

ABSTRACT
Organs-on-chips are broadly defined as microfabricated surfaces or devices designed to engineer cells into microscale tissues with native-like features and then extract physiologically relevant readouts at scale. Because they are generally compatible with patient-derived cells, these technologies can address many of the human relevance limitations of animal models. As a result, organs-on-chips have emerged as a promising new paradigm for patient-specific disease modeling and drug development. Because neuromuscular diseases span a broad range of rare conditions with diverse etiology and complex pathophysiology, they have been especially challenging to model in animals and thus are well suited for organ-on-chip approaches. In this Review, we first briefly summarize the challenges in neuromuscular disease modeling with animal models. Next, we describe a variety of existing organ-on-chip approaches for neuromuscular tissues, including a survey of cell sources for both muscle and nerve, and two- and three-dimensional neuromuscular tissue-engineering techniques. Although researchers have made tremendous advances in modeling neuromuscular diseases on a chip, the remaining challenges in cell sourcing, cell maturity, tissue assembly and readout capabilities limit their integration into the drug development pipeline today. However, as the field advances, models of healthy and diseased neuromuscular tissues on a chip, coupled with animal models, have vast potential as complementary tools for modeling multiple aspects of neuromuscular diseases and identifying new therapeutic strategies.
KEY WORDS: Skeletal muscle, Motor neurons, Amyotrophic lateral sclerosis, Induced pluripotent stem cells, Tissue engineering, Microfluidic devices
Summary: Modeling neuromuscular diseases is challenging due to their complex etiology and pathophysiology. Here, we review the cell sources and tissue-engineering procedures that are being integrated as emerging neuromuscular disease models.
Introduction
Neuromuscular diseases collectively affect 160 per 100,000 people worldwide and are generally characterized by progressive motor impairment and muscular atrophy (Deenen et al., 2015). Although these conditions have diverse etiologies, they each affect one or more components of the motor unit (see Box 1, Fig. 1). For decades, animal models, especially humanized mice (De Giorgio et al., 2019; Nair et al., 2019; Aartsma-Rus and van Putten, 2020), have been the gold standard for neuromuscular disease modeling. More recently, non-mammalian models, such as fruit flies (Lloyd and Taylor, 2010), Caenorhabditis elegans (Sleigh and Sattelle, 2010) and zebrafish (Babin et al., 2014), have also been used for neuromuscular disease modeling. Although these simpler models are limited by their lower conservation with human genetics, anatomy and physiology compared to mice, they are beneficial because of their lower cost, rapid growth rate, tractable anatomy and ease of genetic manipulation. In general, animal models capture important hallmarks of their human disease counterparts and thus are invaluable for understanding disease progression on an organ- and organism-level scale. However, disease phenotypes in animals can vary widely from humans in terms of progression, severity and other characteristics (De Giorgio et al., 2019; Aartsma-Rus and van Putten, 2020; Babin et al., 2014).
Box 1. Structure and physiology of the motor unit.
All voluntary movements are controlled by a collection of motor units, each of which comprises a single motor neuron and all the muscle fibers that it innervates (Fig. 1). Motor neurons have a soma that resides in the motor cortex, brain stem or spinal cord, and a single myelinated axon that forms specialized synapses, known as neuromuscular junctions (NMJs), on muscle fibers. Muscle fibers are elongated multi-nucleated cells that are packed with myofibrils, each of which is an interconnected chain of contractile sarcomere units. Multiple muscle fibers are bundled together and wrapped in connective tissue to form a muscle.
Contraction of a motor unit begins when signals from the central nervous system trigger an action potential in the motor neuron, which induces the axon to release the neurotransmitter acetylcholine into the synaptic cleft of the NMJ. Acetylcholine binds to acetylcholine receptors on the membrane of the muscle fiber, which depolarizes the membrane and initiates an action potential. The muscle fiber then propagates this action potential along its length, triggering the entry of extracellular calcium through voltage-sensitive ion channels in the membrane and subsequently a large release of calcium from the sarcoplasmic reticulum. This increase in cytosolic calcium enables the heads of myosin filaments to pull on actin filaments, shortening the sarcomere and ultimately contracting the muscle fiber in an ATP-demanding process. Depending on the frequency of the action potential transmitted by the motor neuron, the muscle fiber undergoes either a singular or sustained contraction, referred to as twitch or tetanus, respectively. Lastly, the free acetylcholine in the NMJ is broken down by acetylcholinesterase, cytosolic calcium is transported back into the sarcoplasmic reticulum, and the membrane potential of the muscle fiber returns to resting levels, thus causing muscle relaxation (reviewed by Hall and Hall, 2015).
Fig. 1.
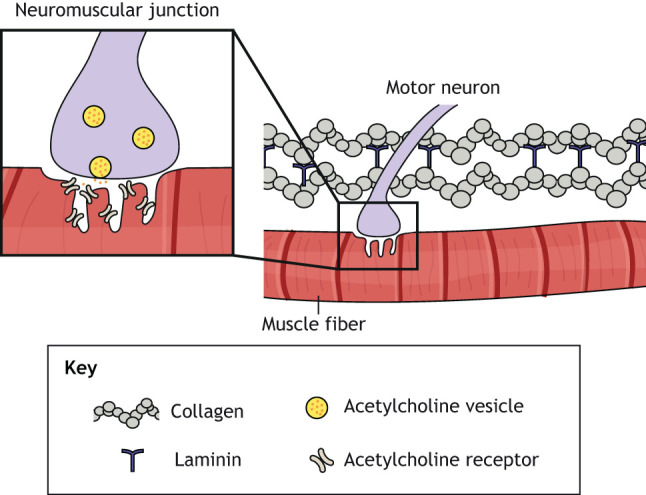
Schematic of the neuromuscular junction. Multi-nucleated muscle fibers are innervated by myelinated motor neurons at neuromuscular junctions (NMJs). At the NMJ, motor neurons release acetylcholine vesicles. The neurotransmitter acetylcholine binds to acetylcholine receptors on the membrane of the muscle fiber, causing membrane depolarization and muscle contraction.
Another limitation of animal models is that it is difficult, if not impossible, to recapitulate the genotypic heterogeneity and allelic variation observed in individuals with neuromuscular diseases without generating an unreasonable number of animal strains (Juneja et al., 2019; Morrice et al., 2018). Even monogenic neuromuscular diseases, such as spinal muscular atrophy (SMA), are difficult to model in animals due to patient-specific genotypic features. SMA is an autosomal recessive disease caused by inactivating mutations in the SMN1 gene, which encodes the survival of motor neuron (SMN) protein (Li, 2017). SMN plays a role in protein homeostasis, cytoskeletal assembly, endocytosis, metabolism and many other processes in motor neurons (Chaytow et al., 2018). SMN shortage or dysfunction causes deficits in axonogenesis, migration, electrophysiology and many other features, leading to neuromuscular junction (NMJ) degeneration and motor neuron death (Laird et al., 2016; McGovern et al., 2015). A second gene, SMN2, also produces SMN, but at ∼20% of the levels transcribed from fully functional SMN1 (Bowerman et al., 2017; Jedrzejowska et al., 2009). SMA has been modeled in mice (Hsieh-Li et al., 2000), Drosophila (Spring et al., 2019), zebrafish (McWhorter et al., 2003) and C. elegans (Briese et al., 2009) by deleting the endogenous Smn gene and overexpressing the human SMN2 gene. However, the severity and progression of SMA largely depends on the number of SMN2 copies in a patient (Bowerman et al., 2017; Jedrzejowska et al., 2009), a patient-specific feature of the disease that is nearly impossible to faithfully recapitulate in animals. The only treatment options for SMA are the gene therapy drugs Spinraza (Dangouloff and Servais, 2019) and Zolgensma (Zuroske, 2019), both of which are extremely expensive and thus impractical for many individuals.
Compared to SMA, several neuromuscular diseases have a more heterogeneous genetic etiology, which is even more challenging to model in animals. For example, Charcot-Marie-Tooth (CMT) diseases have been linked to 870 mutations in over 80 genes (McCorquodale et al., 2016), such as PMP22, MPZ, GJB1 or MFN2 (Morena et al., 2019; Saporta et al., 2011). This genetic heterogeneity partially explains the wide range of age of onset and disease symptoms, which usually involve involuntary contraction of limbs and loss of sensation due to axon demyelination. CMT has been modeled in zebrafish and other animal models by introducing a mutation in a single gene known to cause a specific subtype of CMT disease, such as mfn2 (Chapman et al., 2013) or prps1 (Pei et al., 2016). However, owing to the vast genetic heterogeneity of CMT diseases, it is infeasible to generate animal models that represent all mutations (Juneja et al., 2019). Largely due to a lack of modular model systems, CMT diseases still lack clinical data supporting any effective treatment beyond physical therapy and pain management (McCorquodale et al., 2016). Mouse models of CMT have also demonstrated that impaired development of the NMJ precedes synaptic deficits (Sleigh et al., 2013; Spaulding et al., 2016), suggesting that microscale models of the motor unit might be useful for elucidating the pathophysiology of this broad group of diseases.
Amyotrophic lateral sclerosis (ALS) is another neuromuscular disease that introduces unique challenges for modeling in animals because it can be either inherited (10%) or sporadic (90%) (Boylan, 2015). In ALS, over 50 genes either directly cause motor neuron death or alter key functions, such as vesicle trafficking, axonal structure and cytoskeletal stability (Boylan, 2015; Seminary et al., 2018; Shi et al., 2018). The most commonly affected genes are C9ORF72, SOD1, TARDBP and FUS, usually occurring in some kind of combination (Lattante et al., 2015; Nguyen et al., 2018). Animal models of ALS have been generated by expressing a mutated version of one of these human genes in mice (Ripps et al., 1995; Zhang et al., 1997; Devoy et al., 2017), Drosophila (Şahin et al., 2017; Watson et al., 2008; Perry et al., 2017; Xu et al., 2013), zebrafish (Shaw et al., 2018; Lissouba et al., 2018) and C. elegans (Oeda et al., 2001; Wang et al., 2009). Additionally, environmental factors, such as pesticides, flame retardants and military-related trauma, have been correlated to ALS (Su et al., 2016). However, the small number of clinical cases and limited model systems make assigning causality from environmental factors very difficult. The natural process of aging has also been tied to ALS, probably because of the aggregation of misfolded proteins and oxidative stress (Jang and Van Remmen, 2011; Turner et al., 2012). Owing, in large part, to the many causes and complex pathophysiologies of ALS, the only therapies are the anti-glutamatergic compound riluzole and the antioxidant edaravone, both of which only assuage symptoms and extend survival for a few months (Nguyen et al., 2018).
Neuromuscular diseases can also be caused by factors external to the motor unit. For example, myasthenia gravis (MG) is a sporadic autoimmune disease in which auto-antibodies selectively destroy acetylcholine receptors, causing a reduction in NMJ signal transmission (Phillips and Vincent, 2016). A mouse model of MG has been developed by injecting rat acetylcholine receptors into mice, which then triggered the development of auto-antibodies to their own acetylcholine receptors (Granato et al., 1976). However, MG cannot be modeled in simple organisms such as Drosophila and C. elegans because they lack an adaptive immune system. The causes of MG are still mostly unknown and treatment is limited to acetylcholinesterase inhibitors, immunosuppressants or a thymectomy (Gilhus, 2016), highlighting the need for additional predictive model systems to develop more targeted therapies.
Collectively, these examples highlight that neuromuscular diseases are very diverse and are characterized by many complex genetic and non-genetic etiologies and pathophysiologies. These complexities introduce many challenges for developing comprehensive animal models. Thus, new disease models that are more efficient and predictive are essential for accelerating our mechanistic knowledge of these diseases, as well as the discovery of effective therapies. Integrating patient-derived cells with microfabricated in vitro platforms, known as organs-on-chips, is an emerging solution to fill the gaps of animal models and holds promise for patient-specific neuromuscular disease modeling and drug development. As discussed below, these platforms are often developed using animal cells or cell lines that are easy to scale, and that can provide important proof-of-concept and basic physiological information. To address issues of human relevance, animal cells or cell lines can then be replaced with patient-derived cells, which can be acquired from a variety of sources. In the next section, we describe the cell sources for neuromuscular disease models, which can ultimately be integrated into the two- (2D) and three-dimensional (3D) engineered tissue platforms described in the following sections.
Cell sources for in vitro models of neuromuscular tissues
In vitro models can mitigate many of the limitations of animal models described above, such as human relevance and scalability. However, the usefulness of any in vitro model is highly dependent on the source and structural and functional maturity of its cells. This is especially complex when modeling neuromuscular tissues, which consist of both muscle cells and motor neurons. In this section, we will describe the types of muscle and motor neuron cell types available today for in vitro models, and weigh up their advantages and disadvantages.
Skeletal muscle cells
Generating skeletal muscle tissue in vitro is a multi-step process. First, mononuclear skeletal myoblasts are seeded on a standard culture surface that is often coated with extracellular matrix (ECM) proteins, such as collagen or laminin. The myoblasts are then expanded in a high-serum growth medium until they reach confluence. The medium is then substituted with a low-serum differentiation medium that triggers the fusion of myoblasts into multi-nucleated myotubes, the in vitro surrogate to muscle fibers (Neville et al., 1997). As illustrated in Fig. 2, several different sources of myoblasts are currently available, each with distinct advantages and disadvantages that are important to consider when engineering neuromuscular disease models.
Fig. 2.

