biomarkers8–11. The goal of this study was to determine whether crizotinib (1) could also be adapted into an effective PET ligand ([18F]-4a). If successful, [18F]-4a could serve as a PET ligand for cancer imaging, improving diagnostic evaluation of individual ALK variations in patients by enabling identification of the appropriate patient population most likely to benefit from crizotinib therapy. Furthermore, long-term efficacy could be assessed through monitoring the level of resistance within the patient population receiving the therapy.
The de novo synthesis of crizotinib has been published12,13 and newer methods have been developed to make synthesis more efficient14. The chemistry developed herein focused on a two-step synthesis of the final tracer ([18F]-4a) through direct modification of the commercially available parent compound (1). While the attractiveness of replacing the 19F at the 3-phenyl position of 1 with 18F was readily apparent, thereby maintaining the nascent drug structure, radiofluorination of aromatic rings containing electron-donating groups can be synthetically demanding15. Incorporation of carbon-11 (11C) at C-3 was also considered. While this would also preserve the native crizotinib structure, the 20.4 min half-life of 11C would limit broad utility of the tracer. The choice was thus made to incorporate 18F (109.4 min half-life) at another site on the molecule, with minimal modification of the parent compound.
Prior structure-activity studies of the crizotinib 3-benzyloxy-2-aminopyridine scaffold have shown tolerance of acetyl functionalities at the 5-piperidine nitrogen16. Accordingly, derivitization of this nitrogen with a 2-fluoroacetyl moiety was considered, with the thought that the non-radioactive (19F) analog 4a would potentially demonstrate ALK-selectivity similar to its parent (1). One of the advantages of this group is the robustness of its radiosynthesis, due in part to its bromoacetyl precursor group that readily undergoes nucleophilic radiofluorination17. Moreover, our lab has previously demonstrated that radiolabeling of small molecules in this manner is reliable and straightforward11.
Synthesis of 4a was achieved in a single, high-yielding step starting from commercially available crizotinib (1) by acylation of the 5-piperidine nitrogen with fluoroacetyl chloride (Scheme 1). Similarly, the N-bromoacetyl piperidine radiofluorination precursor (4b) was accessible using bromoacetyl bromide (Scheme 1).
Compound 4a was biochemically characterized and compared to 1 in vitro in two separate assays1. The first was a kinase assay that involved treatment of ALK mutant cell line H3122 lung cancer cells containing the EML-4-ALK E13; A20 fusion with increasing concentrations of compounds 1 and 4a for one hour. Subsequent western blot analysis showed a comparable, if not improved, inhibition of ALK phosphorylation by 4a, particularly at a concentration of 500 nM (Figure 2a). A cell viability assay was also performed, in which cells were seeded in 12-well plates at 25% confluency and treated with compounds 1 and 4a. Seventy-two hours after drug addition, cells were stained with propidium iodide and counted on a FACSanto II machine. Figure 2b shows comparable inhibition of cells harboring fusion variants or activating ALK mutations. Taken together, these data demonstrate that the original biological activity of 1 remained intact in analog 4a, suggesting a strong potential for an imaging probe (5) that would accurately reflect activity in vivo.
