

骨髓增生异常综合征(MDS)是一组起源于造血干细胞的异质性髓系克隆性疾病,多种基因突变在MDS预后判断中有重要价值[1]–[3]。10%~21%的MDS患者可检测到U2AF1基因突变,U2AF1基因突变的患者进展为急性髓系白血病(AML)的风险增高[4]–[6],而这类患者对化疗或异基因造血干细胞移植(allo-HSCT)治疗反应的报道较少。本研究我们回顾性分析59例U2AF1基因突变MDS患者的临床特征,比较化疗及allo-HSCT的疗效,并分析影响移植患者预后的危险因素。
病例与方法
1.病例资料:回顾性分析2016年7月至2018年12月苏州大学附属第一医院进行二代测序(NGS)检测的251例MDS患者资料,MDS诊断均经骨髓形态学分析、骨髓活检等相关检查确诊,诊断符合参考文献[7]标准。其中U2AF1基因突变59例(23.5%)。根据WHO 2016分型标准[8]分类:MDS伴单系病态造血(MLD-SLD)2例,MDS伴多系病态造血(MDS-MLD)19例,MDS伴原始细胞增多-1(MDS-EB-1)21例,MDS伴原始细胞增多-2(MDS-EB-2)17例。
2.NGS方法:提取初诊患者骨髓单个核细胞,抽提基因组DNA,构建51个血液病相关的常见热点基因Ion AmpliSeq文库,使用ABI Ion Torrent S5测序仪进行检测。NGS扩增子平均基因覆盖率98.03%,平均测序深度2 500×,95%以上的目标区域测序深度2 000×。致病性突变位点主要根据COSMIC数据库及文献报道确定。
3.治疗方法:10例患者接受去甲基化治疗,6例接受去甲基联合预激治疗,7例接受支持治疗。36例患者接受allo-HSCT,其中32例采用改良BuCy(白消安+环磷酰胺)±抗胸腺细胞球蛋白(ATG)预处理方案,2例采用FBAA(氟达拉滨+白消安+阿糖胞苷+ATG)预处理方案,1例采用FluCy(氟达拉滨+环磷酰胺)预处理方案,1例采用克拉屈滨+大剂量阿糖胞苷+白消安预处理方案;接受同胞HLA全相合移植的患者使用甲氨蝶呤(MTX)联合环孢素A(CsA)预防移植物抗宿主病(GVHD),接受无关HLA全相合和单倍型移植的患者使用ATG、MTX、CsA及霉酚酸酯(MMF)预防GVHD。
4.造血重建标准:中性粒细胞绝对计数连续3 d>0.5×109/L为粒系造血重建,PLT连续3 d>20×109/L且脱离血小板输注为巨核系造血重建。
5.疗效评价及随访:根据修订的2006年MDS国际工作组(IWG)治疗反应标准[9]进行评价。随访截止日期为2019年5月31日。化疗组总生存(OS)时间定义为确诊日期至患者任何原因死亡或末次随访日,无进展生存(PFS)时间定义为确诊日期至血液学复发、死亡日期或末次随访日;allo-HSCT组OS时间指定义为干细胞回输至死亡或末次随访日,PFS时间定义为干细胞回输至血液学复发、死亡日期或末次随访日。
6.统计学处理:应用SPSS 25.0软件进行统计分析。计量资料以中位数(范围)表示,采用Mann-Witney U检验进行比较。分类资料以例数(构成比)表示,采用卡方检验或Fisher精确概率法进行比较。采用Cox风险模型进行影响OS、PFS的单因素分析,P<0.05为差异有统计学意义。应用R 3.4.2软件绘制统计图。
结果
1.临床特征:59例U2AF1基因突变患者中,男43例(72.9%),女16例(27.1%),中位年龄44(17~72)岁。初诊时中位WBC 4.49(1.05~22.50)×109/L、PLT 71(4~379)×109/L、HGB 87(43~160)g/L。中位骨髓原幼细胞比例为5.9%(0~18%)。IPSS分组:低危3例(5.1%),中危-1 33例(55.9%),中危-2 17例(28.8%),高危6例(10.2%);IPSS-R分组:低危11例(18.6%),中危19例(32.2%),高危21例(35.6%),极高危8例(13.6%)。52例患者接受了化疗或allo-HSCT,其中allo-HSCT组36例,化疗组16例,其临床特征见表1。allo-HSCT组中位年龄低于化疗组(41岁对50岁,P=0.002)。
表1. 52例U2AF1基因突变骨髓增生异常综合征患者临床特征.
