

Abstract
Accumulating evidence indicates that macrophage polarization plays a crucial role in coxsackievirus B3 (CVB3)-induced viral myocarditis (VM). Our previous study demonstrated that long noncoding ribonucleic acid (lncRNA) AK085865 ablation confers susceptibility to VM by regulating macrophage polarization. However, the detailed molecular mechanisms by which AK085865 regulates macrophage polarization remain to be explored. In this study, we found that AK085865 specifically interacts with interleukin enhancer-binding factor 2 (ILF2) and facilitates M2 macrophage polarization by functioning as a negative regulator in the ILF2-ILF3 complex-mediated microRNA (miRNA or miR) processing pathway. miR-192 was downregulated, whereas the levels of pri-miR-192 were significantly increased in bone marrow-derived macrophages (BMDMs) from AK085865−/− mice compared with the BMDMs from wild-type (WT) mice. Conversely, knockdown of ILF2 resulted in elevated levels of mature miR-192 and decreased expression of pri-miR-192 in BMDMs from AK085865−/− mice. Moreover, miR-192 overexpression promoted macrophage M2 polarization in vitro, and interleukin-1 receptor-associated kinase 1 (IRAK1) was identified as a direct target. miR-192 overexpression effectively rescued mice from lethal myocarditis caused by CVB3 infection and switched myocardial-infiltrating macrophages to a predominant M2 phenotype. Collectively, our findings uncover a critical mechanism of AK085865 in the regulation of macrophage polarization in vitro and in vivo and provide a potential, clinically significant therapeutic target.
Keywords: long noncoding RNA, viral myocarditis, macrophage polarization, miR-192, IRAK1
Graphical Abstract
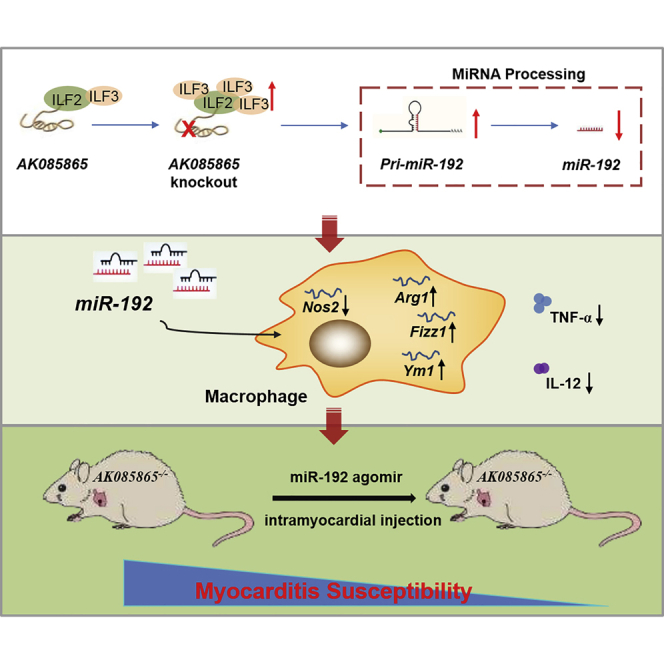
Lv and colleagues demonstrated that lncRNA AK085865 promotes macrophage M2 polarization in CVB3-induced VM by regulating ILF2-ILF3 complex-mediated miR-192 biogenesis, which rescues AK085865 KO mice from enhanced susceptibility to CVB3-induced VM. These observations uncover a critical mechanism of AK085865 in macrophage polarization and provide a potential therapeutic target for VM.
Introduction
Viral myocarditis (VM) is a virus-induced cardiac inflammatory disease that can further develop to chronic VM, dilated cardiomyopathy, and even heart failure. It appears to be a major cause of sudden cardiac death in children and adults.1,2 Coxsackievirus B3 (CVB3) has been identified as the most common pathogen for VM.3,4 Emerging evidence indicates that the overwhelming inflammation is the main pathologic factor in the course of VM.2 However, the regulatory mechanism underlying the inflammation is still poorly defined.