Muscle and motor neuron cell sources for in vitro models. Myoblasts and motor neurons available for in vitro models fall into three categories: immortalized cell lines, primary cells and pluripotent stem cell derivatives. Immortalized cell lines recapitulate the basic properties of the original cell type and are inexpensive and easy to expand in culture. However, the immortalization process causes de-differentiation and a loss of important structural and functional features. Primary cells are usually the most mature and physiologically relevant cell source but are relatively costly and difficult to obtain, and have a limited ability to expand in culture. Pluripotent stem cells can be widely expanded in culture and then differentiated into myoblasts or motor neurons. Induced pluripotent stem cells have the additional advantage of patient specificity. However, pluripotent stem cell derivatives are generally heterogeneous and immature. hESC, human embryonic stem cell; hiPSC, human induced pluripotent stem cell.
Primary myoblasts are harvested from embryonic or adult animals, such as chicks (Urja et al., 2018; Vallette et al., 1986) or mice (Hindi et al., 2017), by excising muscle tissue and either enzymatically digesting it to a cell suspension or collecting the cells that migrate from cultured tissue explants (Vaughan and Lamia, 2019). Myoblasts are then purified using simple pre-plating steps or more sophisticated techniques, such as magnetic cell sorting (Sincennes et al., 2017; Spinazzola and Gussoni, 2017). Human primary myoblasts can be isolated from muscle tissue collected during a surgical procedure or by needle biopsy (Joyce et al., 2012), and similarly processed and purified.
The structural and functional properties of myotubes differentiated from primary myoblasts closely recapitulate those of native muscle, such as a high density of myofibrils and spontaneous contractile behavior (Pimentel et al., 2017). However, primary myoblasts can only be passaged a few times before their growth rate and myogenic capacity decline, leading to limited supply and passage-dependent variability. These supply and variability issues are especially problematic for human primary myoblasts, which are generally isolated from relatively small muscle biopsies. Moreover, primary myoblasts can vary widely in purity and functional maturity depending on the isolation and purification methods, and the characteristics of the subject (Cheng et al., 2014; Soriano-Arroquia et al., 2017). This issue is further exacerbated by data indicating a beneficial role for fibroblasts in myotube function (Rao et al., 2013), raising questions about the ideal purity of primary myoblasts for in vitro studies and adding further variability to the performance of myotubes differentiated from primary myoblasts.
Compared to primary myoblasts, immortalized myoblast cell lines are a more convenient source of cells that are relatively pure and easy to expand. The most common myoblast cell line is C2C12, which was isolated from 2-month-old mouse muscle in 1977 (Yaffe and Saxel, 1977). Another common cell line is L6, which was isolated from newborn rat muscle in 1968 (Yaffe, 1968). Both C2C12 and L6 cells proliferate rapidly with a generation time of ∼24 h and fuse into multi-nucleated contractile myotubes (McMahon et al., 1994; Öberg et al., 2011). Thus, they are a convenient model system for investigating processes such as myotube fusion (Zhao et al., 2015) or degeneration (Menconi et al., 2008). These cells are also compatible with gene transfection (Balcı and Dinçer, 2009), which is useful for establishing the functions of genetic variants in myoblast growth or fusion (Prinsen and Veerkamp, 1998), or introducing disease-relevant mutations (Liang et al., 2016).
Myotubes differentiated from cell lines have lower levels of structural and functional maturity, and distinct metabolic properties compared to myotubes differentiated from primary myoblasts (Abdelmoez et al., 2019; Robinson et al., 2019), probably because the immortalization process causes some amount of de-differentiation and loss of myogenic properties that continue to decline with increasing passage number. Thus, although myoblast cell lines are a reproducible and cost-effective cell source compared to primary myoblasts, myotubes differentiated from cell lines have limited relevance to native muscle tissue. Furthermore, a commercialized human myoblast cell line does not currently exist, raising further concerns about the translatability of data collected using common myoblast cell lines, which are derived from rodents. However, primary human myoblasts have been immortalized by the forced expression of a telomerase subunit and cyclin dependent kinase 4, which blocks a stress pathway (Mamchaoui et al., 2011). Myotubes generated from these cell lines produce relatively mature sarcomeres (Morris et al., 2020) and form NMJs when co-cultured with motor neurons (Saini et al., 2019), suggesting that they could become a promising cell source.
To overcome the limitations of primary myoblasts and myoblast cell lines, several protocols for deriving myogenic cells from human embryonic stem cells (hESCs) or human induced pluripotent stem cells (hiPSCs) have recently emerged (Salani et al., 2012). These cells have the advantages of human origin and essentially limitless supply, as hESCs and hiPSCs can be expanded in culture for many passages without loss of functionality. hESCs and hiPSCs are also compatible with gene editing techniques, such as CRISPR/Cas9, which can be used to introduce or correct select disease-relevant mutations (Shi et al., 2018; Young et al., 2016). Because hiPSCs are reprogrammed from somatic cells, such as skin fibroblasts, hiPSC-derived myogenic cells can be used to generate patient-specific myotubes, which makes them especially desirable for modeling inherited neuromuscular diseases.
One approach for generating myogenic progenitors from hiPSCs is to overexpress master regulators of myogenic differentiation, such as MYOD1 (Abujarour et al., 2014; Rao et al., 2018) or PAX7 (Darabi et al., 2012). A similar reprogramming process has also been used to directly transdifferentiate other cell types, such as fibroblasts, into myogenic progenitors (Boularaoui et al., 2018; Ito et al., 2017; Lattanzi et al., 1998). A second approach, known as directed differentiation, guides hiPSCs through native-like myogenic developmental pathways by sequentially adding small molecules that activate or suppress specific signaling pathways (Chal et al., 2016, 2015; Maffioletti et al., 2015; Shelton et al., 2016; van der Wal et al., 2018; Xi et al., 2017). Directed differentiation is generally slower than transdifferentiation, but the resulting myogenic progenitors are thought to be a closer match to native myoblasts because they follow a more natural differentiation process (Jiwlawat et al., 2018).
Although impressive progress has been made in deriving myogenic progenitors from hiPSCs, most current protocols generally suffer from wide variability and low efficiency (Jiwlawat et al., 2018). These issues limit cell yield and purity. However, protocols for cryopreserving and expanding hiPSC-derived myogenic progenitors are being developed (van der Wal et al., 2018), which helps mitigate issues with differentiation variability and throughput. Despite these practical limitations related to cell differentiation, hESC- and hiPSC-derived myogenic progenitors successfully fuse into myotubes that contain myofibrils and exhibit key functional behaviors, such as calcium cycling and contractility (Rao et al., 2018; Skoglund et al., 2014). However, myofibrils in hESC- and hiPSC-derived myotubes still have immature features compared to native muscle fibers or myotubes derived from primary myoblasts (Lainé et al., 2018).
The structural and functional immaturity of myotubes probably contributes to the stunted maturation of NMJs that form between hiPSC-derived myotubes and motor neurons. However, several approaches for maturing hiPSC-derived myotubes are under development, such as identifying small molecules that boost maturation (Selvaraj et al., 2019) or applying other strategies discussed below. Additionally, the maturation of C2C12 myotubes has been improved by applying biophysical cues, such as mechanical stretch (Chang et al., 2016; Heher et al., 2015) or electrical stimulation (Ito et al., 2014; Nedachi et al., 2008), which might have similar benefits for hiPSC-derived myotubes. Thus, although hiPSC-derived myotubes have significant potential as an essentially limitless source of patient-specific myotubes, researchers need to enhance the differentiation efficiency and maturity of these cells to improve their throughput and relevance for modeling neuromuscular diseases in vitro.
Motor neurons
Compared to myoblasts, fewer sources of motor neurons exist for in vitro models. Because most motor neurons stem from the spinal cord and project onto muscle fibers, it is not possible to isolate intact primary motor neurons from humans. However, primary motor neurons can be isolated from embryonic or adult mice by extracting and digesting spinal cord tissue and using density gradient separation to isolate motor neurons from supporting cell types, such as astrocytes and other glial cells (Beaudet et al., 2015; Gingras et al., 2007). Although rodents are a viable source of primary motor neurons, these cells are not human, which limits their relevance for neuromuscular disease modeling. Furthermore, the cell yield is relatively low and cannot be increased with passaging because motor neurons are terminally differentiated and non-proliferative.
Because motor neurons do not proliferate, true motor neuron cell lines do not exist. However, a hybrid mouse cell line (NSC-34) has been generated by fusing neuroblastoma cells with embryonic motor neurons. NSC-34 cells retain the proliferative properties of the tumor cells while also exhibiting select neuronal properties, such as acetylcholine synthesis, neurotransmitter release and neurofilament proteins (Cashman et al., 1992). This cell line has been used to measure the neurotoxicity of drugs (Maier et al., 2013) and receptor trafficking (Matusica et al., 2008), and has also been transfected to introduce mutations relevant to ALS (Gomes et al., 2010; Pinto et al., 2017). Similar to myoblast cell lines, the disadvantage of these cells is their non-human origin and limited relevance to native motor neurons. For example, these cells do not replicate glutamate-mediated excitotoxicity (Madji Hounoum et al., 2016), questioning their ability to replicate key features of neuromuscular diseases.
Human motor neurons can also be derived from hESCs and hiPSCs via reprogramming or directed differentiation. hESCs and hiPSCs have been reprogrammed into motor neurons by overexpressing NGN2 (also known as NEUROG2), ISL1 and LHX3 (Goto et al., 2017; Hester et al., 2011; Lee et al., 2012). Human fibroblasts have also been transdifferentiated into motor neurons by overexpressing eight genes (Son et al., 2011). Several directed differentiation methods have also been established, which entail dosing hESCs or hiPSCs with a combination of neurotrophic factors, retinoic acid, sonic hedgehog and Notch inhibitors (Du et al., 2015; Hu and Zhang, 2009; Li et al., 2008b; Qu et al., 2014; Shimojo et al., 2015). Motor neurons have also been differentiated from the human fetal spinal cord stem cell line NSI-566RSC (Guo et al., 2010), which serves as another relatively accessible source of human motor neurons.
Similar to other stem cell derivatives, stem cell-derived motor neurons can be limited by cell heterogeneity, varying differentiation efficiency and stunted maturation (Ichida et al., 2018). Although mouse motor neurons derived from transdifferentiated fibroblasts or directly differentiated iPSCs have a transcriptome that is similar to primary motor neurons (Ichida et al., 2018), how the structural and functional properties of these cells compare to their primary counterpart is mostly unknown. Despite the limited functional characterization of these cells, hiPSC-derived motor neurons have already been shown to be a promising cell source for patient-specific modeling of neuromuscular diseases such as ALS (Dimos et al., 2008; Sances et al., 2016; Sareen et al., 2013; Shi et al., 2018) and SMA (Fuller et al., 2016; Murdocca et al., 2016). Thus, hiPSC-derived motor neurons are likely to contribute to the development of new therapies for these diseases that account for the genotype of the patient.
Engineered in vitro models of neuromuscular tissues
In addition to cell source, another important consideration for in vitro model development is the configuration of the cells, such that the cultured tissue is anatomically relevant and integrated with assays to measure functional phenotypes. Initial approaches for engineering neuromuscular tissues in vitro entailed simply seeding dissociated motor neurons (Daniels et al., 2000; Das et al., 2010; Guo et al., 2011; Kengaku et al., 1991; Son et al., 2011; Umbach et al., 2012) or spinal cord explants (Askanas et al., 1987; Braun et al., 1997) on top of a 2D layer of myotubes attached to a conventional culture surface. Over the course of several days, the neurons extend axons and form NMJs with the myotubes that successfully exhibit functional post-synaptic potentials. However, image analysis has revealed blotchy colocalization of pre- and post-synaptic markers and poor acetylcholine receptor clustering in these simple co-cultures compared to native NMJs (Das et al., 2010; Umbach et al., 2012). This limited synaptic maturity brings into question the ability of these culture systems to accurately model disease-relevant phenotypes.
The relatively stunted NMJ development in conventional co-cultures could be attributed to many factors. First, without spatial organization cues, myoblasts fuse into branched myotubes with random orientations that poorly recapitulate the architecture of native muscle fibers (Bettadapur et al., 2016; Denes et al., 2019), which can limit the formation of elongated myofibrils and mature sarcomeres. Second, myotubes often delaminate from conventional culture surfaces within ∼2 weeks as they generate increasing amounts of mechanical stress (Wang et al., 2012; Sun et al., 2013). This can probably be attributed to both the high stiffness of conventional culture substrates and the limited number of cell-adhesive molecules presented on their surfaces (Bettadapur et al., 2016). Limited culture lifetime is especially problematic for engineering neuromuscular tissues because NMJ maturation probably requires longer than 2 weeks. Third, in situ, motor neuron soma are located in the spinal cord and only the axons of motor neurons physically interact with muscle fibers. Thus, seeding motor neurons on top of myotubes is not anatomically relevant and might alter the physiology of one or both cell types.
Simple mixed co-cultures also suffer from technical problems that limit data collection. For example, measuring forces generated by cells is not possible on most, if not all, conventional culture substrates, precluding quantitative assessment of muscle force production due to motor neuron stimulation. This is a key functional readout in animal models, with high relevance to the severity of neuromuscular disease (Bonetto et al., 2015). A second limitation is that mixed co-cultures afford minimal independent control or analysis of each cell type, as the cells are cultured in the same medium and any electrical stimulation or drug treatment reaches both cell types simultaneously. Similarly, isolating material from each cell type independently to measure changes in gene or protein expression, which is often important for establishing disease mechanisms as well as drug effects, is challenging.
