

Abstract
Recent technological advancements have enabled the profiling of a large number of genome-wide features in individual cells. However, single-cell data present unique challenges that require the development of specialized methods and software infrastructure to successfully derive biological insights. The Bioconductor project has rapidly grown to meet these demands, hosting community-developed open-source software distributed as R packages. Featuring state-of-the-art computational methods, standardized data infrastructure, and interactive data visualization tools, we present an overview and online book (https://osca.bioconductor.org) of single-cell methods for prospective users.
Editorial summary:
This Perspective highlights open-source software for single-cell analysis released as part of the Bioconductor project, providing an overview for users and developers.
Introduction
Since 2001, the Bioconductor project [1] has attracted a rich community of developers and users from diverse scientific fields, driving the development of open-source software packages using the R language for the analysis of high-throughput biological data [2–6]. While bulk profiling technologies have yielded important scientific insights and methods [7–9], recent advancements in technologies to profile samples at single-cell resolution have emerged that can answer previously inaccessible scientific questions [10–20]. Bioconductor has been home to a wide range of software packages used in analyzing bulk profiling data, and more recently it has expanded significantly into the realm of single-cell data analysis with a rapidly growing list of community contributed software packages (Figure 1).
Figure 1: Number of Bioconductor packages for the analysis of high-throughput sequencing data over ten years.
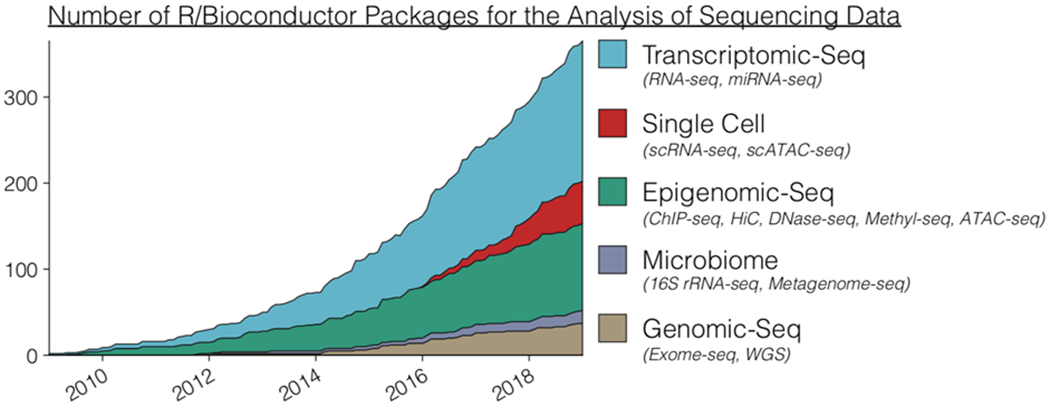
Bioconductor software packages associated with the analysis of sequencing data were tracked by date of submission over the course of ten years. Software packages were uniquely defined by their primary sequencing technology association, with examples of specific terms used for annotation in parentheses.
Current single-cell assays can be both high-throughput, measuring thousands to millions of cells, and high dimensional, measuring thousands of features within each individual cell. Compared to bulk assays, there are two defining characteristics of single-cell data that must be specially handled to achieve biological insight: (i) the increased scale of the number of observations (i.e., cells) that are assayed in large compendiums such as those from the Human Cell Atlas [21, 22] and the Mouse Cell Atlas [23], and (ii) the increased sparsity of the data due to biological fluctuations in the measured traits or limited sensitivity for quantifying small numbers of molecules [13, 24–26]. These unique characteristics have motivated the development of statistical methods tailored for single-cell data analysis [27–30]. Furthermore, as single-cell technologies mature, the increasing complexity and volume of data require fundamental changes in data access, management, and infrastructure alongside specialized methods to facilitate scalable analyses.
To address these challenges, software packages developed for the analysis of single-cell data have become an integral part of the Bioconductor project. Herein we primarily focus on the analysis of single-cell RNA-seq (scRNA-seq) data, much of the concepts mentioned herein are also generalizable to other types of single-cell assays. We cover (1) data import, (2) common data containers for storing single-cell assay data (3) fast and robust methods for transforming raw single-cell data into processed data suitable for downstream analyses, (4) interactive data visualization, and (5) downstream analyses. To help users leverage this robust and scalable framework, we describe selected packages and present an online book (https://osca.bioconductor.org) covering installation, sources of help, specialized topics pertaining to specific aspects of scRNA-seq analysis, and complete workflows analyzing various scRNA-seq datasets. The references for all packages are available at http://bioconductor.org/packages/.
Data Infrastructure
One of Bioconductor’s strongest advantages is the availability of common representations and infrastructure for complex, highly interdependent data sets [1]. Bioconductor uses standardized data containers to enable modularity and interoperability of diverse packages while maintaining robust end-user accessibility. To this end, Bioconductor employs a flexible object-oriented paradigm called S4 [31] that enables encapsulation of multiple object components into a single instance with a rich and user-friendly interface. Such an approach is especially important for biological analysis, as there are often many links between primary data and metadata that need to be preserved throughout an analysis.
The SingleCellExperiment container
Bioconductor uses the SingleCellExperiment class for storing single-cell assay data and metadata (Figure 2). Primary data, such as count matrices, are stored in the assays component as one or more matrices, where rows represent features (e.g. genes, transcripts) and columns represent cells. In addition, low-dimensional representations of the primary data, and metadata describing cell or feature characteristics can also be stored in the SingleCellExperiment object. Through the SingleCellExperiment class, all pertinent data and results relevant to a scRNA-seq experiment can be stored in a single instance. By standardizing the storage of single cell data and results, Bioconductor fosters interoperability between single-cell analysis packages and facilitates the development and usage of complex analysis workflows.
Figure 2: Overview of the SingleCellExperiment class.
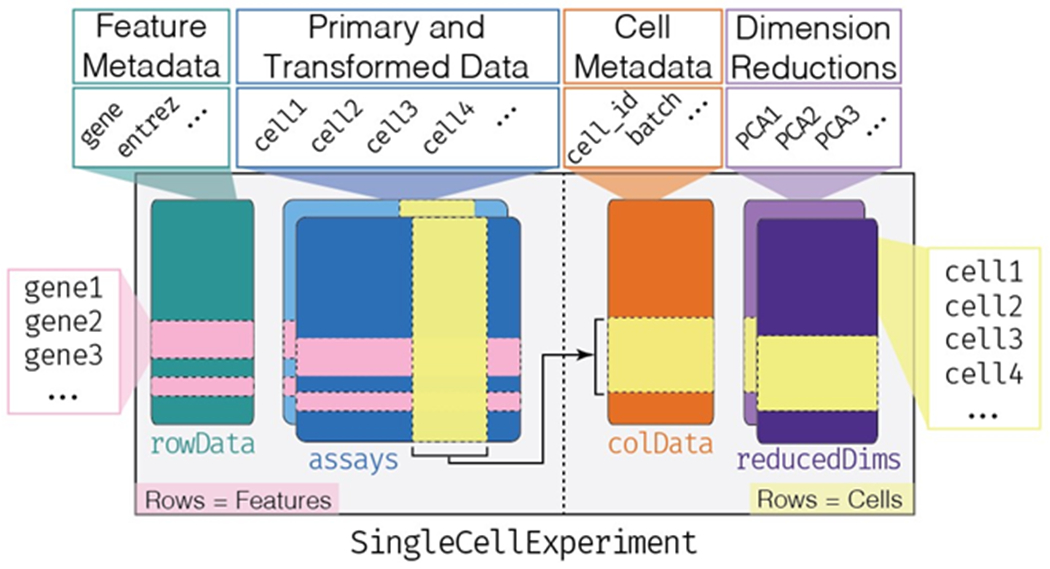
The SingleCellExperiment class instantiates an object (SingleCellExperiment herein abbreviated sce) capable of storing various datatypes associated with single-cell assays. A sce object is organized into components (e.g. rowData, assays, colData, reducedDims). In the assays component the rows represent features such as genes (horizontal pink bands), and the columns represent cells (vertical yellow band). The rowData and colData components can hold information (such as metadata) about the features and cells, respectively. Note that in the colData and reducedDims components, cells are represented as rows (horizontal yellow bands) and the number of columns in the assays component must match the number of rows in the colData and reducedDims components.
Data Processing
The aim of this section is to describe the precursor steps that are common to most scRNA-seq analyses. These preliminary steps follow a general workflow (Figure 3): (1) preprocessing raw sequencing data to produce a per-gene (or transcript) per-cell expression count matrix, followed by creating a SingleCellExperiment object, (2) applying quality control metrics and subsequent removal of low quality cells that would otherwise interfere with downstream analyses, (3) converting counts into normalized expression values to eliminate cell and gene-specific biases, (4) performing feature selection to pick a subset of biologically relevant genes for downstream analyses, (5) applying dimensionality reduction methods to compact the data and reduce noise, and (6) if applicable, integrating multiples batches of scRNA-seq data.
Figure 3: Bioconductor workflow for analyzing single-cell data.