As the pivotal inflammatory cell subset, macrophages have been found to be enriched in heart tissues as early as day 3 post-CVB3 infection.5 Macrophages, as master regulators of inflammation, are highly plastic and heterogeneous. Mirroring the T helper 1 (Th1)/Th2 nomenclature, macrophages can be polarized into M1 (classically activated macrophages) or M2 (alternatively activated macrophages), depending on the cardiac microenvironment.6 M1 macrophages, induced by lipopolysaccharide (LPS) and interferon-γ (IFN-γ), typically promote myocardial inflammation and tissue destruction by producing proinflammatory cytokines. In contrast, M2 macrophages, induced by interleukin 4 (IL-4) or IL-13, secrete anti-inflammatory cytokines associated with tissue repair.7, 8, 9 Our previous studies and other studies have shown that M1 macrophages significantly aggravate myocarditis, whereas M2 macrophages alleviate myocardial inflammation.10, 11, 12, 13, 14, 15, 16 Despite the importance of macrophage polarization to the inflammation in VM, its regulatory mechanisms are still not fully understood.
Long noncoding ribonucleic acids (lncRNAs) are a large class of noncoding protein transcripts that are greater than 200 bases in length.17 They are involved in many physiological and pathological processes, including genomic imprinting, embryonic development, cell differentiation, tumor metastasis, and cell-cycle regulation.18, 19, 20 Although a number of lncRNAs have been reported to have crucial functions in diverse processes and diseases, only a few lncRNAs have been shown to regulate the immune system.21, 22, 23, 24
Our previous study identified the lncRNA AK085865 as a functionally important regulator during macrophage polarization, and we demonstrated that AK085865 ablation confers susceptibility to VM by regulating macrophage polarization.10 In the present study, we aimed to investigate the detailed molecular mechanisms by which AK085865 regulates macrophage polarization in VM.
Results
Identification of IL Enhancer-Binding Factor 2 (ILF2) As a Binding Partner of lncRNA AK085865
To explore the potential function of lncRNA AK085865 in the regulation of macrophage polarization, we attempted to identify the protein partners of AK085865. To this end, we performed RNA-protein binding assays by incubating in vitro-transcribed biotinylated AK085865 or its antisense control RNA with whole-cell extracts from bone marrow-derived macrophages (BMDMs). RNA-protein complexes were captured using streptavidin magnetic beads and resolved via sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and protein bands (35–50 kD) that were specifically enriched in AK085865 pull-down assays were subjected to mass spectrometry (MS) for identification (Figure 1A). This approach identified three RNA-binding proteins that exhibited higher enrichment in AK085865 pull-down assays relative to antisense controls. The data of MS analysis of the proteins pulled down by lncRNA AK085865 are shown in Table S1.
Figure 1.

Identification of ILF2 As a Binding Partner of AK085865
(A) SDS-PAGE analysis of nuclear extracts purified from the BMDM in vitro binding assay using biotinylated AK085865 or antisense control RNA. The highlighted protein bands were subjected to mass spectrometry analysis. (B) Western blot confirms the interaction of AK085865 and ILF2 in vitro. (C) RIP was performed using antibodies against ILF2 in BMDMs. RNAs interacting with ILF2 were eluted, reverse transcribed, and quantified by qRT-PCR. (D) Schematic of AK085865 deletion mutants (Muts) used in the RNA-protein binding assays. (E) ILF2 binds the 3′-region of AK085865. The RNA-protein binding assay was performed using biotinylated full-length or deletion Muts of AK085865, and the nuclear extracts were isolated from BMDMs, captured using streptavidin beads, and subjected to western blot against ILF2. Experiments were repeated three times, and a representative result is shown.