To overcome these diverse biological and technical challenges, researchers have developed several types of tunable culture surfaces and microfabricated devices to engineer more sophisticated neuromuscular tissues in vitro. These surfaces and devices are usually also integrated with assays for quantifying structural and functional tissue phenotypes. In particular, when coupled with the hiPSC-derived cell sources described above, these platforms, known as organs-on-chips or microphysiological systems, have immense potential for advancing neuromuscular disease modeling and drug development. In this section, we describe both 2D and 3D engineered tissue models.
Engineered 2D models of neuromuscular tissues
Engineering a neuromuscular tissue in vitro depends on first culturing mature and stable myotubes. In native skeletal muscle fibers, the ECM plays a key role in tissue development and physiology by binding to integrin receptors and providing biomechanical support as the muscle fibers contract (Gillies and Lieber, 2011). The ECM is also a rich source of biochemical cues that regulate behaviors such as adhesion, proliferation and differentiation. Furthermore, the ECM plays an active role in skeletal muscle disease, injury and aging, with fibrosis and subsequent tissue stiffening contributing to diminished muscle function (Mann et al., 2011). Owing to the documented importance of the ECM in native muscle fibers, several types of tunable culture substrates that mimic aspects of native muscle ECM have been developed, as described below. However, another important feature of in vitro models of neuromuscular tissues is the ability to measure muscle contractility in response to motor neuron stimulation. Thus, we also describe later in this section how engineered substrates have been integrated with contractility assays.
Natural biomaterial substrates
Hydrogels synthesized from natural polymers are popular culture substrates due to their biocompatibility, although they can suffer from batch-to-batch variability. Collagen hydrogels are routinely used as culture substrates for myoblasts and myotubes due to their intrinsic bioactivity (Palade et al., 2019), as we also discuss above. Aligned myotubes have been fabricated on collagen hydrogels by embedding topographical features into the hydrogel (Kim et al., 2017) or using ultrasound to pattern the cells acoustically (Armstrong et al., 2018). Gelatin, a partially hydrolyzed form of collagen, is also crosslinked into thermostable hydrogels by either mixing gelatin polymers with enzymatic crosslinking agents (Bettadapur et al., 2016; Denes et al., 2019; Suh et al., 2017) or methacrylating gelatin polymers such that they are compatible with photopolymerization techniques (Hosseini et al., 2012; Sun et al., 2018). Crosslinking increases the stiffness of the hydrogel and reduces its degradability (Sun et al., 2018), which can be advantageous for in vitro neuromuscular models that need to be stable for several weeks.
To promote myotube alignment on gelatin hydrogels, the surface can be micromolded with polydimethylsiloxane (PDMS) (a silicone elastomer) stamps with ridges several micrometers in size (Bettadapur et al., 2016; Chal et al., 2016; Denes et al., 2019; Hosseini et al., 2012). PDMS stamps are fabricated by casting PDMS on silicon wafer templates made using photolithography, which can generate feature sizes of ∼1 µm (Suh et al., 2017). Probably due to their enhanced bioactivity, micromolded gelatin hydrogels can extend the culture lifetime and maturation of C2C12 myotubes compared to synthetic culture surfaces (Bettadapur et al., 2016; Denes et al., 2019). Carbon nanotubes have also been embedded into methacrylated gelatin hydrogels to enhance myotube maturation by increasing electrical conductivity (Ahadian et al., 2015; Ramón-Azcón et al., 2013). To better mimic the basement membrane of muscle, micromolded gelatin hydrogels have also been crosslinked with a layer of laminin, which improves the adherence, morphology and electrophysiology of myotubes and neural cells (Besser et al., 2020).
Synthetic biomaterial substrates
Synthetic biomaterials are advantageous culture substrates because their mechanical and biochemical properties are highly controllable and reproducible. For example, polyethylene glycol and polyacrylamide (PA) are both biologically inert hydrophilic polymers that can be crosslinked into hydrogels with elastic moduli tuned to match the developing, healthy or fibrotic muscle tissue matrices (Engler et al., 2004). Another synthetic biomaterial that is implemented as a culture surface is the aforementioned PDMS. The elasticity of PDMS can be easily tuned to physiological or pathological values by altering the ratio of base to crosslinker (Wang et al., 2014) or blending different formulations of PDMS (Palchesko et al., 2012).
To achieve consistent cell adhesion, researchers must functionalize the synthetic substrate with ECM proteins. This is generally considered an advantage because ECM ligand type and concentration can be specified. Because collagen accounts for up to 10% of the dry weight of muscle (Gillies and Lieber, 2011), several studies have fabricated substrates for C2C12 cultures by transferring collagen onto PDMS (Duffy et al., 2016) or PA hydrogels (Engler et al., 2004; Li et al., 2008a). Because the basement membrane of muscle fibers is enriched in laminin and fibronectin, synthetic substrates functionalized with either of these glycoproteins also promote myoblast adhesion and fusion into myotubes (Duffy et al., 2016; Gilbert et al., 2010; Palchesko et al., 2012; Ziemkiewicz et al., 2018).
Synthetic biomaterials are also compatible with many micropatterning techniques that can be used to introduce microscale features on the surface to spatially control cell adhesion and alignment (Falconnet et al., 2006), as shown in Fig. 3. For example, PDMS stamps generated using the same photolithography techniques described above can be used to transfer ECM proteins onto a surface in a process known as microcontact printing (Qin et al., 2010). This process has been used to prescribe myotube alignment on Petri dishes (Bajaj et al., 2011), PDMS-coated surfaces (Bettadapur et al., 2016; Jiwlawat et al., 2019; Nesmith et al., 2016; Palchesko et al., 2012; Sun et al., 2013) and PA hydrogels (Li et al., 2008a). Photolithography has also been used to selectively expose strips of a PA hydrogel to UV light, which activates only the exposed regions for collagen binding and thus myoblast adhesion (Engler et al., 2004). Myotubes have also been aligned on substrates with nanoscale ridges fabricated using electron beam lithography, in which electrons are scanned in a defined pattern on a wafer coated with a light-sensitive photoresist (Wang et al., 2012). Another alternative is solvent-assisted capillary force lithography, in which a polymer solution is molded on a silicon wafer with features at the hundreds of nanometers scale (Yang et al., 2014).
Fig. 3.

Engineered 2D neuromuscular tissues. Conventional approaches for engineering neuromuscular tissues in vitro entailed mixed co-cultures (center). New advances to improve the architecture and assaying capabilities of 2D neuromuscular tissues include microfabricated surfaces (top), compartmentalized culture devices (right), and integration of optogenetics (left). ChR2, channelrhodopsin-2; ECM, extracellular matrix; PA, polyacrylamide; PDMS, polydimethylsiloxane; UV, ultraviolet.
Integrated contractility assays
Micropatterned synthetic culture substrates are especially compatible with assays that quantify myotube contractility because the mechanical properties of the substrate are well defined, and myotube architecture can be controlled to increase the magnitude and reproducibility of contractile force production. Micropatterned PA gels are widely used as a substrate for traction force microscopy, a technique that quantifies forces generated by cells by tracking the displacement of fluorescent beads embedded in the hydrogel. Although traction force microscopy is more commonly used for cardiac myocytes (Ariyasinghe et al., 2017; McCain et al., 2012; Pasqualini et al., 2018; Ribeiro et al., 2015), it has also been used to quantify forces generated by micropatterned C2C12 myotubes (Li et al., 2008a).
Contractility can also be quantified by culturing myotubes on flexible cantilevers, as in the muscular thin film (MTF) assay. The MTF assay entails first spin coating a glass coverslip with a layer of poly(N-isopropylacrylamide) (PNIPAAm), a temperature-sensitive polymer, followed by a layer of PDMS (Feinberg et al., 2007). The PDMS is then laser-cut into arrays of cantilevers with dimensions ranging from 1 mm to 5 mm (Agarwal et al., 2013), microcontact printed with lines of fibronectin, and used to culture myotubes. After the desired culture period, the muscle-PDMS cantilevers, referred to as MTFs, are released by reducing the temperature from 37°C to 25°C to solubilize the PNIPAAm. Electrodes are then used to stimulate myotube contraction, which causes cantilever bending. Contractile stress is calculated based on the radius of curvature of each MTF (Grosberg et al., 2011). The MTF assay has been successfully used to measure twitch and tetanus forces generated by C2C12 myotubes (Sun et al., 2013) and primary human myotubes (Nesmith et al., 2016). Microfabricated silicon cantilevers with dimensions of <1 mm have also been used as a culture substrate for primary rat myotubes, and the contractile stresses in this system are measured based on the myotube-induced deflection of laser light (Smith et al., 2014a; Wilson et al., 2010). These compact laser systems are advantageous for multiplexing, which increases testing throughput and scalability for drug screening applications (Smith et al., 2014b).
Engineered co-cultures of skeletal muscle and motor neurons
Microfabricated surfaces have also been developed to improve mixed co-cultures of myotubes and motor neurons. For example, photopolymerization techniques have been used to fabricate PA hydrogels with alternating soft and stiff stripes that mimic the rigidity of nervous tissue and muscle tissue, respectively (Happe et al., 2017). On these surfaces, myoblasts preferentially migrate onto the stiffer stripes and fuse into aligned myotubes. When co-cultured with motor neurons, myotubes on mechanically patterned hydrogels exhibited increased acetylcholine receptor clustering compared to myotubes co-cultured on uniform hydrogels (Happe et al., 2017).
NMJ maturation has also been achieved in mixed co-cultures by applying electrical stimulation using a bioreactor (Charoensook et al., 2017). This approach could be further refined by transfecting one or both cell types with channelrhodopsin, a membrane channel that is activated by blue light (Fig. 3). Because chronic optogenetic stimulation of myotubes can improve maturity (Rangarajan et al., 2014), a similar strategy applied to co-cultures could be a relatively non-invasive approach for maturation. Transfecting motor neurons with channelrhodopsin is also a powerful experimental tool for mixed co-cultures because it enables users to stimulate only motor neurons and therefore more clearly identify responses in the muscle that are driven specifically by motor neurons (Lin et al., 2019; Steinbeck et al., 2016).
Compartmentalized culture devices have also been microfabricated to physically isolate motor neurons and myotubes into separate chambers (Fig. 3). These chambers are connected by microchannels that are permissive to axons, but not cell bodies, to allow the controlled formation of NMJs in the myotube chamber (Santhanam et al., 2018; Taylor et al., 2003). Because the chambers are chemically isolated, these devices allow each cell type to be cultured in its own medium, which may boost viability. Furthermore, drugs or other small molecules can be selectively added to one or both chambers (Santhanam et al., 2018), which can be useful for establishing drug mechanisms. Structural and functional analyses are also easier in these devices compared to mixed co-cultures because NMJs form in relatively prescribed locations and cells in each chamber can be electrically stimulated independently.
Engineered 3D models of neuromuscular tissues
Although engineered 2D neuromuscular tissues have many advantages from an assay perspective, they fundamentally lack the bundle-like architecture and cell-ECM interactions of native muscle fibers. To address this, researchers have developed several approaches to engineer miniature 3D muscle bundles (Fig. 4). These types of approaches were first reported in the late 1990s and entailed injecting primary myoblasts mixed in an ECM pre-polymer solution into a rectangular chamber with patches of stainless-steel screening (Shansky et al., 1997) or Velcro (Powell et al., 1999) at its longitudinal ends. As the myoblasts fused into myotubes, they detached from the bottom surface but remained embedded in the ECM and anchored by the screening or Velcro, forming an elongated 3D muscle bundle with aligned myotubes.
Fig. 4.

Engineered 3D neuromuscular tissues. (A) Aligned 3D muscle bundles are engineered by mixing myoblasts in an ECM pre-polymer solution of Matrigel, thrombin and fibrinogen, and casting it into a microfabricated support structure, such as a Velcro frame. Scale bars: 50 mm (left image); 50 µm (right image). Adapted from Madden et al. (2015). (B) Compartmentalized fluidic devices have been microfabricated to controllably co-culture 3D muscle bundles and motor neuron spheroids, and generated NMJs after 14 days in culture. D7, day 7; D14, day 14; DAPI, 4′,6-diamidino-2-phenylindole; nAChR, nicotinic acetylcholine receptor; SAA, sarcomeric alpha-actinin; Tuj1, neuron-specific class III β-tubulin. Scale bars: 2 mm (left image); 10 µm (right image). Adapted with permission from Osaki et al. (2018) and Uzel et al. (2016). The images in this figure are not published under the terms of the CC-BY license of this article. For permission to reuse, please see Madden et al. (2015), Osaki et al. (2018) and Uzel et al. (2016).
Over the past two decades, approaches for engineering 3D muscle bundles have been advanced and refined. Several different types of culture chambers with anchor points have been fabricated (Costantini et al., 2017a; Smith et al., 2016), including Velcro and nylon frames (Davis et al., 2017, 2019; Madden et al., 2015; Rao et al., 2018; Smith et al., 2016; Zhang et al., 2018) and microfabricated chambers with pillars (Osaki et al., 2018, 2020; Uzel et al., 2016). Testing of multiple ECM solutions has also revealed that fibrin hydrogels are optimal for encapsulating myotubes due to their strength (Hinds et al., 2011; Pollot et al., 2018), although these hydrogel compositions are not necessarily physiological. To improve assay capabilities, contractile forces have been measured in 3D muscle bundles with custom force transducers (Davis et al., 2019; Madden et al., 2015; Rao et al., 2018) or by tracking the displacement of pillars (Osaki et al., 2018; Uzel et al., 2016). Similar to 2D tissues, biophysical cues, such as mechanical stretch (Powell et al., 2002) and optogenetic stimulation (Mills et al., 2019), or addition of fibroblasts (Dennis et al., 2001) have also been shown to mature 3D muscle bundles.
