

Abstract
Alzheimer’s Disease (AD) is the most common cause of dementia, affecting 25 million people worldwide. Accumulation of Amyloid-β (Aβ) in the mitochondria has been shown to adversely affect key enzymes including pyruvate dehydrogenase (PDH), succinate dehydrogenase (SDH), oxoglutarate dehydrogenase (OGDH). Accumulation of Aβ is also believed to increase Tau expression and pathology. Tau, in its toxic state, results in synaptic damage causing memory and cognitive dysfunction. We are developing a drug to treat AD namely AmyTrap. The active pharmacological ingredient is a retro inverso, Aβ-binding peptide which sequesters Aβ. We wanted to examine the effect of AmyTrap peptide on Aβ-induced mitochondrial dysfunction and Tau phosphorylation. Therefore, we treated SH-SY5Y neuroblastoma cells with wild-type Aβ, a mutant AβY10A, AmyTrap peptide (RI-peptide), or Aβ and RI-peptide for 72 hours. The mutant AβY10A is known to impact the self-aggregating property of Aβ as this Tyr10 is essential for self-aggregation. As expected, AβY10A reversed PDH, OGDH and SDH dysfunction to near normal levels. Further, AβY10A successfully reversed Tau phosphorylation, suggesting that Tyr10 is also associated with Aβ-induced cytotoxicity. RI-peptide was able to significantly reverse SDH dysfunction with limited effect on PDH and Tau phosphorylation. The findings are suggestive that the Tyr10 on Aβ plays a critical role in the self-aggregation. Further studies are warranted to expand these findings.
Keywords: Alzheimer’s Disease, Amyloid, Mitochondria, Retro-inverso peptide, SH-SY5Y neuroblastoma cells, AmyTrap
1. Introduction
Alzheimer’s Disease (AD) is the most common cause of dementia, affecting about 25 million people worldwide and regarded as the fourth most common cause of death. The incidence of AD and associated costs are predicted to increase due to increasing life expectancy and average age [Alzheimer’s Association, 2012; Wilson et al., 2011], while the number of AD patients is expected to double over the next 20 years [Alzheimer’s Association 2006]. Biochemical, neuropathological and genetic data available till-date suggests that beta-amyloid (Aβ) accumulation in the neurons precedes its deposit in the extracellular space. Upon accumulation in the intracellular space, Aβ was reported to disrupt the normal functioning of neurons leading to oxidative injury and apoptosis [Chen and Yan, 2007; A Massad, 2011], even before the formation of senile plaques extracellularly and neurofibrillary tangles. Therefore, it is hypothesized that the intra-neuronal Aβ accumulation is the first step of a fatal cascade of events leading to neurodegeneration in AD [Wirths et al., 2004]. Further, it has been reported that mitochondrial abnormalities such as dystrophic neurites, the loss of dendritic branches and the pathological alteration of the dendritic spines in the brains of AD cases [Palop and Mucke, 2010; Parthsarathy et al., 2013; Rijal et al., 2013; Xie et al., 2013] are correlated with intra-neuronal accumulation of Aβ. Based on these observations, the mitochondrial cascade hypothesis was proposed [Xie et al., 2013] to explain late-onset, sporadic AD, stating that Aβ deposition, neurofibrillary tangle formation and neurodegeneration are consequent to mitochondrial malfunctioning.
Research has shown that intracellular Aβ is imported into the mitochondria disrupting several key enzymes, resulting in oxidative stress, mitochondrial dysfunction, and finally apoptosis of neuronal cells [Chen and Yan, 2007; A Massad, 2011; Manyevitch et al., 2018]. In addition to Aβ, intracellular accumulation of Tau is also noted. Intracellular accumulation of hyper-phosphorylated Tau negatively regulates the binding of Tau to microtubules, resulting in microtubule destabilization and impaired axonal transport. Aβ accumulation precedes Tau, thus placing Tau downstream of Aβ in the patho-cascade of AD, providing support for the amyloid cascade hypothesis [Fein et al., 2008]. An important observation favoring Aβ is the promising efficacy of antibodies in a set of patient population. In trials with three different Aβ monoclonal antibodies including Solanezumab, Crenezumab, and Aducanumab that have been investigated for the treatment of AD by Eli Lilly, Roche, and Biogen, respectively [Gandbhir and Sundaram et al., 2019], post-hoc analysis suggested a slowing of cognitive decline in mild AD subjects. Especially interesting, a clinical study with Aducanumab (Biogen) that included patients with mild to moderate AD showed a cognitive benefit with a significant reduction in Aβ burden in the brain. Furthermore, a phase 2 Alzheimer’s trial once nearly added to the number of disappointing attempts against the disease has re-emerged with new positive results, showing that an anti-amyloid beta protofibril antibody can slow clinical symptom decline, and reduce the accumulation of plaque in the brain [Kresge, 2018]. As stated in the article, “the prospect of being able to offer meaningful disease-modifying therapies to individuals suffering from this terrible disease is both exciting and humbling” [Kresge, 2018]. These positive results provide motivation and support for targeting and treating Aβ for AD. Based on the foregoing facts, it is important to understand the role of mitochondrial enzymes in the stimulation of processing and accumulation of intracellular Aβ in health and disease. As a first step, we set out to study the effect of Aβ (wild type and mutant) incubation on a clinically relevant neuroblastoma cell line and the role of our amyloid binding drug AmyTrap peptide in altering any observed abnormalities.
Import and accumulation of Aβ in the mitochondria has been shown to effect key enzymes including pyruvate dehydrogenase (PDH), isocitrate dehydrogenase, and oxoglutarate dehydrogenase (OGDH), which undergo enzymatic dysfunction in AD [Chen and Yan, 2007]. These dysfunctions prevent the production of ATP, depleting cellular energy leading to synaptic abnormalities that result in neuronal degeneration [Pagani and Eckert, 2011; Bubber et al., 2005]. Both PDH and OGDH have been thoroughly studied along while other key enzymes have known dysfunction but are not as focused upon. While PDH and OGDH activity have been shown to decrease, metabolic enzymes succinate dehydrogenase (SDH) and malate dehydrogenase (MDH) activity have been shown to increase in the presence of AD [Pagani and Eckert 2011]. Though the first half of enzymes in the tricarboxylic acid (TCA) cycle decreases in activity other pathways bypass PDH and OGDH to feed directly into SDH and MDH reactions. Bypassing the initial steps of TCA cycle may increase the enzymatic activity of SDH and MDH. While there are many treatments being tested against AD there is no treatment available in AD that reverses this metabolic dysfunction. Thus, drugs that can be used to treat AD by targeting and sequestering Aβ may be able to reverse enzymatic activity to normal levels.
