

Abstract
Cytochrome P450 46A1 encoded by CYP46A1 catalyzes cholesterol 24-hydroxylation and is a CNS-specific enzyme that controls cholesterol removal and turnover in the brain. Accumulating data suggest that increases in cytochrome P450 46A1 activity in mouse models of common neurodegenerative diseases affect various, apparently unlinked biological processes and pathways. Yet, the underlying reason for these multiple enzyme activity effects is currently unknown. Herein, we tested the hypothesis that cytochrome P450 46A1-mediated sterol flux alters physico-chemical properties of the plasma membranes and thereby membrane-dependent events. We used 9-month-old 5XFAD mice (an Alzheimer’s disease model) treated for 6 months with the anti-HIV drug efavirenz. These animals have previously been shown to have improved behavioural performance, increased cytochrome P450 46A1 activity in the brain, and increased sterol flux through the plasma membranes. We further examined 9-month-old Cyp46a1−/− mice, which have previously been observed to have cognitive deficits and decreased sterol flux through brain membranes. Synaptosomal fractions from the brain of efavirenz-treated 5XFAD mice had essentially unchanged cholesterol levels as compared to control 5XFAD mice. However with efavirenz treatment in these mice, there were changes in the membrane properties (increased cholesterol accessibility, ordering, osmotic resistance and thickness) as well as total glutamate content and ability to release glutamate in response to mild stimulation. Similarly, the cholesterol content in synaptosomal fractions from the brain of Cyp46a1−/− mice was essentially the same as in wild-type mice but knockout of Cyp46a1 was associated with changes in membrane properties and glutamate content and its exocytotic release. Changes in Cyp46a1−/− mice were in the opposite direction to those observed in efavirenz-treated versus control 5XFAD mice. Incubation of synaptosomal fractions with the inhibitors of glycogen synthase kinase 3, cyclin-dependent kinase 5, protein phosphatase 1/2 A, and protein phosphatase 2B revealed that increased sterol flux in efavirenz-treated versus control 5XFAD mice affected the ability of all four enzymes to modulate glutamate release. In contrast, in Cyp46a1−/− versus wild-type mice, decreased sterol flux altered the ability of only cyclin-dependent kinase 5 and protein phosphatase 2B to regulate the glutamate release. Collectively, our results support cytochrome P450 46A1-mediated sterol flux as an important contributor to the fundamental properties of the membranes, protein phosphorylation and synaptic transmission. Also, our data provide an explanation of how one enzyme, cytochrome P450 46A1, can affect multiple pathways and processes and serve as a common potential target for several neurodegenerative disorders.
Keywords: Alzheimer’s disease, brain, cholesterol, CYP46A1, membrane properties
Cytochrome P450 46A1 controls cholesterol removal and turnover in the brain. Evidence is provided that the P450-mediated sterol flux alters physico-chemical properties of synaptosomal membranes and thereby membrane-dependent processes. Thus, we found how one enzyme can affect multiple pathways and serve as a common potential target for different brain disorders.
Graphical Abstract
Graphical Abstract.
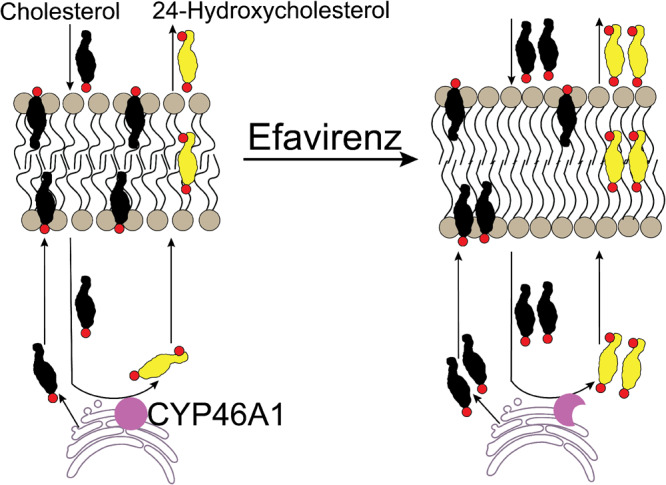
Introduction
Cytochrome P450 46A1 (CYP46A1) is a CNS-specific enzyme, normally residing in the endoplasmic reticulum of multiple types of neurons (Ramirez et al., 2008). CYP46A1 converts cholesterol to 24-hydroxycholesterol (24HC) and thereby disposes the majority of the brain cholesterol excess due to rapid 24HC flux into the systemic circulation (Lutjohann et al., 1996; Bjorkhem et al., 1998; Lund et al., 1999; Meaney et al., 2002; Lund et al., 2003). Cyp46a1−/− mice have the same steady-state levels of brain cholesterol as wild-type animals but a decreased rate of brain cholesterol biosynthesis as a compensatory mechanism to counteract the lack of the major cholesterol elimination pathway (Lund et al., 2003). Accordingly, cholesterol turnover and sterol flux through the membranes are decreased in the brain of Cyp46a1−/− mice. Similarly, the cholesterol levels are unchanged in the brain of wild-type mice with CYP46A1 activation by pharmacologic means (Mast et al., 2014). Yet, their brain cholesterol biosynthesis is enhanced, thus leading to increases in cholesterol turnover and sterol flux through the membranes.
Studies in mice revealed a broad therapeutic potential of increased CYP46A1 activity for various neurodegenerative disorders such as Alzheimer’s and Huntington’s diseases, Niemann-Pick disease type C and spinocerebellar ataxia (Hudry et al., 2010; Burlot et al., 2015; Boussicault et al., 2016; Mast et al., 2017b; Kacher et al., 2019; Mitroi et al., 2019; Nóbrega et al., 2019; Petrov et al., 2019b). Even glioblastoma, an aggressive brain tumour, was found to be inhibited in mice by pharmacologic CYP46A1 activation (Han et al., 2020). Mechanistically, increases in CYP46A1 activity affected multiple processes and led to improvements in neurotransmission; enhancement in vesicular and endosomal trafficking; stimulation of proteasome and autophagy machineries; reduction of neuronal atrophy; and decreases in microgliosis (Hudry et al., 2010; Burlot et al., 2015; Boussicault et al., 2016; Kacher et al., 2019; Mitroi et al., 2019; Nóbrega et al., 2019). Conversely, lack of CYP46A1 was found to elicit severe deficits in memory and learning, likely due to impaired long-term potentiation as a result of decreased protein prenylation (Kotti et al., 2006, 2008). Furthermore, region-specific decreases in CYP46A1 expression by genetic means led to the manifestations of Alzheimer’s and Huntington’s diseases (Djelti et al., 2015; Boussicault et al., 2016), spinocerebellar ataxia (Nóbrega et al., 2019) and seizure-like discharges (Chali et al., 2015). These detrimental effects were associated with neuronal degeneration and apoptosis; impairment of the lysosomal pathway; aberrant recruitment of the amyloid precursor protein to lipid rafts; endoplasmic reticulum stress; and abnormal tau phosphorylation (Djelti et al., 2015; Boussicault et al., 2016; Nóbrega et al., 2019).
Recently we discovered that in 5XFAD mice, a model of rapid amyloidogenesis (Oakley et al., 2006), CYP46A1 can be activated by a very low dose of efavirenz (EFV), an anti-HIV drug (Mast et al., 2017b; Petrov et al., 2019a). EFV increased the brain cholesterol 24-hydroxylation and turnover, improved mouse performance in behavioural tests and altered the activation of glial cells as well as the expression of essential synaptic proteins (Mast et al., 2017b; Petrov et al., 2019a). The effects on the amyloid β levels (Aβ) were the treatment paradigm-specific, either Aβ decreasing or not changing. Studies by the omics approaches documented altered protein phosphorylation in EFV-treated 5XFAD mice and pointed to the effects on synaptic function, inflammation, microglia phenotype, oxidative stress, cellular hypoxia, ubiquitin-proteasome system, autophagy and apoptosis (Petrov et al., 2019b). Protein phosphorylation was also altered in Cyp46a1−/− mice (Mast et al., 2017a), and the affected processes overlapped in part with those in EFV-treated 5XFAD mice (synaptic transmission, ubiquitination and cytoskeleton maintenance). Yet the set of the differentially phosphorylated proteins mostly differed (Mast et al., 2017a). Subsequent studies by others provided additional evidence that EFV-induced CYP46A1 activation is beneficial by showing that it elicits therapeutic effects under the conditions of depression (Patel et al., 2017), reduces tau phosphorylation in early Alzheimer’s disease neurons (van der Kant et al., 2019), and suppresses glioblastoma proliferation in the brain (Han et al., 2020). CYP46A1 is now being tested as a drug target in the two clinical trials (NCT03706885 and NCT03650452, ClinicalTrials.gov), one in patients with early Alzheimer’s disease and the other in children with frequent seizures due to rare genetic diseases.