Microfluidic devices have also been fabricated to engineer and maintain 3D muscle bundles (Agrawal et al., 2017; Shimizu et al., 2015). These systems are advantageous because they continuously perfuse fresh media to the engineered tissues, which probably improves viability compared to static culture. Furthermore, microfluidic devices can be used to screen drugs at a higher throughput and can be linked to other microfluidic organ-on-chip systems to capture organ-organ interactions, and mimic organism-level responses (Novak et al., 2020).
One common approach to innervate 3D muscle bundles is to directly seed them with spheroids of motor neurons (Afshar Bakooshli et al., 2019; Morimoto et al., 2013; Smith et al., 2016). These systems have demonstrated that the resulting NMJs are functional but still have relatively diffuse acetylcholine receptor clustering (Morimoto et al., 2013). Acetylcholine clustering in 3D muscle bundles has been advanced by adding the basement membrane components agrin and laminin (Wang et al., 2013), which could help improve NMJ formation in these co-cultures. Despite their limited maturity, these 3D neuromuscular tissues show reduced contractility in response to sera from MG patients (Morimoto et al., 2013), recapitulating the pathological response in MG and demonstrating their promise for modeling complex neuromuscular diseases.
Similar to 2D models, compartmentalized microdevices have been developed to culture motor neuron spheroids and engineered muscle bundles in separate compartments connected by axon-permissive channels (Fig. 4) (Osaki et al., 2018, 2020; Uzel et al., 2016). In these studies, the muscle bundles were attached to flexible pillars and the motor neurons were optogenetically modified. With this combination of technologies, the users could quantify muscle contractility as a function of motor neuron stimulation, a key readout of NMJ function. This type of device was also used to capture NMJ degeneration in tissues generated using hiPSC-derived motor neurons from an ALS patient. Importantly, the application of two ALS drug candidates, bosutinib and rapamycin, to this model reduced muscle atrophy and dysfunction (Osaki et al., 2018), demonstrating how this type of approach has the potential for patient-specific disease modeling and drug screening.
Conclusions
Recently developed approaches to model healthy and diseased neuromuscular tissues on a chip have the potential to capture the vastly heterogeneous genotypes and phenotypes of individuals with a variety of neuromuscular disorders. Newer technologies, such as 3D bioprinting, which is a form of additive manufacturing that uses cells and other biomaterials as ‘inks’ to print living structures (Choi et al., 2016; Costantini et al., 2017b; Kang et al., 2016; Kim et al., 2020), will probably further advance these models. However, in vitro models of neuromuscular tissues are far from achieving adult-like maturity, especially when based on hiPSC-derived muscle cells and motor neurons. Furthermore, in vitro models of neuromuscular tissues lack the supporting cells known to be important regulators of NMJs in health and disease, such as Schwann cells (Santosa et al., 2018). Most in vitro models also currently lack immune cells, despite the established role of neuroinflammation in many neuromuscular diseases (Mäurer et al., 2002; Thonhoff et al., 2018). However, researchers have begun developing models that integrate immune cells, such as macrophages (Juhas et al., 2018), to probe the role of the immune system in muscle injury and repair. Given these limitations, in vitro models are most powerful when implemented hand-in-hand with animal models, which have less human relevance but more advanced motor unit structure and physiology. Together, these complementary model systems are likely to pave the way for more effective and personalized therapies for these debilitating diseases.
Footnotes
Competing interests
The authors declare no competing or financial interests.
Funding
This project was supported by the University of Southern California Viterbi School of Engineering, Women in Science and Engineering, University of Southern California, an Amyotrophic Lateral Sclerosis Association Starter Grant (18-IIA-401 to M.L.M.), a Rose Hills Foundation Innovator Grant to M.L.M., and a National Science Foundation Graduate Research Fellowship Grant (DGE 1418060 to J.W.S.).
References
- Aartsma-Rus A. and van Putten M. (2020). The use of genetically humanized animal models for personalized medicine approaches. Dis. Model. Mech. 13, dmm041673 10.1242/dmm.041673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelmoez A. M., Sardón Puig L., Smith J. A. B., Gabriel B. M., Savikj M., Dollet L., Chibalin A. V., Krook A., Zierath J. R. and Pillon N. J. (2019). Comparative profiling of skeletal muscle models reveals heterogeneity of transcriptome and metabolism. Am. J. Physiol. Cell Physiol. 318, C615-C626. 10.1152/ajpcell.00540.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abujarour R., Bennett M., Valamehr B., Lee T. T., Robinson M., Robbins D., Le T., Lai K. and Flynn P. (2014). Myogenic differentiation of muscular dystrophy-specific induced pluripotent stem cells for use in drug discovery. Stem Cells Transl. Med. 3, 149-160. 10.5966/sctm.2013-0095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afshar Bakooshli M., Lippmann E. S., Mulcahy B., Iyer N., Nguyen C. T., Tung K., Stewart B. A., van den Dorpel H., Fuehrmann T., Shoichet M. et al. (2019). A 3D culture model of innervated human skeletal muscle enables studies of the adult neuromuscular junction. eLife 8, e44530 10.7554/eLife.44530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agarwal A., Goss J. A., Cho A., McCain M. L. and Parker K. K. (2013). Microfluidic heart on a chip for higher throughput pharmacological studies. Lab. Chip 13, 3599-3608. 10.1039/c3lc50350j [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal G., Aung A. and Varghese S. (2017). Skeletal muscle-on-a-chip: an in vitro model to evaluate tissue formation and injury. Lab. Chip 17, 3447-3461. 10.1039/C7LC00512A [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahadian S., Ramón-Azcón J., Estili M., Liang X., Ostrovidov S., Shiku H., Ramalingam M., Nakajima K., Sakka Y., Bae H. et al. (2015). Hybrid hydrogels containing vertically aligned carbon nanotubes with anisotropic electrical conductivity for muscle myofiber fabrication. Sci. Rep. 4, 4271 10.1038/srep04271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ariyasinghe N. R., Reck C. H., Viscio A. A., Petersen A. P., Lyra-Leite D. M., Cho N. and McCain M. L. (2017). Engineering micromyocardium to delineate cellular and extracellular regulation of myocardial tissue contractility. Integr. Biol. 9, 730-741. 10.1039/C7IB00081B [DOI] [PMC free article] [PubMed] [Google Scholar]
- Armstrong J. P. K., Puetzer J. L., Serio A., Guex A. G., Kapnisi M., Breant A., Zong Y., Assal V., Skaalure S. C., King O. et al. (2018). Engineering anisotropic muscle tissue using acoustic cell patterning. Adv. Mater. 30, 1802649 10.1002/adma.201802649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Askanas V., Kwan H., Alvarez R. B., Engel W. K., Kobayashi T., Martinuzzi A. and Hawkins E. F. (1987). De novo neuromuscular junction formation on human muscle fibres cultured in monolayer and innervated by foetal rat spinal cord: ultrastructural and ultrastructural-cytochemical studies. J. Neurocytol. 16, 523-537. 10.1007/BF01668506 [DOI] [PubMed] [Google Scholar]
- Babin P. J., Goizet C. and Raldúa D. (2014). Zebrafish models of human motor neuron diseases: advantages and limitations. Prog. Neurobiol. 118, 36-58. 10.1016/j.pneurobio.2014.03.001 [DOI] [PubMed] [Google Scholar]
- Bajaj P., Reddy B. Jr, Millet L., Wei C., Zorlutuna P., Bao G. and Bashir R. (2011). Patterning the differentiation of C2C12 skeletal myoblasts. Integr. Biol. 3, 897-909. 10.1039/c1ib00058f [DOI] [PubMed] [Google Scholar]
- Balci B. and Dinçer P. (2009). Efficient transfection of mouse-derived C2C12 myoblasts using a matrigel basement membrane matrix. Biotechnol. J. 4, 1042-1045. 10.1002/biot.200800269 [DOI] [PubMed] [Google Scholar]
- Beaudet M.-J., Yang Q., Cadau S., Blais M., Bellenfant S., Gros-Louis F. and Berthod F. (2015). High yield extraction of pure spinal motor neurons, astrocytes and microglia from single embryo and adult mouse spinal cord. Sci. Rep. 5, 16763 10.1038/srep16763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Besser R. R., Bowles A. C., Alassaf A., Carbonero D., Claure I., Jones E., Reda J., Wubker L., Batchelor W., Ziebarth N. et al. (2020). Enzymatically crosslinked gelatin–laminin hydrogels for applications in neuromuscular tissue engineering. Biomaterials Sci. 8, 591-606. 10.1039/C9BM01430F [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettadapur A., Suh G. C., Geisse N. A., Wang E. R., Hua C., Huber H. A., Viscio A. A., Kim J. Y., Strickland J. B. and McCain M. L. (2016). Prolonged culture of aligned skeletal myotubes on micromolded gelatin hydrogels. Sci. Rep. 6, 28855 10.1038/srep28855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonetto A., Andersson D. C. and Waning D. L. (2015). Assessment of muscle mass and strength in mice. BoneKEy Rep. 4, 732 10.1038/bonekey.2015.101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boularaoui S. M., Abdel-Raouf K. M. A., Alwahab N. S. A., Kondash M. E., Truskey G. A., Teo J. C. M. and Christoforou N. (2018). Efficient transdifferentiation of human dermal fibroblasts into skeletal muscle. J. Tissue Eng. Regen. Med. 12, e918-e936. 10.1002/term.2415 [DOI] [PubMed] [Google Scholar]
- Bowerman M., Becker C. G., Yáñez-Muñoz R. J., Ning K., Wood M. J. A., Gillingwater T. H., Talbot K. and Consortium U. S. R. (2017). Therapeutic strategies for spinal muscular atrophy: SMN and beyond. Dis. Model. Mech. 10, 943-954. 10.1242/dmm.030148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boylan K. (2015). Familial amyotrophic lateral sclerosis. Neurol. Clin. 33, 807-830. 10.1016/j.ncl.2015.07.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun S., Croizat B., Lagrange M.-C., Poindron P. and Warter J.-M. (1997). Degeneration of cocultures of spinal muscular atrophy muscle cells and rat spinal cord explants is not due to secreted factors and cannot be prevented by neurotrophins. Muscle Nerve 20, 953-960. [DOI] [PubMed] [Google Scholar]
- Briese M., Esmaeili B., Fraboulet S., Burt E. C., Christodoulou S., Towers P. R., Davies K. E. and Sattelle D. B. (2009). Deletion of smn-1, the Caenorhabditis elegans ortholog of the spinal muscular atrophy gene, results in locomotor dysfunction and reduced lifespan. Hum. Mol. Genet. 18, 97-104. 10.1093/hmg/ddn320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cashman N. R., Durham H. D., Blusztajn J. K., Oda K., Tabira T., Shaw I. T., Dahrouge S. and Antel J. P. (1992). Neuroblastoma×spinal cord (NSC) hybrid cell lines resemble developing motor neurons. Dev. Dyn. 194, 209-221. 10.1002/aja.1001940306 [DOI] [PubMed] [Google Scholar]
- Chal J., Oginuma M., Al Tanoury Z., Gobert B., Sumara O., Hick A., Bousson F., Zidouni Y., Mursch C., Moncuquet P. et al. (2015). Differentiation of pluripotent stem cells to muscle fiber to model Duchenne muscular dystrophy. Nat. Biotechnol. 33, 962-969. 10.1038/nbt.3297 [DOI] [PubMed] [Google Scholar]
- Chal J., Al Tanoury Z., Hestin M., Gobert B., Aivio S., Hick A., Cherrier T., Nesmith A. P., Parker K. K. and Pourquié O. (2016). Generation of human muscle fibers and satellite-like cells from human pluripotent stem cells in vitro. Nat. Protoc. 11, 1833-1850. 10.1038/nprot.2016.110 [DOI] [PubMed] [Google Scholar]
- Chang Y.-J., Chen Y.-J., Huang C.-W., Fan S.-C., Huang B.-M., Chang W.-T., Tsai Y.-S., Su F.-C. and Wu C.-C. (2016). Cyclic stretch facilitates myogenesis in C2C12 myoblasts and rescues thiazolidinedione-inhibited myotube formation. Front. Bioeng. Biotechnol. 4, 27 10.3389/fbioe.2016.00027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman A. L., Bennett E. J., Ramesh T. M., De Vos K. J. and Grierson A. J. (2013). Axonal transport defects in a mitofusin 2 loss of function model of charcot-marie-tooth disease in zebrafish. PLoS ONE 8, e67276 10.1371/journal.pone.0067276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charoensook S. N., Williams D. J., Chakraborty S., Leong K. W. and Vunjak-Novakovic G. (2017). Bioreactor model of neuromuscular junction with electrical stimulation for pharmacological potency testing. Integr. Biol. 9, 956-967. 10.1039/C7IB00144D [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaytow H., Huang Y.-T., Gillingwater T. H. and Faller K. M. E. (2018). The role of survival motor neuron protein (SMN) in protein homeostasis. Cell. Mol. Life Sci. 75, 3877-3894. 10.1007/s00018-018-2849-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C. S., El-Abd Y., Bui K., Hyun Y.-E., Hughes R. H., Kraus W. E. and Truskey G. A. (2014). Conditions that promote primary human skeletal myoblast culture and muscle differentiation in vitro. Am. J. Physiol. Cell Physiol. 306, C385-C395. 10.1152/ajpcell.00179.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi Y.-J., Kim T. G., Jeong J., Yi H.-G., Park J. W., Hwang W. and Cho D.-W. (2016). 3D cell printing of functional skeletal muscle constructs using skeletal muscle-derived bioink. Adv. Healthc. Mater. 5, 2636-2645. 10.1002/adhm.201600483 [DOI] [PubMed] [Google Scholar]