Figure 2:
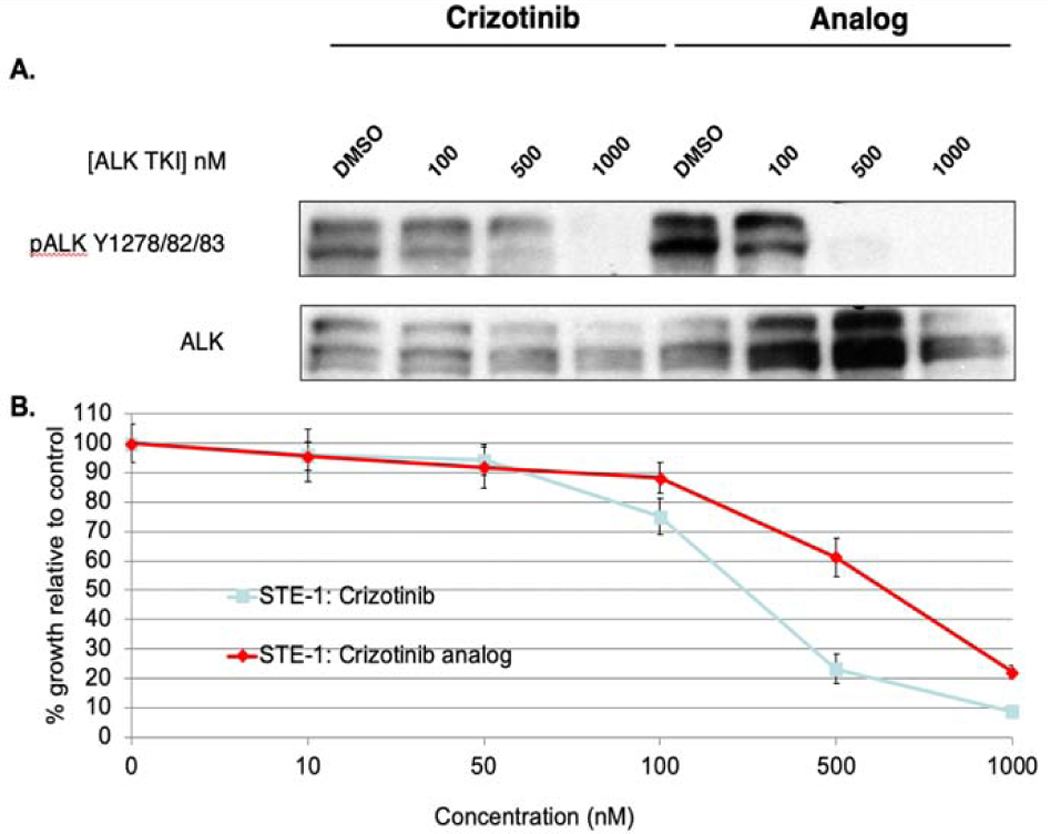
A. Comparative Western blot analysis of H3122 (EML4-ALK E13; A20) lung cancer cells treated with varying concentrations of crizotinib (1) and analog (4a). B. Comparative tumor cell growth inhibition study of crizotinib (1) and analog (4a).
Microfluidic-radiolabeling approaches were initially applied to ascertain labeling feasibility of precursor 4b. Microfluidic approaches enable rapid optimization and evaluation of various radiochemical conditions, which may subsequently inform the feasibility of larger scale production. This was done using a commercial microfluidic module (NanoTek®) that enabled carefully controlled stoichiometry between [18F]fluoride and 4b, highly controlled reaction temperatures and precisely controlled reaction timeframes. The controllable flow rates of the mixed reactants passing through known lengths of heated reactors enabled pinpoint reaction times to be set and/or varied to systematically workout optimized production parameters. Using this technology, we carried out multiple, small-scale, sequential radiolabelings in a relatively short amount of time, using minimal precursor, reagents, and [18F]fluoride. This information was then used to better inform the transition to the GE TRACERlab™ FX N synthesizer module for larger, preclinical scale productions. Using cyclotron-generated [18F]fluoride and K+-K2.2.2/K2CO3, a series of reaction temperatures and solvents were investigated and monitored by radioTLC for product formation. Figure 3A shows a comparison of [18F]fluoride incorporation using two different anhydrous solvents (DMF, DMSO) and in a reaction temperature range (80–180 °C). Of the solvents, DMSO gave higher [18F]fluoride incorporation, with nearly 20% at 180 °C, compared to the maximum of ≥5% with DMF at 120 °C. The data generated from these experiments also highlighted the thermal stability (up to 180 °C) of precursor 4b.