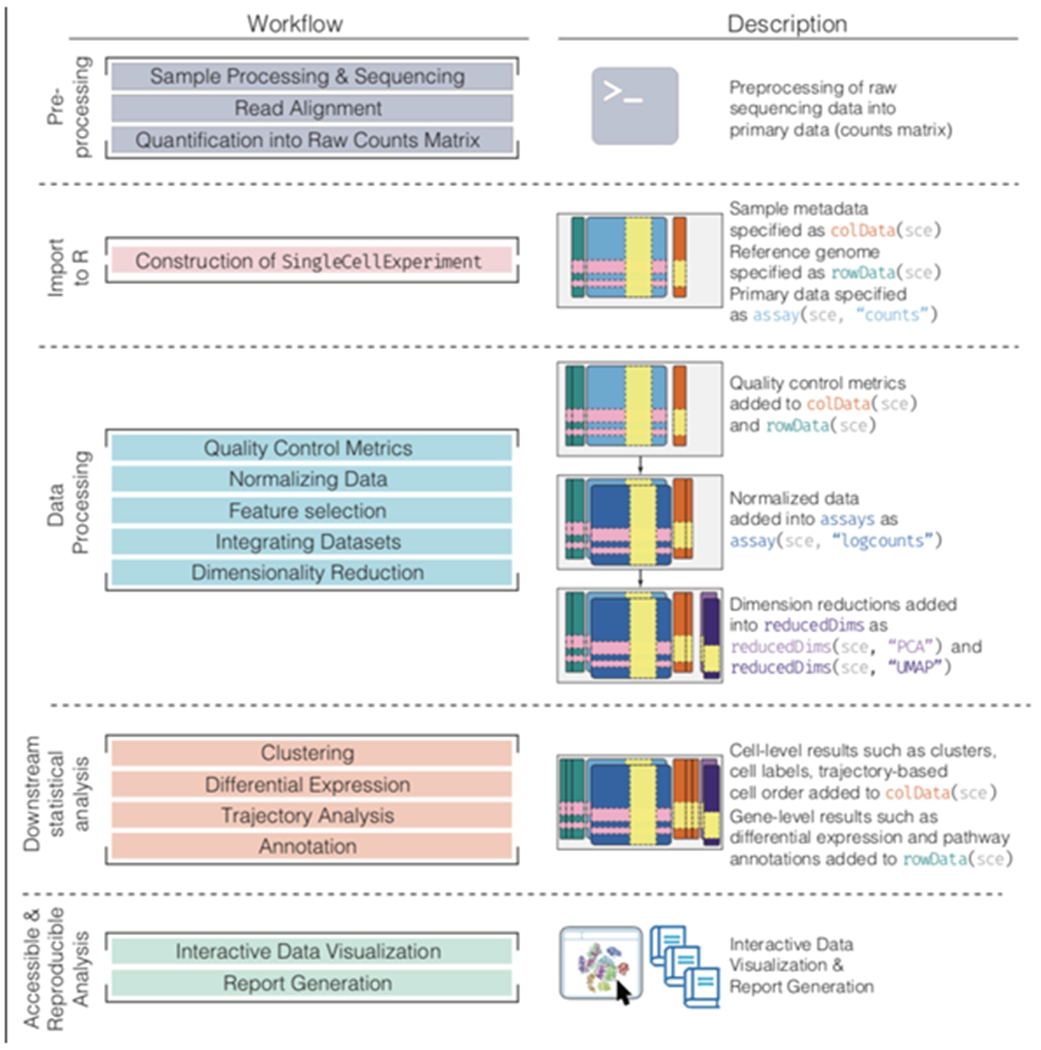
A typical analytical workflow using Bioconductor leads to the creation and evolution of a SingleCellExperiment (or sce) object during data processing and downstream statistical analysis (left column). An example of a sce object evolving throughout the course of a workflow is shown, including visualization, analysis, and annotation (right column).
Preprocessing
For scRNA-seq data, preprocessing involves the alignment of sequencing reads to a reference transcriptome and quantification into a per-cell and per-gene count matrix of expression values. While various preprocessing methods are available as command line software, Bioconductor packages such as scPipe [32] and scruff [33] provide a preprocessing workflow that is entirely written in R. For preprocessing workflows utilizing command line software, the DropletUtils [34] and tximeta Bioconductor packages can import the results from various tools including Cell Ranger [35] (10X Genomics), Kallisto-Bustools [36], and Alevin [37]. Notably, pseudo-alignment methods such as Alevin and Kallisto significantly reduce compute time and memory usage.
In all the above workflows, the end result is the import of a count matrix into R and creation of a SingleCellExperiment object. For specific file formats, we can use dedicated methods from the DropletUtils (for 10X data) or tximeta (for pseudo-alignment methods) packages.
Quality Control
Low-quality libraries in scRNA-seq data can arise from a variety of sources such as cell damage during dissociation or failure in library preparation (e.g., inefficient reverse transcription or PCR amplification). These usually manifest as “cells” with low total counts, few expressed genes and high mitochondrial read proportions. These low-quality libraries are problematic as they can contribute to misleading results in downstream analyses.
For droplet-based protocols, it is common to exclude data from droplets that did not contain exactly one cell. The DropletUtils [34] package distinguishes between empty – ambient RNA-containing – and cell-containing droplets, based on the frequency of each droplet barcode observed and a comparison of their respective expression profile with that of the ambient solution. It can also remove artificial cells generated by barcode swapping in droplet-based experiments [38]. Similarly, droplets that likely contain more than one cell (doublets) can be identified using the scran [28] or scds [39] packages, which compare the droplets in question against the expression profile of simulated doublets.
After excluding empty droplets and identifying potential doublets, droplets containing potentially damaged cells or exhibiting poor read coverage are filtered out. The library size - defined as the total sum of counts across all relevant features for each cell - is an oft-used metric for filtering. Cells with small library sizes are more likely to be of low quality as the RNA has been lost at some point during library preparation, either due to cell lysis or inefficient cDNA capture and amplification. Another metric is the number of expressed features in each cell - defined as the number of endogenous genes with non-zero counts for that cell. Cells with very few expressed genes are likely to be of poor quality as the diverse transcript population has not been successfully captured. The proportion of reads mapped to genes in the mitochondrial genome can also be used, as high proportions indicate the possible loss of cytoplasmic RNA due to cell damage, wherein the mitochondria - being larger than individual transcript molecules - are less likely to escape through holes in the cell membrane [40]. The scater [41] package simplifies the calculation of these various metrics.
Normalization
Systematic differences in coverage between libraries are often observed in scRNA-seq data, such as differences due to sequencing depth [25, 28, 42]. This typically arises from differences in cDNA capture or PCR amplification efficiency across cells, attributable to the difficulty of achieving consistent library preparation with minimal starting material. Normalization aims to remove these systematic differences such that they do not interfere with comparisons of the expression profiles between cells, for example during clustering or differential expression analyses.
Here we consider methods that moderate systematic differences within a single scRNA-seq experiment that bias all genes in a similar manner. This includes, for example, a change in sequencing depth that scales the expected coverage of all genes by a certain factor. Library size normalization is the simplest strategy for performing scaling normalization, as implemented in scater [41]. While this approach makes the assumption that there is no imbalance in the differentially expressed genes (DEG) between any pair of cells, normalization accuracy is usually not a major consideration for exploratory scRNA-seq analysis, as there are minimal effects on cluster separation.
Accurate normalization however is important for procedures that involve estimation and interpretation of per-gene statistics as in DEG. Composition biases that systematically shift log-fold changes are most often observed when multiple cell types are present in a given scRNA-seq dataset. Normalization by deconvolution overcomes this by pooling counts from many cells to increase the size of the counts for accurate size factor estimation, followed by deconvolution into cell-based factors for normalization per-cell, as implemented in scran [28].
Alternatively, BASiCS [43], zinbwave [30], and MAST [27] provide model-based approaches to normalization that can not only handle such library size or composition biases, but also can adjust for known covariates or other intrinsic technical factors that could conceal biologically meaningful variation [25]. These methods enable more complex scaling strategies such as non-linear transformations of the data. For reviews on this topic, see [42].
Imputation
Imputation methods have been proposed to address the challenge of data sparsity in single-cell assays [44, 45]. As scRNA-seq experiments frequently fail to measure expression for some genes, leading to an overabundance of zero-values [46], zero-inflated models have been developed. However, there are differences in the degree of zero-inflation depending on type of assay or protocol [46–48], suggesting that the optimal method is assay dependent. Furthermore, imputation methods for scRNA-seq data have been shown to generate false-positive results and decrease the reproducibility of cell-type specific markers [49].
Feature Selection
Exploratory analyses of scRNA-seq data is often directed to characterize heterogeneity across cells. Procedures such as clustering and dimensionality reduction compare cells based on their gene expression profiles. However, the choice of genes to use in these calculations has a major impact on the behavior and performance of such downstream methods. Feature selection methods aim to identify genes that contain useful information about the biology of the system while removing genes that contain random noise. By limiting analyses to such genes, interesting biological structure is preserved minus the variance that obscures that structure. Furthermore, focusing on such a subset of the transcriptome can significantly reduce the size of the dataset, improving the computational efficiency of downstream analyses. See references [50, 51] for reviews in feature selection methods.
The simplest approach to feature selection is to select the most variable genes based on their expression across the population. This assumes that genuine biological differences will manifest as increased variation in the affected genes, compared to other genes that are only affected by technical noise or a baseline level of uninteresting biological variation (e.g., from transcriptional bursting). However, the log-transformation does not achieve perfect variance stabilization. This means that the variance of a gene is more affected by its abundance than the underlying biological heterogeneity. Thus, calculation of the per-gene variance for feature selection requires modelling of the mean-variance relationship. Packages such as scran [52], BASiCS [43], and scFeatureFilter adopt this approach.
Alternate metrics to variance have also been proposed, such as selecting genes based on their deviance, a metric that quantifies how well each gene fits a null model of constant expression across cells [48]. Unlike variance based feature selection approaches, calculating the deviance is done on raw UMI counts, thus making the approach less sensitive to errors brought on by normalization. The deviance can be calculated using the glmpca package.
Dimensionality Reduction
Dimensionality reduction aims to reduce the number of separate dimensions in the data. This is possible because different genes are correlated if they are affected by the same biological process. Thus, we do not need to store separate information for individual genes, but can instead compress multiple features into a single dimension. Dimensionality reduction approaches thus create low-dimensional representations that aim to preserve the most meaningful structures in the dataset. This has the additional benefit of reducing noise by averaging across multiple genes to obtain a more precise representation of patterns in the data (e.g. related to a specific pathway). Computational work in downstream analyses is also reduced, as calculations only need to be performed for a few dimensions rather than thousands of genes. More aggressive dimensionality reduction schemes yield two- or three-dimensional representations that can be directly visualized to assist in the interpretation of the results.