As the pathophysiology of AD is very complex, other mechanisms and targets beyond Aβ and Tau (e.g. inflammation, ER stress, and autophagy) are hypothesized to be the causation of AD. The future of AD treatment will likely be in combination therapies that include targeting amyloid, Tau, and modulators of additional mechanisms for maximum efficacy. Therefore, even if anti-amyloid therapies individually may not cure AD, they are very likely to show disease modifying properties and thus are expected to remain as part of the combination therapies in future.
It is understandable that since Aβ accumulates in cells and initiates AD pathology even before the earliest manifestation of extracellular oligomerization and deposition of Aβ in brain, an ideal drug should be able to bind and inhibit intracellular Aβ effectively [Matharu et al., 2010]. We have developed an Aβ-binding peptide AmyTrap and demonstrated its ability to bind Aβ in vitro by enzyme -linked immunosorbent assay (ELISA) and in vivo by restoring memory functions in AD mice when systemically administered [Gandbhir and Sundaram, 2019; Sundaram et al., 2008, 2012]. Our active pharmacological ingredient (API) is AmyTrap, an Aβ-capturing retro-inverso (RI) peptide (Fig. 1). The API was designed as a tetrameric peptide with a high affinity for a specific GXXXG motif in Aβ. We envisage that binding of intracellular Aβ to AmyTrap will inhibit Aβ induced mitochondrial dysfunction, Tau accumulation, and apoptosis of neurons. By intracellular binding and inhibition, the drug could prevent Aβ’s interaction with the mitochondrial proteins which in turn could strengthen the mitochondrial function. Thus, the intracellular amyloid binding drug can achieve double the benefit such as amyloid removal and thereby relieving the load on the mitochondrial function. Therefore, in the present study we attempted to examine the effect of AmyTrap on Aβ induced cytotoxicity and mitochondrial enzymes. We used the well-established neuroblastoma cell culture model, SH-SY5Y [Agholme et al., 2010], to answer these questions. The model expresses neurospecific markers along with apparent neuronal morphology [Agholme et al., 2010]. The model has been utilized in many in vitro studies for toxicity studies in AD and other neurodegenerative diseases to address similar questions. The results presented here represent the extended pharmacological findings published recently [Gandbhir and Sundaram et al., 2019] on AmyTrap. Inhibition assays were performed to identify peak concentrations of AmyTrap peptide that inhibited Aβ42 cytotoxicity [Gandbhir and Sundaram et al., 2019]. In vivo safety, pharmacokinetics, and efficacy were also performed to evaluate AmyTrap function [Gandbhir and Sundaram et al., 2019].
Fig 1. The Amytrap peptide.
An Aβ binding retro-inverso tetrameric peptide consisting of 4 chains of the same synthetic peptide that are linked to Lysine and Cysteine
2. Methods
2.1. Structure of AmyTrap peptide
AmyTrap peptide was designed as a tetrameric retro-inverso peptide as presented in a recently performed study [Gandbhir and Sundaram et al., 2019]. The peptide was synthesized with D-amino acids in the sequence WKGEWTGR at LifeTein (NJ, USA). The tetrameric peptide consists of 4 chains of the above sequence that are linked together by Lysine and Cysteine [Fig. 1]. Purity of AmyTrap was evaluated and confirmed by HPLC and mass spectrometry. Functional binding of Aβ by AmyTrap was tested by ELISA and cellular-based assays.
2.2. Cell viability Assay
SH-SY5Y neuroblastoma cells purchased from ATCC (VA, USA) were grown, sub-cultured and treated with different concentrations (1, 5, 10, 20, 50, 100 μM) of pre-aggregated Aβ42 with or without the highest concentration of API that was not cytotoxic [Gandbhir and Sundaram et al., 2019]. Cell cultures were incubated for 72 hrs. and cell viability was assessed using CellTiterGlo (CTG) reagent from Promega. The cell growth inhibition from the cells that are exposed only to pre-aggregated Aβ42 was used to estimate the concentration of Aβ42 that results in 50% inhibition (IC50). The IC50 for Aβ42 was derived by dividing the mean cell growth by the mean blanked control cell growth for each concentration, then multiplied by 100. The percent toxicity data was fitted for the 4-Parameter (4P) curve, with interpolation set to 50. The concentration of the Aβ42 that results in 50% inhibition was calculated from this curve. A second experiment was done where SH-SY5Y cells were incubated with Aβ42 at IC50 and with different concentrations (1, 5, 10, 20, 50, 100, 200 μM) of API. Cell viability was measured, and IC50 for ATC is estimated as described [Gandbhir and Sundaram et al., 2019].
2.3. Mitochondrial Dysfunction
Derived from the cell viability data, 1 × 106 cells were seeded into wells and treated with AmyTrap peptide (RI-peptide; 10 μM), wild-type Aβ42 (4 μM), mutant Aβ42Y10A (4 μM) or a combination of RI-peptide (10 μM) and wild-type Aβ42 (4 μM). Wild-type and mutant Aβ42 were purchased from Anaspec [CA] for these experiments. Previous studies performed with the tyrosine mutant has identified a necessity for tyrosine in ROS formation which is observed in wild-type Aβ42 [Coskuner & Uversky, 2017]. Cells were treated for 72 hours in a 37°C, CO2 incubator. Cell viability was measured at the end of the experiment for cytotoxicity assessment by CTG reagent. All the treatments were done in triplicate and total cell extracts of SH-SY5Y cells were prepared. Following the treatments and incubations, cells were washed with phosphate buffered saline (PBS) and lysed in mammalian protein extraction reagent (M-PER; ThermoFisher Scientific, MO). Debris was removed by centrifugation (12,000 × g for 3 minutes). Protein estimation was performed on lysates through bicinchoninic acid (BCA) assay as per manufacturer’s instructions (Thermo Scientific; Pierce™ BCA Protein Assay kit). Equal concentration of protein extracts (20 μg) were prepared and resolved on 4 – 12 % SDS-PAGE gel. Resolved bands were transferred on to a polyvinylidene difluoride (PVDF) membrane and blocked in 5% non-fat milk in TBST for one hour. The membrane was then washed three times, 5 minutes each with TBST, and probed with the primary antibody (1:1000 diluted in 0.1% bovine serum albumin (BSA) in TBST) overnight. The primary antibodies - PDH, ACL, and OGDH were purchased from Cell Signal Technologies, MA. USA. After incubation, the blots were washed three times, 5 minutes each with TBST, and an HRP-tagged secondary antibody (1:10,000 diluted in 0.1% BSA in TBST; Cell Signal Technologies) was used to probe for one hour, then washed three times, 5 minutes each with TBST. ECL Western Blotting Substrate (Pierce™) was used to detect chemiluminescence and captured the signal on film. An internal control utilizing glyceraldehyde 3-phosphate dehydrogenase (GAPDH) was analyzed by stripping the PVDF membranes and probing with anti-GAPDH antibody (1:10,000; Proteintech®). The membranes were washed and probed with an HRP-tagged anti-mouse antibody. Bands were imaged and captured as previously stated. Bands corresponding to the predicted molecular weight size were processed and analyzed through ImageJ software (National Institutes of Health and the Laboratory for Optical and Computational instrumentation). Once the area under the curve and band intensity was determined, the percent difference was calculated against the untreated value for each mitochondrial enzyme and graphed.