The underlying reason for multiple CYP46A1 activity effects is currently unknown. However, there is a common event in EFV-treated 5FAD mice and mice with Cyp46a1 ablation. This common event is altered cholesterol turnover in the brain and therefore altered sterol flux through the plasma membranes and in particular lipid rafts, i.e. ordered, cholesterol- and sphingolipid-enriched microdomains of the plasma membranes (Pike, 2003). Increased sterol flux from the cytosol to the plasma membranes makes more cholesterol enter and leave the plasma membranes per unit of time. Hence more of the newly synthesized cholesterol enters the plasma membranes and is then selectively recruited to lipid rafts (Kannan et al., 2007; Olsen et al., 2013; Javanainen et al., 2017). As a result, both membranes and lipid rafts may become affected. Conversely, a decreased sterol flux could have the opposite effects. Thus, both increased and decreased sterol fluxes can trigger in turn various changes in the membrane-associated cellular processes, for example vesicular trafficking and synaptic transmission (Suzuki et al., 2007; Sebastião et al., 2013; Egawa et al., 2016). These sterol flux effects could be similar to those of the altered membrane cholesterol content, which is known to affect the physico-chemical properties of the membranes, formation of lipid rafts as well as conformation and membrane distribution of integral membrane proteins (Simons and Ehehalt, 2002; Pike, 2003; Yang et al., 2016). Herein, we tested our sterol flux hypothesis by evaluating the properties of synaptosomal fractions isolated from the brain of mice with increased and decreased sterol fluxes. We also investigated the sterol flux effects on the activity of the two lipid raft-associated protein kinases and two protein phosphatases that regulate the exocytotic glutamate release. We obtained experimental support for our hypothesis and linked the CYP46A1-mediated sterol flux, fundamental membrane properties, protein phosphorylation, synaptic transmission and behaviour in a model. By introducing and initial testing the sterol flux hypothesis, this study created the basis for the major conceptual advance in our understanding of the role of CYP46A1 and cholesterol homeostasis in the brain and paved the way towards development of new, disease-modifying treatments for Alzheimer’s disease and other disorders of the brain.
Materials and methods
Animal models
Five models were used, all of which were 9 months old: (i) EFV-treated 5XFAD mice—a model of CYP46A1 activation and increased sterol flux in the brain affected by Aβ deposition; (ii) control (vehicle-treated) 5XFAD mice—a model of basal CYP46A1 activity and sterol flux in the brain affected by Aβ deposition; (iii) Cyp46a1−/− mice—a model of full CYP46A1 inhibition by genetic means and decreased sterol flux; (iv) B6SJL mice, the background strain for 5XFAD animals; and (v) C57BL/6J; 129S6/SvEv mice, the background strain for Cyp46a1−/− mice. The latter two groups served as models of basal CYP46A1 activity and sterol flux in healthy brains of different genetic backgrounds. 5XFAD mice co-express two human proteins, amyloid precursor protein and presenelin 1, which carry a total of five mutations underlying familial Alzheimer’s disease. These transgenic animals represent a rapid-onset, severe amyloid plaque model recapitulating the major features of Alzheimer’s disease (amyloid plaques, gliosis, neurodegeneration and memory deficits) (Oakley et al., 2006; Kimura and Ohno, 2009; Crouzin et al., 2013). Only F1 generation of the hemizygous mice was used after crossing 5XFAD hemizygous males with wild-type B6SJL females (The Jackson Laboratory). Gender-matched B6SJL littermates served as a control. The Pde6brd1 mutation leading to blindness was bred out of our colony. 5XFAD mice were put on S-EFV (Toronto Research Chemicals Inc.) dissolved in drinking water as described (Petrov et al., 2019a). The treatment started at 3 months of age and continued for 6 months. The Cyp46a1+/+ and Cyp46a1−/− colonies were obtained from Cyp46a1+/− mice on the mixed C57BL/6J; 129S6/SvEv background (Lund et al., 2003), which were provided by Dr D. Russell (University of Texas Southwestern, Dallas, TX). All animals were maintained in a temperature and humidity-controlled environment with 12-h light–dark cycle with standard rodent chow and water provided ad libitum. All animal experiments were approved by the Institutional Animal Care and Use Committee and conformed to recommendations of the American Veterinary Association Panel on Euthanasia.
Isolation of synaptosomal fractions
Mice were fasted overnight and sacrificed the following morning. The brains were quickly isolated, dissected from the cerebellum, which was discarded, and rinsed in ice-cold phosphate-buffered saline. Synaptosomal fractions were obtained by centrifugation from 10% brain homogenates in Syn-PER™ Synaptic Protein Extraction Reagent (TermoFisher Scientific). See Supplementary material for details. Synaptosomal fractions were enriched with the pre- and postsynaptic proteins (e.g. synaptophysin and postsynaptic density-95, PSD-95, respectively), lacked nuclear proteins (e.g. histone deacetylase 2), and contained the marker proteins for astrocytes (e.g. glutamine synthetase and excitotoxic amino acid transporter 2) (Supplementary Fig. 1). Mostly the same preparations of synaptosomal fractions were used in all of the experiments described in the present work.
Western blots
Protein separation by SDS-PAGE and membrane processing were as described (Petrov et al., 2019a). The source, clonality and dilutions of primary and secondary antibodies are indicated in Supplementary Table 1 of the Supplementary material.
Sterol quantifications
Total cholesterol, free lathosterol, desmosterol and 24HC were measured by gas chromatography-mass spectroscopy using deuterated sterol analogues as internal standards (Mast et al., 2011).
Cholesterol availability in the membranes
Domain 4 (D4) of perfringolysin O (PFO), a cholesterol-dependent cytolysin, was used, which contained the D434S and C459A mutations to increase the D4 sensitivity for cholesterol detection (Johnson et al., 2012). This mutant D4 also had the superfolder green fluorescent protein (GFP) and glutathione S-transferase (GST) at the C-terminus. See Supplementary material for the preparation of the PFO-D4-GFP-GST construct, its expression and purification as well as the conditions for membrane binding. The PFO-D4-GFP-GST fluorescence was quantified at the emission peak of 510 nm after excitation at 485 nm.
Membrane ordering
Two probes, di-4-ANEPPDHQ and F2N12S (TermoFisher Scientific) were used (Shynkar et al., 2007; Ashdown and Owen, 2015), which were individually incubated with synaptosomal fractions as described in details in Supplementary material. Di-4-ANEPPDHQ was excited at 473 nm and its general polarization (GP) was calculated as a ratio of (I560−I650) to (I560+I650), where I560 and I650 represents the fluorescence intensity at 560 and 650 nm, respectively. F2N12S was excited at 405 nm, and the ratio between the intensity of its short- and long-wavelength emission bands was calculated at 485 and 575 nm, respectively.
Osmotic resistance
Synaptosomal fractions were assessed for light scattering at 520 nm at different (0–0.4 M) NaCl concentrations. See Supplementary material for details.
Transmission electron microscopy
Synaptosomal fractions were post-fixed with 1% osmium tetroxide, 1% tannic acid and 1% p-phenylenediamine to preserve and enhance the visualization of membranous structures and osmium-treated neutral lipids (Guyton and Klemp, 1988). See Supplementary material for details. Images were acquired by a FEI Tecnai Spirit T12 electron microscope (ThermoFisher Scientific) with a Gatan US4000 4kx4k CCD camera (Gatan). ImageJ software (NIH) was used to measure the membrane thickness.
Glutamate content
The Glutamate Assay Kit (BioVision) was used, in which glutamate is enzymatically oxidized, thus leading to absorbance at 450 nm. See Supplementary material for details.
Inhibition of protein kinases and protein phosphatases
Synaptosomal fractions were incubated for 20 min at 37°C either with the inhibitor dissolved in DMSO or DMSO alone followed by induction of the glutamate release by 117 mM KCl and the measurements of the glutamate content. The inhibitors and final inhibitor concentrations were as follows: 20 μM R-roscovitive or 10 μM butyrolactone I (Enzo Life Sciences) to inhibit cyclin-dependent kinase 5 (CDK5); 10 μM SB 216763 (Tocris) to inhibit glycogen synthase kinase 3 (GSK3); 1 μM okadaic acid (Tocris) to inhibit protein phosphatases 1 and 2 A (PP1/2A), and 30 μM cyclosporin A (Tocris) to inhibit protein phosphatase 2B or calcineurin (PP2B) inhibition (Bauerfeind et al., 1997; Marks and McMahon, 1998; Hosaka et al., 1999; Tomizawa et al., 2002; Chergui et al., 2004; Lee et al., 2004; Chen et al., 2007; Wei et al., 2010; Xue et al., 2011; Miranda-Barrientos et al., 2014). See Supplementary material for details.
Statistical analysis
Data represent the mean ± SD; the sample size is indicated in each figure or in the figure legend. Data from all available brains were used in experiments. There were no exclusions of statistical outliers but for some experiments there was not enough sample remaining from some animals. Randomization and blinding were not used in experiments. Either a two-tailed, unpaired Student’s t-test or a two-way ANOVA followed by Bonferroni post hoc comparisons were used for data analyses. The GraphPad Prism (GraphPad) and Origin Pro (OriginLab) software were used. Statistical significance was defined as *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Data availability
The authors confirm that the data supporting the findings of this study are available upon request.