A common first step to dimensionality reduction of scRNA-seq data is principal components analysis (PCA). PCA discovers axes (principal components, PCs) in high-dimensional space that capture the largest amount of variation. The top PCs capture the dominant factors of heterogeneity in the data set, and thus can be used to efficiently perform dimensionality reduction. This takes advantage of the well-studied theoretical properties of the PCA - namely, that a low-rank approximation formed from the top PCs is the optimal approximation of the original data for a given matrix rank. Given this property, calculations performed using the top PCs (or any similar low-rank approximation) takes advantage of data compression and denoising, which includes downstream analyses such as clustering.
No matter the approach, dimensionality reduction for visualization necessarily involves discarding information and distorting the distances between cells. Thus, it is ill-advised to directly analyze the low-dimensional coordinates used for plotting. Rather, these plots should only be used to interpret or communicate the results of quantitative analyses based on a more accurate, higher-rank representation of the data. This ensures that analyses make use of the information that was lost during compression into two dimensions. For example, given a discrepancy between the visible clusters on a 2-dimensional plot and those identified by clustering using the top PCs, one would be inclined to favor the latter.
The SingleCellExperiment class has a dedicated component, reducedDims, for storing lower dimensional representations of the assay data (Figure 2). The scater [41] package provides convenience wrapper functions for dimensionality reduction algorithms including Principal Components Analysis (PCA), t-Distributed Stochastic Neighbor Embedding (t-SNE) [53], and Uniform Manifold Approximation and Projection (UMAP) [54]. Diffusion map methods are available via the destiny [55] package. The zinbwave [30] and glmpca [48] packages use a zero-inflated negative binomial model and a multinomial model, respectively, for model-based dimensionality reduction approaches that can account for confounding factors.
Integrating Datasets
Large scRNA-seq projects usually need to generate data across multiple batches due to logistical constraints. However, the processing of different batches is often subject to uncontrollable differences, e.g., changes in operator, differences in reagent quality. This results in systematic differences in the observed expression in cells from different batches. Furthermore, as the prevalence of scRNA-seq data expands and reference datasets become available, encountering such confounding variables will become inevitable in meta-analysis contexts. Such batch effects are problematic as they can be major drivers of heterogeneity in the data, masking relevant biological differences and complicating the interpretation of results.
While generalized linear modeling frameworks can be used to integrate disparate data sets [6], these frameworks may be sub-optimal in the scRNA-seq context. This is often due to the underlying assumption that the composition of cell populations is either known or identical across batches of cells. To overcome these limitations, bespoke methods have been developed for batch correction of single-cell data [56, 57] that do not require a priori knowledge about the composition of the population. This enables exploratory analyses of scRNA-seq data where such knowledge is usually unavailable.
Before performing any correction, it is worth examining whether any batch are present in a dataset. This can be examined by performing PCA on the log-expression values of select genes, followed by graph based clustering to obtain a summary of the population structure. Ideally, clusters should consist of cells from replicate scRNA-seq datasets. However, if instead clusters are comprised of cells from a single batch, this indicates that cells of the same type are artificially separated due to technical differences. Approaches such as t-SNE and UMAP will also typically show a strong separation between cells from different batches that are consistent with such clustering results. Notably, such a diagnostic that relies on the degree of intermingling may not be effective when the batches involved may indeed contain unique subpopulations, but is nonetheless a useful first approximation.
Supervised integration via the labeling of cells a priori (see Annotation) can be used via packages such as scMerge [57] and scmap [58] to guide the application of any batch correction on the gene expression values or to adjust lower dimensional representations. On the other hand, unsupervised approaches such as mutual nearest neighbours (MNN) identify pairs of cells from different batches that belong in each other’s set of nearest neighbours. Thus, the difference between cells in MNN pairs can be used as an estimate of the batch effect, the subtraction of which yields batch-corrected values [56]. Vitally, by altering the number of k-nearest neighbors that are considered, the aggressiveness of the batch correction can be tuned, wherein a higher k results in more generous matching of subpopulations across batches. This MNN-based approach is implemented in the batchelor package.
The success of the batch correction is contingent on the preservation of biological heterogeneity, as one could envision a correction method simply aggregating all cells together, which would achieve perfect mixing but also discard the biology of interest. To this end, the CellMixS package can be used to evaluate the degree of cell mixing across batches. Another useful heuristic is to compare clusters identified in the merged data against those identified per batch. Ideally, we should see a many-to-1 mapping where the across-batch clustering is nested inside the within-batch clustering, indicating that any within-batch structure was preserved post-correction. A summary statistic such as the Rand index can then be calculated, where larger Rand indices are more desirable.
Downstream Statistical Analysis
The choice of methods and workflows can differ greatly depending on the specific goals of the investigation and the experimental protocol used. Following data processing, Bioconductor can be used to generate new biological insights from single-cell data, using tools that are interoperable with the SingleCellExperiment class and that scale with cell number. Our online book (https://osca.bioconductor.org) provides prospective users workflows and case studies for downstream analyses and visualizations (Figure 4).
Figure 4: Select visualizations derived from various Bioconductor workflows.
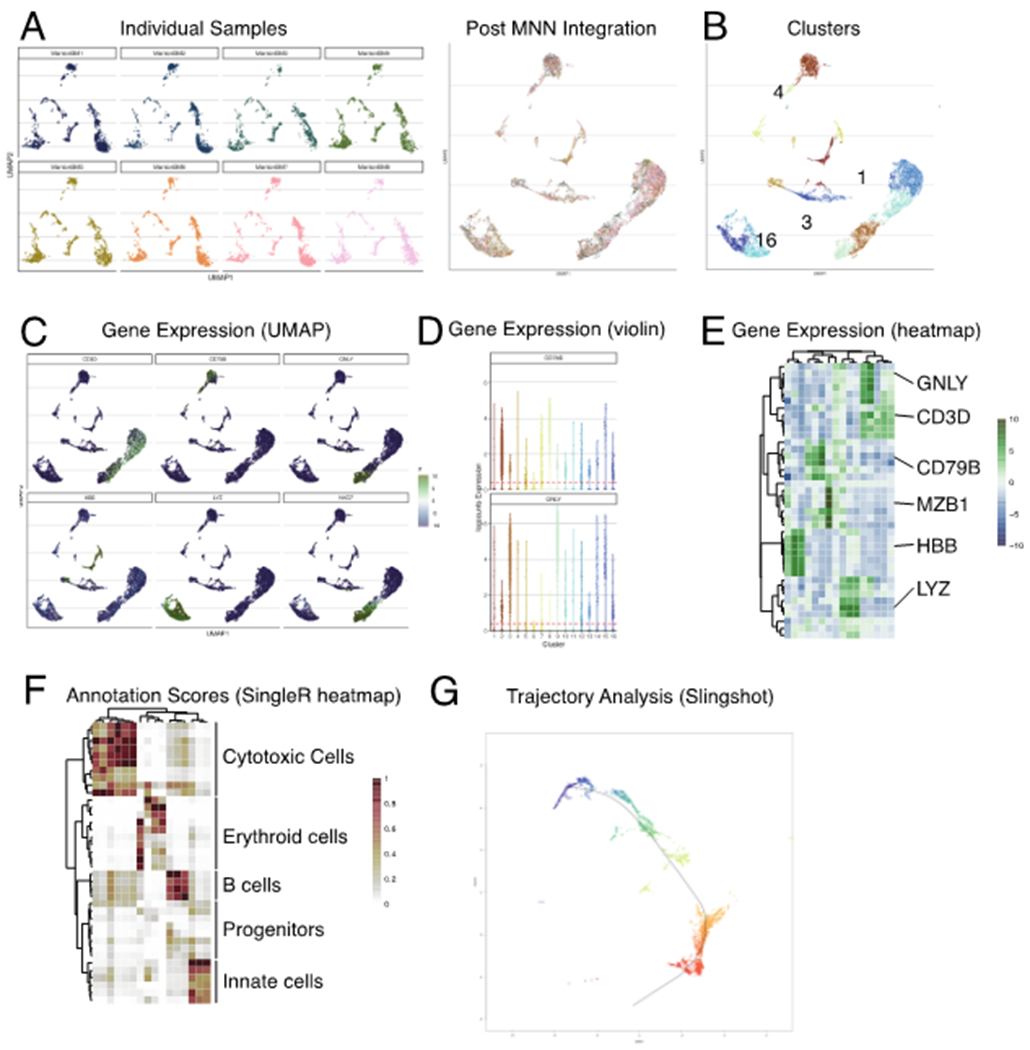
Various visualizations associated with preprocessing (blue boxes) and downstream statistical analyses (orange boxes). The example data set used throughout was generated as part of the Human Cell Atlas [21]). Details on the generation of these figures are described in our online companion book (https://osca.bioconductor.org)
Clustering
Clustering is used in scRNA-seq data analysis to empirically define groups of cells with similar expression profiles. This allows us to describe population heterogeneity in terms of discrete labels that can be more easily understood, rather than attempting to comprehend the high-dimensional manifold on which the cells truly reside. After annotation based on differentially expressed marker genes, the clusters can be treated as proxies for more abstract biological concepts such as cell types or states.
It is worth highlighting the distinction between clusters and cell types. The former is an empirical construct while the latter is a biological truth (albeit a vaguely defined one). Thus, it is helpful to realize that clustering, like a microscope, is simply a tool to explore the data. One can zoom in and out by changing the resolution of the clustering parameters, and experiment with different clustering algorithms to obtain alternative perspectives of the data.