2.4. Tau Phosphorylation
Tau phosphorylation in treated cells was detected through western blot which was performed as described in section 2.3. Neuroblastoma cell extracts of equal amounts (20 μg) were resolved on an SDS-PAGE gel, transferred to a PVDF membrane and probed with a monoclonal antibody specific for phosphorylated Tau (phospho-Tau) protein (Cell Signal Technologies, MA, USA). The blot was probed with an HRP-tagged secondary antibody as described in section 2.3. Bands corresponding to the predicted molecular weight were processed and analyzed through ImageJ software as described in section 2.3. Once the area under the curve and band intensity was determined, the percent difference was calculated against the untreated value for each mitochondrial enzyme.
2.5. AmyTrap peptide Cellular Penetration
AmyTrap cellular penetration was analyzed by Western blot. 1 × 106 cells were seeded into wells and treated with AmyTrap peptide (RI-peptide) (10 μM) or the combination of RI-peptide and wild-type Aβ42 as described in section 2.3. Each peptide was reconstituted in filtered distilled water. Cells were treated for 72 hours in a 37°C, CO2 incubator. Cell viability was measured at the end of the experiment for cytotoxicity assessment by CTG reagent. All the treatments were done in triplicate and total cell extracts of SH-SY5Y cells were prepared. Following the treatments and incubations, cells were washed with phosphate buffered saline (PBS) and lysed in mammalian protein extraction reagent (M-PER; ThermoFisher Scientific, MO). Debris was removed by centrifugation (12,000 × g for 3 minutes). Protein estimation was performed on lysates through bicinchoninic acid (BCA) assay as per manufacturer’s instructions. Western blotting was carried out as in section 2.3 with whole cell protein extracts (20 μg) The membrane was probed with a high titer rabbit derived anti-AmyTrap peptide primary antibody (1:5,000 in 0.1% BSA in TBST; 17) and a secondary HRP conjugated anti-rabbit antibody (1:10,000 in 0.1% BSA in TBST). The values obtained from ImageJ analysis were graphed as the average for each treatment from triplicate experiments.
3. Results
Preliminary studies had established that the API (active pharmacological ingredient from AmyTrap peptide) was capable of binding and inhibiting Aβ42 both in vitro and in vivo. The main goal of the current study was to assess the ability of the AmyTrap peptide in protecting against the Aβ induced mitochondrial dysfunction via a cellular toxicity assay. To address this, SH-SY5Y cells, a well-studied neuroblastoma cell line, were treated with pre-aggregated Aβ42, and the API at non-toxic concentrations. As a control, the highest concentration of AmyTrap peptide was tested for inhibitory activity, if any, on the induced toxicity by pre-aggregated Aβ42. This was performed to examine the maximum drug dose the cells can tolerate. At the end of the experiment, cell lysates were prepared. Metabolic enzymes were measured in cell lysates. In addition, analysis was performed on lysates to detect any changes in Tau phosphorylation status. If there was any observed inhibition of Tau phosphorylation, more support would be generated for the targeting of Aβ as the primary target in AD.
3.1. Expression of Metabolic enzymes
The ability of AmyTrap to protect against Aβ induced mitochondrial dysfunction was assessed in SH-SY5Y neuroblastoma cells. Cells were treated with pre-aggregated Aβ42, mutant Aβ42Y10A and the API at non-toxic concentrations. Extracts were tested for expression of key mitochondrial enzyme proteins by western blot as follows: pyruvate dehydrogenase (PDH), ATP-citrate lyase (ACL), and oxoglutarate dehydrogenase (OGDH), succinate dehydrogenase (SDH) and fumarase. The results from this test including the images are presented in figure 2 and indicate expression as band intensity. Enzyme levels for the Untreated cells were used to calculate changes in each treatment group and are presented below as a percentage. Compared to the control, pyruvate dehydrogenase expression was significantly higher after treatment with Aβ (↑ 73 %, “*” p < 0.05) or Aβ and AmyTrap peptide (RI-peptide) combination (↑ 72 %, “*” p < 0.05). The mutant AβY10A treatment significantly reduced expression of PDH (↓ 20 %, “*” p < 0.05). ATP citrate lyase expression was significantly reduced after treatment with AβY10A (↓ 17 %, “**” p < 0.01) or the combination treatment (↓ 20 %, “***” p < 0.001) in comparison with the untreated group. Wild-type Aβ treatment did not significantly affect the expression of ATP citrate lyase. Succinate dehydrogenase expression was significantly elevated after exposure to Aβ (↑ 250 %, “**” p < 0.01) and the combination treatment reversed this expression to near control levels (↑ 57 %, “*” p < 0.05). Interestingly, exposure to the mutant AβY10A produced near normal levels of SDH. OGDH expression was significantly elevated after cells were exposed to Aβ (↑ 11.6 %, “*” p < 0.05) and further elevated by the combination treatment (↑ 53 %, “**” p < 0.01) in comparison with the Untreated group. Mutant AβY10A treatment did not have any significant change on OGDH expression.
Fig 2. Analysis of Expression of Mitochondrial Enzymes.