Results
Sterol profiles of synaptosomal fractions
Synaptosomes or isolated nerve terminals have the molecular machinery necessary for the uptake, storage and release of neurotransmitter and carry the morphological features as well as most of the chemical properties of the original nerve terminals (Nicholls and Sihra, 1986; Schrimpf et al., 2005; Bai and Witzmann, 2007). Four sterols were quantified: cholesterol as CYP46A1 substrate; 24HC as CYP46A1 product; lathosterol and desmosterol as markers of cholesterol biosynthesis in neurons and astrocytes, respectively (Pfrieger and Ungerer, 2011). In EFV-treated 5XFAD versus control mice, the levels of cholesterol in synaptosomal fractions were the same in male mice and only slightly decreased (by ∼12%) in female mice (Fig. 1A). In the corresponding brain homogenates, the cholesterol levels were unchanged in mice of both sexes (Supplementary Fig. 2A). The 24HC content was decreased in synaptosomal fractions from EFV-treated 5XFAD mice of both sexes (Fig. 1B) but increased in their brain homogenates (Supplementary Fig. 2B). This difference was likely due to a poor oxysterol retention inside cells (Meaney et al., 2002) and hence removal when synaptosomes were washed during the isolation. In contrast, the levels of cholesterol precursors lathosterol and desmosterol were increased in both synaptosomal fractions and brain homogenates from EFV-treated 5XFAD mice of both sexes (Fig. 1C and D, Supplementary. Fig. 2C and D). Thus, in synaptosomal fractions and brain homogenates from male and female 5XFAD mice, EFV treatment led to increases in the cholesterol precursor levels with essentially no effect on cholesterol content, a result consistent with increases in cholesterol turnover and sterol flux through the membranes in both types of samples.
Figure 1.

Sterol profiles in synaptosomal fractions. (A–D) The levels of cholesterol, 24HC, lathosterol and desmosterol, respectively, in synaptosomal fractions isolated from the brain of vehicle-treated (Cntr, black circles) and EFV-treated (Tx, green circles) 5XFAD mice as well as wild-type (Cyp46a1+/+) mice on the mixed (C57BL/6J; 129S6/SvEv) background (black circles) and Cyp46a1−/− mice (magenta circles) on the same background. The results are the mean ± SD of the measurements in individual animals (n = 3–9 mice per group and gender). Statistical analysis: a two-way ANOVA followed by Bonferroni post hoc comparisons. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001.
Figure 2.

Cholesterol availability and ordering in the membranes of synaptosomal fractions. (A) Cholesterol availability as assessed by binding of PFO-D4-GFP-GST and its fluorescence intensity (I) at 510 nm. The PFO-D4-GFP-GST fluorescence was corrected for the non-specific PFO-D4-GFP-GST binding as described in the main text and shown in Supplementary Fig. 3B and C. The averaged emission spectra of PFO-D4-GFP-GST are shown on the right. (B) Membrane ordering as assessed by binding of di-4-ANEPPDHQ and its emission spectra, which were used to calculate the dye GP using the following equation: , where I560 nm and I650 nm are the fluorescence intensity at 560 and 650 nm, respectively. (C) Membrane ordering and asymmetry as assessed by binding of F2N12S and its emission spectra (on the right), which were used to calculate the ratio of the fluorescence intensity at 485 and 575 nm. All results are the mean ± SD of the measurements in individual animals (n = 3–8 mice per group and gender). Statistical analysis: a two-way ANOVA followed by Bonferroni post hoc comparisons. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. Cntr, control or vehicle-treated 5XFAD mice (black circles and traces); Tx, EFV-treated 5XFAD mice (green circles and traces); Cyp46a1+/+ mice (black circles and traces) and Cyp46a1−/− (magenta circles and traces) on the mixed (C57BL/6J; 129S6/SvEv) background.
In Cyp46a1−/− mice versus Cyp46a1+/+ animals, the cholesterol levels in synaptosomal fractions were unchanged and moderately decreased (by ∼21%) in male and female mice, respectively (Fig. 1A). Similarly, there was no change and a decrease (by 14%) in male and female Cyp46a1−/− mice, respectively, in the cholesterol levels in brain homogenates (Supplementary Fig. 2A). 24HC was not detected either in synaptosomal fractions or brain homogenates of Cyp46a1−/− mice due to lack of CYP46A1 which produces this oxysterol (Fig. 1B, Supplementary Fig. 2B). The levels of lathosterol and desmosterol were decreased in Cyp46a1−/− mice at least 5-fold in both synaptosomal fractions and brain homogenates (Fig. 1C and D, Supplementary Fig. 2C and D). Thus, Cyp46a1 ablation had the sex-specific effect on the cholesterol levels but not the levels of other sterols in brain homogenates and synaptosomal fractions. Hence, both males and females had a decrease in cholesterol turnover and sterol flux through the membranes.
Cholesterol accessibility in the synaptosomal fraction membranes
PFO-D4 is a non-lytic derivative of the θ-toxin, which binds cholesterol in a concentration-dependent manner and selectively recognizes accessible membrane cholesterol (Ohno-Iwashita et al., 2010). PFO-D4 has been a common probe for analysing the distribution and dynamics of membrane cholesterol in lipid rafts (Johnson et al., 2017). Methyl-β-cyclodextrin (MCD) is an efficient cholesterol-extracting agent (Zidovetzki and Levitan, 2007). The PFO-D4-GFP-GST construct containing the fluorescently labelled PFO-D4 was generated, expressed in Escherichia coli and purified (see Supplementary material for details). This construct was then investigated for binding to the membranes of intact synaptosomal fractions (pre-MCD samples, Supplementary Fig. 3A) and from the fractions, in which ∼95% of total cholesterol was removed by MCD (post-MCD samples). In intact synaptosomal fractions, more cholesterol was accessible to PFO-D4 in EFV-treated 5XFAD mice of both sexes than control animals, whereas less cholesterol was accessible to PFO-D4 in Cyp46a1−/− than Cyp46a1+/+ mice (Supplementary Fig. 3B). In contrast, in post-MCD samples, the pattern of changes was the opposite, probably due to increased (EFV-treated versus control 5XFAD mice) and decreased (Cyp46a1−/− versus Cyp46a1+/+ mice) amounts of cholesterol available for extraction with MCD (Supplementary Fig. 3C). Hence, when corrected for the fluorescence due to non-specific PFO-D4 binding to cholesterol-depleted synaptosomal fractions, the PFO-D4-GFP-GST fluorescence remained higher in EFV-treated versus control 5XFAD mice (by 31% and 39% in males and females, respectively) and lower in Cyp46a1−/− versus Cyp46a1+/+ mice (by 51% and 19% in males and females, respectively) (Fig. 2A). Thus, increased sterol flux in EFV-treated 5XFAD mice seemed to increase cholesterol accessibility in the synaptosomal membranes, whereas the lack of CYP46A1 or decreased sterol flux had an opposite effect.
Membrane ordering
Di-4-ANEPPDHQ is a dye, which has emission maxima at 500–580 nm and 620–750 nm when it binds to the ordered and disordered membrane phases, respectively (Ashdown and Owen, 2015). Interaction of di-4-ANEPPDHQ with samples from control 5XFAD mice of both sexes and female Cyp46a1−/− mice produced a typical two-peak emission spectra, whereas those from EFV-treated 5XFAD mice and Cyp46a1+/+ mice of both sexes generated only one-peak emission spectra with a maximum at ∼600 nm (Fig. 2B). These spectra were then used to calculate the di-4-ANEPPDHQ GP, a quantitative indicator of membrane ordering. In both sexes, GP was higher upon di-4-ANEPPDHQ binding to synaptosomal fractions from EFV-treated than control 5XFAD mice and lower in the samples from Cyp46a1−/− than Cyp46a1+/+ mice, thus suggesting increased (in EFV-treated 5XFAD mice) and decreased (in Cyp46a1−/− mice) membrane ordering. To confirm this result, F2N12S, the second lipid-sensitive dye was used, whose short- (485 nm) and long-wavelength (575 nm) emission maxima correspond to the probe binding to the membrane disordered and ordered phases, respectively (Shynkar et al., 2007). The 485 nm band is also sensitive to the phospholipid distribution between the outer and inner membrane leaflets (Shynkar et al., 2007). In both sexes, EFV treatment decreased the intensity of the 485 nm band and the ratio of intensities of the 485 nm to 575 nm bands (Fig. 2C). Conversely, Cyp46a1 ablation increased the 485 nm band intensity and the 485 nm to 575 nm band ratio (Fig. 2C). Thus, changes in the emission spectra of both dyes were consistent and suggested an increase in membrane ordering in animals with increased sterol flux (EVF-treated 5XFAD mice) and a decrease in membrane ordering in animals with decreased sterol flux (Cyp46a1−/− mice).
Osmotic resistance
Membrane resistance to osmotic changes (a reflection of water permeability and ability to swell or shrink) is known to depend on the cholesterol content (Yang et al., 2016) and was estimated by light scattering (Keen and White, 1970), which is increased and decreased when biological particles shrink and swell, respectively (Koch, 1961). In the absence of NaCl, all synaptosomal preparations seemed to scatter the light similarly (Fig. 3), probably because they were swollen to the same extent. Yet, increasing osmotic pressure due to increased NaCl concentrations always led to a higher light scattering of the samples from control 5XFAD and Cyp46a1−/− mice of both sexes than the samples from EFV-treated and Cyp46a1+/+ animals of both sexes, respectively. These results suggested that EFV treatment (or increased sterol flux) increased synaptosomal fraction resistance to osmotic stress, whereas the lack of CYP46A1 (or decreased sterol flux) made the membranes more susceptible to the osmotic stress.
Figure 3.