Graph-based clustering is a flexible and scalable technique for clustering large scRNA-seq datasets. A graph is constructed where each node is a cell that is connected to its nearest neighbours (NN) in the high-dimensional space. Edges are weighted based on the similarity between the cells involved, with higher weight given to cells that are more closely related. Algorithms such as louvain and leiden [59] can then be used to identify clusters of cells.
BiocNeighbors provides an engine for both exact and approximate nearest neighbor detection, with scran building the actual graph. Notably, for large scRNA-seq datasets, approximate NN methods trade an acceptable loss in accuracy for vastly improved run times, with the added advantage of smoothing over noise and sparsity. Alternative approaches include the SIMLR package [60], which uses multiple kernels to learn a distance metric between cells that best fits the data and can then be used for clustering and dimension reduction. For large data, the mbkmeans package implements a scalable version of the k-means algorithm. Finally, the SC3 [61] and clusterExperiment [62] packages calculate consensus clusters derived from multiple parameterizations.
Many of these packages allow quantitative and visual evaluation of the clustering results, alongside external packages designed solely for data visualization and evaluation (e.g., clustree). Clusters can also be evaluated independently by assessing metrics such as cluster modularity or the silhouette coefficient.
Differential Expression
Differential gene expression (DGE) analysis can be used to identify marker genes that drive the separation between clusters. These marker genes allow us to assign biological meaning to each cluster based on their functional annotation. In the most obvious case, the marker genes for each cluster are a priori associated with particular cell types, allowing for clustering to serve as a proxy for cell type identity. The same principle can be applied to detect more subtle differences such as activation status or differentiation state. An alternative to DGE analysis for cell type annotation is gene set enrichment analysis, which groups genes into pre-specified gene modules or biological pathways to facilitate biological interpretation. We discuss this topic in the annotation section.
DGE can also be used to compare individual cells within a given population across conditions such as time or treatment, while adjusting for covariates (e.g. patient id, batch effects).
Across differential expression methods, two general approaches stand out. The first approach retrofits well supported and long-standing DE analysis frameworks initially designed for bulk RNA-sequencing (edgeR [2], DESeq2 [5], limma-voom [6]) that have made the transition to scRNA-seq through various approaches, such as by creating pseudo-bulk RNA-seq profiles. Alternatively, approaches such as zinbwave [30] can be used to downweight excess zeros observed in scRNA-seq data during the dispersion estimation and model fitting steps prior to assessing DE, and consequently further enabling the adaptation of bulk RNA-seq based DE methods for use with scRNA-seq data [63].
The second class of approaches is uniquely tailored for single-cell data because the statistical methods proposed directly model the zero-inflation component, frequently observed in scRNA-seq data. These methods explicitly separate gene expression into two components: the discrete component, which describes the frequency of a discrete component (zero versus non-zero expression), and the continuous component, where the level of gene expression is quantified. While all the methods mentioned herein can test for differences in the continuous component, only this second class of approaches can explicitly model the discrete component, and thus test for differences in the frequency of expression. To do this, the MAST [27] package utilizes a hurdle model framework, whereas the scDD [64], BASiCS [43], and SCDE [14] use Bayesian mixture and hierarchical models, respectively. Together, these methods are able to provide a broader suite of testing functionality and can be directly utilized on scRNA-seq data contained within the SingleCellExperiment class.
For more details regarding DE analysis and the benchmarking of the various packages mentioned above, see [65–67].
Trajectory Analysis
Heterogeneity may also be modeled as a continuous spectrum arising from biological processes, such as cell differentiation. A specialized application of dimension reduction specific to single-cell analysis - trajectory analysis or pseudotime inference - uses phylogenetic methods to order cells along a (often time continuous) trajectory, such as development over time. Inferred trajectories can identify transition between cell states, a differentiation process, or events responsible for bifurcations in a dynamic cellular process [68].
Modern approaches for trajectory inference have minimized the need for extensive parameterization and can test for differential gene expression across various topologies (e.g., Monocle [69], LineagePulse, and switchde [70]). Moreover, several Bioconductor packages for trajectory inference (e.g., slingshot [71], TSCAN [29], Monocle [69], cellTree [72], and MFA [73]) were recently demonstrated to have excellent performance [74]. As different methods can produce drastically different results for the same dataset, a suite of methods and parameterizations must be tested to assess robustness. Bioconductor facilitates such testing by providing standardized data representation such as the SingleCellExperiment class objects. See [74] for further discussion.
Annotation
The most challenging task in scRNA-seq data analysis is arguably the interpretation of the results. Obtaining clusters of cells is fairly straightforward, but it is more difficult to determine what biological state is represented by each of those clusters. Doing so requires bridging the gap between the current dataset and prior biological knowledge, and the latter is not always available in a consistent and quantitative manner. As such, interpretation of scRNA-seq data is often manual and a common bottleneck in the analysis workflow.
To expedite this step, various computational approaches can be applied that exploit prior information to assign meaning to an uncharacterized scRNA-seq dataset. The most obvious sources of prior information are curated gene sets associated with particular biological processes (e.g., from the Gene Ontology (GO) or the Kyoto Encyclopedia of Genes and Genomes (KEGG) collections).
An alternative approach involves directly comparing expression profiles to published reference datasets where each sample or cell has already been annotated with its putative biological state by domain experts.
Gene Signature Enrichment
Classical gene set enrichment (GSE) approaches have the advantage of not requiring reference expression values. This is particularly useful when dealing with gene sets derived from the literature or other qualitative forms of biological knowledge. In the context of cell annotation, GSE is typically performed on a group of cells (or cluster) to identify the gene set (or pathway) that is enriched in these cells. The enriched pathway can then be used to deduce a cell type (or state).
Bioconductor provides dedicated packages to programmatically access predefined gene signatures from databases such as MSigDB [75], KEGG [76], Reactome [77], and Gene Ontology (GO) [78]. EnrichmentBrowser [79]) simplifies the compilation of gene signature collections from such repositories. This prior knowledge is used to test for the enrichment of specific gene modules in scRNA-seq data, often adapting existing gene set analysis methods originally developed for bulk data. The EnrichmentBrowser [79], EGSEA [80], and fgsea packages each provide some version of classical gene set enrichment analysis (GSEA). Alternative approaches to testing for gene set enrichment are implemented in MAST [27], AUCell [81], and slalom [82].
Automated Classification of Cells
A conceptually straightforward annotation approach is to compare the single-cell expression profiles with previously annotated reference datasets. Labels can then be assigned to each cell in an uncharacterized dataset based on the most similar reference sample(s), based on some similarity metric. This is a standard classification challenge that can be tackled by standard machine learning techniques such as random forests and support vector machines. Any published and labelled RNA-seq dataset (bulk or single-cell) can be used as a reference, though its reliability depends greatly on the domain expertise of the original authors who assigned the labels in the first place.
The SingleR method [83] provides one such automated method for cell type annotation assignment. SingleR labels cells based on the reference samples with the highest Spearman rank correlations, and thus can be considered a rank-based variant of k-nearest-neighbor classification. To reduce noise, SingleR identifies marker genes between pairs of labels and computes the correlation using only those markers. A number of built-in reference datasets are included with the package that are derived from a variety of sources and tissues, including Immunological Genome project (ImmGen), ENCODE, and the Database for Immune Cell Expression (DICE).
Accessible Analysis
With the increased interest in data from single-cell assays, Bioconductor has developed not only the methods and software to analyze the data, but also has prioritized making the data itself and the data analysis tools more easily accessible to both users and developers. Specifically, the community has contributed data packages, containing both publicly available published data and simulated data, and interactive data visualization tools. Making single-cell data and data analysis tools more accessible allows researchers to leverage these resources in their own work and democratizes data analysis.
Benchmarking
As new single-cell assays, statistical methods, and corresponding software are developed, it is increasingly important to facilitate the publication of data sets, to reproduce existing analyses as well as to enable comparisons across new and existing tools. Bioconductor houses a collection of data packages focused on providing accessible and well-annotated versions of data ready for analysis, alongside vignettes that can be used to reproduce manuscript figures and showcase data characteristics.
To facilitate querying of published data packages on Bioconductor, the ExperimentHub package enables programmatic access of published data sets using a standardized interface. Of note, the scRNAseq package provides direct access to a curated selection of high-quality scRNA-seq data from various contexts. In addition, simulated data are useful for benchmarking methods.
Alternately, the splatter package [84] can simulate scRNA-seq data that contains multiple cell types, batch effects, varying levels of dropout events, differential gene expression, and trajectories. The splatter package uses both its own simulation framework and wraps around other simulation frameworks with differing generative models to provide a comprehensive resource for single-cell data simulation.
To promote the reproducibility of benchmark comparisons assessing the performance of single-cell methods, software packages have been developed that provide infrastructure to compute and store the results of applying different methods to a data set. The SummarizedBenchmark [85] and CellBench [86] packages provide interfaces for which to store metadata (method parameters, package versions) and evaluation metrics.
Interactive Data Visualization
The maturation of web technologies has opened new avenues for interactive data exploration, aided by shiny, an R package facilitating development of rich graphical user interfaces. The iSEE [87] and singleCellTK packages provide full-featured applications for interactive visualization of scRNA-seq datasets through an internet browser, eliminating the need for programming experience if the instance is hosted on the web. Both packages directly interface with the SingleCellExperiment data container to enable scRNA-seq analysis results.