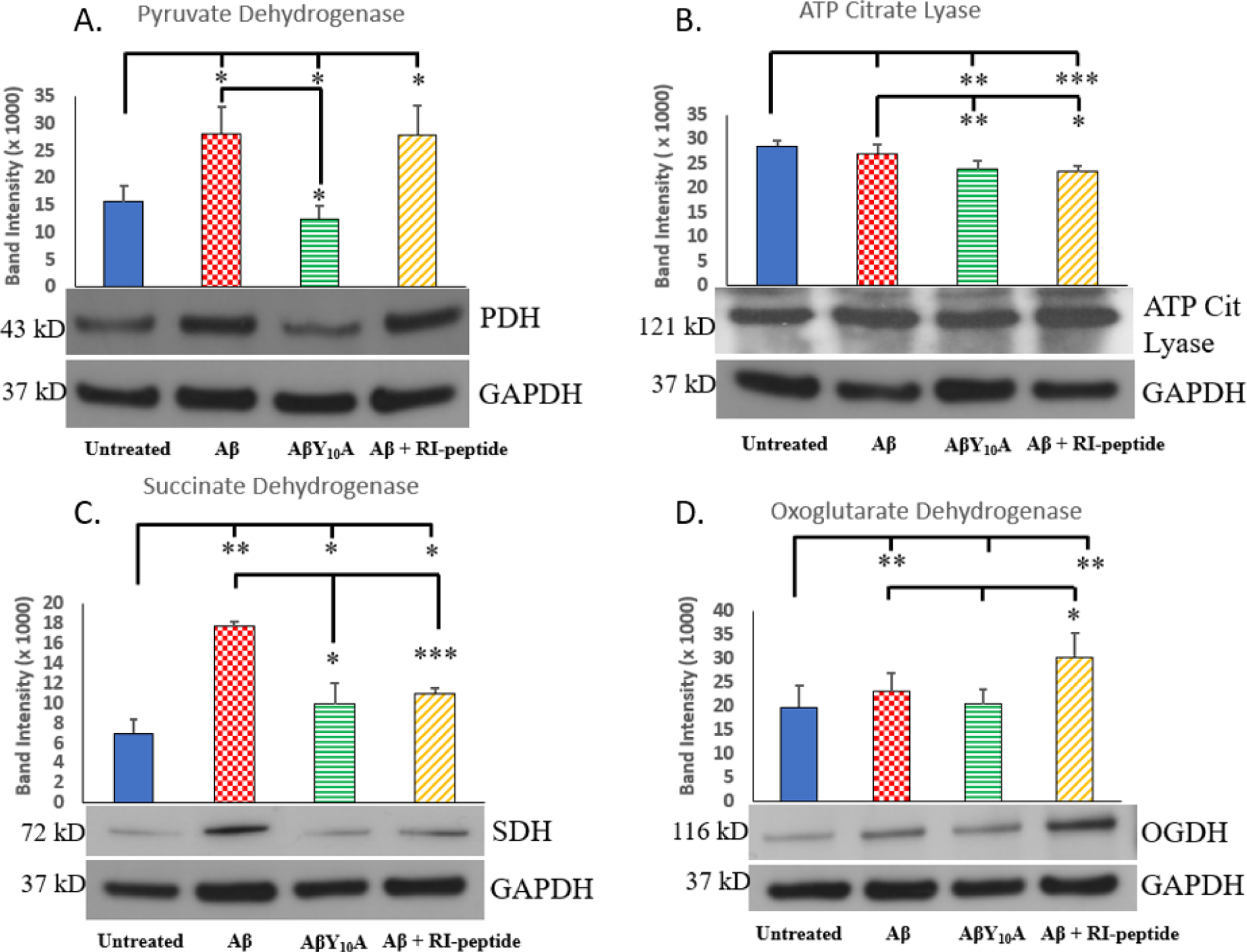
Each enzyme is represented by a representative immunoblot and graph of the band intensity (A) Pyruvate Dehydrogenase (43 kDa), (B) ATP Citrate Lyase (121 kDa), (C) Succinate Dehydrogenase (72 kDa), (D) Oxoglutarate Dehydrogenase (116 kDa). Treatment groups are Untreated, wild-type Aβ42 (4 μM), mutant AβY10A (4 μM) or a combination of Aβ42 (4 μM) and RI-peptide (10 μM). Band intensities are from 3 different experiments and values are represented as Mean ± SD. Significance was calculated by Student’s t-test against the untreated neuroblastoma group for all treatments and then between Aβ and AβY10A or Aβ and Aβ + RI-peptide. A * indicates p < 0.05, ** indicates p < 0.01 and *** indicates p < 0.001.
3.2. Tau expression
To examine if intracellularly targeted AmyTrap has the ability to inhibit intracellular Aβ-induced Tau phosphorylation, the levels of phosphorylated and non-phosphorylated forms of Tau protein (pSer400-Tau, and pThr231-Tau) in harvested cells were analyzed. SH-SY5Y cells were treated with Aβ, Aβ Y10A mutant or Aβ and RI-peptide combination for 72 h and assessed the phosphorylation of Tau by performing western blotting using an antibody specific to Tau (Tau-Ser400) phosphorylation. Expression of phosphorylated Tau-Ser400 was significantly higher after treatment with Aβ (↑ 12.3 %, “*” p < 0.05; Fig. 3). Treatment with AβY10A reversed Tau phosphorylation suggesting Tyr10 is somehow involved in aiding the phosphorylation of Ser-400. After the combination treatment, phospho-Tau was elevated (↑ 10 %, “**” p < 0.01) but was not greater than the wild-type Aβ treatment in neuroblastoma cells. This observation suggested that the mechanism of action of our RI-peptide may not greatly influence Aβ-induced Tau phosphorylation.
Fig 3. Expression of phosphorylated Tau.