The ability of RNA-binding protein to bind AK085865 was confirmed by western blotting. We confirmed that AK085865 specifically interacted with ILF2 but not with eukaryotic translation elongation factor 1 alpha 1 (EEF1A1) or RBMX-like 1 (RBMXL1) (Figure 1B). We also confirmed the AK085865-ILF2 interaction in vivo. ILF2 RNA-binding protein immunoprecipitation (RIP) in noncrosslinked BMDMs, followed by qRT-PCR analysis of copurified RNAs, showed that AK085865 was specifically enriched in ILF2 immunoprecipitates (Figure 1C). These results indicate that AK085865 specifically interacts with ILF2, both in vitro and in vivo.
We also took advantage of a series of deletion mutants of AK085865 to map the ILF2 binding region. The results from in vitro binding assays indicated that ILF2 interacted with the 3′-466 nucleotide region of AK085865 (Figures 1D and 1E). This region was both necessary and sufficient to bind ILF2 (Figure 1E). These data indicate that AK085865 interacts with ILF2 via its 3′-466 region, which is essential for ILF2 binding.
lncRNA AK085865 Regulates ILF2 Functions
Previous studies have shown that the ILF2 and ILF3 proteins always form a heterodimer, and the ILF2-ILF3 complex negatively regulates the pri-microRNA (miRNA) processing step, resulting in a reduction of mature miRNA production.25,26 The direct binding of AK085865 to ILF2 raised the possibility that AK085865 may regulate ILF2-ILF3 protein levels and/or functions. We first compared the protein levels of ILF2 and ILF3 using western blot analysis in BMDMs of AK085865−/− and wild-type (WT) mice, and we found that AK085865 deletion caused an increase in ILF2 and ILF3 protein levels (Figure 2A). Moreover, a coimmunoprecipitation (coIP) assay was used to analyze the interaction between endogenous ILF2 and ILF3 using an antibody against ILF2 in BMDMs from AK085865−/− and WT mice. coIP analysis revealed that there was a direct interaction between ILF2 and ILF3, and AK085865 deletion enhanced the binding of ILF2 and ILF3 (Figure 2B).
Figure 2.

lncRNA AK085865 Regulates ILF2-ILF3 Complex Functions and miRNA Biogenesis
BMDMs were prepared from AK085865−/− (KO) and wild-type (WT) mice. (A) ILF2 and ILF3 protein levels were detected by western blot. β-actin was used as a loading control. (B) Coimmunoprecipitation was performed using antibodies against ILF2 for the pull-down assay, and the proteins interacting with ILF2 were eluted and quantified by western blot. (C) Volcano plots of miRNAs differentially expressed in BMDMs from AK085865−/− and WT mice. Upregulated and downregulated genes are shown as red and blue dots, respectively; n = 3 for each group. (D) Validation of the selected miRNAs differentially expressed in BMDMs from AK085865−/− and WT mice (n = 6–8/group). (E) Knockdown of ILF2 decreases pri-miRNA and increases mature miRNA. BMDMs from WT mice were transfected with ILF2 siRNAs, and RNAs were isolated and analyzed for the amount of pri-miRNAs and mature miRNAs by qRT-PCR with specific primers. GAPDH and snRNA U6 were used as an internal control and for normalization of the data. (F and G) Knockdown of ILF2 rescues the accumulation of pri-miRNA processing mediated by AK085865 deletion. BMDMs from AK085865−/− mice were transfected with ILF2 siRNAs. RNAs were isolated and analyzed for (F) the amount of pri-miRNAs and (G) mature miRNAs by qRT-PCR with specific primers. The data are expressed as the mean ± SEM of three independent experiments. NC, negative control. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Recently, it was reported that the ILF2-ILF3 complex suppressed miRNA processing through binding to pri- or pre-miRNAs.25 To examine whether miRNA processing is regulated by AK085865, we analyzed and compared the miRNA expression profiles of BMDMs from AK085865−/− and WT mice. The miRNA profile data were submitted to the Gene Expression Omnibus (GEO) database, and the accession number is GEO: GSE125574. In total, 24 miRNAs were differentially expressed between the two groups listed above (Figure 2C). The differential expression of the selected miRNAs was validated by qRT-PCR. Among them, microRNA (miR)-7a was upregulated (fold change > 2 and p < 0.05), whereas miR-139, miR-149-3p, and miR-192 were downregulated (fold change < −2 and p < 0.05) in BMDMs from AK085865−/− mice compared with BMDMs from WT mice (Figure 2D).