Discussion
Since the early days of genomics, the Bioconductor project has embraced the development of open-source and open-development software through the R statistical programming language. Bioconductor has established best practices for coordinated package versioning and code review. Alongside community-contributed packages, a core developer team (https://www.bioconductor.org/about/core-team) implements and maintains the essential infrastructure, and reviews contributed packages to ensure they satisfy a set of guidelines to ensure interoperability across packages. These packages are organized into BiocViews, an ontology of topics that classify packages by task or technology. For example, topics in single-cell analysis are labeled under the view SingleCell. Most importantly, the broader Bioconductor community - accessible through various means including forums, Slack, or mailing lists - is a model of altruism in code sharing and technical help. Together, these practices produce high-quality, well maintained packages, contributing to a unified and stable environment for biological research.
Most recently, the Bioconductor community has developed state-of-the-art computational methods, infrastructure, and interactive data visualization tools available as software packages for the analysis of data derived from single-cell experiments. Emerging single-cell technologies in epigenomics, T-cell and B-cell repertoires, spatial profiling, and sequencing-based protein profiling [88–95] promise to continue driving advances in computational biology. In particular, technologies enabling multimodal profiling are rapidly developing, and Bioconductor has laid the groundwork necessary to support statistical methodologies that fully leverage such approaches.
In addition, Bioconductor’s standardized data containers enable interoperability within and between Bioconductor packages as well as other software. Analysis stored in a SingleCellExperiment can be converted to formats usable with Seurat [96], Monocle [69], and Python’s scanpy [97], enabling the use of the tools that best serve the objective at hand. Indeed, R has a long history of interoperability with other programming languages. Four examples are the Rcpp [98] package for integrating C++ compiled code into R, the rJava package to call Java code from within R, the .Fortran() function in base R to call Fortran code, and the reticulate CRAN package for interfacing with Python. This interoperability enables common machine learning frameworks such as TensorFlow/Keras to be used directly in R.
To the newcomer, the wealth of single-cell analyses possible in Bioconductor can be daunting. To address the rapid growth of contributed packages within the single-cell analysis space, we have summarized and highlighted state-of-the-art data infrastructure (Figure 2), methods and software, and organized the packages along a typical workflow (Figure 3) for the most common single-cell analyses (Figure 4). Finally, we have developed an online companion book that provides more details on focused topics as well as complete coding workflows (https://osca.bioconductor.org). This effort will be continuously updated and maintained with new packages as they emerge, which increases discoverability of Bioconductor resources.
Supplementary Material
Acknowledgements
Bioconductor is supported by the National Human Genome Research Institute (NHGRI) and National Cancer Institute (NCI) of the National Institutes of Health (NIH) (U41HG004059, U24CA180996), the European Union (EU) H2020 Personalizing Health and Care Program Action (contract number 633974), and the SOUND Consortium. In addition, MM, SCH, RG, WH, ATLL, and DR are supported by the Chan Zuckerberg Initiative (CZI) DAF (2018-183201, 2018-183560), an advised fund of Silicon Valley Community Foundation. DR, WH, MM and SCH are supported by 2019-002443 from the CZI. SCH is supported by the NIH/NHGRI (R00HG009007). RAA and RG are supported by the Integrated Immunotherapy Research Center at Fred Hutch. MM is supported by the NCI/NHGRI (U24CA232979). LG is supported by a research fellowship from the German Research Foundation (GE3023/1-1). LW and VJC are supported by the NCI (U24CA18099). VJC is additionally supported by NCI U01 CA214846 and Chan Zuckerberg Initiative DAF (2018-183436). ATLL received support from CRUK (A17179) and the Wellcome Trust (WT/108437/Z/15). FM is supported by the German Federal Ministry of Education and Research (BMBF 01EO1003). MLS is supported by the German Network for Bioinformatics Infrastructure (031A537B). DR is supported by the Programma per Giovani Ricercatori Rita Levi Montalcini from the Italian Ministry of Education, University, and Research. HP is supported by the NIH Bioconductor grant (U41HG004059).
Footnotes
Competing Financial Interests
RG declares ownership in CellSpace Biosciences.
References
- [1].Huber Wolfgang, Vincent J Carey Robert Gentleman, Anders Simon, Carlson Marc, Benilton S Carvalho Hector Corrada Bravo, Davis Sean, Gatto Laurent, Girke Thomas, Gottardo Raphael, Hahne Florian, Hansen Kasper D, Irizarry Rafael A, Lawrence Michael, Michael I Love, MacDonald James, Obenchain Valerie, Oleś Andrzej K, Pagès Hervé, Reyes Alejandro, Shannon Paul, Smyth Gordon K, Tenenbaum Dan, Waldron Levi, and Morgan Martin. Orchestrating high-throughput genomic analysis with Bioconductor. Nat Methods, 12(2):115–21, 02 2015. doi: 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Robinson Mark D, McCarthy Davis J, and Smyth Gordon K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics, 26(1):139–40, 2010. doi: 10.1093/bioinformatics/btp616. URL https://bioconductor.org/packages/edgeR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Lawrence Michael, Huber Wolfgang, Hervé Pagès Patrick Aboyoun, Carlson Marc, Gentleman Robert, Morgan Martin T, and Carey Vincent J. Software for computing and annotating genomic ranges. PLoS Comput Biol, 9(8):e1003118, 2013. doi: 10.1371/journal.pcbi.1003118. URL https://bioconductor.org/packages/IRanges. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Aryee Martin J, Jaffe Andrew E, Corrada-Bravo Hector, Ladd-Acosta Christine, Feinberg Andrew P, Hansen Kasper D, and Irizarry Rafael A. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics, 30(10):1363–9, 2014. doi: 10.1093/bioinformatics/btu049. URL https://bioconductor.org/packages/minfi. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Love Michael I, Huber Wolfgang, and Anders Simon. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol, 15(12):550, 2014. doi: 10.1186/s13059-014-0550-8. URL https://bioconductor.org/packages/DESeq2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Ritchie Matthew E, Phipson Belinda, Wu Di, Hu Yifang, Law Charity W, Shi Wei, and Smyth Gordon K. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res, 43(7):e47, 2015. doi: 10.1093/nar/gkv007. URL https://bioconductor.org/packages/limma. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Serratì Simona, De Summa Simona, Pilato Brunella, Petriella Daniela, Lacalamita Rosanna, Tommasi Stefania, and Pinto Rosamaria. Next-generation sequencing: advances and applications in cancer diagnosis. Onco Targets Ther, 9:7355–7365, 2016. doi: 10.2147/OTT.S99807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Nakato Ryuichiro and Shirahige Katsuhiko. Recent advances in ChIP-seq analysis: from quality management to whole-genome annotation. Brief Bioinform, 18(2):279–290, 2017. doi: 10.1093/bib/bbw023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Kukurba Kimberly R and Montgomery Stephen B. RNA sequencing and analysis. Cold Spring Harb Protoc, 2015(11):951–69, 2015. doi: 10.1101/pdb.top084970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Kolodziejczyk Aleksandra A, Kim Jong Kyoung, Svensson Valentine, Marioni John C, and Teichmann Sarah A. The technology and biology of single-cell RNA sequencing. Mol Cell, 58(4):610–20, 05 2015. doi: 10.1016/j.molcel.2015.04.005. [DOI] [PubMed] [Google Scholar]