Figure 3:
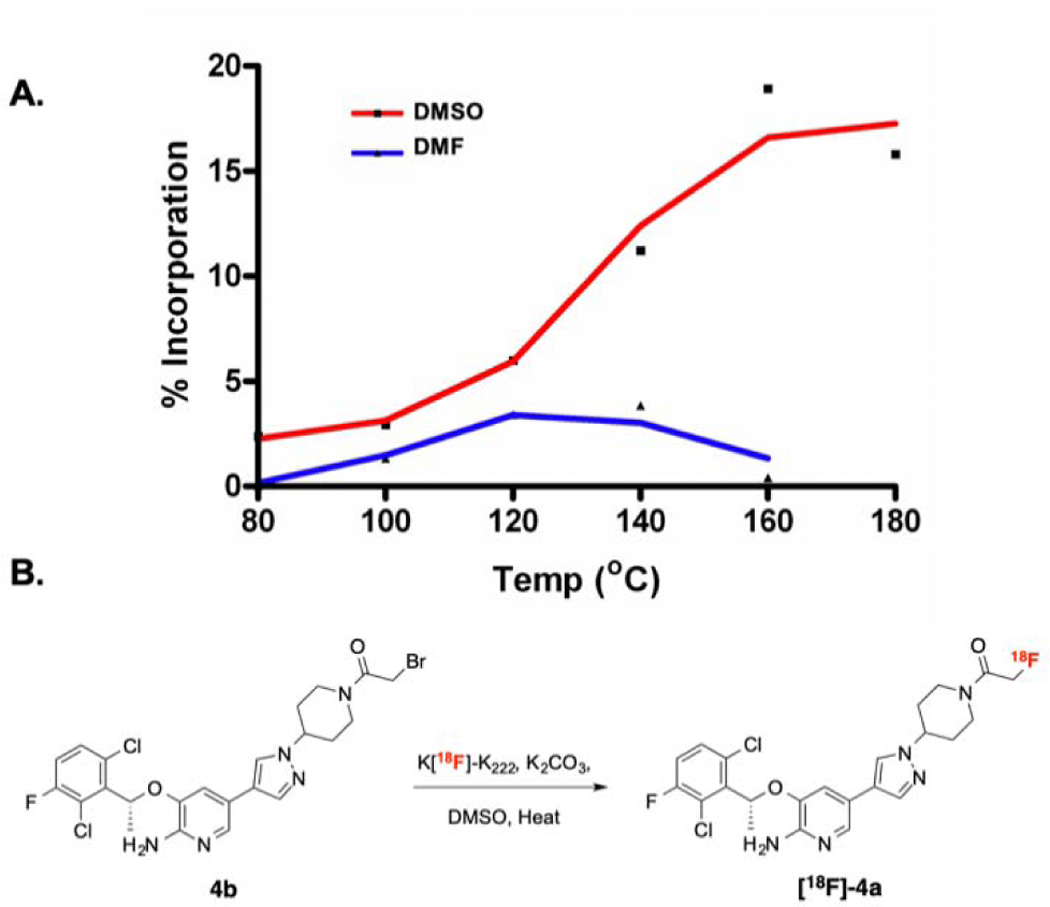
A. Comparison of [18F]-fluoride incorporation in different solvents using the Advion NanoTek. B. Preclinical production of [18F]-4a utilizing DMSO as the solvent.
Adaptation of these reaction conditions allowed preclinical production of [18F]-4a in the TRACERlab™ with labeling conditions of 120 °C for 15 min (Figure 3B). Purification of [18F]-4a was carried out with preparative HPLC using an ethanol:water (1:1) mobile phase on a C18 semi-preparative column. The retention time of [18F]-4a was 23 min according to gamma detection and corresponded to the UV retention time of nonradioactive 4a. Radiochemical purity was consistently greater than 95% (n=4), with molar activities in the range of 500–2700 Ci/mmol (18.5–100 TBq/mmol) (n=4).
[18F]-4a was evaluated in vivo in athymic nude mice (n=5) (Foxn1nu/nu; Harlan Laboratories, Indianapolis, IN). Following retro-orbital administration of 550 µCi (20.4 MBq) of [18F]-4a, dynamic PET images were acquired (90 minutes) on a Siemens Focus 220 microPET (Knoxville, TN), followed by CT (micro-CAT II; Siemens Preclinical Solutions) for attenuation correction.
For biodistribution studies, approximately 200 µCi of [18F]-4a were injected retro-orbitally in the mice (n=6). Three mice per group were sacrificed at 30 and 90 minutes. Organs of interest (brain, heart, liver, spleen, lungs, kidney, blood, colon) were immediately harvested, weighed, and counted for 1 minute in a well counter. Radioactivity obtained from each organ was calculated as percentage of the injected dose per gram of the tissue (%ID/g).