- Costantini M., Testa S., Fornetti E., Barbetta A., Trombetta M., Cannata S. M., Gargioli C. and Rainer A. (2017a). Engineering muscle networks in 3D gelatin methacryloyl hydrogels: influence of mechanical stiffness and geometrical confinement. Front. Bioeng. Biotechnol. 5, 22 10.3389/fbioe.2017.00022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costantini M., Testa S., Mozetic P., Barbetta A., Fuoco C., Fornetti E., Tamiro F., Bernardini S., Jaroszewicz J., Święszkowski W. et al. (2017b). Microfluidic-enhanced 3D bioprinting of aligned myoblast-laden hydrogels leads to functionally organized myofibers in vitro and in vivo. Biomaterials 131, 98-110. 10.1016/j.biomaterials.2017.03.026 [DOI] [PubMed] [Google Scholar]
- Dangouloff T. and Servais L. (2019). Clinical evidence supporting early treatment of patients with spinal muscular atrophy: current perspectives. Ther. Clin. Risk Management 15, 1153-1161. 10.2147/TCRM.S172291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniels M. P., Lowe B. T., Shah S., Ma J., Samuelsson S. J., Lugo B., Parakh T. and Uhm C.-S. (2000). Rodent nerve-muscle cell culture system for studies of neuromuscular junction development: refinements and applications. Microsc. Res. Tech. 49, 26-37. [DOI] [PubMed] [Google Scholar]
- Darabi R., Arpke R. W., Irion S., Dimos J. T., Grskovic M., Kyba M. and Perlingeiro R. C. R. (2012). Human ES- and iPS-derived myogenic progenitors restore DYSTROPHIN and improve contractility upon transplantation in dystrophic mice. Cell Stem Cell 10, 610-619. 10.1016/j.stem.2012.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das M., Rumsey J. W., Bhargava N., Stancescu M. and Hickman J. J. (2010). A defined long-term in vitro tissue engineered model of neuromuscular junctions. Biomaterials 31, 4880-4888. 10.1016/j.biomaterials.2010.02.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis B. N. J., Santoso J. W., Walker M. J., Cheng C. S., Koves T. R., Kraus W. E. and Truskey G. A. (2017). Human, tissue-engineered, skeletal muscle myobundles to measure oxygen uptake and assess mitochondrial toxicity. Tissue Eng. C Methods 23, 189-199. 10.1089/ten.tec.2016.0264 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis B. N. J., Santoso J. W., Walker M. J., Oliver C. E., Cunningham M. M., Boehm C. A., Dawes D., Lasater S. L., Huffman K., Kraus W. E. et al. (2019). Modeling the effect of TNF-α upon drug-induced toxicity in human, tissue-engineered myobundles. Ann. Biomed. Eng. 47, 1596-1610. 10.1007/s10439-019-02263-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Giorgio F., Maduro C., Fisher E. M. C. and Acevedo-Arozena A. (2019). Transgenic and physiological mouse models give insights into different aspects of amyotrophic lateral sclerosis. Dis. Model. Mech. 12, dmm037424 10.1242/dmm.037424 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deenen J. C. W., Horlings C. G. C., Verschuuren J. J. G. M., Verbeek A. L. M. and van Engelen B. G. M. (2015). The epidemiology of neuromuscular disorders: a comprehensive overview of the literature. J. Neuromuscul. Dis. 2, 73-85. 10.3233/JND-140045 [DOI] [PubMed] [Google Scholar]
- Denes L. T., Riley L. A., Mijares J. R., Arboleda J. D., McKee K., Esser K. A. and Wang E. T. (2019). Culturing C2C12 myotubes on micromolded gelatin hydrogels accelerates myotube maturation. Skelet. Muscle 9, 17 10.1186/s13395-019-0203-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dennis R. G., Kosnik P. E., Gilbert M. E. and Faulkner J. A. (2001). Excitability and contractility of skeletal muscle engineered from primary cultures and cell lines. Am. J. Physiol. Cell Physiol. 280, C288-C295. 10.1152/ajpcell.2001.280.2.C288 [DOI] [PubMed] [Google Scholar]
- Devoy A., Kalmar B., Stewart M., Park H., Burke B., Noy S. J., Redhead Y., Humphrey J., Lo K., Jaeger J. et al. (2017). Humanized mutant FUS drives progressive motor neuron degeneration without aggregation in ‘FUSDelta14’ knockin mice. Brain 140, 2797-2805. 10.1093/brain/awx248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimos J. T., Rodolfa K. T., Niakan K. K., Weisenthal L. M., Mitsumoto H., Chung W., Croft G. F., Saphier G., Leibel R., Goland R. et al. (2008). Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science 321, 1218-1221. 10.1126/science.1158799 [DOI] [PubMed] [Google Scholar]
- Du Z.-W., Chen H., Liu H., Lu J., Qian K., Huang C. T.-L., Zhong X., Fan F. and Zhang S.-C. (2015). Generation and expansion of highly pure motor neuron progenitors from human pluripotent stem cells. Nat. Commun. 6, 6626 10.1038/ncomms7626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duffy R. M., Sun Y. and Feinberg A. W. (2016). Understanding the role of ECM protein composition and geometric micropatterning for engineering human skeletal muscle. Ann. Biomed. Eng. 44, 2076-2089. 10.1007/s10439-016-1592-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engler A. J., Griffin M. A., Sen S., Bönnemann C. G., Sweeney H. L. and Discher D. E. (2004). Myotubes differentiate optimally on substrates with tissue-like stiffness: pathological implications for soft or stiff microenvironments. J. Cell Biol. 166, 877-887. 10.1083/jcb.200405004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falconnet D., Csucs G., Grandin H. M. and Textor M. (2006). Surface engineering approaches to micropattern surfaces for cell-based assays. Biomaterials 27, 3044-3063. 10.1016/j.biomaterials.2005.12.024 [DOI] [PubMed] [Google Scholar]
- Feinberg A. W., Feigel A., Shevkoplyas S. S., Sheehy S., Whitesides G. M. and Parker K. K. (2007). Muscular thin films for building actuators and powering devices. Science 317, 1366-1370. 10.1126/science.1146885 [DOI] [PubMed] [Google Scholar]
- Fuller H. R., Mandefro B., Shirran S. L., Gross A. R., Kaus A. S., Botting C. H., Morris G. E. and Sareen D. (2016). Spinal muscular atrophy patient iPSC-derived motor neurons have reduced expression of proteins important in neuronal development. Front. Cell. Neurosci. 9, 506 10.3389/fncel.2015.00506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert P. M., Havenstrite K. L., Magnusson K. E. G., Sacco A., Leonardi N. A., Kraft P., Nguyen N. K., Thrun S., Lutolf M. P. and Blau H. M. (2010). Substrate elasticity regulates skeletal muscle stem cell self-renewal in culture. Science 329, 1078-1081. 10.1126/science.1191035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilhus N. E. (2016). Myasthenia gravis. N. Engl. J. Med. 375, 2570-2581. 10.1056/NEJMra1602678 [DOI] [PubMed] [Google Scholar]
- Gillies A. R. and Lieber R. L. (2011). Structure and function of the skeletal muscle extracellular matrix. Muscle Nerve 44, 318-331. 10.1002/mus.22094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gingras M., Gagnon V., Minotti S., Durham H. D. and Berthod F. (2007). Optimized protocols for isolation of primary motor neurons, astrocytes and microglia from embryonic mouse spinal cord. J. Neurosci. Methods 163, 111-118. 10.1016/j.jneumeth.2007.02.024 [DOI] [PubMed] [Google Scholar]
- Gomes C., Escrevente C. and Costa J. (2010). Mutant superoxide dismutase 1 overexpression in NSC-34 cells: effect of trehalose on aggregation, TDP-43 localization and levels of co-expressed glycoproteins. Neurosci. Lett. 475, 145-149. 10.1016/j.neulet.2010.03.065 [DOI] [PubMed] [Google Scholar]
- Goto K., Imamura K., Komatsu K., Mitani K., Aiba K., Nakatsuji N., Inoue M., Kawata A., Yamashita H., Takahashi R. et al. (2017). Simple derivation of spinal motor neurons from ESCs/iPSCs using Sendai virus vectors. Mol. Ther. Methods Clin. Dev. 4, 115-125. 10.1016/j.omtm.2016.12.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Granato D. A., Fulpius B. W. and Moody J. F. (1976). Experimental myasthenia in Balb/c mice immunized with rat acetylcholine receptor from rat denervated muscle. Proc. Natl. Acad. Sci. USA 73, 2872-2876. 10.1073/pnas.73.8.2872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosberg A., Alford P. W., McCain M. L. and Parker K. K. (2011). Ensembles of engineered cardiac tissues for physiological and pharmacological study: heart on a chip. Lab. Chip 11, 4165-4173. 10.1039/c1lc20557a [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X., Johe K., Molnar P., Davis H. and Hickman J. (2010). Characterization of a human fetal spinal cord stem cell line, NSI-566RSC, and its induction to functional motoneurons. J. Tissue Eng. Regen. Med. 4, 181-193. 10.1002/term.223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X., Gonzalez M., Stancescu M., Vandenburgh H. H. and Hickman J. J. (2011). Neuromuscular junction formation between human stem cell-derived motoneurons and human skeletal muscle in a defined system. Biomaterials 32, 9602-9611. 10.1016/j.biomaterials.2011.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall J. E. and Hall M. E. (2015). Guyton and Hall Textbook of Medical Physiology, 13th edn. Saunders. [Google Scholar]
- Happe C. L., Tenerelli K. P., Gromova A. K., Kolb F. and Engler A. J. (2017). Mechanically patterned neuromuscular junctions-in-a-dish have improved functional maturation. Mol. Biol. Cell 28, 1950-1958. 10.1091/mbc.e17-01-0046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heher P., Maleiner B., Prüller J., Teuschl A. H., Kollmitzer J., Monforte X., Wolbank S., Redl H., Rünzler D. and Fuchs C. (2015). A novel bioreactor for the generation of highly aligned 3D skeletal muscle-like constructs through orientation of fibrin via application of static strain. Acta Biomater. 24, 251-265. 10.1016/j.actbio.2015.06.033 [DOI] [PubMed] [Google Scholar]
- Hester M. E., Murtha M. J., Song S. W., Rao M., Miranda C. J., Meyer K., Tian J., Boulting G., Schaffer D. V., Zhu M. X. et al. (2011). Rapid and efficient generation of functional motor neurons from human pluripotent stem cells using gene delivered transcription factor codes. Mol. Ther. 19, 1905-1912. 10.1038/mt.2011.135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hindi L., McMillan J. D., Afroze D., Hindi S. M. and Kumar A. (2017). Isolation, culturing, and differentiation of primary myoblasts from skeletal muscle of adult mice. Bio Protoc. 7, e2248 10.21769/BioProtoc.2248 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinds S., Bian W., Dennis R. G. and Bursac N. (2011). The role of extracellular matrix composition in structure and function of bioengineered skeletal muscle. Biomaterials 32, 3575-3583. 10.1016/j.biomaterials.2011.01.062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosseini V., Ahadian S., Ostrovidov S., Camci-Unal G., Chen S., Kaji H., Ramalingam M. and Khademhosseini A. (2012). Engineered contractile skeletal muscle tissue on a microgrooved methacrylated gelatin substrate. Tissue Eng. Part A 18, 2453-2465. 10.1089/ten.tea.2012.0181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh-Li H. M., Chang J.-G., Jong Y.-J., Wu M.-H., Wang N. M., Tsai C. H. and Li H. (2000). A mouse model for spinal muscular atrophy. Nat. Genet. 24, 66-70. 10.1038/71709 [DOI] [PubMed] [Google Scholar]
- Hu B.-Y. and Zhang S.-C. (2009). Differentiation of spinal motor neurons from pluripotent human stem cells. Nat. Protoc. 4, 1295-1304. 10.1038/nprot.2009.127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichida J. K., Staats K. A., Davis-Dusenbery B. N., Clement K., Galloway K. E., Babos K. N., Shi Y., Son E. Y., Kiskinis E., Atwater N. et al. (2018). Comparative genomic analysis of embryonic, lineage-converted and stem cell-derived motor neurons. Development 145, dev168617 10.1242/dev.168617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito A., Yamamoto Y., Sato M., Ikeda K., Yamamoto M., Fujita H., Nagamori E., Kawabe Y. and Kamihira M. (2014). Induction of functional tissue-engineered skeletal muscle constructs by defined electrical stimulation. Sci. Rep. 4, 4781 10.1038/srep04781 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito N., Kii I., Shimizu N., Tanaka H. and Takeda S. (2017). Direct reprogramming of fibroblasts into skeletal muscle progenitor cells by transcription factors enriched in undifferentiated subpopulation of satellite cells. Sci. Rep. 7, 8097 10.1038/s41598-017-08232-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang Y. C. and Van Remmen H. (2011). Age-associated alterations of the neuromuscular junction. Exp. Gerontol. 46, 193-198. 10.1016/j.exger.2010.08.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jedrzejowska M., Milewski M., Zimowski J., Borkowska J., Kostera-Pruszczyk A., Sielska D., Jurek M. and Hausmanowa-Petrusewicz I. (2009). Phenotype modifiers of spinal muscular atrophy: the number of SMN2 gene copies, deletion in the NAIP gene and probably gender influence the course of the disease. Acta Biochim. Pol. 56, 103-108. 10.18388/abp.2009_2521 [DOI] [PubMed] [Google Scholar]