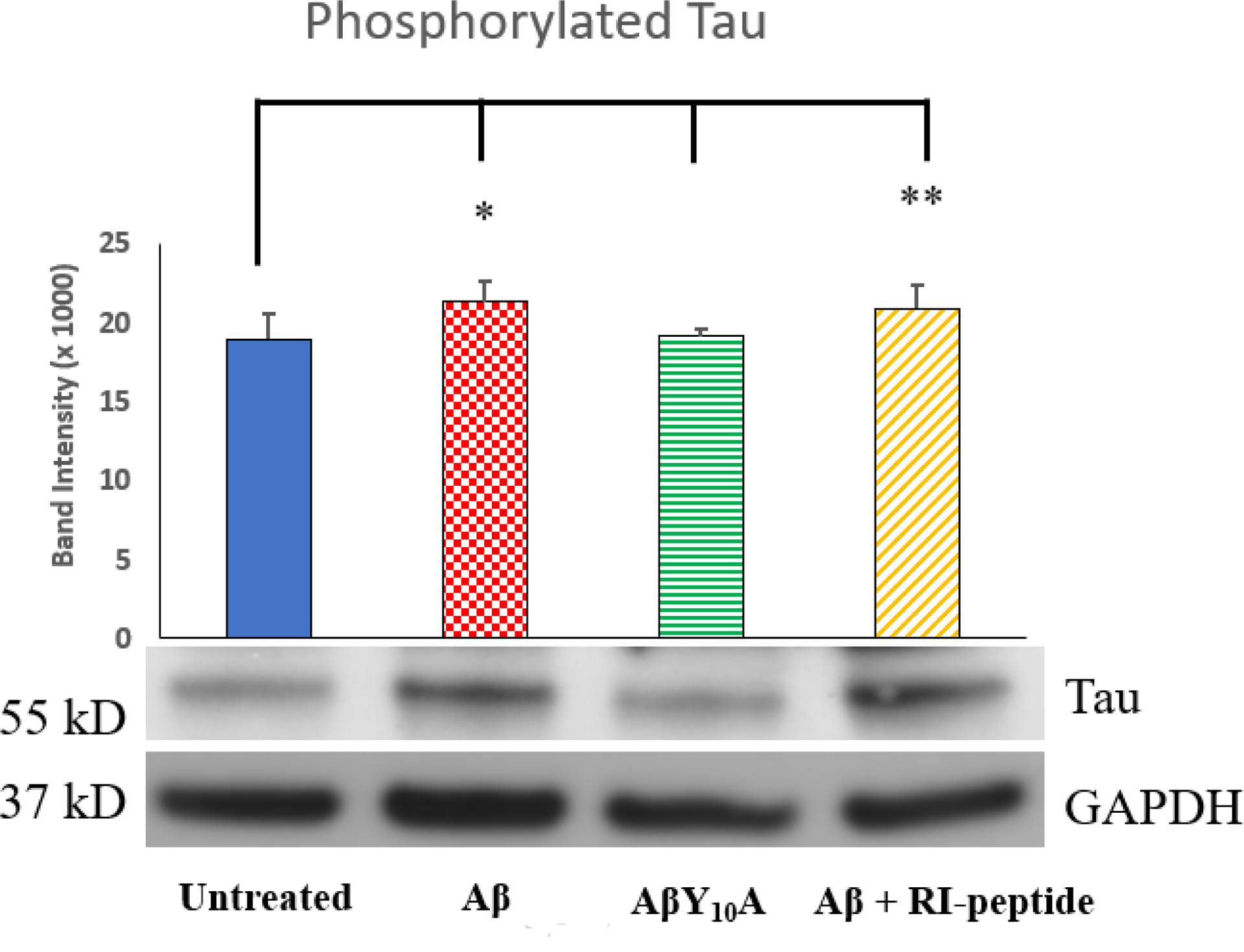
Analysis of phosphorylated Tau was performed through ImageJ analysis. The images of the immunoblot of Tau is shown below the graph. Treatment groups are Untreated, wild-type Aβ42 (4 μM), mutant AβY10A (4 μM) or a combination of Aβ42 (4 μM) and RI-peptide (10 μM). Significance was calculated against Untreated Neuroblastoma group by Student’s t-test. Experiments were performed in triplicate. Values are represented as Mean ± SD. A * indicates p < 0.05, ** indicates p < 0.01.
3.3. Intracellular Accumulation of AmyTrap peptide
To examine if AmyTrap peptide enters the cell, extracts were analyzed by Western Blot utilizing a home-made, high titer anti-AmyTrap antibody [Gandbhir and Sundaram et al., 2019]. Harvested SH-SY5Y cells treated with AmyTrap peptide or the combination treatment of AmyTrap peptide and Aβ42 were probed. AmyTrap peptide (100 ng) loaded directly served as the positive control. Intracellular AmyTrap peptide was detectable in cells treated with AmyTrap peptide alone or the combination treatment. Cells treated with AmyTrap peptide alone were found to have elevated levels of the peptide that were less than 100 ng when compared with the positive control. Greater levels of AmyTrap peptide were detected in the combination treatment and was observed to be significantly higher than the treatment with AmyTrap peptide alone (Fig 4. “**” p < 0.01).
Fig 4. Intracellular Accumulation of Amytrap (RI-Peptide).
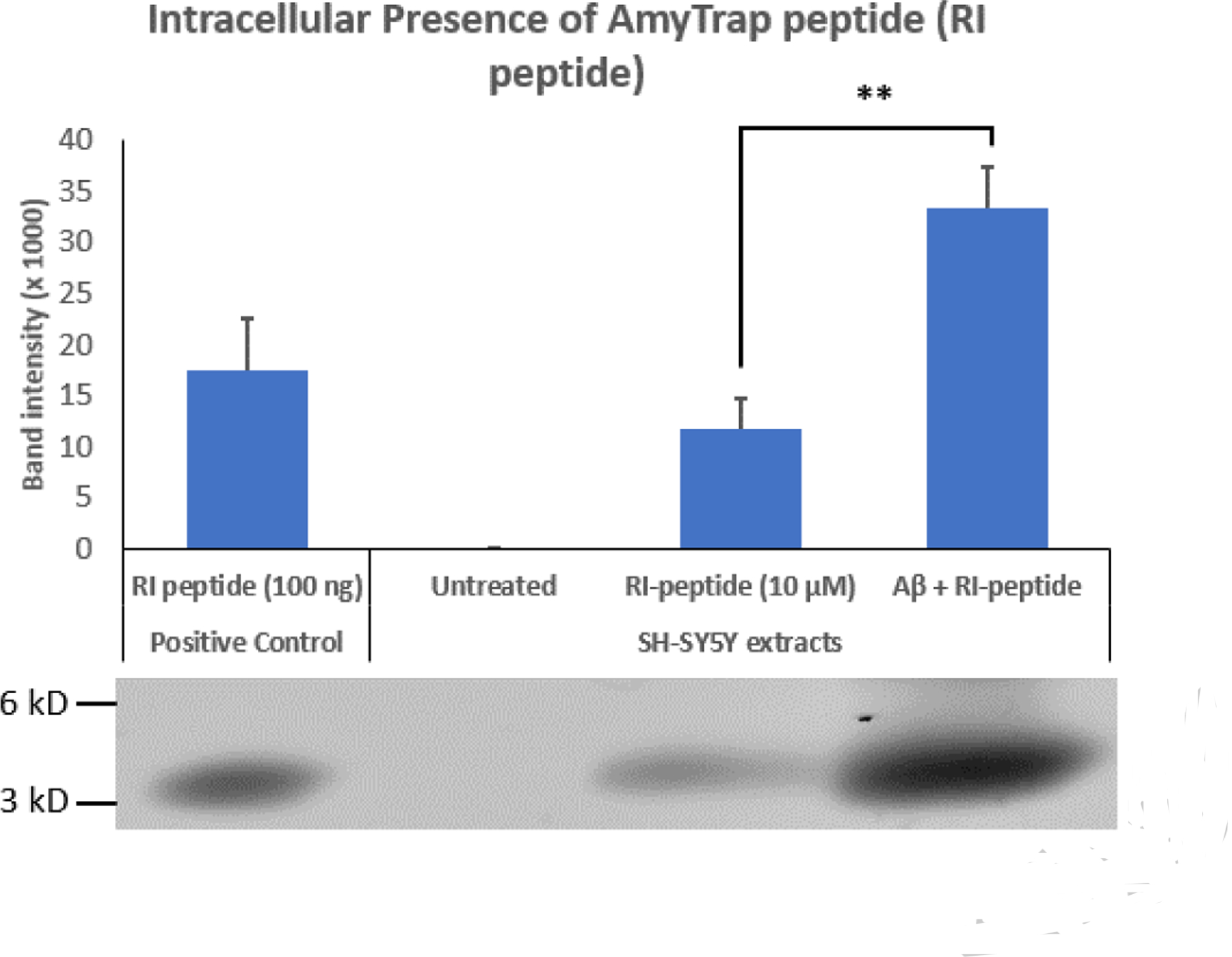
Analysis of the presence of intracellular RI-peptide was performed through ImageJ. Treatment groups are Untreated, RI-peptide (10 μM), or a combination of Aβ42 (4 μM) and RI-peptide (10 μM). A positive control consisting of a known concentration of RI-peptide (100 ng) was utilized to evaluate the intracellular concentration of peptide observed between treatment groups. The image of the immunoblot of RI-peptide is shown below the graph. Significance was calculated against Untreated Neuroblastoma group by Student’s t-test. Experiments were performed in triplicate. Values are represented as Mean ± SD. A ** indicates p < 0.01.
4. Discussion
Aβ plays a major role in the pathogenesis of AD causing cognitive and neuronal damage. Intracellular Aβ has been shown to induce severe damage to the mitochondria through Tau accumulation and dysfunction of metabolic enzymes. We have been working on developing a drug to treat AD, namely, AmyTrap. Our proprietary AmyTrap peptide binds and removes Aβ from the system as observed in a recent study [Gandbhir and Sundaram et al., 2019]. The API, AmyTrap peptide, has been shown to recover cognitive function in vivo [Gandbhir and Sundaram et al., 2019]. Besides, we observed that AmyTrap up to 20 μM concentration significantly countered the toxic effects of Aβ42 in vitro. These results are consistent with studies performed on different retro-inverso peptides in reversing Aβ42 toxicity [Parthsarathy et al., 2013; Coskuner and Uverskty 2017]. The present study is a logic extension of this recent study. We set out to test our drug to see if it is able to reverse mitochondrial dysfunction associated with Aβ42 exposure. We chose to address these questions utilizing an established neuroblastoma cell culture system. By inhibiting Aβ and reducing toxicity, AmyTrap may be able to reverse the effect on mitochondrial dysfunction, Tau phosphorylation and Aβ accumulation. Aβ accumulation within the mitochondria has been shown to induce dysfunction of many key mitochondrial enzymes [Chen and Yan 2007; Wirths et al., 2004; Pagani and Eckert, 2011; Mossmann et al., 2014; Swerdlow et al., 2017]. In order to test if the AmyTrap peptide was able to recover mitochondrial enzyme function and reverse phosphorylation of Tau, the neuroblastoma cell line, SH-SY5Y was used. Treatments were performed with wild type Aβ, a mutant AβY10A, AmyTrap and a combination of Aβ and AmyTrap. The mutant AβY10A was tested to examine the effect of Tyrosine10 in Tau phosphorylation. Tyr10 has been implicated in the folding and beta sheet formation of Aβ [Coskuner and Uverskty 2017].