Thirty minutes after injection, the liver (14.95 %ID/g) was the organ of major [18F]-4a accumulation, with the kidney being the next highest at 1.93 %ID/g (Table 1). The brain (0.2633 %ID/g) was the organ of least accumulation. All other organs ranged from 0.6775 to 1.237 %ID/g. A similar trend was seen 90 minutes after injection, though with lower overall %ID/g values. The liver showed an average %ID/g of 5.547 and the brain 0.2273. The spleen replaced the kidney as next highest at 0.5668 %ID/g, with the rest of the organs ranging from 0.299 to 0.5456. In vivo microPET imaging of [18F]-4a biodistribution in athymic nude mice (Figure 4) was found to agree with the cut and count values.
Table 1.
%ID/g of [18F)-4a in Harvested Tissues
| Tissue | 30 minutes | 90 minutes |
|---|---|---|
| Brain | 0.2633±0.066 | 0.2273±0.072 |
| Liver | 14.95±4.631 | 5.547±1.385 |
| Spleen | 0.6775±0.331 | 0.5668±0.469 |
| Heart | 0.8972±0.242 | 0.299±0.072 |
| Blood | 1.237±0.359 | 0.4511±0.085 |
| Kidney | 1.933±1.063 | 0.5456±0.182 |
| Lung | 0.7859±0.142 | 0.4682±0.4361 |
| Colon | 0.6965±0.175 | 0.3096±0.105 |
Figure 4:
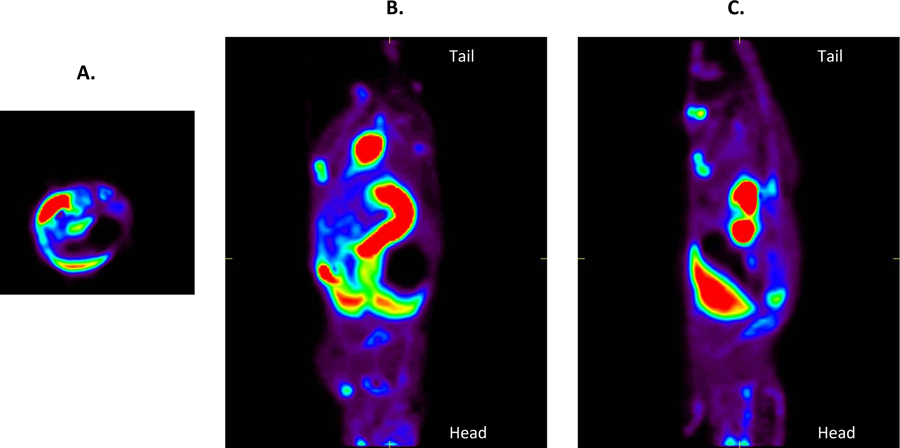
Summation of a 90-minute dynamic PET imaging study showing the biodistribution of [18F]-4a in a healthy athymic nude mouse. (A) Transverse (B) Coronal and (C) Axial views are shown.
Recent studies have shown that analogues of ALK-inhibiting drugs, such as crizotinib, that have been radiolabeled with 18F could prove to be viable PET tracers18. These tracers can then be used to gauge the levels of ALK protein in tumors, determine which patients would benefit from clinical trials, and in pharmacokinetic/pharmacodynamic analyses, all while retaining their specificity for ALK receptors18.
In this study, we successfully adapted an approved lung cancer, small-molecule therapeutic (crizotinib, 1) into a PET tracer ([18F]-4a) with in vivo biological activity analogous to the parent compound. PET imaging studies and biodistribution studies of ([18F]-4a) suggest typical accumulation of the tracer in the major organs within mice. The utility of PET within this context holds numerous possibilities for the research and development of pharmaceuticals, ranging from informing on the prospective pharmacokinetics of a drug about to enter into clinical trial to monitoring pharmacokinetics once a trial has begun, with the latter being of particular note in the event of addition of a second therapy or drug resistance19.