| 化疗组(16例) | allo-HSCT组(36例) | 统计量 | P值 | ||
| 性别(例,男/女) | 11/5 | 26/10 | 0.729 | 0.69 | |
| 年龄>45岁a[例数(%)] | 13(81.3) | 14(38.9) | 8.156 | 0.02 | |
| WBC[×109/L,M(范围)] | 3.96(1.30~22.50) | 5.05(1.05~18.02) | 2.003 | 0.05 | |
| HGB[g/L,M(范围)] | 83(45~160) | 91(51~137) | 1.401 | 0.38 | |
| PLT[×109/L,M(范围)] | 51(4~240) | 60(4~355) | 0.297 | 0.65 | |
| 骨髓原幼细胞比例[%,M(范围)] | 6.1(0~16.0) | 6.4(0~18.0) | 0.050 | 0.86 | |
| IPSS-R[例数(%)] | 4.939 | 0.53 | |||
| 低危 | 2(12.5) | 7(19.4) | |||
| 中危 | 6(37.5) | 10(27.8) | |||
| 高危 | 4(25.0) | 15(41.7) | |||
| 极高危 | 4(25.0) | 4(11.1) | |||
| 染色体核型[例数(%)] | |||||
| 正常核型 | 8(50.0) | 15(41.7) | 0.317 | 0.85 | |
| +8 | 3(18.8) | 11(30.6) | 0.790 | 0.67 | |
| -7/7q- | 3(18.8) | 1(2.8) | 5.049 | 0.08 | |
| 基因突变数目≥4个[例数(%)] | 6(37.5) | 12(33.3) | 1.256 | 0.53 | |
| ASXL1突变[例数(%)] | 3(18.8) | 14(38.9) | 3.569 | 0.16 | |
| RUNX1突变[例数(%)] | 6(37.5) | 6(16.7) | 2.759 | 0.25 | |
| SETBP1突变[例数(%)] | 2(12.5) | 5(13.9) | 0.022 | 0.98 | |
| U2AF1突变类型[例数(%)] | 3.991 | 0.14 | |||
| S34 | 10(62.5) | 31(86.1) | |||
| 其他 | 6(37.5) | 5(13.9) | |||
| U2AF1 VAF[%,M(范围)] | 37.9(4.6~49.7) | 37.6(5.5~47.4) | 0.268 | 0.79 | |
注:a:allo-HSCT组为患者移植时年龄;IPSS-R:修订的国际预后积分系统;VAF:等位基因变异频率
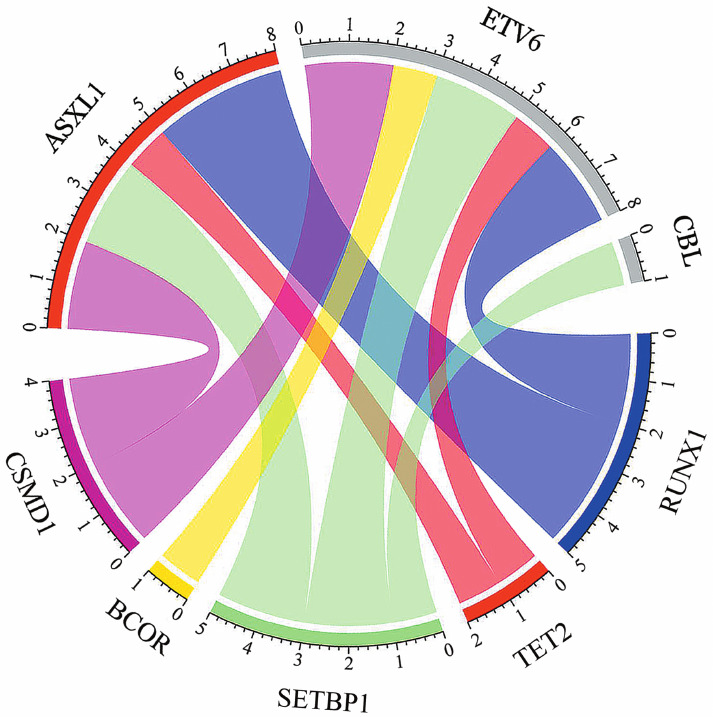
2.突变基因检测:251例患者中共检出165个基因突变,患者平均基因突变数为2.8个(范围1~6个)。48例(81.4%)患者合并其他基因突变。与U2AF1基因突变共存的突变频率较高的基因依次为ASXL1(25.6%,21/59)、RUNX1(23.7%,14/59)、ETV6(18.6%,11/42)、SETBP1(13.6%,8/59)、CBL(8.5%,5/59)、TET2(6.8%,4/59)等(图1)。59例U2AF1突变的患者中检出63个U2AF1基因突变位点,其分布如下:2号外显子突变49例(83.1%),其中S34F 30例(50.8%),S34Y 17例(28.8%),R35L及R35W各1例(1.7%);6号外显子突变5例(8.5%),其中Q157R 3例(5.1%),Q157P 2例(3.4%);4例(6.8%)患者同时检出S34F和Q157R突变;1例患者为G159插入突变(c.472_477dup TATGAG,p.Glu159_Met160insTyrGlu),63个突变位点的中位等位基因变异频率(VAF)为37.9%(4.6%~49.7%)。
图1. 59例伴U2AF1突变骨髓增生异常综合征患者基因突变谱.
3.生存分析:截至2019年5月31日,中位随访时间为25.2(0.3~34.9)个月,化疗组中位OS时间25.3(1.1~27.1)个月,而allo-HSCT组中位OS时间未达到。allo-HSCT组2年OS及PFS率较化疗组显著增加[OS:(73.2±10.5)%对(51.1±13.9)%,P=0.004;PFS:(70.9±11.4)%对(43.6±14.1)%,P=0.008]。在allo-HSCT组患者中,未合并RUNX1基因突变的患者2年PFS率较合并该突变的患者显著增高(80.0%对50.0%,P=0.002)。12例(33.3%)移植患者存在4个以上的突变基因,突变基因≥4个的患者移植后1年PFS率较突变基因<4个患者显著降低[(61.7±15.6)%对(94.7±5.1)%,P=0.015],而接受化疗治疗的两组患者1年PFS率差异无统计学意义[(60.0±15.5)%对(66.7±15.2)%,P=0.735]。