To address the cause of the alteration in the levels of these miRNAs, we measured the levels of the corresponding pri-miRNAs in the BMDMs from AK085865−/− and WT mice. The levels of pri-miR-139 and pri-miR-192 examined were significantly increased in the BMDMs from AK085865−/− mice compared to those of WT mice (Figure 2E). Conversely, knockdown of ILF2 resulted in elevated levels of mature miR-139 and miR-192 and decreased expression of pri-miR-139 and pri-miR-192 in BMDMs from AK085865−/− mice (Figures 2F and 2G). Therefore, these results raise the possibility that the reductions of miR-139 and miR-192 levels in the BMDMs from AK085865−/− mice are due to a suppression of pri-miRNA processing by enhanced binding of ILF2 and ILF3.
miR-192 Plays a Role in the Development of Macrophage Polarization In Vitro
As mentioned above, we concluded that lncRNA AK085865 facilitates M2 macrophage polarization by functioning as a negative regulator in the ILF2-ILF3 complex-mediated miRNA processing pathway. Therefore, we next evaluated the effect of miR-139 and miR-192 regulated by the AK085865/ILF2/ILF3 axis on macrophage polarization. We found that miR-139 and miR-192 knockdown diminished the phenotypical expression of M2 macrophages while promoting polarization to the M1 phenotype in BMDMs from WT mice (Figure 3A). We selected miR-192 with higher function for further study. We found that miR-192 overexpression decreased tumor necrosis factor (TNF)-α and IL-12 levels in the supernatant collected from the WT mice BMDMs (Figures 3B and 3C). Additionally, the mRNA level of M1 marker gene inducible nitric oxide synthase (iNOS) also decreased (Figure 3D), whereas the mRNA levels of M2 marker genes Arg1, FIZZ1, and YM-1 were increased in miR-192 overexpression BMDMs compared to the control (Figure 3E). These results indicated that overexpression of miR-192 promoted macrophage M2 polarization in vitro.
Figure 3.

The Effect of miR-139 and miR-192 on Macrophage Polarization In Vitro
(A) BMDMs were transfected with 100 nM scramble control or inhibitors against miR-139 or miR-192 for 48 h, followed by stimulation with LPS (100 ng/mL) plus IFN-γ (20 ng/mL) or IL-4 (20 ng/mL) for an additional 48 h. The mRNA levels of iNOS and Arg1 were determined by qRT-PCR. (B–E) BMDMs were transfected with 100 nM control or miR-192 mimic for 48 h, followed by stimulation with LPS (100 ng/mL) plus IFN-γ (20 ng/mL) or IL-4 (20 ng/mL) for an additional 48 h. (B and C) Supernatants from the two groups of cells were analyzed for TNF-α (B) and IL-12 (C) by ELISA. (D and E) RNA was extracted from the two groups of cells and subjected to qRT-PCR analysis with primers specific to iNOS (D) for M1 polarization or Arg1, YM1, and FIZZ1 (E) for M2 polarization. GAPDH was set as the endogenous control. The data are expressed as the mean ± SEM of three independent experiments. NC, negative control. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
IL-1 Receptor-Associated Kinase 1 (IRAK1) Is a Functional Target of miR-192
To ascertain the mechanism by which miR-192 affects macrophage polarization, we analyzed the predicted target genes of miR-192 involved in macrophage activation and polarization. IRAK1 contains a predicted miR-192 target sequence in its 3′ untranslated region (UTR) (Figure 4A). IRAK1 is the main mediator of Toll-like receptor 4 (TLR4) activation, and it is critical to the LPS-mediated macrophage inflammatory response through its activation of the nuclear factor κB (NF-κB), p38, and c-Jun N-terminal protein kinase (JNK) signaling pathways.27,28
Figure 4.