Resistance of synaptosomal fractions to osmotic stress. Optical density (OD) at 520 nm of synaptosomal fractions resuspended in Locke’s solution containing different NaCl concentrations. All results are the mean ± SD of the measurements in individual animals (n = 3–7 mice per group and sex). Statistical analysis: a two-way ANOVA followed by Bonferroni post hoc comparisons. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. Cntr, control or vehicle-treated 5XFAD mice (black open circles and black lines); Tx, EFV-treated 5XFAD mice (green open circles and green lines); Cyp46a1+/+ mice (black open circles and black lines) and Cyp46a1−/− (magenta open circles and magenta lines) on the mixed (C57BL/6J; 129S6/SvEv) background.
Membrane thickness
This was evaluated by transmission electron microscopy (TEM) and focused on the measurements of the pre- and postsynaptic membranes. Only male mice were evaluated because the directionality of changes observed in Figs 1–3 was the same in both sexes either upon EFV treatment or Cyp46a1 ablation. The thickness of both pre- and postsynaptic membranes was always higher (by 16% and 19%, respectively) in EFV-treated versus control 5XFAD mice (Fig. 4A and B). In addition, electron density adjacent to the postsynaptic membranes (the so-called postsynaptic density) was more pronounced in EFV-treated relative to control 5XFAD mice. Conversely, the pre- and postsynaptic membrane thickness as well as postsynaptic density were lower in Cyp46a1−/− versus Cyp46a1+/+ mice (by 11% and 13%, respectively, Fig. 4C and D). Thus, pharmacological CYP46A1 activation and subsequent increase in sterol flux appeared to increase both membrane thickness at the synapses and postsynaptic density, while Cyp46a1 ablation exerted the reverse effects.
Figure 4.

TEM of synaptosomal fractions. (A and C) Representative electron micrographs of the synaptic regions (n = 3 male mice per group). Coloured rectangles in the panels on the left and right indicate the regions that are shown at higher magnifications in the middle panels. Orange font and arrows indicate the pre- and postsynaptic parts, postsynaptic density (PSD) and synaptic cleft; yellow asterisks indicate some of the synaptic vesicles; and yellow lines show membrane thickness. (B and D) The quantification of the pre- and postsynaptic membrane thickness. All results are the mean ± SD of the measurements in individual animals (n = 3 male mice per group), 8–22 synaptosomes/mouse and 5–10 measurements/membrane. Statistical analysis: a two-tailed, unpaired Student’s t-test. *P ≤ 0.05. Cntr, control or vehicle-treated 5XFAD mice; Tx, EFV-treated 5XFAD mice; Cyp46a1+/+ and Cyp46a1−/−, wild-type and knockout mice on the C57BL/6J; 129S6/SvEv background.
Sucrose- and KCl-releasable as well as total glutamate pools
Glutamate release occurs under stimulation of synaptic vesicle exocytosis, which can be evoked by sucrose or KCl (Nicholls and Sihra, 1986; Rosenmund and Stevens, 1996). The sucrose-evoked (≤500 mM) exocytosis usually reflects the neurotransmitter release from a small population of synaptic vesicles constituting the readily releasable pool; the size of this pool determines the efficiency of synaptic transmission (Rosenmund and Stevens, 1996; Darios et al., 2009; Kaeser and Regehr, 2017). Conversely, KCl (>50 mM) stimulates robust synaptic vesicle exocytosis from synaptic vesicles constituting recycling and reserve pools, which can contribute to excitotoxicity (Nicholls and Sihra, 1986; Budd et al., 1996; Sodero et al., 2012). Herein, glutamate release was assessed under the conditions of mild (35 mM sucrose) and stronger (117 mM KCl) stimulation of synaptic vesicle exocytosis (Valencia et al., 2013), and the total glutamate content was measured as well. Experiments also included the characterization of synaptosomal fractions from the brain of male B6SJL mice, a background strain for the male 5XFAD mice characterized by TEM (Fig. 4). In control 5XFAD versus B6SJL male mice, the glutamate release evoked by sucrose was decreased (Fig. 5A and D), while the KCl-induced glutamate release was increased (Fig. 5B and D). Remarkably, EFV treatment made the glutamate release in 5XFAD male mice similar to that in B6SJL male mice under both stimulation conditions, i.e. normalized the glutamate release. Similarly, the glutamate release was increased and decreased in female EFV-treated versus control 5XFAD mice under the mild and stronger exocytosis stimulation, respectively (Fig. 5A, B and D). The opposite changes, a decrease and increase in the glutamate release under the sucrose- and KCl-induced stimulation, respectively, were observed in Cyp46a1−/− versus Cyp46a1+/+ mice of both sexes (Fig. 5A, B and D). The quantification of the total glutamate pool in synaptosomal fractions (Fig. 5C and D) revealed that this glutamate pool is: decreased in control 5XFAD versus B6SJL male mice but is increased in EFV-treated versus control 5XFAD mice of both sexes; and is decreased in Cyp46a1−/− versus Cyp46a1+/+ mice. Thus, EFV treatment counteracted the changes in the sucrose-, KCl-releasable and total glutamate pools in control 5XFAD versus B6SJL mice and exerted the effects opposite to those observed in Cyp46a1−/− versus Cyp46a1+/+ mice.
Figure 5.

Glutamate (Glu) release and total Glu pools in synaptosomal fractions. (A and B) Glu release evoked by 35 mM sucrose and 117 mM KCl, respectively. (C) The total Glu pools. (D) A pie chart representation of the sucrose and KCl-releasable Glu pools (blue and yellow sectors, respectively) relative to the total Glu pool. The size of the circles reflects the size of the total Glu pools. The percentages of the sucrose- and KCl-releasable Glu pools relative to the total Glu pool are also shown. All results are the mean ± SD of the measurements in individual animals (n = 3–7 mice per group and gender). Statistical analysis: a two-way ANOVA followed by Bonferroni post hoc comparisons. ***P ≤ 0.001 of control (Cntr, or vehicle-treated 5XFAD mice) versus EFV-treated 5XFAD (Tx) mice on the B6SJL background or Cyp46a1+/+ and Cyp46a1−/− (wild-type and knockout mice on the C57BL/6J; 129S6/SvEv background, respectively). B6SJL, mice on the B6SJL background.
Effects of protein kinase and protein phosphatase inhibition on KCl-induced glutamate release
Previously, we found that EFV treatment of 5XFAD mice and Cyp46a1 ablation affect protein phosphorylation and could modulate the activity of the same protein kinases (e.g. CDK5 and GSK3) (Mast et al., 2017a; Petrov et al., 2019b). Consequently, the activity of PP1/2A and PP2B, the most abundant brain phosphatases, which dephosphorylate >90% of neuronal phosphoproteins (Mansuy and Shenolikar, 2006), could be affected as well. Hence, we investigated the effect of the CDK5, GSK3, PP1/2A and PP2B inhibition on the KCI-induced glutamate release as the amount of the neurotransmitter released in this protocol was higher than in that utilizing sucrose. In both EVF-treated 5XFAD mice and Cyp46a1−/− mice, the inhibition of GSK3, CDK5 and PP2B did not alter the glutamate release, and only the inhibition of PP1/2A had an effect, namely led to a decrease in the neurotransmitter release (Figs 6 and 7). This result suggests that only the activity of PP1/2A seems to contribute to the glutamate release in these mouse models. Yet, when EFV-treated and Cyp46a1−/− mice were compared to control 5XFAD and Cyp46a1+/+ mice, respectively, their inhibition patterns were different. Specifically, in EFV-treated versus control 5XFAD mice, EFV treatment modulated the activity of GSK3, CDK5 as well as PP2B and engaged PP1/2A in the regulation of the glutamate release. In Cyp46a1−/− versus Cyp46a1+/+ mice, lack of CYP46A1 also affected the activity of CDK5 and PP2B but had no effect on the contribution of GSK3 and PP1/2A to the glutamate release. Thus, both CYP46A1 activation with EFV and Cyp46a1 ablation altered the activity of protein kinase CDK5 and protein phosphatase PP2B and had the model-specific effects on the engagement of the protein kinase GSK3 and protein phosphatase PP1/2A. These model-specific effects were probably due to different inhibition patterns of control 5XFAD mice and Cyp46a1+/+ mice. In B6SJL mice and Cyp46a1+/+ mice on the C57BL/6J; 129S6/SvEv background, the glutamate release was similarly affected by the inhibition of CDK5, PP1/2A and PP2B but not GSK3. Yet, in control 5XFAD mice, the activity of not only CDK5 and PP2B but also of GSK3 contributed to the glutamate release, and PP1/2A was not involved. Thus, a different inhibition pattern of EFV-treated versus control 5XFAD mice and Cyp46a1−/− versus Cyp46a1+/+ mice is likely do due to the differences in the basal activity of the enzymes in the AD model versus wild-type mice.
Figure 6.