4.移植疗效及预后因素分析:36例allo-HSCT患者中,15例移植前获得完全缓解(CR),4例部分缓解(PR),3例处于疾病进展状态(PD),3例转化为AML,11例去甲基治疗桥接移植。HLA全相合移植13例(同胞供者7例,无关供者6例),单倍型移植23例。回输单个核细胞(MNC)9.44(4.35~17.59)×108/kg、CD34+细胞4.76(2.50~10.55)×106/kg。粒系重建、巨核系重建中位时间分别为12(9~28)d、13.5(9~29)d。11例(30.6%)患者发生急性GVHD(aGVHD),中位发生时间35(13~72)d,其中Ⅰ、Ⅱ度6例(16.7%),Ⅲ、Ⅳ度3例(8.4%),不典型2例(5.6%)。9例(25%)患者发生慢性GVHD,中位发病时间140(93~262)d,其中局限型7例(19.4%),广泛型2例(5.6%)。单因素分析显示,突变基因≥4个、RUNX1基因突变为影响患者PFS(HR=2.917,95% CI 0.969~8.787,P=0.057;HR=3.503,95% CI 1.173~10.460,P=0.025)的危险因素。
讨论
近年来随着NGS的广泛应用,在大部分MDS患者中发现了多种涉及RNA剪接机制的重现性体细胞突变[10]–[11],目前已有大量研究报道这些突变的临床意义,Thol等[12]研究表明SRSF2是MDS患者总体预后不良及高转白风险的独立危险因素,Li等[13]发现突变组总体OS时间较阴性组显著缩短。Wu等[14]表明在中低危组的MDS患者中,U2AF1基因突变与较短的转化为AML(转白)时间相关,类似结论在较早的文献中也有报道[12]。关于allo-HSCT对U2AF1突变的MDS患者预后影响的文献较少,本研究我们观察到allo-HSCT组较化疗组2年OS及PFS率显著增高,伴有RUNX1基因突变是影响患者移植后PFS的危险因素。
本研究中59例患者检测出63个U2AF1突变位点,共4种突变类型,86.4%的基因突变位于S34,14.3%位于Q157,与文献[13]–[14]报道相似。Yip等[15]研究发现U2AF1 S34F突变促进了红细胞的凋亡,提示S34F突变能够破坏细胞生长并增强其凋亡,从而导致骨髓细胞病态造血。本研究中未发现S34F突变患者与其他位点突变患者的临床特征差异具有统计学意义,由于其他位点的突变频率较低,若扩大样本量可能得出更有意义的结论。
本研究中23.5%的患者伴有U2AF1基因突变。分析该组患者突变谱,与U2AF1突变合并出现频率最高的是ASXL1基因(25.6%),而与U2AF1作用机制相同的剪接体基因突变发生率为2.4%,是由于剪接因子的突变通常相互排斥,而与表观遗传修饰因子的突变共存,与文献[16]相一致。RUNX1基因突变检出率(23.7%)仅次于ASXL1基因,已有文献报道MDS患者伴有RUNX1基因突变往往提示预后不良,在髓系肿瘤中RUNX1基因突变型患者(13个月)较野生型(21个月)中位OS时间显著降低[17]。值得注意的是,我们发现伴RUNX1基因突变的患者移植后2年OS率显著低于RUNX1基因突变阴性的患者,同样表示携带该基因的患者预后较差,提示移植可能改善此类患者的预后。此外,MDS患者伴随突变基因的数量是预测疾病进展的危险因素,随着突变数目的增加,患者转白的风险显著增高[18]。本研究中存在4个以上突变基因的患者allo-HSCT后预后不佳,而在化疗组患者中,以上危险因素差异均无统计学意义。
本研究结果显示,allo-HSCT可以改善U2AF1基因突变MDS患者预后,基因突变≥4个、RUNX1基因突变是影响移植疗效的危险因素。
Funding Statement
基金项目: 国家科技重大专项课题 (2017ZX09304021); 国家重点研发计划 (2017YFA0104502); 江苏省医学杰出人才项目 (JCRCA2016002); 江苏省科教强卫工程-临床医学中心 (YXZXA2016002)
References