In an earlier study we have demonstrated that AmyTrap peptide up to 20 μM is capable of reversing cellular cytotoxicity. Therefore, in the present study we wanted to determine the mechanism of this reversal. Hence, the presence of intracellular AmyTrap peptide was analyzed by immunoblotting. The results indicate that AmyTrap peptide was able to penetrate and accumulate within SH-SY5Y cells, either alone or in combination treatment. Interestingly, in the combination treatment, it seems that the presence of amyloid may drive the intracellular penetration of AmyTrap peptide. This is likely because of AmyTrap’s high affinity towards Aβ. These results are consistent with previous reports of improved cellular penetration of D-peptides due to stability and protease resistance [Najjar et al., 2017]. Under these circumstances, pretreatment with AmyTrap may be beneficial in preventing long-term intracellular accumulation of Aβ42 and associated cytotoxicity.
For the analyses of mitochondrial enzyme dysfunction, the following four enzymes were tested: pyruvate dehydrogenase (PDH), oxoglutarate dehydrogenase (OGDH), ATP citrate lyase (ACL) and succinate dehydrogenase (SDH). These enzymes serve as indicators of mitochondrial health status, and hence are altered in AD pathology [Chen and Yan, 2007; Mossmann et al., 2014; Glabe and Kayed, 2006]. Expression of PDH, SDH and OGDH were elevated significantly upon wild type Aβ treatment, and this was reversed by the mutant AβY10A (Fig. 2). Overexpression of these enzymes may be due to a possible modified TCA cycle wherein an alternate pathway utilizes OGDH instead of PDH due to low pyruvate concentration [Manyevitch et al., 2018; Bubber et al., 2005]. The shunt further utilizes SDH, Fumarase and MDH, with oxaloacetate and glutamate acting as substrates, to produce energy. However, expression of these enzymes (PDH and OGDH) were only partially affected by the combination treatment. AmyTrap and the mutant AβY10A were able to reverse expression of SDH to near normal levels. In addition, evidence from other studies have shown that increased SDH expression is a major cause of oxidative stress which further damages neuronal cells [Farshbaf and Kiani-Esfahani, 2017]. Due to Aβ42 induced cytotoxicity, the cells may have initiated oxidative stress responses which in turn further induced SDH dysfunction, but AmyTrap sequestering Aβ42 may have inhibited this stress response by inhibiting SDH over expression. Cells treated with the mutant or combination treatment reversed expression to near normal levels. PDH expression and OGDH expression did not seem to be reversed by AmyTrap peptide in the combination treatment. It is possible that AmyTrap’s activity may be to lessen the impact of amyloid cytotoxicity on the cells, thus allowing cellular recovery of mitochondrial dysfunction. Further analyses may be necessary to determine if AmyTrap in the combination treatment benefits the cells at regulatory sites for these specific enzymes. These results support that AmyTrap is able to partially reverse Aβ-induced mitochondrial dysfunction. AmyTrap may likely disrupt amyloid folding and aggregation which in turn reduces the cytotoxicity.