Effects of protein kinase and protein phosphatase inhibition on KCl-induced glutamate (Glu) release. Traces of the KCl-induced Glu release after synaptosomal fractions were incubated with or no DMSO (black and grey traces, respectively), R-roscovitine (magenta traces), butyrolactone I (orange traces), SB 216763 (green traces), okadaic acid (ink traces) and cyclosporin A (blue traces) or co-inhibited with R-roscovitive plus okadaic acid (olive traces) and R-roscovitive plus cyclosporin A (teal traces). All traces are the mean ± SD of the measurements in individual animals (n = 4–8 mice per group and gender). Statistical analysis: a two-way ANOVA followed by Bonferroni post hoc comparisons. *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001. B6SJL, mice on the B6SJL background; Cntr, control or vehicle-treated 5XFAD mice, and Tx, EFV-treated 5XFAD mice on the B6SJL background; Cyp46a1+/+ and Cyp46a1−/−, wild-type and knockout mice on the C57BL/6J; 129S6/SvEv background.
Figure 7.

Schematic summary of the data in Fig. 6. The number in the middle of each circle represents the KCl-releasable Glu pool (nmol/mg). Blunt-ends denote enzyme inhibition; upwards (↑), downwards (↓) and left-right (↔) arrows indicate increases, decreases, and no change in Glu release, respectively, after enzyme inhibition. Fold-changes in Glu release (inhibitor versus vehicle) are shown near the arrows; the colour code for the inhibitors is the same as in Fig. 6.
Discussion
Herein, we characterized synaptosomal fractions isolated from the brain of mice with the long-term pharmacological activation and genetic ablation of CYP46A1. This P450 enzyme is the CNS-specific cholesterol 24-hydroxylase, whose activity modulation affects multiple biological processes and could be beneficial for treatment of different brain disorders (Petrov and Pikuleva, 2019). First, we confirmed that sterol flux is indeed increased and decreased, respectively, through the membranes of the evaluated mouse models relative to their corresponding controls (Fig. 1). Then we established that PFO-D4, a specific probe for accessible cholesterol (Ohno-Iwashita et al., 2010), detected more cholesterol in the membranes of EFV-treated than control 5XFAD mice and less cholesterol in the samples from the Cyp46a1−/− than Cyp46a1+/+ mice (Fig. 2A). Since PFO-D4 mainly interacts with cholesterol in lipid rafts (Olsen et al., 2013) and detergent-resistant membranes from the ordered membrane microdomains (Waheed et al., 2001; Shimada et al., 2002; Ohno-Iwashita et al., 2004; Nelson et al., 2010), we next used di-4-ANEPPDHQ and F2N12S, the two dyes sensitive to lipid environment (Shynkar et al., 2007; Ashdown and Owen, 2015). Binding of both dyes to the synaptosomal membranes was consistent and indicated increased ordering in the membranes with increased sterol flux (from EFV-treated 5XFAD mice) and decreased membrane ordering in the membranes with decreased sterol flux (Cyp46a1−/−) (Fig. 2B and C).
Membrane ordering alters membrane rigidity and compressibility (Dufourc, 2008; Barriga et al., 2016) and can consequently affect membrane resistance to osmotic stress (Foretz et al., 2011). This property was investigated as well by measuring the changes in the membrane light scattering as a function of increased osmolarity (Fig. 3). CYP46A1 activation or increased sterol flux increased osmotic resistance of synaptosomal fractions, while the Cyp46a1 knockout (or decreased sterol flux) made synaptosomal fractions more susceptible to changes in external osmotic pressure.
Changes in lipid ordering could alter the membrane thickness (Lingwood and Simons, 2010; Saita et al., 2016). Hence, we used TEM and estimated the thickness of the pre- and postsynaptic membranes, which was increased and decreased as a result of CYP46A1 activation and absence, respectively (Fig. 4). Notably, the TEM analysis suggested that postsynaptic density becomes more prominent upon CYP46A1 activation and less prominent when the enzyme is lacking (Fig. 4A and C). This result is consistent with a positive correlation between membrane ordering and the formation of postsynaptic density (Tulodziecka et al., 2016) as well as increased expression of PSD-95, a main postsynaptic density component (Cheng et al., 2006). Also, previously, we and others showed that the PSD-95 expression is increased in EFV-treated versus control 5XFAD and transgenic mice overexpressing CYP46A1 (Maioli et al., 2013; Petrov et al., 2019a, b). Thus, by using different approaches, we documented that there are changes in cholesterol availability, ordering, thickness, osmotic resistance and postsynaptic density in the membranes from EFV-treated versus control 5XFAD mice and Cyp46a1−/− versus Cyp46a1+/+ mice. These changes were always in the opposite direction in EFV-treated and Cyp46a1−/− mice, i.e. correlated with the rate of sterol flux.
Membrane-dependent processes include synaptic transmission, which involves the exocytotic neurotransmitter release from synaptic vesicles (Jahn and Sudhof, 1994). Therefore, synaptosomal fractions were assessed for release of glutamate, the major excitatory neurotransmitter in the CNS. The glutamate release was induced either by sucrose or KCl to monitor the sterol flux effects on the ready releasable and other (recycling and reserve) neurotransmitter pools, respectively. In both animal models of sterol flux, the glutamate release was affected when evoked either by sucrose or KCl (Fig. 5A and B) with the direction of changes being always the opposite for the increased and decreased sterol fluxes. This result is consistent with the available data suggesting that perturbing the membrane properties (thickness, availability of cholesterol and cholesterol-dependent formation of membrane nanodomains) could directly contribute to several processes. These processes include exocytotic neurotransmitter release (Puchkov and Haucke, 2013; Lauwers et al., 2016); synaptic vesicle fusion (Zhu and Stevens, 2008); vesicular docking and trafficking (Zhang et al., 2009). Moreover, an increase in the sucrose-induced glutamate release, which usually determines synaptic strength (Kaeser and Regehr, 2017), suggests an improvement in synaptic communication in EFV-treated 5XFAD mice and is consistent with improvements in behavioural tests in this model as compared to control 5XFAD mice (Mast et al., 2017b; Petrov et al., 2019a). Conversely, a decrease in the sucrose releasable glutamate pool in Cyp46a1−/− mice indicates an impairment in synaptic communication and is supported by significant behavioural deficiencies in this genotype (Kotti et al., 2006).
As for the KCl-inducible glutamate pool, a decrease in this pool was suggested to be protective against excitotoxicity and thereby a loss of presynaptic terminal and postsynaptic density (Waataja et al., 2008; Akanuma et al., 2013). A decrease in this pool could be beneficial in EFV-treated 5XFAD mice (Fig. 5C) as are increases in their postsynaptic density and total glutamate pool (Figs 4A and 5C). Consequently, an increase in the KCl-induced glutamate release in Cyp46a1−/− mice and decreases in their total glutamate pool and postsynaptic density could be detrimental for this genotype (Figs 4C and 5C). Thus, in both EFV-treated 5XFAD and Cyp46a1−/− mice, the directionality of the sterol flux effects was the opposite for sucrose- and KCl-induced glutamate release, possibly a reflection of the effect on different pools of synaptic vesicles releasing the glutamate. Indeed, in the ready releasable pool, synaptic vesicles are already docked to the presynaptic membranes, whereas in the recycling and reserve neurotransmitter pools, synaptic vesicles should still be delivered to the exocytotic sites.
Changes in membrane properties caused by an altered sterol flux can affect glutamate release indirectly via changes in protein phosphorylation. The latter could be a result of the altered targeting of the membrane-associated protein kinases and phosphatases to their protein substrates and vice versa as suggested by the altered phosphoproteome in EFV-treated versus control 5XFAD mice and Cyp46a1−/− versus Cyp46a1+/+ mice. Also, our data pointed to changes in the activity of the overlapping proteins kinases in mice with increased and decreased sterol fluxes (Mast et al., 2017a; Petrov et al., 2019b). Of these kinases, we chose to further study CDK5 and GSK3 as their activity depends on the membrane microdomains (Sui et al., 2006; Hernandez et al., 2009; Valencia et al., 2010; Nishikawa et al., 2016). CDK5 and GSK3 are also involved in regulation of synaptic vesicle exocytosis (Tomizawa et al., 2002; Chergui et al., 2004; Kim and Ryan, 2010; Zhu et al., 2010). In addition, we investigated PP1/2A and PP2B as protein phosphorylation reflects the activities of not only kinases but phosphatases as well. PP1/2A and PP2B participate in the control of synaptic vesicle cycling (Guatimosim et al., 2002; Kumashiro et al., 2005; Marra et al., 2012).
We found that both CDK5 and PP2B lost their ability to regulate the KCl-induced glutamate release in response to the changes in sterol flux, i.e. in EFV-treated 5XFAD mice and Cyp46a1−/− mice (Figs 6 and 7). We also established that the CDK5 inhibition had opposite effects on the glutamate release in control 5XFAD versus B6SJL mice (Figs 6 and 7), possibly a reflection of the aberrant CDK5 hyperactivity in the brain in Alzheimer's disease (Shukla et al., 2012; Liu et al., 2016), and hence control 5XFAD mice as a disease model. As for GSK3, we documented that this kinase is engaged in the regulation of the KCl-induced glutamate exocytosis in control 5XFAD but not EFV-treated 5XFAD and B6SJL mice (Figs 6 and 7). Conversely, PP1/2A were not engaged in control 5XFAD mice but contributed to the glutamate release in EFV-treated 5XFAD and B6SJL mice. These results likely mirror the GSK3 hyperactivation and PP2A hypoactivation shown to occur in Alzheimer’s disease (Hu et al., 2009; Javadpour et al., 2019). Accordingly, increased sterol flux in EFV-treated 5XFAD mice seems to reverse the effects of the putative CDK5 and GSK3 hyperactivation and PP2A hypoactivation on the KCl-induced glutamate release in control 5XFAD mice. In addition, the CDK5 activity was also shown to be aberrantly increased in response to many neurotoxic stimulus and ischaemia (Liu et al., 2016). Therefore, pharmacologic CYP46A1 activation by EFV could be a general therapeutic tool to rescue the pattern of aberrant changes in CDK5, GSK3, PP2A and PP2B in different pathologic conditions.