- Jiwlawat N., Lynch E., Jeffrey J., Van Dyke J. M. and Suzuki M. (2018). Current progress and challenges for skeletal muscle differentiation from human pluripotent stem cells using transgene-free approaches. Stem Cells Int. 2018, 6241681 10.1155/2018/6241681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiwlawat N., Lynch E. M., Napiwocki B. N., Stempien A., Ashton R. S., Kamp T. J., Crone W. C. and Suzuki M. (2019). Micropatterned substrates with physiological stiffness promote cell maturation and Pompe disease phenotype in human induced pluripotent stem cell-derived skeletal myocytes. Biotechnol. Bioeng. 116, 2377-2392. 10.1002/bit.27075 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce N. C., Oskarsson B. and Jin L.-W. (2012). Muscle biopsy evaluation in neuromuscular disorders. Phys. Med. Rehabil. Clin. N Am. 23, 609-631. 10.1016/j.pmr.2012.06.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juhas M., Abutaleb N., Wang J. T., Ye J., Shaikh Z., Sriworarat C., Qian Y. and Bursac N. (2018). Incorporation of macrophages into engineered skeletal muscle enables enhanced muscle regeneration. Nat. Biomed. Eng. 2, 942-954. 10.1038/s41551-018-0290-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juneja M., Burns J., Saporta M. A. and Timmerman V. (2019). Challenges in modelling the Charcot-Marie-Tooth neuropathies for therapy development. J. Neurol. Neurosurg. Psychiatr. 90, 58-67. 10.1136/jnnp-2018-318834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H.-W., Lee S. J., Ko I. K., Kengla C., Yoo J. J. and Atala A. (2016). A 3D bioprinting system to produce human-scale tissue constructs with structural integrity. Nat. Biotechnol. 34, 312-319. 10.1038/nbt.3413 [DOI] [PubMed] [Google Scholar]
- Kengaku M., Kawata A., Kawashima S. and Nakane M. (1991). Role of fibronectin in the inhibitory effect of TGF-beta on choline acetyltransferase activity in co-cultures of spinal cord neurons and myotubes. Brain Res. Dev. Brain Res. 61, 281-284. 10.1016/0165-3806(91)90144-8 [DOI] [PubMed] [Google Scholar]
- Kim M., Kim W. J. and Kim G. H. (2017). Topologically micropatterned collagen and poly(ε-caprolactone) struts fabricated using the poly(vinyl alcohol) fibrillation/leaching process to develop efficiently engineered skeletal muscle tissue. ACS Appl. Material. Interfaces 9, 43459-43469. 10.1021/acsami.7b14192 [DOI] [PubMed] [Google Scholar]
- Kim W. J., Lee H., Lee J. U., Atala A., Yoo J. J., Lee S. J. and Kim G. H. (2020). Efficient myotube formation in 3D bioprinted tissue construct by biochemical and topographical cues. Biomaterials 230, 119632 10.1016/j.biomaterials.2019.119632 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lainé J., Skoglund G., Fournier E. and Tabti N. (2018). Development of the excitation-contraction coupling machinery and its relation to myofibrillogenesis in human iPSC-derived skeletal myocytes. Skelet. Muscle 8, 1 10.1186/s13395-017-0147-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laird A. S., Mackovski N., Rinkwitz S., Becker T. S. and Giacomotto J. (2016). Tissue-specific models of spinal muscular atrophy confirm a critical role of SMN in motor neurons from embryonic to adult stages. Hum. Mol. Genet. 25, 1728-1738. 10.1093/hmg/ddw044 [DOI] [PubMed] [Google Scholar]
- Lattante S., Ciura S., Rouleau G. A. and Kabashi E. (2015). Defining the genetic connection linking amyotrophic lateral sclerosis (ALS) with frontotemporal dementia (FTD). Trends Genet. 31, 263-273. 10.1016/j.tig.2015.03.005 [DOI] [PubMed] [Google Scholar]
- Lattanzi L., Salvatori G., Coletta M., Sonnino C., Cusella De Angelis M. G., Gioglio L., Murry C. E., Kelly R., Ferrari G., Molinaro M. et al. (1998). High efficiency myogenic conversion of human fibroblasts by adenoviral vector-mediated MyoD gene transfer. An alternative strategy for ex vivo gene therapy of primary myopathies. J. Clin. Invest. 101, 2119-2128. 10.1172/JCI1505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Cuvillier J. M., Lee B., Shen R., Lee J. W. and Lee S.-K. (2012). Fusion protein Isl1-Lhx3 specifies motor neuron fate by inducing motor neuron genes and concomitantly suppressing the interneuron programs. Proc. Natl. Acad. Sci. USA 109, 3383-3388. 10.1073/pnas.1114515109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. (2017). How do SMA-linked mutations of SMN1 lead to structural/functional deficiency of the SMA protein?. PLoS ONE 12, e0178519 10.1371/journal.pone.0178519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B., Lin M., Tang Y., Wang B. and Wang J. H.-C. (2008a). A novel functional assessment of the differentiation of micropatterned muscle cells. J. Biomech. 41, 3349-3353. 10.1016/j.jbiomech.2008.09.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X.-J., Hu B.-Y., Jones S. A., Zhang Y.-S., LaVaute T., Du Z.-W. and Zhang S.-C. (2008b). Directed differentiation of ventral spinal progenitors and motor neurons from human embryonic stem cells by small molecules. Stem Cells 26, 886-893. 10.1634/stemcells.2007-0620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang R., Dong W., Shen X., Peng X., Aceves A. G. and Liu Y. (2016). Modeling myotonic dystrophy 1 in C2C12 myoblast cells. J. Vis. Exp., e54078 10.3791/54078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin C.-Y., Yoshida M., Li L.-T., Ikenaka A., Oshima S., Nakagawa K., Sakurai H., Matsui E., Nakahata T. and Saito M. K. (2019). iPSC-derived functional human neuromuscular junctions model the pathophysiology of neuromuscular diseases. JCI Insight 4, e124299 10.1172/jci.insight.124299 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lissouba A., Liao M., Kabashi E. and Drapeau P. (2018). Transcriptomic analysis of zebrafish TDP-43 transgenic lines. Front. Mol. Neurosci. 11, 463 10.3389/fnmol.2018.00463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd T. E. and Taylor J. P. (2010). Flightless flies: Drosophila models of neuromuscular disease. Ann. N. Y. Acad. Sci. 1184, e1-e20. 10.1111/j.1749-6632.2010.05432.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madden L., Juhas M., Kraus W. E., Truskey G. A. and Bursac N. (2015). Bioengineered human myobundles mimic clinical responses of skeletal muscle to drugs. eLife 4, e04885 10.7554/eLife.04885 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madji Hounoum B., Vourc'h P., Felix R., Corcia P., Patin F., Guéguinou M., Potier-Cartereau M., Vandier C., Raoul C., Andres C. R. et al. (2016). NSC-34 motor neuron-like cells are unsuitable as experimental model for glutamate-mediated excitotoxicity. Front. Cell. Neurosci. 10, 118 10.3389/fncel.2016.00118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maffioletti S. M., Gerli M. F. M., Ragazzi M., Dastidar S., Benedetti S., Loperfido M., VandenDriessche T., Chuah M. K. and Tedesco F. S. (2015). Efficient derivation and inducible differentiation of expandable skeletal myogenic cells from human ES and patient-specific iPS cells. Nat. Protoc. 10, 941-958. 10.1038/nprot.2015.057 [DOI] [PubMed] [Google Scholar]
- Maier O., Böhm J., Dahm M., Brück S., Beyer C. and Johann S. (2013). Differentiated NSC-34 motoneuron-like cells as experimental model for cholinergic neurodegeneration. Neurochem. Int. 62, 1029-1038. 10.1016/j.neuint.2013.03.008 [DOI] [PubMed] [Google Scholar]
- Mamchaoui K., Trollet C., Bigot A., Negroni E., Chaouch S., Wolff A., Kandalla P. K., Marie S., Di Santo J., St Guily J. L. et al. (2011). Immortalized pathological human myoblasts: towards a universal tool for the study of neuromuscular disorders. Skelet. Muscle 1, 34 10.1186/2044-5040-1-34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mann C. J., Perdiguero E., Kharraz Y., Aguilar S., Pessina P., Serrano A. L. and Muñoz-Cánoves P. (2011). Aberrant repair and fibrosis development in skeletal muscle. Skelet. Muscle 1, 21 10.1186/2044-5040-1-21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matusica D., Fenech M. P., Rogers M.-L. and Rush R. A. (2008). Characterization and use of the NSC-34 cell line for study of neurotrophin receptor trafficking. J. Neurosci. Res. 86, 553-565. 10.1002/jnr.21507 [DOI] [PubMed] [Google Scholar]
- Mäurer M., Toyka K. V. and Gold R. (2002). Immune mechanisms in acquired demyelinating neuropathies: lessons from animal models. Neuromuscul. Disord. 12, 405-414. 10.1016/S0960-8966(01)00302-9 [DOI] [PubMed] [Google Scholar]
- McCain M. L., Desplantez T., Geisse N. A., Rothen-Rutishauser B., Oberer H., Parker K. K. and Kleber A. G. (2012). Cell-to-cell coupling in engineered pairs of rat ventricular cardiomyocytes: relation between Cx43 immunofluorescence and intercellular electrical conductance. Am. J. Physiol. Heart Circ. Physiol. 302, H443-H450. 10.1152/ajpheart.01218.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCorquodale D., Pucillo E. M. and Johnson N. E. (2016). Management of Charcot-Marie-Tooth disease: improving long-term care with a multidisciplinary approach. J. Multidiscip. Healthc. 9, 7-19. 10.2147/JMDH.S69979 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGovern V. L., Iyer C. C., Arnold W. D., Gombash S. E., Zaworski P. G., Blatnik A. J. III, Foust K. D. and Burghes A. H. M. (2015). SMN expression is required in motor neurons to rescue electrophysiological deficits in the SMNΔ7 mouse model of SMA. Hum. Mol. Genet. 24, 5524-5541. 10.1093/hmg/ddv283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMahon D. K., Anderson P. A., Nassar R., Bunting J. B., Saba Z., Oakeley A. E. and Malouf N. N. (1994). C2C12 cells: biophysical, biochemical, and immunocytochemical properties. Am. J. Physiol. 266, C1795-C1802. 10.1152/ajpcell.1994.266.6.C1795 [DOI] [PubMed] [Google Scholar]
- McWhorter M. L., Monani U. R., Burghes A. H. M. and Beattie C. E. (2003). Knockdown of the survival motor neuron (Smn) protein in zebrafish causes defects in motor axon outgrowth and pathfinding. J. Cell Biol. 162, 919-931. 10.1083/jcb.200303168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menconi M., Gonnella P., Petkova V., Lecker S. and Hasselgren P.-O. (2008). Dexamethasone and corticosterone induce similar, but not identical, muscle wasting responses in cultured L6 and C2C12 myotubes. J. Cell. Biochem. 105, 353-364. 10.1002/jcb.21833 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mills R. J., Parker B. L., Monnot P., Needham E. J., Vivien C. J., Ferguson C., Parton R. G., James D. E., Porrello E. R. and Hudson J. E. (2019). Development of a human skeletal micro muscle platform with pacing capabilities. Biomaterials 198, 217-227. 10.1016/j.biomaterials.2018.11.030 [DOI] [PubMed] [Google Scholar]
- Morena J., Gupta A. and Hoyle J. C. (2019). Charcot-marie-tooth: from molecules to therapy. Int. J. Mol. Sci. 20, 3419 10.3390/ijms20143419 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morimoto Y., Kato-Negishi M., Onoe H. and Takeuchi S. (2013). Three-dimensional neuron–muscle constructs with neuromuscular junctions. Biomaterials 34, 9413-9419. 10.1016/j.biomaterials.2013.08.062 [DOI] [PubMed] [Google Scholar]
- Morrice J. R., Gregory-Evans C. Y. and Shaw C. A. (2018). Animal models of amyotrophic lateral sclerosis: A comparison of model validity. Neural Regener. Res. 13, 2050-2054. 10.4103/1673-5374.241445 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morris T. A., Naik J., Fibben K. S., Kong X., Kiyono T., Yokomori K. and Grosberg A. (2020). Striated myocyte structural integrity: Automated analysis of sarcomeric z-discs. PLoS Comput. Biol. 16, e1007676 10.1371/journal.pcbi.1007676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murdocca M., Ciafrè S. A., Spitalieri P., Talarico R. V., Sanchez M., Novelli G. and Sangiuolo F. (2016). SMA human iPSC-derived motor neurons show perturbed differentiation and reduced miR-335-5p expression. Int. J. Mol. Sci. 17, 1231 10.3390/ijms17081231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nair R. R., Corrochano S., Gasco S., Tibbit C., Thompson D., Maduro C., Ali Z., Fratta P., Arozena A. A., Cunningham T. J. et al. (2019). Uses for humanised mouse models in precision medicine for neurodegenerative disease. Mamm. Genome 30, 173-191. 10.1007/s00335-019-09807-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nedachi T., Fujita H. and Kanzaki M. (2008). Contractile C2C12 myotube model for studying exercise-inducible responses in skeletal muscle. Am. J. Physiol. Endocrinol. Metab. 295, E1191-E1204. 10.1152/ajpendo.90280.2008 [DOI] [PubMed] [Google Scholar]