- [11].Patel Anoop P, Tirosh Itay, Trombetta John J, Shalek Alex K, Gillespie Shawn M, Wakimoto Hiroaki, Cahill Daniel P, Nahed Brian V, Curry William T, Martuza Robert L, Louis David N, RozenblattRosen Orit, Suvà Mario L, Regev Aviv, and Bernstein Bradley E. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science, 344(6190):1396–401, 2014. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Tirosh Itay, Izar Benjamin, Prakadan Sanjay M, Wadsworth Marc H 2nd, Treacy Daniel, Trombetta John J, Rotem Asaf, Rodman Christopher, Lian Christine, MSurphy George, Fallahi-Sichani Mohammad, Dutton-Regester Ken, Lin Jia-Ren, Cohen Ofir, Shah Parin, Lu Diana, Genshaft Alex S, Hughes Travis K, Ziegler Carly G K, S Kazer Samuel W, GaSillSard Aleth, KSolb Kellie E, Villani Alexandra-Chloé, Johannessen Cory M, AndSreSeSv Aleksandr Y, Van Allen Eliezer M, Bertagnolli Monica, Sorger Peter K, Sullivan Ryan J, Flaherty Keith T, Frederick Dennie T, Jané-Valbuena Judit, Yoon Charles H, Rozenblatt-Rosen Orit, Shalek Alex K, Regev Aviv, and Garraway Levi A. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell RNA-seq. Science, 352(6282):189–96, 2016. doi: 10.1126/science.aad0501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Karaayvaz Mihriban, Cristea Simona, Gillespie Shawn M, Patel Anoop P, Mylvaganam Ravindra, Luo Christina C, Specht Michelle C, Bernstein Bradley E, Michor Franziska, and Ellisen Leif W. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat Commun, 9(1):3588, 2018. doi: 10.1038/s41467-018-06052-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Fan Jean, Lee Hae-Ock, Lee Soohyun, Ryu Da-Eun, Lee Semin, Xue Catherine, Kim Seok Jin, Kim Kihyun, Barkas Nikolaos, Park Peter J, Park Woong-Yang, and Kharchenko Peter V. Linking transcriptional and genetic tumor heterogeneity through allele analysis of single-cell RNA-seq data. Genome Res, 28(8): 1217–1227, 2018. doi: 10.1101/gr.228080.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Levitin Hanna Mendes, Yuan Jinzhou, and Sims Peter A. Single-Cell Transcriptomic Analysis of Tumor Heterogeneity. Trends Cancer, 4(4):264–268, 2018. doi: 10.1016/j.trecan.2018.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Paulson KG Voillet V, McAfee MS, Hunter DS, Wagener FD, Perdicchio M, Valente WJ, Koelle SJ, Church CD, Vandeven N, Thomas H, Colunga AG, Iyer JG, Yee C, Kulikauskas R, Koelle DM, Pierce RH, Bielas JH, Greenberg PD, Bhatia S, Gottardo R, Nghiem P, and Chapuis AG. Acquired cancer resistance to combination immunotherapy from transcriptional loss of class I HLA. Nat Commun, 9(1): 3868, 09 2018. doi: 10.1038/s41467-018-06300-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Zeisel Amit, Muñoz-Manchado Ana B, Codeluppi Simone, Lönnerberg Peter, La Manno Gioele, Juréus Anna, Marques Sueli, Munguba Hermany, He Liqun, Betsholtz Christer, Rolny Charlotte, Gonçalo Castelo-Branco, Hjerling-Leffler Jens, and Linnarsson Sten. Brain structure. Cell types in the mouse cortex and hippocampus revealed by single-cell RNA-seq. Science, 347(6226):1138–42, 03 2015. doi: 10.1126/science.aaa1934. [DOI] [PubMed] [Google Scholar]
- [18].Deng Qiaolin, Daniel Ramsköld Björn Reinius, and Sandberg Rickard. Single-cell RNA-seq reveals dynamic, random monoallelic gene expression in mammalian cells. Science, 343(6167):193–6, 2014. doi: 10.1126/science.1245316. [DOI] [PubMed] [Google Scholar]
- [19].Vladimir Yu Kiselev Tallulah S. Andrews, and Hemberg Martin. Challenges in unsupervised clustering of single-cell RNA-seq data. Nature Reviews Genetics, 2019. doi: 10.1038/s41576-018-0088-9. [DOI] [PubMed] [Google Scholar]
- [20].Cannoodt Robrecht, Saelens Wouter, and Saeys Yvan. Computational methods for trajectory inference from single-cell transcriptomics. Eur J Immunol, 46(11):2496–2506, 2016. doi: 10.1002/eji.201646347. [DOI] [PubMed] [Google Scholar]
- [21].Regev Aviv, Teichmann Sarah A, Lander Eric S, Amit Ido, Benoist Christophe, Birney Ewan, Bodenmiller Bernd, Campbell Peter, Carninci Piero, Clatworthy Menna, Clevers Hans, Deplancke Bart, Dunham Ian, Eberwine James, Eils Roland, Enard Wolfgang, Farmer Andrew, Fugger Lars, Göttgens Berthold, Hacohen Nir, Haniffa Muzlifah, Hemberg Martin, Kim Seung, Klenerman Paul, Kriegstein Arnold, Lein Ed, Linnarsson Sten, Lundberg Emma, Lundeberg Joakim, Majumder Partha, Marioni John C, Merad Miriam, Mhlanga Musa, Nawijn Martijn, Netea Mihai, Nolan Garry, Pe’er Dana, Phillipakis Anthony, Ponting Chris P, Stephen Quake, Reik Wolf, Rozenblatt-Rosen Orit, Sanes Joshua, Satija Rahul, Schumacher Ton N, Shalek, Shapiro Ehud, Sharma Padmanee, Shin Jay W, Stegle Oliver, Stratton Michael, Stubbington Michael J T, Theis Fabian J, Uhlen Matthias, van Oudenaarden Alexander, Wagner Allon, Watt Fiona, Weissman Jonathan, Wold Barbara, Xavier Ramnik, Yosef Nir, and Human Cell Atlas Meeting Participants. The Human Cell Atlas. Elife, 6, 2017. doi: 10.7554/eLife.27041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Rozenblatt-Rosen Orit, Stubbington Michael J T, Regev Aviv, and Teichmann Sarah A. The human cell atlas: from vision to reality. Nature, 550(7677):451–453, 10 2017. doi: 10.1038/550451a. [DOI] [PubMed] [Google Scholar]
- [23].Han Xiaoping, Wang Renying, Zhou Yincong, Fei Lijiang, Sun Huiyu, Lai Shujing, Saadatpour Assieh, Zhou Ziming, Chen Haide, Ye Fang, Huang Daosheng, Xu Yang, Huang Wentao, Jiang Mengmeng, Jiang Xinyi, Mao Jie, Chen Yao, Lu Chenyu, Xie Jin, Fang Qun, Wang Yibin, Yue Rui, Li Tiefeng, Huang He, Stuart H Orkin Guo-Cheng Yuan, Chen Ming, and Guo Guoji. Mapping the Mouse Cell Atlas by Microwell-Seq. Cell, 173(5):1307, 05 2018. doi: 10.1016/j.cell.2018.05.012. [DOI] [PubMed] [Google Scholar]
- [24].McDavid Andrew, Finak Greg, Chattopadyay Pratip K, Dominguez Maria, Lamoreaux Laurie, Ma Steven S, Roederer Mario, and Gottardo Raphael. Data exploration, quality control and testing in single-cell qPCR-based gene expression experiments. Bioinformatics, 29(4):461–7, 2013. doi: 10.1093/bioinformatics/bts714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Hicks Stephanie C, Townes F William, Teng Mingxiang, and Irizarry Rafael A. Missing data and technical variability in single-cell RNA-sequencing experiments. Biostatistics, 19(4):562–578, 2018. doi: 10.1093/biostatistics/kxx053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kharchenko Peter V, Silberstein Lev, and Scadden David T. Bayesian approach to single-cell differential expression analysis. Nat Methods, 11(7):740–2, 2014. doi: 10.1038/nmeth.2967. URL https://bioconductor.org/packages/scde. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Finak G, McDavid A, Yajima M, and others. MAST: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol, 16: 278, 2015. doi:s13059-015-0844-5. URL https://bioconductor.org/packages/MAST. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Lun Aaron T L, Bach Karsten, and Marioni John C. Pooling across cells to normalize single-cell RNA sequencing data with many zero counts. Genome Biol, 17:75, 2016. doi: 10.1186/s13059-016-0947-7. URL https://bioconductor.org/packages/scran. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ji Zhicheng and Ji Hongkai. TSCAN: Pseudo-time reconstruction and evaluation in single-cell RNA-seq analysis. Nucleic Acids Res, 44(13):e117, 2016. doi: 10.1093/nar/gkw430. URL https://bioconductor.org/packages/TSCAN. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Risso Davide, Perraudeau Fanny, Gribkova Svetlana, Dudoit Sandrine, and Vert Jean-Philippe. A general and flexible method for signal extraction from single-cell RNA-seq data. Nat Commun, 9(1):284, 2018. doi: 10.1038/s41467-017-02554-5. URL https://bioconductor.org/packages/zinbwave. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Chambers John M. Object-oriented programming, functional programming and R. Statistical Science, 29 (2):167–180, 2014. [Google Scholar]
- [32].Tian Luyi, Su Shian, Dong Xueyi, Amann-Zalcenstein Daniela, Biben Christine, Seidi Azadeh, Hilton Douglas J, Naik Shalin H, and Ritchie Matthew E. scPipe: A flexible R/Bioconductor preprocessing pipeline for single-cell RNA-sequencing data. PLoS Comput Biol, 14(8):e1006361, 2018. doi: 10.1371/journal.pcbi.1006361. URL https://bioconductor.org/packages/scPipe. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wang Zhe, Hu Junming, Johnson W Evan, and Campbell Joshua D. scruff: an R/Bioconductor package for preprocessing single-cell RNA-sequencing data. BMC Bioinformatics, 20(1):222, May 2019. doi: 10.1186/s12859-019-2797-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Lun Aaron T. L., Riesenfeld Samantha, Andrewsand Tomas Gomes Tallulah The Phuong Dao, participants in the 1st Human Cell Atlas Jamboree, and John C. Marioni. Emptydrops: distinguishing cells from empty droplets in droplet-based single-cell rna sequencing data. Genome Biol, 20:63, 2019. doi: 10.1186/s13059019-1662-y. URL https://bioconductor.org/packages/DropletUtils. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zheng Grace X Y, Terry Jessica M, Belgrader Phillip, Ryvkin Paul, Bent Zachary W, Wilson Ryan, Ziraldo Solongo B, Wheeler Tobias D, McDermott Geoff P, Zhu Junjie, Gregory Mark T, Shuga Joe, Montesclaros Luz, Underwood Jason G, Masquelier Donald A, Nishimura Stefanie Y, Schnall-Levin Michael, Wyatt Paul W, Hindson Christopher M, Bharadwaj Rajiv, Wong Alexander, Ness Kevin D, Beppu Lan W, Deeg H Joachim, McFarland Christopher, Loeb Keith R, Valente William J, Ericson Nolan G, Stevens Emily A, Radich Jerald P, Mikkelsen Tarjei S, Hindson Benjamin J, and Bielas Jason H. Massively parallel digital transcriptional profiling of single cells. Nat Commun, 8:14049, 2017. doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Páll Melsted A Booeshaghi Sina, Gao Fan, Beltrame Eduardo, Lu Lambda, Hjorleifsson Kristján Eldjárn, Gehring Jase, and Pachter Lior Modular and efficient pre-processing of single-cell rna-seq. bioRxiv, page 673285, 2019. doi: 10.1101/673285. [DOI] [Google Scholar]