We propose the following model to unify all the data obtained (Fig. 8). EFV treatment activated CYP46A1 in 5XFAD mice and increased sterol flux in the brain, whereas Cyp46a1 ablation abolished CYP46A1 activity and decreased sterol flux in brain. As a result, the properties of the synaptosomal fraction membranes were altered, and the observed changes correlated with an increase or decrease in sterol flux. The altered membrane properties directly or indirectly (via the alterations in protein phosphoproteome) affected the synaptic function, in particular the neurotransmitter release, and ultimately led to behavioural improvements in EFV-treated 5XFAD mice and behavioural impairments in Cyp46a1−/− mice.
Figure 8.

Proposed model unifying the effects of increased (in green) and decreased (in magenta) sterol fluxes. Biological processes are in bold; the properties of synaptosomal fractions are in regular font; arrowheads indicate the links between biological processes; upwards (↑), downwards (↓) and left-right (↔) arrows indicate increase, decrease and no change. Cntr, control or vehicle-treated 5XFAD mice; Tx, EFV-treated 5XFAD mice; Cyp46a1+/+ and Cyp46a1−/−, wild-type and knockout mice, respectively. See Discussion section for explanation.
In summary, by using different mouse models and experimental approaches, we obtained evidence in support of the CYP46A1-mediated sterol flux as a general mechanism in the brain, which affects the fundamental properties of the membranes as well as synaptic vesicle exocytosis and activity of some of the lipid raft-associated protein kinases and phosphatases. Changes in this sterol flux allow to explain the multiple effects of the CYP46A1 activity modulation in the brain and the enzyme’s therapeutic potential for the treatment of different disorders of the brain. Thus, CYP46A1 and cholesterol metabolism seem to integrate different important processes in the brain and hence play roles far beyond those in cholesterol elimination.
Supplementary material
Supplementary material is available at Brain Communications online.
Supplementary Material
Acknowledgements
The authors thank the Case Western Reserve University Visual Sciences Research Center Core Facilities [supported by the U.S. National Institutes of Health Grant P30 EY11373] for assistance with mouse breeding (Heather Butler and Kathryn Franke). The authors are also grateful to Dr. Hisashi Fujioka (Electron Microscopy Core facility) for his help with studies of synaptosomal ultrastructure, and Dr. Paul Park for providing access to the spectrofluorimeter.
Funding
This work was supported in part in by the U.S. National Institute of Health Grant AG067552 (to I.A.P.).
Competing interests
The authors report no competing interests.
Glossary
- 24HC =
24-hydroxycholesterol
- AD =
Alzheimer’s disease
- Aβ =
amyloid β peptide
- CDK5 =
cyclin-dependent kinase 5
- CYP46A1 =
cytochrome P450 46A1
- EFV =
efavirenz
- GP =
general polarization
- GSK3 =
glycogen synthase kinase 3
- MCD =
methyl-β-cyclodextrin
- PFO-D4-GFP-GST =
domain D4 of perfringolysin O labelled with superfolder green fluorescent protein and glutathione S-transferase
- PP1/2A =
protein phosphatase 1/2A
- PP2B =
protein phosphatase 2B or calcineurin
- PSD-95 =
postsynaptic density-95
- TEM =
transmission electron microscopy
References
- Akanuma S-I, Hirose S, Tachikawa M, Hosoya K-I.. Localization of organic anion transporting polypeptide (Oatp) 1a4 and Oatp1c1 at the rat blood-retinal barrier. Fluids Barriers CNS 2013; 10: 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashdown GW, Owen DM.. Imaging membrane order using environmentally sensitive fluorophores In: Owen DM, editor. Methods in membrane lipids. New York, NY: Springer New York; 2015. p. 115–22. [DOI] [PubMed] [Google Scholar]
- Bai F, Witzmann FA.. Synaptosome proteomics. Subcell Biochem 2007; 43: 77–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barriga HMG, Law RV, Seddon JM, Ces O, Brooks NJ.. The effect of hydrostatic pressure on model membrane domain composition and lateral compressibility. Phys Chem Chem Phys 2016; 18: 149–55. [DOI] [PubMed] [Google Scholar]
- Bauerfeind R, Takei K, De Camilli P.. Amphiphysin I is associated with coated endocytic intermediates and undergoes stimulation-dependent dephosphorylation in nerve terminals. J Biol Chem 1997; 272: 30984–92. [DOI] [PubMed] [Google Scholar]
- Bjorkhem I, Lutjohann D, Diczfalusy U, Stahle L, Ahlborg G, Wahren J.. Cholesterol homeostasis in human brain: turnover of 24S-hydroxycholesterol and evidence for a cerebral origin of most of this oxysterol in the circulation. J Lipid Res 1998; 39: 1594–600. [PubMed] [Google Scholar]
- Boussicault L, Alves S, Lamazière A, Planques A, Heck N, Moumné L, et al. CYP46A1, the rate-limiting enzyme for cholesterol degradation, is neuroprotective in Huntington’s disease. Brain 2016; 139: 953–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Budd DC, May GR, Nicholls DG, McCormack JG.. Inhibition by lifarizine of intracellular Ca2+ rises and glutamate exocytosis in depolarized rat cerebrocortical synaptosomes and cultured neurones. Br J Pharmacol 1996; 118: 162–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burlot M-A, Braudeau J, Michaelsen-Preusse K, Potier B, Ayciriex S, Varin J, et al. Cholesterol 24-hydroxylase defect is implicated in memory impairments associated with Alzheimer-like Tau pathology. Hum Mol Genet 2015; 24: 5965–76. [DOI] [PubMed] [Google Scholar]
- Chali F, Djelti F, Eugene E, Valderrama M, Marquer C, Aubourg P, et al. Inhibiting cholesterol degradation induces neuronal sclerosis and epileptic activity in mouse hippocampus. Eur J Neurosci 2015; 41: 1345–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Gu Z, Liu W, Yan Z.. Glycogen synthase kinase 3 regulates N-methyl-D-aspartate receptor channel trafficking and function in cortical neurons. Mol Pharmacol 2007; 72: 40–51. [DOI] [PubMed] [Google Scholar]
- Cheng D, Hoogenraad CC, Rush J, Ramm E, Schlager MA, Duong DM, et al. Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol Cell Proteomics 2006; 5: 1158–70. [DOI] [PubMed] [Google Scholar]
- Chergui K, Svenningsson P, Greengard P.. Cyclin-dependent kinase 5 regulates dopaminergic and glutamatergic transmission in the striatum. Proc Natl Acad Sci USA 2004; 101: 2191–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crouzin N, Baranger K, Cavalier M, Marchalant Y, Cohen-Solal C, Roman FS, et al. Area-specific alterations of synaptic plasticity in the 5XFAD mouse model of Alzheimer’s disease: dissociation between somatosensory cortex and hippocampus. PLoS One 2013; 8: e74667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darios F, Wasser C, Shakirzyanova A, Giniatullin A, Goodman K, Munoz-Bravo JL, et al. Sphingosine facilitates SNARE complex assembly and activates synaptic vesicle exocytosis. Neuron 2009; 62: 683–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Djelti F, Braudeau J, Hudry E, Dhenain M, Varin J, Bieche I, et al. CYP46A1 inhibition, brain cholesterol accumulation and neurodegeneration pave the way for Alzheimer’s disease. Brain 2015; 138: 2383–98. [DOI] [PubMed] [Google Scholar]
- Dufourc EJ. Sterols and membrane dynamics. J Chem Biol 2008; 1: 63–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egawa J, Pearn ML, Lemkuil BP, Patel PM, Head BP.. Membrane lipid rafts and neurobiology: age-related changes in membrane lipids and loss of neuronal function. J Physiol 2016; 594: 4565–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foretz M, Hébrard S, Guihard S, Leclerc J, Cruzeiro MD, Hamard G, et al. The AMPKgamma1 subunit plays an essential role in erythrocyte membrane elasticity, and its genetic inactivation induces splenomegaly and anemia. FASEB J 2011; 25: 337–47. [DOI] [PubMed] [Google Scholar]
- Guatimosim C, Hull C, Von Gersdorff H, Prado MAM.. Okadaic acid disrupts synaptic vesicle trafficking in a ribbon-type synapse. J Neurochem 2002; 82: 1047–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyton JR, Klemp KF.. Ultrastructural discrimination of lipid droplets and vesicles in atherosclerosis: value of osmium-thiocarbohydrazide-osmium and tannic acid-paraphenylenediamine techniques. J Histochem Cytochem 1988; 36: 1319–28. [DOI] [PubMed] [Google Scholar]
- Han M, Wang S, Yang N, Wang X, Zhao W, Saed HS, et al. Therapeutic implications of altered cholesterol homeostasis mediated by loss of CYP46A1 in human glioblastoma. EMBO Mol Med 2020; 12: e10924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez P, Lee G, Sjoberg M, Maccioni RB.. Tau phosphorylation by cdk5 and Fyn in response to amyloid peptide Abeta (25-35): involvement of lipid rafts. J Alzheimers Dis 2009; 16: 149–56. [DOI] [PubMed] [Google Scholar]