Supplementary Material
Acknowledgments
This work was supported by the Vanderbilt Center for Molecular Probes, the Kleberg Foundation, U24 CA220325, P50 CA236733, and NIH 1S10 OD016245. We would like to thank Adria Payne and Allison S. Cohen for their support through the revissions process.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
References and notes
- 1.Lovly CM; Heuckmann JM; de Stanchina E; Chen H; Thomas RK; Liang C; Pao W Cancer Res 2011, 71, 4920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bray F; Ferlay J; Soerjomataram I; Siegel RL; Torre LA; Jemal A CA Cancer J. Clin 2018, 68, 394. [DOI] [PubMed] [Google Scholar]
- 3.Ou SH Expert Rev. Anticancer Ther 2012, 12, 151. [DOI] [PubMed] [Google Scholar]
- 4.Curran MP Drugs 2012, 72, 99. [DOI] [PubMed] [Google Scholar]
- 5.Du X; Shao Y; Qin HF; Tai YH; Gao HJ Thoracic Cancer 2018, 9, 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.National Comprehensive Cancer Network. Metastatic Lung Cancer 2019. https://www.nccn.org/patients/guidelines/content/PDF/lung-metastatic-patient.pdf. Accessed March 10, 2019.
- 7.ClinicalTrials.gov. Bethesda, MD: National Library of Medicine (US) Identifier https://clinicaltrials.gov/ct2/show/NCT00722592, Platinum resistant ovarian cancer evaluation of Doxil and EC145 combination therapy (PRECEDENT). https://clinicaltrials.gov/ct2/show/NCT00722592. [Google Scholar]
- 8.Deane NG; Manning HC; Foutch AC; Washington MK; Aronow BA; Bornhop DJ; Coffey RJ Mol. Cancer Res 2007, 5, 341. [DOI] [PubMed] [Google Scholar]
- 9.Wyatt SK; Manning HC; Bai M; Bailey SN; Gallant P; Ma G; McIntosh L; Bornhop DJ Mol. Imaging Biol 2010, 12, 349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tang D; Hight MR; McKinley ET; Fu A; Buck JR; Smith RA; Tantawy MN; Peterson TE; Colvin DC; Ansari MS; Nickels M; Manning HC J. Nucl. Med 2012, 53, 287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Buck JR; McKinley ET; Hight MR; Fu A; Tang D; Smith RA; Tantawy MN; Peterson TE; Colvin D; Ansari MS; Baldwin RM; Zhao P; Guleryuz S; Manning HC J. Nucl. Med 2011, 52, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cui JJ; Tran-Dube M; Shen H; Nambu M; Kung PP; Pairish M; Jia L; Meng J; Funk L; Botrous I; McTigue M; Grodsky N; Ryan K; Padrique E; Alton G; Timofeevski S; Yamazaki S; Li Q; Zou H; Christensen J; Mroczkowski B; Bender S; Kania RS; Edwards MP J. Med. Chem 2011, 54, 6342. [DOI] [PubMed] [Google Scholar]
- 13.de Koning PD; McAndrew D; Moore D; Moses IB; Boyles DC; Kissick K; Stanchina CL; Cuthbertson T; Kamatani A; Rahman L; Rodriguez R; Urbina A; Sandoval A; Rose PR Org. Process Res. Dev 2011, 15, 1018. [Google Scholar]
- 14.Fussell SJ; Luan A; Peach P; Scotney G Tetrahedron Letters 2012, 53, 948. [Google Scholar]
- 15.Bolton RJ Labelled Compounds & Radiopharmaceuticals 2002, 45, 485. [Google Scholar]
- 16.Cui J, Jean et al. , Pfizer I, Ed. (USA, 2006). [Google Scholar]
- 17.Briard E; Zoghbi SS; Simeon FG; Imaizumi M; Gourley JP; Shetty HU; Lu S; Fujita M; Innis RB; Pike VW J. Med. Chem 2009, 52, 688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Radaram B; Pisaneschi F; Rao Y; Yang P; Piwnica-Worms D; Alauddin MM Eur. J Med. Chem 2019, 182, 111571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Doebele RC; Pilling AB; Aisner DL; Kutateladze TG; Le AT; Weickhardt AJ; Kondo KL; Linderman DJ; Heasley LE; Franklin WA; Varella-Garcia M; Camidge DR Clin. Cancer. Res 2012, 18, 1472. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.