- 1.Tefferi A, Lasho TL, Patnaik MM, et al. Targeted next-generation sequencing in myelodysplastic syndromes and prognostic interaction between mutations and IPSS-R[J] Am J Hematol. 2017;92(12):1311–1317. doi: 10.1002/ajh.24901. [DOI] [PubMed] [Google Scholar]
- 2.Lindsley RC, Saber W, Mar BG, et al. Prognostic mutations in myelodysplastic syndrome after stem-cell transplantation[J] N Engl J Med. 2017;376(6):536–547. doi: 10.1056/NEJMoa1611604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.李 冰, 王 静雅, 刘 晋琴, et al. 靶向测序检测511例骨髓增生异常综合征患者基因突变[J] 中华血液学杂志. 2017;38(12):1012–1016. doi: 10.3760/cma.j.issn.0253-2727.2017.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Graubert TA, Shen D, Ding L, et al. Recurrent mutations in the U2AF1 splicing factor in myelodysplastic syndromes[J] Nat Genet. 2011;44(1):53–57. doi: 10.1038/ng.1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Qian J, Yao DM, Lin J, et al. U2AF1 mutations in Chinese patients with acute myeloid leukemia and myelodysplastic syndrome[J] PLoS One. 2012;7(9):e45760. doi: 10.1371/journal.pone.0045760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.王 继英, 马 娇, 蔺 亚妮, et al. 118例骨髓增生异常综合征及相关疾病患者RNA剪接体复合物蛋白编码基因SF3B1、U2AF1和SRSF2突变分析[J] 中华血液学杂志. 2017;38(3):192–197. doi: 10.3760/cma.j.issn.0253-2727.2017.03.004. [DOI] [Google Scholar]
- 7.中华医学会血液学分会. 骨髓增生异常综合征中国诊断与治疗指南(2019年版)[J] 中华血液学杂志. 2019;40(2):89–97. doi: 10.3760/cma.j.issn.0253-2727.2019.02.001. [DOI] [Google Scholar]
- 8.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia[J] Blood. 2016;127(20):2391–2405. doi: 10.1182/blood-2016-06-721662. [DOI] [PubMed] [Google Scholar]
- 9.Cheson BD, Greenberg PL, Bennett JM, et al. Clinical application and proposal for modification of the International Working Group(IWG)response criteria in myelodysplasia[J] Blood. 2006;108(2):419–425. doi: 10.1182/blood-2005-10-4149. [DOI] [PubMed] [Google Scholar]
- 10.Yoshida K, Sanada M, Shiraishi Y, et al. Frequent pathway mutations of splicing machinery in myelodysplasia[J] Nature. 2011;478(7367):64–69. doi: 10.1038/nature10496. [DOI] [PubMed] [Google Scholar]