- Hosaka M, Hammer RE, Südhof TC.. A phospho-switch controls the dynamic association of synapsins with synaptic vesicles. Neuron 1999; 24: 377–87. [DOI] [PubMed] [Google Scholar]
- Hu S, Begum AN, Jones MR, Oh MS, Beech WK, Beech BH, et al. GSK3 inhibitors show benefits in an Alzheimer’s disease (AD) model of neurodegeneration but adverse effects in control animals. Neurobiol Dis 2009; 33: 193–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudry E, Van Dam D, Kulik W, De Deyn PP, Stet FS, Ahouansou O, et al. Adeno-associated virus gene therapy with cholesterol 24-hydroxylase reduces the amyloid pathology before or after the onset of amyloid plaques in mouse models of Alzheimer’s disease. Mol Ther 2010; 18: 44–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jahn R, Sudhof TC.. Synaptic vesicles and exocytosis. Annu Rev Neurosci 1994; 17: 219–46. [DOI] [PubMed] [Google Scholar]
- Javadpour P, Dargahi L, Ahmadiani A, Ghasemi R.. To be or not to be: PP2A as a dual player in CNS functions, its role in neurodegeneration, and its interaction with brain insulin signaling. Cell Mol Life Sci 2019; 76: 2277–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javanainen M, Martinez-Seara H, Vattulainen I.. Nanoscale membrane domain formation driven by cholesterol. Sci Rep 2017; 7: 1143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BB, Breña M, Anguita J, Heuck AP.. Mechanistic insights into the cholesterol-dependent binding of perfringolysin O-based probes and cell membranes. Sci Rep 2017; 7: 13793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson BB, Moe PC, Wang D, Rossi K, Trigatti BL, Heuck AP.. Modifications in perfringolysin O domain 4 alter the cholesterol concentration threshold required for binding. Biochemistry 2012; 51: 3373–82. [DOI] [PubMed] [Google Scholar]
- Kacher R, Lamazière A, Heck N, Kappes V, Mounier C, Despres G, et al. CYP46A1 gene therapy deciphers the role of brain cholesterol metabolism in Huntington’s disease. Brain 2019; 142: 2432–50. [DOI] [PubMed] [Google Scholar]
- Kaeser PS, Regehr WG.. The readily releasable pool of synaptic vesicles. Curr Opin Neurobiol 2017; 43: 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannan KB, Barlos D, Hauser CJ.. Free cholesterol alters lipid raft structure and function regulating neutrophil Ca2+ entry and respiratory burst: correlations with calcium channel raft trafficking. J Immunol 2007; 178: 5253–61. [DOI] [PubMed] [Google Scholar]
- Keen P, White TD.. A light-scattering technique for the study of the permeability of rat brain synaptosomes in vitro. J Neurochem 1970; 17: 565–71. [DOI] [PubMed] [Google Scholar]
- Kim SH, Ryan TA.. CDK5 serves as a major control point in neurotransmitter release. Neuron 2010; 67: 797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura R, Ohno M.. Impairments in remote memory stabilization precede hippocampal synaptic and cognitive failures in 5XFAD Alzheimer mouse model. Neurobiol Dis 2009; 33: 229–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch AL. Some calculations on the turbidity of mitochondria and bacteria. Biochim Biophys Acta 1961; 51: 429–41. [DOI] [PubMed] [Google Scholar]
- Kotti T, Head DD, McKenna CE, Russell DW.. Biphasic requirement for geranylgeraniol in hippocampal long-term potentiation. Proc Natl Acad Sci USA 2008; 105: 11394–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotti TJ, Ramirez DMO, Pfeiffer BE, Huber KM, Russell DW.. Brain cholesterol turnover required for geranylgeraniol production and learning in mice. Proc Natl Acad Sci USA 2006; 103: 3869–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumashiro S, Lu Y-F, Tomizawa K, Matsushita M, Wei F-Y, Matsui H.. Regulation of synaptic vesicle recycling by calcineurin in different vesicle pools. Neurosci Res 2005; 51: 435–43. [DOI] [PubMed] [Google Scholar]
- Lauwers E, Goodchild R, Verstreken P.. Membrane lipids in presynaptic function and disease. Neuron 2016; 90: 11–25. [DOI] [PubMed] [Google Scholar]
- Lee SY, Wenk MR, Kim Y, Nairn AC, De Camilli P.. Regulation of synaptojanin 1 by cyclin-dependent kinase 5 at synapses. Proc Natl Acad Sci USA 2004; 101: 546–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lingwood D, Simons K.. Lipid rafts as a membrane-organizing principle. Science 2010; 327: 46–50. [DOI] [PubMed] [Google Scholar]
- Liu S-L, Wang C, Jiang T, Tan L, Xing A, Yu J-T.. The role of Cdk5 in Alzheimer’s disease. Mol Neurobiol 2016; 53: 4328–42. [DOI] [PubMed] [Google Scholar]
- Lund EG, Guileyardo JM, Russell DW.. cDNA cloning of cholesterol 24-hydroxylase, a mediator of cholesterol homeostasis in the brain. Proc Natl Acad Sci USA 1999; 96: 7238–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lund EG, Xie C, Kotti T, Turley SD, Dietschy JM, Russell DW.. Knockout of the cholesterol 24-hydroxylase gene in mice reveals a brain-specific mechanism of cholesterol turnover. J Biol Chem 2003; 278: 22980–8. [DOI] [PubMed] [Google Scholar]
- Lutjohann D, Breuer O, Ahlborg G, Nennesmo I, Siden A, Diczfalusy U, et al. Cholesterol homeostasis in human brain: evidence for an age-dependent flux of 24S-hydroxycholesterol from the brain into the circulation. Proc Natl Acad Sci USA 1996; 93: 9799–804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maioli S, Båvner A, Ali Z, Heverin M, Ismail M-A-M, Puerta E, et al. Is it possible to improve memory function by upregulation of the cholesterol 24S-hydroxylase (CYP46A1) in the brain? PLoS One 2013; 8: e68534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansuy IM, Shenolikar S.. Protein serine/threonine phosphatases in neuronal plasticity and disorders of learning and memory. Trends Neurosci 2006; 29: 679–86. [DOI] [PubMed] [Google Scholar]
- Marks B, McMahon HT.. Calcium triggers calcineurin-dependent synaptic vesicle recycling in mammalian nerve terminals. Curr Biol 1998; 8: 740–9. [DOI] [PubMed] [Google Scholar]
- Marra V, Burden JJ, Thorpe JR, Smith IT, Smith SL, Häusser M, et al. A preferentially segregated recycling vesicle pool of limited size supports neurotransmission in native central synapses. Neuron 2012; 76: 579–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Li Y, Linger M, Clark M, Wiseman J, Pikuleva IA.. Pharmacologic stimulation of cytochrome P450 46A1 and cerebral cholesterol turnover in mice. J Biol Chem 2014; 289: 3529–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Lin JB, Anderson KW, Bjorkhem I, Pikuleva IA.. Transcriptional and post-translational changes in the brain of mice deficient in cholesterol removal mediated by cytochrome P450 46A1 (CYP46A1). PLoS One 2017a; 12: e0187168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Reem R, Bederman I, Huang S, DiPatre PL, Bjorkhem I, et al. Cholestenoic acid is an important elimination product of cholesterol in the retina: comparison of retinal cholesterol metabolism with that in the brain. Invest Ophthalmol Vis Sci 2011; 52: 594–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mast N, Saadane A, Valencia-Olvera A, Constans J, Maxfield E, Arakawa H, et al. Cholesterol-metabolizing enzyme cytochrome P450 46A1 as a pharmacologic target for Alzheimer’s disease. Neuropharmacology 2017b; 123: 465–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meaney S, Bodin K, Diczfalusy U, Björkhem I.. On the rate of translocation in vitro and kinetics in vivo of the major oxysterols in human circulation: critical importance of the position of the oxygen function. J Lipid Res 2002; 43: 2130–5. [DOI] [PubMed] [Google Scholar]
- Miranda-Barrientos J, Nieto-Mendoza E, Hernandez-Echeagaray E.. The Cdk5 inhibitor Roscovitine increases LTP induction in corticostriatal synapses. ASN Neuro 2014; 6: AN20140006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitroi DN, Pereyra‐Gómez G, Soto‐Huelin B, Senovilla F, Kobayashi T, Esteban JA, et al. NPC1 enables cholesterol mobilization during long-term potentiation that can be restored in Niemann-Pick disease type C by CYP46A1 activation. EMBO Rep 2019; 20: e48143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson LD, Chiantia S, London E.. Perfringolysin O association with ordered lipid domains: implications for transmembrane protein raft affinity. Biophys J 2010; 99: 3255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls DG, Sihra TS.. Synaptosomes possess an exocytotic pool of glutamate. Nature 1986; 321: 772–3. [DOI] [PubMed] [Google Scholar]