- Nesmith A. P., Wagner M. A., Pasqualini F. S., O'Connor B. B., Pincus M. J., August P. R. and Parker K. K. (2016). A human in vitro model of Duchenne muscular dystrophy muscle formation and contractility. J. Cell Biol. 215, 47-56. 10.1083/jcb.201603111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neville C., Rosenthal N., McGrew M., Bogdanova N. and Hauschka S. (1997). Chapter 5 Skeletal muscle cultures. Methods Cell Biol. 52, 85-116. 10.1016/S0091-679X(08)60375-1 [DOI] [PubMed] [Google Scholar]
- Nguyen H. P., Van Broeckhoven C. and van der Zee J. (2018). ALS genes in the genomic Era and their implications for FTD. Trends Genet. 34, 404-423. 10.1016/j.tig.2018.03.001 [DOI] [PubMed] [Google Scholar]
- Novak R., Ingram M., Marquez S., Das D., Delahanty A., Herland A., Maoz B. M., Jeanty S. S. F., Somayaji M. R., Burt M. et al. (2020). Robotic fluidic coupling and interrogation of multiple vascularized organ chips. Nat. Biomed. Eng. 4, 407-420. 10.1038/s41551-019-0497-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Öberg A. I., Dehvari N. and Bengtsson T. (2011). β-Adrenergic inhibition of contractility in L6 skeletal muscle cells. PLoS ONE 6, e22304 10.1371/journal.pone.0022304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oeda T., Shimohama S., Kitagawa N., Kohno R., Imura T., Shibasaki H. and Ishii N. (2001). Oxidative stress causes abnormal accumulation of familial amyotrophic lateral sclerosis-related mutant SOD1 in transgenic Caenorhabditis elegans. Hum. Mol. Genet. 10, 2013-2023. 10.1093/hmg/10.19.2013 [DOI] [PubMed] [Google Scholar]
- Osaki T., Uzel S. G. M. and Kamm R. D. (2018). Microphysiological 3D model of amyotrophic lateral sclerosis (ALS) from human iPS-derived muscle cells and optogenetic motor neurons. Sci. Adv. 4, eaat5847 10.1126/sciadv.aat5847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osaki T., Uzel S. G. M. and Kamm R. D. (2020). On-chip 3D neuromuscular model for drug screening and precision medicine in neuromuscular disease. Nat. Protoc. 15, 421-449. 10.1038/s41596-019-0248-1 [DOI] [PubMed] [Google Scholar]
- Palade J., Pal A., Rawls A., Stabenfeldt S. and Wilson-Rawls J. (2019). Molecular analysis of muscle progenitor cells on extracellular matrix coatings and hydrogels. Acta Biomater. 97, 296-309. 10.1016/j.actbio.2019.08.019 [DOI] [PubMed] [Google Scholar]
- Palchesko R. N., Zhang L., Sun Y. and Feinberg A. W. (2012). Development of polydimethylsiloxane substrates with tunable elastic modulus to study cell mechanobiology in muscle and nerve. PLoS ONE 7, e51499 10.1371/journal.pone.0051499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualini F. S., Agarwal A., O'Connor B. B., Liu Q., Sheehy S. P. and Parker K. K. (2018). Traction force microscopy of engineered cardiac tissues. PLoS ONE 13, e0194706 10.1371/journal.pone.0194706 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pei W., Xu L., Varshney G. K., Carrington B., Bishop K., Jones M. P., Huang S. C., Idol J., Pretorius P. R., Beirl A. et al. (2016). Additive reductions in zebrafish PRPS1 activity result in a spectrum of deficiencies modeling several human PRPS1-associated diseases. Sci. Rep. 6, 29946 10.1038/srep29946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perry S., Han Y., Das A. and Dickman D. (2017). Homeostatic plasticity can be induced and expressed to restore synaptic strength at neuromuscular junctions undergoing ALS-related degeneration. Hum. Mol. Genet. 26, 4153-4167. 10.1093/hmg/ddx304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips W. D. and Vincent A. (2016). Pathogenesis of myasthenia gravis: update on disease types, models, and mechanisms. F1000Research 5, 1513 10.12688/f1000research.8206.1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pimentel M. R., Falcone S., Cadot B. and Gomes E. R. (2017). In vitro differentiation of mature myofibers for live imaging. J. Vis. Exp. e55141 10.3791/55141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto S., Cunha C., Barbosa M., Vaz A. R. and Brites D. (2017). Exosomes from NSC-34 cells transfected with hSOD1-G93A are enriched in miR-124 and drive alterations in microglia phenotype. Front. Neurosci. 11, 213 10.3389/fnins.2017.00273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollot B. E., Rathbone C. R., Wenke J. C. and Guda T. (2018). Natural polymeric hydrogel evaluation for skeletal muscle tissue engineering. J. Biomed. Mater. Res. B Appl. Biomater. 106, 672-679. 10.1002/jbm.b.33859 [DOI] [PubMed] [Google Scholar]
- Powell C., Shansky J., Del Tatto M., Forman D. E., Hennessey J., Sullivan K., Zielinski B. A. and Vandenburgh H. H. (1999). Tissue-engineered human bioartificial muscles expressing a foreign recombinant protein for gene therapy. Hum. Gene. Ther. 10, 565-577. 10.1089/10430349950018643 [DOI] [PubMed] [Google Scholar]
- Powell C. A., Smiley B. L., Mills J. and Vandenburgh H. H. (2002). Mechanical stimulation improves tissue-engineered human skeletal muscle. Am. J. Physiol. Cell Physiol. 283, C1557-C1565. 10.1152/ajpcell.00595.2001 [DOI] [PubMed] [Google Scholar]
- Prinsen F. M. C. and Veerkamp H. J. (1998). Transfection of L6 myoblasts with adipocyte fatty acid-binding protein cDNA does not affect fatty acid uptake but disturbs lipid metabolism and fusion. Biochem. J. 329, 265-273. 10.1042/bj3290265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin D., Xia Y. and Whitesides G. M. (2010). Soft lithography for micro- and nanoscale patterning. Nat. Protoc. 5, 491-502. 10.1038/nprot.2009.234 [DOI] [PubMed] [Google Scholar]
- Qu Q., Li D., Louis K. R., Li X., Yang H., Sun Q., Crandall S. R., Tsang S., Zhou J., Cox C. L. et al. (2014). High-efficiency motor neuron differentiation from human pluripotent stem cells and the function of Islet-1. Nat. Commun. 5, 3449 10.1038/ncomms4449 [DOI] [PubMed] [Google Scholar]
- Ramón-Azcón J., Ahadian S., Estili M., Liang X., Ostrovidov S., Kaji H., Shiku H., Ramalingam M., Nakajima K., Sakka Y. et al. (2013). Dielectrophoretically aligned carbon nanotubes to control electrical and mechanical properties of hydrogels to fabricate contractile muscle myofibers. Adv. Mater. 25, 4028-4034. 10.1002/adma.201301300 [DOI] [PubMed] [Google Scholar]
- Rangarajan S., Madden L. and Bursac N. (2014). Use of flow, electrical, and mechanical stimulation to promote engineering of striated muscles. Ann. Biomed. Eng. 42, 1391-1405. 10.1007/s10439-013-0966-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao N., Evans S., Stewart D., Spencer K. H., Sheikh F., Hui E. E. and Christman K. L. (2013). Fibroblasts influence muscle progenitor differentiation and alignment in contact independent and dependent manners in organized co-culture devices. Biomed. Microdevices 15, 161-169. 10.1007/s10544-012-9709-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao L., Qian Y., Khodabukus A., Ribar T. and Bursac N. (2018). Engineering human pluripotent stem cells into a functional skeletal muscle tissue. Nat. Commun. 9, 126 10.1038/s41467-017-02636-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribeiro A. J. S., Ang Y.-S., Fu J.-D., Rivas R. N., Mohamed T. M. A., Higgs G. C., Srivastava D. and Pruitt B. L. (2015). Contractility of single cardiomyocytes differentiated from pluripotent stem cells depends on physiological shape and substrate stiffness. Proc. Natl. Acad. Sci. USA 112, 12705-12710. 10.1073/pnas.1508073112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ripps M. E., Huntley G. W., Hof P. R., Morrison J. H. and Gordon J. W. (1995). Transgenic mice expressing an altered murine superoxide dismutase gene provide an animal model of amyotrophic lateral sclerosis. Proc. Natl. Acad. Sci. USA 92, 689-693. 10.1073/pnas.92.3.689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson M. M., Sather B. K., Burney E. R., Ehrlicher S. E., Stierwalt H. D., Franco M. C. and Newsom S. A. (2019). Robust intrinsic differences in mitochondrial respiration and H2O2 emission between L6 and C2C12 cells. Am. J. Physiol. Cell Physiol. 317, C339-C347. 10.1152/ajpcell.00343.2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Şahin A., Held A., Bredvik K., Major P., Achilli T.-M., Kerson A. G., Wharton K., Stilwell G. and Reenan R. (2017). Human SOD1 ALS mutations in a Drosophila knock-in model cause severe phenotypes and reveal dosage-sensitive gain- and loss-of-function components. Genetics 205, 707-723. 10.1534/genetics.116.190850 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saini J., Faroni A., Abd Al Samid M., Reid A. J., Lightfoot A. P., Mamchaoui K., Mouly V., Butler-Browne G., McPhee J. S., Degens H. et al. (2019). Simplified in vitro engineering of neuromuscular junctions between rat embryonic motoneurons and immortalized human skeletal muscle cells. Stem Cells Cloning Adv. Appl. 12, 1-9. 10.2147/SCCAA.S187655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salani S., Donadoni C., Rizzo F., Bresolin N., Comi G. P. and Corti S. (2012). Generation of skeletal muscle cells from embryonic and induced pluripotent stem cells as an in vitro model and for therapy of muscular dystrophies. J. Cell. Mol. Med. 16, 1353-1364. 10.1111/j.1582-4934.2011.01498.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sances S., Bruijn L. I., Chandran S., Eggan K., Ho R., Klim J. R., Livesey M. R., Lowry E., Macklis J. D., Rushton D. et al. (2016). Modeling ALS with motor neurons derived from human induced pluripotent stem cells. Nat. Neurosci. 19, 542-553. 10.1038/nn.4273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santhanam N., Kumanchik L., Guo X., Sommerhage F., Cai Y., Jackson M., Martin C., Saad G., McAleer C. W., Wang Y. et al. (2018). Stem cell derived phenotypic human neuromuscular junction model for dose response evaluation of therapeutics. Biomaterials 166, 64-78. 10.1016/j.biomaterials.2018.02.047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santosa K. B., Keane A. M., Jablonka-Shariff A., Vannucci B. and Snyder-Warwick A. K. (2018). Clinical relevance of terminal Schwann cells: an overlooked component of the neuromuscular junction. J. Neurosci. Res. 96, 1125-1135. 10.1002/jnr.24231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saporta A. S. D., Sottile S. L., Miller L. J., Feely S. M. E., Siskind C. E. and Shy M. E. (2011). Charcot-Marie-Tooth disease subtypes and genetic testing strategies. Ann. Neurol. 69, 22-33. 10.1002/ana.22166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sareen D., O'Rourke J. G., Meera P., Muhammad A. K. M. G., Grant S., Simpkinson M., Bell S., Carmona S., Ornelas L., Sahabian A. et al. (2013). Targeting RNA foci in iPSC-derived motor neurons from ALS patients with a C9ORF72 repeat expansion. Sci. Transl. Med. 5, 208ra149 10.1126/scitranslmed.3007529 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selvaraj S., Mondragon-Gonzalez R., Xu B., Magli A., Kim H., Lainé J., Kiley J., McKee H., Rinaldi F., Aho J. et al. (2019). Screening identifies small molecules that enhance the maturation of human pluripotent stem cell-derived myotubes. eLife 8, e47970, 10.7554/eLife.47970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seminary E. R., Sison S. L. and Ebert A. D. (2018). Modeling protein aggregation and the heat shock response in ALS iPSC-derived motor neurons. Front. Neurosci. 12, 86 10.3389/fnins.2018.00086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shansky J., Del Tatto M., Chromiak J. and Vandenburgh H. (1997). A simplified method for tissue engineering skeletal muscle organoids in vitro. In Vitro Cell. Dev. Biol. Anim. 33, 659-661. 10.1007/s11626-997-0118-y [DOI] [PubMed] [Google Scholar]
- Shaw M. P., Higginbottom A., McGown A., Castelli L. M., James E., Hautbergue G. M., Shaw P. J. and Ramesh T. M. (2018). Stable transgenic C9orf72 zebrafish model key aspects of the ALS/FTD phenotype and reveal novel pathological features. Acta Neuropathol. Commun. 6, 125 10.1186/s40478-018-0629-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton M., Kocharyan A., Liu J., Skerjanc I. S. and Stanford W. L. (2016). Robust generation and expansion of skeletal muscle progenitors and myocytes from human pluripotent stem cells. Methods 101, 73-84. 10.1016/j.ymeth.2015.09.019 [DOI] [PubMed] [Google Scholar]