- 11.Visconte V, Makishima H, Maciejewski JP, et al. Emerging roles of the spliceosomal machinery in myelodysplastic syndromes and other hematological disorders[J] Leukemia. 2012;26(12):2447–2454. doi: 10.1038/leu.2012.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Thol F, Kade S, Schlarmann C, et al. Frequency and prognostic impact of mutations in SRSF2, U2AF1, and ZRSR2 in patients with myelodysplastic syndromes[J] Blood. 2012;119(15):3578–3584. doi: 10.1182/blood-2011-12-399337. [DOI] [PubMed] [Google Scholar]
- 13.Li B, Liu J, Jia Y, et al. Clinical features and biological implications of different U2AF1 mutation types in myelodysplastic syndromes[J] Genes Chromosomes Cancer. 2018;57(2):80–88. doi: 10.1002/gcc.22510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu SJ, Tang JL, Lin CT, et al. Clinical implications of U2AF1 mutation in patients with myelodysplastic syndrome and its stability during disease progression[J] Am J Hematol. 2013;88(11):E277–282. doi: 10.1002/ajh.23541. [DOI] [PubMed] [Google Scholar]
- 15.Yip BH, Steeples V, Repapi E, et al. The U2AF1S34F mutation induces lineage-specific splicing alterations in myelodysplastic syndromes[J] J Clin Invest. 2017;127(6):2206–2221. doi: 10.1172/JCI91363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Haferlach T, Nagata Y, Grossmann V, et al. Landscape of genetic lesions in 944 patients with myelodysplastic syndromes[J] Leukemia. 2014;28(2):241–247. doi: 10.1038/leu.2013.336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gaidzik VI, Teleanu V, Papaemmanuil E, et al. RUNX1 mutations in acute myeloid leukemia are associated with distinct clinico-pathologic and genetic features[J] Leukemia. 2016;30(11):2160–2168. doi: 10.1038/leu.2016.126. [DOI] [PubMed] [Google Scholar]
- 18.Papaemmanuil E, Gerstung M, Malcovati L, et al. Clinical and biological implications of driver mutations in myelodysplastic syndromes[J] Blood. 2013;122(22):3616–3627. doi: 10.1182/blood-2013-08-518886. [DOI] [PMC free article] [PubMed] [Google Scholar]