- Nishikawa T, Takahashi T, Nakamori M, Hosomi N, Maruyama H, Miyazaki Y, et al. The identification of raft-derived tau-associated vesicles that are incorporated into immature tangles and paired helical filaments. Neuropathol Appl Neurobiol 2016; 42: 639–53. [DOI] [PubMed] [Google Scholar]
- Nóbrega C, Mendonça L, Marcelo A, Lamazière A, Tomé S, Despres G, et al. Restoring brain cholesterol turnover improves autophagy and has therapeutic potential in mouse models of spinocerebellar ataxia. Acta Neuropathol 2019; 138: 837–58. [DOI] [PubMed] [Google Scholar]
- Oakley H, Cole SL, Logan S, Maus E, Shao P, Craft J, et al. Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J Neurosci 2006; 26: 10129–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno-Iwashita Y, Shimada Y, Hayashi M, Iwamoto M, Iwashita S, Inomata M.. Cholesterol-binding toxins and anti-cholesterol antibodies as structural probes for cholesterol localization. Subcell Biochem 2010; 51: 597–621. [DOI] [PubMed] [Google Scholar]
- Ohno-Iwashita Y, Shimada Y, Waheed AA, Hayashi M, Inomata M, Nakamura M, et al. Perfringolysin O, a cholesterol-binding cytolysin, as a probe for lipid rafts. Anaerobe 2004; 10: 125–34. [DOI] [PubMed] [Google Scholar]
- Olsen BN, Bielska AA, Lee T, Daily MD, Covey DF, Schlesinger PH, et al. The structural basis of cholesterol accessibility in membranes. Biophys J 2013; 105: 1838–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel TK, Patel VB, Rana DG.. Possible anti-depressant effect of efavirenz and pro-depressive-like effect of voriconazole in specified doses in various experimental models of depression in mice. Pharmacol Rep 2017; 69: 1082–7. [DOI] [PubMed] [Google Scholar]
- Petrov AM, Lam M, Mast N, Moon J, Li Y, Maxfield E, et al. CYP46A1 activation by efavirenz leads to behavioral improvement without significant changes in amyloid plaque load in the brain of 5XFAD mice. Neurotherapeutics 2019a; 16: 710–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov AM, Mast N, Li Y, Pikuleva IA.. The key genes, phosphoproteins, processes, and pathways affected by efavirenz-activated CYP46A1 in the amyloid-decreasing paradigm of efavirenz treatment. FASEB J 2019b; 33: 8782–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrov AM, Pikuleva IA.. Cholesterol 24-hydroxylation by CYP46A1: benefits of modulation for brain diseases. Neurotherapeutics 2019; 16: 635–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pfrieger FW, Ungerer N.. Cholesterol metabolism in neurons and astrocytes. Prog Lipid Res 2011; 50: 357–71. [DOI] [PubMed] [Google Scholar]
- Pike LJ. Lipid rafts: bringing order to chaos. J Lipid Res 2003; 44: 655–67. [DOI] [PubMed] [Google Scholar]
- Puchkov D, Haucke V.. Greasing the synaptic vesicle cycle by membrane lipids. Trends Cell Biol 2013; 23: 493–503. [DOI] [PubMed] [Google Scholar]
- Ramirez DMO, Andersson S, Russell DW.. Neuronal expression and subcellular localization of cholesterol 24-hydroxylase in the mouse brain. J Comp Neurol 2008; 507: 1676–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenmund C, Stevens CF.. Definition of the readily releasable pool of vesicles at hippocampal synapses. Neuron 1996; 16: 1197–207. [DOI] [PubMed] [Google Scholar]
- Saita E, Albanesi D, de Mendoza D.. Sensing membrane thickness: lessons learned from cold stress. Biochim Biophys Acta 2016; 1861: 837–46. [DOI] [PubMed] [Google Scholar]
- Schrimpf SP, Meskenaite V, Brunner E, Rutishauser D, Walther P, Eng J, et al. Proteomic analysis of synaptosomes using isotope-coded affinity tags and mass spectrometry. Proteomics 2005; 5: 2531–41. [DOI] [PubMed] [Google Scholar]
- Sebastião AM, Colino-Oliveira M, Assaife-Lopes N, Dias RB, Ribeiro JA.. Lipid rafts, synaptic transmission and plasticity: impact in age-related neurodegenerative diseases. Neuropharmacology 2013; 64: 97–107. [DOI] [PubMed] [Google Scholar]
- Shimada Y, Maruya M, Iwashita S, Ohno-Iwashita Y.. The C-terminal domain of perfringolysin O is an essential cholesterol-binding unit targeting to cholesterol-rich microdomains. Eur J Biochem 2002; 269: 6195–203. [DOI] [PubMed] [Google Scholar]
- Shukla V, Skuntz S, Pant HC.. Deregulated Cdk5 activity is involved in inducing Alzheimer’s disease. Arch Med Res 2012; 43: 655–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shynkar VV, Klymchenko AS, Kunzelmann C, Duportail G, Muller CD, Demchenko AP, et al. Fluorescent biomembrane probe for ratiometric detection of apoptosis. J Am Chem Soc 2007; 129: 2187–93. [DOI] [PubMed] [Google Scholar]
- Simons K, Ehehalt R.. Cholesterol, lipid rafts, and disease. J Clin Invest 2002; 110: 597–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sodero AO, Vriens J, Ghosh D, Stegner D, Brachet A, Pallotto M, et al. Cholesterol loss during glutamate-mediated excitotoxicity. EMBO J 2012; 31: 1764–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sui Z, Kovács AD, Maggirwar SB.. Recruitment of active glycogen synthase kinase-3 into neuronal lipid rafts. Biochem Biophys Res Commun 2006; 345: 1643–8. [DOI] [PubMed] [Google Scholar]
- Suzuki S, Kiyosue K, Hazama S, Ogura A, Kashihara M, Hara T, et al. Brain-derived neurotrophic factor regulates cholesterol metabolism for synapse development. J Neurosci 2007; 27: 6417–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tomizawa K, Ohta J, Matsushita M, Moriwaki A, Li S-T, Takei K, et al. Cdk5/p35 regulates neurotransmitter release through phosphorylation and downregulation of P/Q-type voltage-dependent calcium channel activity. J Neurosci 2002; 22: 2590–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulodziecka K, Diaz-Rohrer BB, Farley MM, Chan RB, Di Paolo G, Levental KR, et al. Remodeling of the postsynaptic plasma membrane during neural development. Mol Biol Cell 2016; 27: 3480–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valencia A, Reeves PB, Sapp E, Li X, Alexander J, Kegel KB, et al. Mutant huntingtin and glycogen synthase kinase 3-beta accumulate in neuronal lipid rafts of a presymptomatic knock-in mouse model of Huntington’s disease. J Neurosci Res 2010; 88: 179–90. [DOI] [PubMed] [Google Scholar]
- Valencia A, Sapp E, Kimm JS, McClory H, Ansong KA, Yohrling G, et al. Striatal synaptosomes from Hdh140Q/140Q knock-in mice have altered protein levels, novel sites of methionine oxidation, and excess glutamate release after stimulation. J Huntingtons Dis 2013; 2: 459–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Kant R, Langness VF, Herrera CM, Williams DA, Fong LK, Leestemaker Y, et al. Cholesterol metabolism is a druggable axis that independently regulates tau and amyloid-beta in iPSC-derived Alzheimer’s disease neurons. Cell Stem Cell 2019; 24: 363–75.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waataja JJ, Kim HJ, Roloff AM, Thayer SA.. Excitotoxic loss of post-synaptic sites is distinct temporally and mechanistically from neuronal death. J Neurochem 2008; 104: 364–75. [DOI] [PubMed] [Google Scholar]
- Waheed AA, Shimada Y, Heijnen HFG, Nakamura M, Inomata M, Hayashi M, et al. Selective binding of perfringolysin O derivative to cholesterol-rich membrane microdomains (rafts). Proc Natl Acad Sci USA 2001; 98: 4926–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Liu W, Yan Z.. Regulation of AMPA receptor trafficking and function by glycogen synthase kinase 3. J Biol Chem 2010; 285: 26369–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue J, Graham ME, Novelle AE, Sue N, Gray N, McNiven MA, et al. Calcineurin selectively docks with the dynamin Ixb splice variant to regulate activity-dependent bulk endocytosis. J Biol Chem 2011; 286: 30295–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang S-T, Kreutzberger AJB, Lee J, Kiessling V, Tamm LK.. The role of cholesterol in membrane fusion. Chem Phys Lipids 2016; 199: 136–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Xue R, Ong W-Y, Chen P.. Roles of cholesterol in vesicle fusion and motion. Biophys J 2009; 97: 1371–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu L-Q, Liu D, Hu J, Cheng J, Wang S-H, Wang Q, et al. GSK-3 beta inhibits presynaptic vesicle exocytosis by phosphorylating P/Q-type calcium channel and interrupting SNARE complex formation. J Neurosci 2010; 30: 3624–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Stevens CF.. Probing synaptic vesicle fusion by altering mechanical properties of the neuronal surface membrane. Proc Natl Acad Sci USA 2008; 105: 18018–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zidovetzki R, Levitan I.. Use of cyclodextrins to manipulate plasma membrane cholesterol content: evidence, misconceptions and control strategies. Biochim Biophys Acta 2007; 1768: 1311–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available upon request.