- Shi Y., Lin S., Staats K. A., Li Y., Chang W.-H., Hung S.-T., Hendricks E., Linares G. R., Wang Y., Son E. Y. et al. (2018). Haploinsufficiency leads to neurodegeneration in C9ORF72 ALS/FTD human induced motor neurons. Nat. Med. 24, 313 10.1038/nm.4490 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu K., Araki H., Sakata K., Tonomura W., Hashida M. and Konishi S. (2015). Microfluidic devices for construction of contractile skeletal muscle microtissues. J. Biosci. Bioeng. 119, 212-216. 10.1016/j.jbiosc.2014.07.003 [DOI] [PubMed] [Google Scholar]
- Shimojo D., Onodera K., Doi-Torii Y., Ishihara Y., Hattori C., Miwa Y., Tanaka S., Okada R., Ohyama M., Shoji M. et al. (2015). Rapid, efficient, and simple motor neuron differentiation from human pluripotent stem cells. Mol. Brain 8, 79 10.1186/s13041-015-0172-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sincennes M. C., Wang Y. X. and Rudnicki M. A. (2017). Primary mouse myoblast purification using magnetic cell separation. Methods Mol. Biol. 1556, 41-50. 10.1007/978-1-4939-6771-1_3 [DOI] [PubMed] [Google Scholar]
- Skoglund G., Laine J., Darabi R., Fournier E., Perlingeiro R. and Tabti N. (2014). Physiological and ultrastructural features of human induced pluripotent and embryonic stem cell-derived skeletal myocytes in vitro. Proc. Natl. Acad. Sci. USA 111, 8275-8280. 10.1073/pnas.1322258111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sleigh J. N. and Sattelle D. B. (2010). C. elegans models of neuromuscular diseases expedite translational research. Transl. Neurosci. 1, 214-227. 10.2478/v10134-010-0032-9 [DOI] [Google Scholar]
- Sleigh J. N., Grice S. J., Burgess R. W., Talbot K. and Cader M. Z. (2013). Neuromuscular junction maturation defects precede impaired lower motor neuron connectivity in Charcot-Marie-Tooth type 2D mice. Hum. Mol. Genet. 23, 2639-2650. 10.1093/hmg/ddt659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. S., Long C. J., McAleer C., Bobbitt N., Srinivasan B. and Hickman J. J. (2014a). Utilization of microscale silicon cantilevers to assess cellular contractile function in vitro. J. Vis. Exp. e51866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. S. T., Long C. J., Pirozzi K., Najjar S., McAleer C., Vandenburgh H. H. and Hickman J. J. (2014b). A multiplexed chip-based assay system for investigating the functional development of human skeletal myotubes in vitro. J. Biotechnol. 185, 15-18. 10.1016/j.jbiotec.2014.05.029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith A. S. T., Passey S. L., Martin N. R. W., Player D. J., Mudera V., Greensmith L. and Lewis M. P. (2016). Creating interactions between tissue-engineered skeletal muscle and the peripheral nervous system. Cells Tissues Organs 202, 143-158. 10.1159/000443634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Son E. Y., Ichida J. K., Wainger B. J., Toma J. S., Rafuse V. F., Woolf C. J. and Eggan K. (2011). Conversion of mouse and human fibroblasts into functional spinal motor neurons. Cell Stem Cell 9, 205-218. 10.1016/j.stem.2011.07.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano-Arroquia A., Clegg P. D., Molloy A. P. and Goljanek-Whysall K. (2017). Preparation and culture of myogenic precursor cells/primary myoblasts from skeletal muscle of adult and aged humans. J. Vis. Exp. 55047 10.3791/51866 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spaulding E. L., Sleigh J. N., Morelli K. H., Pinter M. J., Burgess R. W. and Seburn K. L. (2016). Synaptic deficits at neuromuscular junctions in two mouse models of charcot–marie–tooth type 2d. J. Neurosci. 36, 3254-3267. 10.1523/JNEUROSCI.1762-15.2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinazzola J. M. and Gussoni E. (2017). Isolation of primary human skeletal muscle cells. Bio. Protoc. 7 10.21769/BioProtoc.2591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spring A. M., Raimer A. C., Hamilton C. D., Schillinger M. J. and Matera A. G. (2019). Comprehensive modeling of spinal muscular atrophy in Drosophila melanogaster. Front. Mol. Neurosci. 12, 113 10.3389/fnmol.2019.00113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinbeck J. A., Jaiswal M. K., Calder E. L., Kishinevsky S., Weishaupt A., Toyka K. V., Goldstein P. A. and Studer L. (2016). Functional connectivity under optogenetic control allows modeling of human neuromuscular disease. Cell Stem Cell 18, 134-143. 10.1016/j.stem.2015.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su F.-C., Goutman S. A., Chernyak S., Mukherjee B., Callaghan B. C., Batterman S. and Feldman E. L. (2016). Association of environmental toxins with amyotrophic lateral sclerosis. JAMA Neurol. 73, 803-811. 10.1001/jamaneurol.2016.0594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suh G. C., Bettadapur A., Santoso J. W. and McCain M. L. (2017). Fabrication of micromolded gelatin hydrogels for long-term culture of aligned skeletal myotubes. Methods Mol. Biol. 1668, 147-163. 10.1007/978-1-4939-7283-8_11 [DOI] [PubMed] [Google Scholar]
- Sun Y., Duffy R., Lee A. and Feinberg A. W. (2013). Optimizing the structure and contractility of engineered skeletal muscle thin films. Acta Biomater. 9, 7885-7894. 10.1016/j.actbio.2013.04.036 [DOI] [PubMed] [Google Scholar]
- Sun M., Sun X., Wang Z., Guo S., Yu G. and Yang H. (2018). Synthesis and properties of Gelatin Methacryloyl (GelMA) hydrogels and their recent applications in load-bearing tissue. Polymers 10, 1290 10.3390/polym10111290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A. M., Rhee S. W., Tu C. H., Cribbs D. H., Cotman C. W. and Jeon N. L. (2003). Microfluidic multicompartment device for neuroscience research. Langmuir 19, 1551-1556. 10.1021/la026417v [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thonhoff J. R., Simpson E. P. and Appel S. H. (2018). Neuroinflammatory mechanisms in amyotrophic lateral sclerosis pathogenesis. Curr. Opin. Neurol. 31, 635-639. 10.1097/WCO.0000000000000599 [DOI] [PubMed] [Google Scholar]
- Turner M. R., Barnwell J., Al-Chalabi A. and Eisen A. (2012). Young-onset amyotrophic lateral sclerosis: historical and other observations. Brain 135, 2883-2891. 10.1093/brain/aws144 [DOI] [PubMed] [Google Scholar]
- Umbach J. A., Adams K. L., Gundersen C. B. and Novitch B. G. (2012). Functional neuromuscular junctions formed by embryonic stem cell-derived motor neurons. PLoS ONE 7, e36049 10.1371/journal.pone.0036049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urja V., Khaire K., Balakrishnan S. and Uggini G. K. (2018). Chick embryonic cells as a source for generating in vitro model of muscle cell dystrophy. In Vitro Cell. Dev. Biol. Anim. 54, 756-769. 10.1007/s11626-018-0297-8 [DOI] [PubMed] [Google Scholar]
- Uzel S. G. M., Platt R. J., Subramanian V., Pearl T. M., Rowlands C. J., Chan V., Boyer L. A., So P. T. C. and Kamm R. D. (2016). Microfluidic device for the formation of optically excitable, three-dimensional, compartmentalized motor units. Sci. Adv. 2, e1501429 10.1126/sciadv.1501429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallette F. M., Vigny M. and Massoulié J. (1986). Muscular differentiation of chicken myotubes in a simple defined synthetic culture medium and in serum supplemented media: expression of the molecular forms of acetylcholinesterase. Neurochem. Int. 8, 121-133. 10.1016/0197-0186(86)90109-9 [DOI] [PubMed] [Google Scholar]
- van der Wal E., Herrero-Hernandez P., Wan R., Broeders M., In ‘t Groen S. L. M., van Gestel T. J. M., van Ijcken W. F. J., Cheung T. H., van der Ploeg A. T., Schaaf G. J. et al. (2018). Large-scale expansion of human iPSC-derived skeletal muscle cells for disease modeling and cell-based therapeutic strategies. Stem Cell Rep. 10, 1975-1990. 10.1016/j.stemcr.2018.04.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vaughan M. and Lamia K. A. (2019). Isolation and differentiation of primary myoblasts from mouse skeletal muscle explants. J. Vis. Exp. e60310 10.3791/60310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Farr G. W., Hall D. H., Li F., Furtak K., Dreier L. and Horwich A. L. (2009). An ALS-linked mutant SOD1 produces a locomotor defect associated with aggregation and synaptic dysfunction when expressed in neurons of Caenorhabditis elegans. PLoS Genet. 5, e1000350 10.1371/journal.pgen.1000350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang P.-Y., Thissen H. and Tsai W.-B. (2012). The roles of RGD and grooved topography in the adhesion, morphology, and differentiation of C2C12 skeletal myoblasts. Biotechnol. Bioeng. 109, 2104-2115. 10.1002/bit.24452 [DOI] [PubMed] [Google Scholar]
- Wang L., Shansky J. and Vandenburgh H. (2013). Induced formation and maturation of acetylcholine receptor clusters in a defined 3D bio-artificial muscle. Mol. Neurobiol. 48, 397-403. 10.1007/s12035-013-8412-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z., Volinsky A. A. and Gallant N. D. (2014). Crosslinking effect on polydimethylsiloxane elastic modulus measured by custom-built compression instrument. J. Appl. Polym. Sci. 131 10.1002/app.41050 [DOI] [Google Scholar]
- Watson M. R., Lagow R. D., Xu K., Zhang B. and Bonini N. M. (2008). A drosophila model for amyotrophic lateral sclerosis reveals motor neuron damage by human SOD1. J. Biol. Chem. 283, 24972-24981. 10.1074/jbc.M804817200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson K., Das M., Wahl K. J., Colton R. J. and Hickman J. (2010). Measurement of contractile stress generated by cultured rat muscle on silicon cantilevers for toxin detection and muscle performance enhancement. PLoS ONE 5, e11042 10.1371/journal.pone.0011042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xi H., Fujiwara W., Gonzalez K., Jan M., Liebscher S., Van Handel B., Schenke-Layland K. and Pyle A. D. (2017). In vivo human somitogenesis guides somite development from hPSCs. Cell Rep. 18, 1573-1585. 10.1016/j.celrep.2017.01.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z., Poidevin M., Li X., Li Y., Shu L., Nelson D. L., Li H., Hales C. M., Gearing M., Wingo T. S. et al. (2013). Expanded GGGGCC repeat RNA associated with amyotrophic lateral sclerosis and frontotemporal dementia causes neurodegeneration. Proc. Natl. Acad. Sci. USA 110, 7778-7783. 10.1073/pnas.1219643110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe D. (1968). Retention of differentiation potentialities during prolonged cultivation of myogenic cells. Proc. Natl. Acad. Sci. USA 61, 477-483. 10.1073/pnas.61.2.477 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaffe D. and Saxel O. (1977). Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 270, 725-727. 10.1038/270725a0 [DOI] [PubMed] [Google Scholar]
- Yang H. S., Ieronimakis N., Tsui J. H., Kim H. N., Suh K.-Y., Reyes M. and Kim D.-H. (2014). Nanopatterned muscle cell patches for enhanced myogenesis and dystrophin expression in a mouse model of muscular dystrophy. Biomaterials 35, 1478-1486. 10.1016/j.biomaterials.2013.10.067 [DOI] [PubMed] [Google Scholar]
- Young C. S., Hicks M. R., Ermolova N. V., Nakano H., Jan M., Younesi S., Karumbayaram S., Kumagai-Cresse C., Wang D., Zack J. A. et al. (2016). A single CRISPR-Cas9 deletion strategy that targets the majority of DMD patients restores dystrophin function in hiPSC-derived muscle cells. Cell Stem Cell 18, 533-540. 10.1016/j.stem.2016.01.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang B., Tu P., Abtahian F., Trojanowski J. Q. and Lee V. M.-Y. (1997). Neurofilaments and orthograde transport are reduced in ventral root axons of transgenic mice that express human SOD1 with a G93A mutation. J. Cell Biol. 139, 1307-1315. 10.1083/jcb.139.5.1307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Hong S., Yen R., Kondash M., Fernandez C. E. and Truskey G. A. (2018). A system to monitor statin-induced myopathy in individual engineered skeletal muscle myobundles. Lab. Chip 18, 2787-2796. 10.1039/C8LC00654G [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Q., Yang S. T., Wang J. J., Zhou J., Xing S. S., Shen C. C., Wang X. X., Yue Y. X., Song J., Chen M. et al. (2015). TNF alpha inhibits myogenic differentiation of C2C12 cells through NF-κB activation and impairment of IGF-1 signaling pathway. Biochem. Biophys. Res. Commun. 458, 790-795. 10.1016/j.bbrc.2015.02.026 [DOI] [PubMed] [Google Scholar]
- Ziemkiewicz N., Talovic M., Madsen J., Hill L., Scheidt R., Patel A., Haas G., Marcinczyk M., Zustiak S. P. and Garg K. (2018). Laminin-111 functionalized polyethylene glycol hydrogels support myogenic activity in vitro. Biomed. Mater. 13, 065007 10.1088/1748-605X/aad915 [DOI] [PubMed] [Google Scholar]
- Zuroske T. (2019). Upcoming market catalysts in Q2 2019. Nat. Rev. Drug Discov. 18, 244 10.1038/d41573-019-00048-1 [DOI] [PubMed] [Google Scholar]