Interestingly, ATP citrate lyase expression was not positively affected by any treatment (Fig. 2B), suggesting that ATP citrate lyase dysfunction may be affected downstream or indirectly by Aβ-induced toxicity. Both the mutant and the combination treatments were observed to have less expression of ATP citrate lyase than the wild-type or untreated cells. Furthermore, the effect of Aβ on the mitochondrial enzymes may result in the neuroblastoma cells shifting their source of energy by shifting to alternate pathways. The mutant AβY10A treatment was able to reverse the effect of wild-type Aβ, suggesting that the Tyrosine at position 10 may play a role in mitochondrial enzyme dysfunction. This Tyrosine could also be involved in the folding and self-aggregation of wild type Aβ and that when mutated prevents this structural requirement for its toxicity [Kummer et al., 2011]. Previous studies have established that by mutating this Tyrosine to an alanine, the resultant mutant Aβ42Y10A fibrillation was diminished [Coskuner and Uversky, 2017] Additional studies with varying concentration of RI-peptide may be necessary to determine additional changes associated with Aβ exposure.
Tau phosphorylation was examined by analysis of neuroblastoma cell extracts utilizing a specific anti-phospho-Tau antibody. Tau phosphorylation plays an important role in AD progression, causing microtubule destabilization and impaired axonal transport. In AD, phospho-Tau levels are reported to be high [Gong and Iqbal, 2008]. Significant elevation of Tau phosphorylation was observed in Aβ treated cells, which was completely reversed by the mutant AβY10A (Fig. 3). The single mutation changing Tyrosine to an alanine at position 10 has resulted in normalized Tau phosphorylation suggests that Tyr10 plays a key role in Aβ induced Tau phosphorylation. However, it appears that Aβ induced phosphorylation of Tau is not affected by AmyTrap in the combination treatment. Thus, further analysis would be needed to pinpoint the mechanism of Tau phosphorylation in relation to these treatments. Tau phosphorylation was not significantly reduced by AmyTrap treatment in comparison with Aβ treated cells. This lack of benefit suggests that AmyTrap does not inhibit phosphorylation induced by Aβ cytotoxicity. However, AmyTrap may hinder phosphorylation as cells treated with wild-type Aβ were observed to have slightly higher levels of Tau (12.5 % more than Untreated). It is possible that long-term cell treatment may be necessary to observe a greater inhibition of Tau phosphorylation
4.1. Conclusion
The goal of the study was to establish the influence of our proprietary therapeutic peptide, AmyTrap, on mitochondrial dysfunction and Tau phosphorylation caused by Aβ toxicity. Intracellular presence of AmyTrap peptide is a positive sign that AmyTrap not only penetrates the cell membrane but remains intact and present for up to 72 hours. AmyTrap peptide was able to partially reverse PDH dysfunction and reversed SDH dysfunction to near normal levels but does not reverse Tau phosphorylation. These results indicate that AmyTrap peptide treatment may prevent and or partially reverse Aβ42-induced mitochondrial dysfunction but does not affect Tau phosphorylation. The reduction in SDH expression was a great success as previous studies have supported that SDH expression is increased in AD pathology [Bubber et al., 2005]. Mutant AβY10A presented interesting results on expression of both mitochondrial enzymes and Tau phosphorylation. Changing the Tyrosine at position 10 to an Alanine significantly affected the expression of these key enzymes, suggesting that this Tyrosine may play a key role for Aβ toxicity in AD. Further testing will be necessary to evaluate the role of Tyrosine in both phosphorylation and mitochondrial dysfunction. It appears that alternate pathways of TCA cycle are triggered in response to Aβ cytotoxicity. Phosphorylation is one of many types of translational modifications that occur with enzymes. These studies suggest that AmyTrap has a partial effect on the reversal of mitochondrial dysfunction which encourages further examination of the therapeutic properties that it presents against AD.
5. Acknowledgements
Partial salary support to Dr. Pazhani Sundaram and Omkar Gandbhir was provided by a small business innovative research phase II grant # R44 AG0 50336, from National Institute on Aging, National Institutes of Health, USA. Dr. P.S., the principal investigator and corresponding author thanks NIH for the grant award.
Abbreviations:
- AD
Alzheimer’s Disease
- Aβ
beta-amyloid
- PDH
pyruvate dehydrogenase
- OGDH
oxoglutarate dehydrogenase
- SDH
succinate dehydrogenase
- MDH
malate dehydrogenase
- ELISA
enzyme-linked immunosorbent assay
- API
active pharmacological ingredient
- ACL
ATP-citrate lyase
- PDK1
pyruvate dehydrogenase kinase
- phospho-Tau
phosphorylated Tau
- Tyr
Tyrosine
Footnotes
Publisher's Disclaimer: This Author Accepted Manuscript is a PDF file of a an unedited peer-reviewed manuscript that has been accepted for publication but has not been copyedited or corrected. The official version of record that is published in the journal is kept up to date and so may therefore differ from this version.