- [37].Srivastava Avi, Malik Laraib, Smith Tom, Sudbery Ian, and Patro Rob. Alevin efficiently estimates accurate gene abundances from dscRNA-seq data. Genome Biol, 20(1):65, March 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Griffiths Jonathan A, Richard Arianne C, Bach Karsten, Lun Aaron T L, and Marioni John C. Detection and removal of barcode swapping in single-cell RNA-seq data. Nat Commun, 9(1):2667, 2018. doi: 10.1038/s41467-018-05083-x. URL https://bioconductor.org/packages/DropletUtils. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Bais Abha S and Kostka Dennis. scds: Computational Annotation of Doublets in Single Cell RNA Sequencing Data. bioRxiv, pages 1–27, 02 2019. doi: 10.1101/564021. URL https://bioconductor.org/packages/scds. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Ilicic Tomislav, Kim Jong Kyoung, Kolodziejczyk Aleksandra A, Bagger Frederik Otzen, McCarthy Davis James, Marioni John C, and Teichmann Sarah A. Classification of low quality cells from single-cell RNA-seq data. Genome biology, pages 1–15, February 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].McCarthy Davis J, Campbell Kieran R, Lun Aaron T L, and Wills Quin F. Scater: pre-processing, quality control, normalization and visualization of single-cell RNA-seq data in R. Bioinformatics, 33(8):1179–1186, 2017. doi: 10.1093/bioinformatics/btw777. URL https://bioconductor.org/packages/scater. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Vallejos Catalina A, Risso Davide, Scialdone Antonio, Dudoit Sandrine, and Marioni John C. Normalizing single-cell RNA sequencing data: challenges and opportunities. Nat Methods, 14(6):565–571, 2017. doi: 10.1038/nmeth.4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Vallejos Catalina A, Richardson Sylvia, and Marioni John C. Beyond comparisons of means: understanding changes in gene expression at the single-cell level. Genome Biol, 17:70, 2016. doi: 10.1186/s13059-016-09303. URL https://bioconductor.org/packages/BASiCS. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Huang Mo, Wang Jingshu, Torre Eduardo, Dueck Hannah, Shaffer Sydney, Bonasio Roberto, John I Murray Arjun Raj, Li Mingyao, and Nancy R Zhang. SAVER: gene expression recovery for single-cell RNA sequencing. Nat Methods, 15(7):539–542, 07 2018. doi: 10.1038/s41592-018-0033-z. URL https://github.com/mohuangx/SAVER. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Li Wei Vivian and Jessica Li Jingyi. An accurate and robust imputation method scImpute for singlecell RNA-seq data. Nat Commun, 9(1):997, 03 2018. doi: 10.1038/s41467-018-03405-7. URL https://github.com/Vivianstats/scImpute. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Svensson Valentine. Droplet scRNA-seq is not zero-inflated. bioRxiv, 2019. doi: 10.1101/582064. [DOI] [PubMed] [Google Scholar]
- [47].Vieth Beate, Ziegenhain Christoph, Parekh Swati, Enard Wolfgang, and Hellmann Ines. powsimR: power analysis for bulk and single cell RNA-seq experiments. Bioinformatics, 33(21):3486–3488, 11 2017. doi: 10.1093/bioinformatics/btx435. URL https://github.com/bvieth/powsimR. [DOI] [PubMed] [Google Scholar]
- [48].Townes F William, Hicks Stephanie C, Aryee Martin J, and Irizarry Rafael A. Feature Selection and Dimension Reduction for Single Cell RNA-Seq based on a Multinomial Model. bioRxiv, 2019. doi: 10.1101/574574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Andrews Tallulah and Hemberg Martin. False signals induced by single-cell imputation [version 2; peer review: 4 approved]. F1000Research, 7(1740), 2019. doi: 10.12688/f1000research.16613.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Andrews Tallulah and Hemberg Martin. M3Drop: Dropout-based feature selection for scRNASeq. Bioinformatics, 2018. doi: 10.1093/bioinformatics/bty1044. URL https://bioconductor.org/packages/M3Drop. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Yip Shun H, Chung Sham Pak, and Wang Junwen. Evaluation of tools for highly variable gene discovery from single-cell RNA-seq data. Brief Bioinform, 2018. doi: 10.1093/bib/bby011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lun Aaron T. L., McCarthy Davis J, and Marioni John C.. A step-by-step workflow for low-level analysis of single-cell RNA-seq data with Bioconductor. F1000Res, 5:2122, 2016. doi: 10.12688/f1000research.9501.2. URL https://www.bioconductor.org/packages/simpleSingleCell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].van der Maaten Laurens and Hinton Geoffrey. Visualizing data using t-SNE. Journal of Machine Learning Research, 9:2579–2605, 2008. URL http://www.jmlr.org/papers/v9/vandermaaten08a.html. [Google Scholar]
- [54].McInnes James Melville Leland, Healy John. UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction, 2018. URL https://arxiv.org/abs/1802.03426. [Google Scholar]
- [55].Angerer Philipp, Haghverdi Laleh, Büttner Maren, Theis Fabian J, Marr Carsten, and Buettner Florian. destiny: diffusion maps for large-scale single-cell data in R. Bioinformatics, 32(8):1241–3, 2016. doi: 10.1093/bioinformatics/btv715. URL https://bioconductor.org/packages/destiny. [DOI] [PubMed] [Google Scholar]
- [56].Haghverdi Laleh, Lun Aaron T L, Morgan Michael D, and Marioni John C. Batch effects in single-cell RNA-sequencing data are corrected by matching mutual nearest neighbors. Nat Biotechnol, 36(5):421–427, 2018. doi: 10.1038/nbt.4091. URL https://bioconductor.org/packages/batchelor. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Lin Yingxin, Ghazanfar Shila, Wang Kevin Y X, Gagnon-Bartsch Johann A, Lo Kitty K, Su Xianbin, Han Ze-Guang, Ormerod John T, Speed Terence P, Yang Pengyi, and Yang Jean Yee Hwa. scMerge leverages factor analysis, stable expression, and pseudoreplication to merge multiple single-cell RNA-seq datasets. Proc Natl Acad Sci U S A, 116(20), 05 2019. doi: 10.1073/pnas.1820006116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Kiselev Vladimir Yu, Yiu Andrew, and Hemberg Martin. scmap: projection of single-cell RNA-seq data across data sets. Nat Methods, 15(5):359–362, 2018. doi: 10.1038/nmeth.4644. URL https://bioconductor.org/packages/scmap. [DOI] [PubMed] [Google Scholar]
- [59].Traag VA, Waltman L, and van Eck NJ. From Louvain to Leiden: guaranteeing well-connected communities. Scientific Reports, 9(1):5233, March 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Wang Bo, Zhu Junjie, Pierson Emma, Ramazzotti Daniele, and Batzoglou Serafim. Visualization and analysis of single-cell RNA-seq data by kernel-based similarity learning. Nat Methods, 14(4):414–416, 2017. doi: 10.1038/nmeth.4207. URL https://bioconductor.org/packages/SIMLR. [DOI] [PubMed] [Google Scholar]
- [61].Yu Kiselev Vladimir, Kirschner Kristina, Schaub Michael T, Andrews Tallulah, Yiu Andrew, Chandra Tamir, Natarajan Kedar N, Reik Wolf, Barahona Mauricio, Green Anthony R, and Hemberg Martin. SC3: consensus clustering of single-cell RNA-seq data. Nat Methods, 14(5):483–486, 2017. doi: 10.1038/nmeth.4236. URL https://bioconductor.org/packages/SC3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Risso Davide, Purvis Liam, Fletcher Russell B, Das Diya, Ngai John, Dudoit Sandrine, and Purdom Elizabeth. clusterExperiment and RSEC: A Bioconductor package and framework for clustering of singlecell and other large gene expression datasets. PLoS Computational Biology, 14(9):e1006378–16, 09 2018. URL https://bioconductor.org/packages/clusterExperiment. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Van den Berge Koen, Perraudeau Fanny, Soneson Charlotte, Love Michael I, Risso Davide, Vert Jean-Philippe, Robinson Mark D, Dudoit Sandrine, and Clement Lieven. Observation weights unlock bulk RNA-seq tools for zero inflation and single-cell applications. Genome Biol, 19(1):24, 2018. doi: 10.1186/s13059-0181406-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Korthauer Keegan D, Chu Li-Fang, Newton Michael A, Li Yuan, Thomson James, Stewart Ron, and Kendziorski Christina. A statistical approach for identifying differential distributions in single-cell RNA-seq experiments. Genome Biol, 17(1):222, 2016. doi: 10.1186/s13059-016-1077-y. URL https://bioconductor.org/packages/scDD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Soneson Charlotte and Robinson Mark D. Bias, robustness and scalability in single-cell differential expression analysis. Nat Methods, 15(4):255–261, 2018. doi: 10.1038/nmeth.4612. [DOI] [PubMed] [Google Scholar]
- [66].Wang Tianyu, Li Boyang, Nelson Craig E, and Nabavi Sheida. Comparative analysis of differential gene expression analysis tools for single-cell RNA sequencing data. BMC Bioinformatics, 20:40, 01 2019. doi: 10.1186/s12859-019-2599-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Crowell Helena L, Soneson Charlotte, Germain Pierre-Luc, Calini Daniela, Collin Ludovic, Raposo Catarina, Malhotra Dheeraj, and Robinson Mark D. On the discovery of population-specific state transitions from multi-sample multi-condition single-cell RNA sequencing data. 8:e43803–24, July 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Andrews Tallulah S and Hemberg Martin. Identifying cell populations with scRNASeq. Mol Aspects Med, 59:114–122, 02 2018. doi: 10.1016/j.mam.2017.07.002. [DOI] [PubMed] [Google Scholar]