Declaration of Interest: None
6. References
- 1.A Massaad C Neuronal and vascular oxidative stress in Alzheimer’s disease. Current Neuropharmacol. 2011. December 1;9(4):662–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Agholme L, Lindström T, Kågedal K, Marcusson J, Hallbeck M. An in vitro model for neuroscience: differentiation of SH-SY5Y cells into cells with morphological and biochemical characteristics of mature neurons. J Alzheimers Dis. 2010. January 1;20(4):1069–1082. [DOI] [PubMed] [Google Scholar]
- 3.Alzheimer’s Association, 2006. Early onset dementia: a national challenge, a future crisis Alzheimer’s Association, Washington, DC. [Google Scholar]
- 4.Alzheimer’s Association, 2012. Alzheimer’s Disease Facts and Figures, Available at: http://www.alz.org/downloads/Facts_Figures. [DOI] [PubMed] [Google Scholar]
- 5.Bubber P, Haroutunian V, Fisch G, Blass JP, Gibson GE. Mitochondrial abnormalities in Alzheimer brain: mechanistic implications. Ann Neurol. 2005. May;57(5):695–703. [DOI] [PubMed] [Google Scholar]
- 6.Chen JX, Yan SD. Amyloid-β-induced mitochondrial dysfunction. J Alzheimers Dis. 2007. January 1;12(2):177–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coskuner O, Uversky VN. Tyrosine regulates β-sheet structure formation in amyloid-β42: A new clustering algorithm for disordered proteins. J Chem Inf Model. 2017. May 12;57(6):1342–58. [DOI] [PubMed] [Google Scholar]
- 8.Farshbaf MJ, Kiani-Esfahani A. Succinate dehydrogenase: Prospect for neurodegenerative diseases. Mitochondrion. 2017. December 8. [DOI] [PubMed] [Google Scholar]
- 9.Fein JA, Sokolow S, Miller CA, Vinters HV, Yang F, Cole GM, Gylys KH. Co-localization of amyloid beta and tau pathology in Alzheimer’s disease synaptosomes. AM J Pathol. 2008. June 1;172(6):1683–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gandbhir O, Sundaram P. Pre-clinical Safety and Efficacy Evaluation of Amytrap, a novel therapeutic to treat Alzheimer’s Disease. J Alzheimers Dis Rep 2019. April 16; 1–18. DOI: 10.3233/ADR-190107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Glabe CG, Kayed R. Common structure and toxic function of amyloid oligomers implies a common mechanism of pathogenesis. Neurology. 2006. January 24;66(1 suppl 1): S74–8. [DOI] [PubMed] [Google Scholar]
- 12.Gong CX, Iqbal K. Hyperphosphorylation of microtubule-associated protein tau: a promising therapeutic target for Alzheimer disease. Curr Med Chem. 2008. October 1;15(23):2321–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kresge N 2018. Biogen surges after positive results in Alzheimer’s Trial. Bloomberg. [Internet]. 2018 July 06 [cited 2018 July 06] Available from: https://www.bloomberg.com/news/articles/2018-07-06/eisai-surges-after-positive-results-in-alzheimer-s-drug-trial?in_source=video_page. [Google Scholar]
- 14.Kummer MP, Hermes M, Delekarte A, Hammerschmidt T, Kumar S, Terwel D, Walter J, Pape HC, König S, Roeber S, Jessen F. Nitration of tyrosine 10 critically enhances amyloid β aggregation and plaque formation. Neuron. 2011. September 8;71(5):833–44. [DOI] [PubMed] [Google Scholar]
- 15.Manyevitch R, Protas M, Scarpiello S, Deliso M, Bass B, Nanajian A, Chang M, Thompson SM, Khoury N, Gonnella R, Trotz M. Evaluation of Metabolic and Synaptic Dysfunction Hypotheses of Alzheimer’s Disease (AD): A Meta-Analysis of CSF Markers. Curr Alzheimer Res. 2018. February 1;15(2):164–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Matharu B, El-Agnaf O, Razvi A, Austen BM. Development of retro-inverso peptides as anti-aggregation drugs for β-amyloid in Alzheimer’s disease. Peptides. 2010. October 1;31(10):1866–72. [DOI] [PubMed] [Google Scholar]
- 17.Mossmann D, Vögtle FN, Taskin AA, Teixeira PF, Ring J, Burkhart JM, Burger N, Pinho CM, Tadic J, Loreth D, Graff C. Amyloid-β peptide induces mitochondrial dysfunction by inhibition of preprotein maturation. Cell Metab. 2014. October 7;20(4):662–9. [DOI] [PubMed] [Google Scholar]
- 18.Najjar K, Erazo-Oliveras A, Brock DJ, Wang TY, Pellois JP. An l-to d-amino acid conversion in an endosomolytic analog of the cell-penetrating peptide TAT influences proteolytic stability, endocytic uptake, and endosomal escape. J Biol Chem. 2017. January 20;292(3):847–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pagani L, Eckert A. Amyloid-Beta interaction with mitochondria. International J Alzheimers Dis. 2011;2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Palop JJ, Mucke L. Amyloid-β–induced neuronal dysfunction in Alzheimer’s disease: from synapses toward neural networks. Nat Neurosci. 2010. July;13(7):812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Parthsarathy V, McClean PL, Hölscher C, Taylor M, Tinker C, Jones G, Kolosov O, Salvati E, Gregori M, Masserini M, Allsop D. A novel retro-inverso peptide inhibitor reduces amyloid deposition, oxidation and inflammation and stimulates neurogenesis in the APPswe/PS1ΔE9 mouse model of Alzheimer’s disease. PLoS One. 2013. January 31;8(1): e54769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rijal AU, Scheibe F, Kosterin I, Abramowski D, Gerth J, Kumar S, Liebau S, Yamaguchi H, Walter J, Staufenbiel M, Thal DR. The type of Aβ-related neuronal degeneration differs between amyloid precursor protein (APP23) and amyloid β-peptide (APP48) transgenic mice. Acta Neuropathol Commun. 2013;1(1):77-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sundaram RK, Kasinathan C, Stein S, Sundaram P. detoxification Depot for β-amyloid Peptides. Curr Alzheimer Res. 2008. February 1;5(1):26–32. [DOI] [PubMed] [Google Scholar]
- 24.Sundaram RK, Kasinathan C, Stein S, Sundaram P. Novel Detox Gel Depot sequesters β-Amyloid Peptides in a mouse model of Alzheimer’s Disease. Int J Pept Res Ther. 2012. June 1;18(2):99–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Swerdlow RH, Burns JM, Khan SM. The Alzheimer’s disease mitochondrial cascade hypothesis. J Alzheimers Dis. 2010. June 3;20(s2): S265–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wilson RS, Weir DR, Leurgans SE, Evans DA, Hebert LE, Langa KM, Plassman BL, Small BJ, Bennett DA. Sources of variability in estimates of the prevalence of Alzheimer’s disease in the United States. Alzheimers Dement. 2011. Jan 1;7(1):74–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wirths O, Multhaup G, Bayer TA. A modified β- amyloid hypothesis: intraneuronal accumulation of the β- amyloid peptide–the first step of a fatal cascade. J Neurochem. 2004. November;91(3):513–20. [DOI] [PubMed] [Google Scholar]
- 28.Xie H, Guan J, Borrelli LA, Xu J, Serrano-Pozo A, Bacskai BJ. Mitochondrial alterations near amyloid plaques in an Alzheimer’s disease mouse model. J Neurosci. 2013. October 23;33(43):17042–51. [DOI] [PMC free article] [PubMed] [Google Scholar]