- [69].Qiu Xiaojie, Mao Qi, Tang Ying, Wang Li, Chawla Raghav, Pliner Hannah A, and Trapnell Cole. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods, 14(10):979–982, 2017. doi: 10.1038/nmeth.4402. URL https://bioconductor.org/packages/monocle. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Campbell Kieran R and Yau Christopher. switchde: inference of switch-like differential expression along single-cell trajectories. Bioinformatics, 33(8):1241–1242, 04 2017. doi: 10.1093/bioinformatics/btw798. URL https://bioconductor.org/packages/switchde. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Street Kelly, Risso Davide, Fletcher Russell B, Das Diya, Ngai John, Yosef Nir, Purdom Elizabeth, and Dudoit Sandrine. Slingshot: cell lineage and pseudotime inference for single-cell transcriptomics. BMC Genomics, 19(1):477, 2018. doi: 10.1186/s12864-018-4772-0. URL https://bioconductor.org/packages/slingshot. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].duVerle David A, Yotsukura Sohiya, Nomura Seitaro, Aburatani Hiroyuki, and Tsuda Koji. CellTree: an R/bioconductor package to infer the hierarchical structure of cell populations from single-cell RNA-seq data. BMC Bioinformatics, 17(1):363, 2016. doi: 10.1186/s12859-016-1175-6. URL http://bioconductor.org/packages/cellTree. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Campbell Kieran R and Yau Christopher. Probabilistic modeling of bifurcations in single-cell gene expression data using a bayesian mixture of factor analyzers. Wellcome Open Res, 2:19, 03 2017. doi: 10.12688/wellcomeopenres.11087.1. URL https://bioconductor.org/packages/MFA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Saelens Wouter, Cannoodt Robrecht, Todorov Helena, and Saeys Yvan. A comparison of single-cell trajectory inference methods. Nature Biotechnology, 37(5):547, 2019. doi: 10.1038/s41587-019-0071-9. [DOI] [PubMed] [Google Scholar]
- [75].Subramanian Aravind, Tamayo Pablo, Vamsi K Mootha Sayan Mukherjee, Ebert Benjamin L, Gillette Michael A, Paulovich Amanda, Pomeroy Scott L, Golub Todd R, Lander Eric S, and Mesirov Jill P. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A, 102(43):15545–50, 2005. doi: 10.1073/pnas.0506580102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Kanehisa Minoru, Furumichi Miho, Tanabe Mao, Sato Yoko, and Morishima Kanae. KEGG: new perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res, 45(D1):D353–D361, 2017. doi: 10.1093/nar/gkw1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Fabregat Antonio, Korninger Florian, Hausmann Kerstin, Sidiropoulos Konstantinos, Williams Mark, Garapati Phani, Jupe Steven, Hermjakob Henning, Jassal Bijay, May Bruce, Wu Guanming, Weiser Joel, Rothfels Karen, Milacic Marija, Webber Marissa, Haw Robin, Sheldon McKay Marc Gillespie, Stein Lincoln, Matthews Lisa, Shamovsky Veronica, and Peter D’Eustachio. The Reactome pathway Knowledgebase. Nucleic Acids Research, 44(D1):D481–D487, 12 2015. doi: 10.1093/nar/gkv1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].M Ashburner C Ball A, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, and Sherlock G. Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet, 25(1):25–9, 2000. doi: 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].L Geistlinger G Csaba, and Zimmer R. Bioconductor’s EnrichmentBrowser: seamless navigation through combined results of set and network-based enrichment analysis. BMC Bioinformatics, 17:45, 2016. doi: 10.1186/s12859-016-0884-1. URL https://bioconductor.org/packages/EnrichmentBrowser. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Alhamdoosh Monther, Ng Milica, Wilson Nicholas J, Sheridan Julie M, Huynh Huy, Wilson Michael J, and Ritchie Matthew E. Combining multiple tools outperforms individual methods in gene set enrichment analyses. Bioinformatics, 33(3):414–424, 02 2017. doi: 10.1093/bioinformatics/btw623. URL https://bioconductor.org/packages/EGSEA. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Aibar Sara, González-Blas Carmen Bravo, Moerman Thomas, Huynh-Thu Vân Anh, Imrichova Hana, Hulselmans Gert, Rambow Florian, Marine Jean-Christophe, Geurts Pierre, Aerts Jan, van den Oord Joost, Atak Zeynep Kalender, Wouters Jasper, and Aerts Stein. SCENIC: single-cell regulatory network inference and clustering. Nat Methods, 14(11):1083–1086, 2017. doi: 10.1038/nmeth.4463. URL https://bioconductor.org/packages/AUCell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Buettner Florian, Pratanwanich Naruemon, McCarthy Davis J, Marioni John C, and Stegle Oliver. fscLVM: scalable and versatile factor analysis for single-cell RNA-seq. Genome Biol, 18(1):212, 2017. doi: 10.1186/s13059-017-1334-8. URL https://bioconductor.org/packages/slalom. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Aran Dvir, Agnieszka P Looney Leqian Liu, Wu Esther, Fong Valerie, Hsu Austin, Chak Suzanna, Naikawadi Ram P, Wolters Paul J, Abate Adam R, Butte Atul J, and Bhattacharya Mallar. Reference-based analysis of lung single-cell sequencing reveals a transitional profibrotic macrophage. Nature Immunology, pages 1–15, January 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Zappia Luke, Phipson Belinda, and Oshlack Alicia. Splatter: simulation of single-cell RNA sequencing data. Genome Biol, 18(1):174, 2017. doi: 10.1186/s13059-017-1305-0. URL https://bioconductor.org/packages/splatter. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Kimes Patrick K and Reyes Alejandro. Reproducible and replicable comparisons using SummarizedBenchmark. Bioinformatics, 35(1):137–139, 01 2019. doi: 10.1093/bioinformatics/bty627. URL https://bioconductor.org/packages/SummarizedBenchmark. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Tian Luyi, Dong Xueyi, Freytag Saskia, Lê Cao Kim-Anh, Su Shian, Abolfazl JalalAbadi, AmannZalcenstein Daniela, Weber Tom S, Seidi Azadeh, Jabbari Jafar S, Naik Shalin H, and Ritchie Matthew E. Benchmarking single cell RNA-sequencing analysis pipelines using mixture control experiments. Nat Methods, 16 (6):479–487, 06 2019. doi: 10.1038/s41592-019-0425-8. URL https://www.bioconductor.org/packages/CellBench. [DOI] [PubMed] [Google Scholar]
- [87].K Rue-Albrecht F Marini, Soneson C, and Lun Aaron T L. iSEE: Interactive SummarizedExperiment Explorer. F1000Research, 7:741, 2018. doi: 10.12688/f1000research.14966.1. URL https://bioconductor.org/packages/iSEE. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Peterson Vanessa M, Zhang Kelvin Xi, Kumar Namit, Wong Jerelyn, Li Lixia, Wilson Douglas C, Moore Renee, McClanahan Terrill K, Sadekova Svetlana, and Klappenbach Joel A. Multiplexed quantification of proteins and transcripts in single cells. Nature Biotechnology, 35(10):936–939, October 2017. [DOI] [PubMed] [Google Scholar]
- [89].Dey Siddharth S, Kester Lennart, Spanjaard Bastiaan, Bienko Magda, and van Oudenaarden Alexander. Integrated genome and transcriptome sequencing of the same cell. Nat Biotechnol, 33(3):285–289, 2015. doi: 10.1038/nbt.3129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Macaulay Iain C, Teng Mabel J, Haerty Wilfried, Kumar Parveen, Ponting Chris P, and Voet Thierry. Separation and parallel sequencing of the genomes and transcriptomes of single cells using GT-seq. Nat Protoc, 11(11):2081–103, 2016. doi: 10.1038/nprot.2016.138. [DOI] [PubMed] [Google Scholar]
- [91].Stoeckius Marlon, Hafemeister Christoph, Stephenson William, Houck-Loomis Brian, Chattopadhyay Pratip K, Swerdlow Harold, Satija Rahul, and Smibert Peter. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods, 14(9):865–868, 2017. doi: 10.1038/nmeth.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Shahi Payam, Kim Samuel C, Haliburton John R, Gartner Zev J, and Abate Adam R. Abseq: Ultrahighthroughput single cell protein profiling with droplet microfluidic barcoding. Sci Rep, 7:44447, 2017. doi: 10.1038/srep44447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Angermueller Christof, Clark Stephen J, Lee Heather J, Macaulay Iain C, Teng Mabel J, Hu Tim Xiaoming, Krueger Felix, Smallwood Sebastien, Ponting Chris P, Voet Thierry, Kelsey Gavin, Stegle Oliver, and Reik Wolf. Parallel single-cell sequencing links transcriptional and epigenetic heterogeneity. Nat Methods, 13(3):229–232, 2016. doi: 10.1038/nmeth.3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Cao Junyue, Darren A Cusanovich Vijay Ramani, Aghamirzaie Delasa, Pliner Hannah A, Hill Andrew J, Daza Riza M, McFaline-Figueroa Jose L, Packer Jonathan S, Christiansen Lena, Steemers Frank J, Adey Andrew C, Trapnell Cole, and Shendure Jay. Joint profiling of chromatin accessibility and gene expression in thousands of single cells. Science, 361(6409):1380–1385, 2018. doi: 10.1126/science.aau0730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Clark Stephen J, Argelaguet Ricard, Kapourani Chantriolnt-Andreas, Stubbs Thomas M, Lee Heather J, Alda-Catalinas Celia, Krueger Felix, Sanguinetti Guido, Kelsey Gavin, Marioni John C, Stegle Oliver, and Reik Wolf. scNMT-seq enables joint profiling of chromatin accessibility DNA methylation and transcription in single cells. Nat Commun, 9(1):781, 2018. doi: 10.1038/s41467-018-03149-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Butler Andrew, Hoffman Paul, Smibert Peter, Papalexi Efthymia, and Satija Rahul. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol, 36(5):411–420, 2018. doi: 10.1038/nbt.4096. URL https://CRAN.R-project.org/package=Seurat. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Wolf F Alexander, Angerer Philipp, and Theis Fabian J. SCANPY: large-scale single-cell gene expression data analysis. Genome Biol, 19(1):15, 2018. doi: 10.1186/s13059-017-1382-0. URL https://github.com/theislab/scanpy. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Eddelbuettel Dirk and François Romain. Rcpp: Seamless R and C++ integration. Journal of Statistical Software, 40(8):1–18, 2011. doi: 10.18637/jss.v040.i08. URL https://CRAN.R-project.org/package=Rcpp. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.