IRAK1 Is a Functional Target of miR-192
(A) The predicted miR-192 seed sequence in the IRAK1 3′ UTR region is shown. (B) Luciferase activity of the reporter plasmid containing WT or Mut IRAK1 3′ UTR cotransfected into human embryonic kidney 293T (HEK293T) cells with miR-192 or control. (C) Luciferase activity of the reporter plasmid containing WT or Mut IRAK1 3′ UTR cotransfected into HEK293T cells with scrambled control or miR-192 inhibitor. (D) Expression of IRAK1 in BMDMs transfected with scrambled control or miR-192 inhibitor. The data are expressed as the mean ± SEM of three independent experiments. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Luciferase reporter assays showed that the miR-192 mimic reduced the activity of the reporter plasmid containing a 3′ UTR sequence of IRAK1, whereas the miR-192 inhibitor increased the activity of the reporter plasmid containing a 3′ UTR sequence of IRAK1. A plasmid with a mutation on the binding site was constructed to carry out the luciferase reporter assay. The results showed that the expression change in IRAK1 expression was abolished when the IRAK1 binding site was mutated (Figures 4B and 4C). Moreover, qRT-PCR and western blotting revealed that the miR-192 inhibitor promoted IRAK1 mRNA and protein expression in BMDMs (Figure 4D).
miR-192 Overexpression Rescues AK085865 Knockout (KO) Mice from Enhanced Susceptibility to CVB3-Induced VM
Our previous study demonstrated that AK085865 ablation confers susceptibility to VM by regulating macrophage polarization.10 However, whether the functionality of AK085865 depends on miR-192 is still unknown. Therefore, we next aimed to investigate whether miR-192 overexpression in vivo affected the course of CVB3-induced VM in AK085865 KO mice. The miR-192 agomir was mixed with the Lipofectamine 2000 transfection reagent and administered to 6-week-old male mice via intramyocardial injection at doses of 10 nmol per mouse, 3 days prior to CVB3 infection. On day 7 after CVB3 infection, miR-192 overexpression resulted in the alleviation of myocardial inflammation, as indicated by the restricted foci of inflammation and a decreased inflamed area (Figure 5A).
Figure 5.

miR-192 Overexpression Rescues AK085865 KO Mice from Enhanced Susceptibility to CVB3-Induced VM
AK085865 KO mice received 1 × 105 PFU of CVB3 i.p. on day 0. For in vivo miR-192 treatment, 30 μL Lipofectamine 2000 transfection reagent was mixed with miR-192 agomir or negative control (10 nmol/site), dissolved in a volume of 50 μL PBS, and the liposome complex was injected into the apex of the left ventricle with a 30-gauge needle at six sites per mouse, 3 days prior to CVB3 infection. (A) Heart sections were stained with H&E on day 7 postinfection (scale bars, 50 μm). (B) The parameters of the viral myocarditis were evaluated by heart/body weight ratio on day 7 postinfection. (C) The parameters of the viral myocarditis were evaluated by loss of body weight on day 7 postinfection. (D) The parameters of the viral myocarditis were evaluated by levels of serum cTnI on day 7 postinfection. (E) The survival rate of mice was observed until day 10 postinfection. Experiments were repeated with 8—10 mice per group. The data represent the mean ± SEM. The results are representative of at least three independent experiments with n ≥ 3. ∗p < 0.05. Differences were determined by (B and D) Student’s t test, (C) ANOVA, or (E) Kaplan-Meier survival analysis.
In agreement with this observation, miR-192 overexpression significantly attenuated heart/body weight ratio increases and reduced the body weight loss associated with systemic illness and serum cTnI levels (Figures 5B–5D). Furthermore, miR-192 overexpression significantly increased the survival rate from approximately 50% to 70% after CVB3 infection (Figure 5E). Together, these data indicate that miR-192 overexpression can effectively rescue AK085865 KO mice from enhanced susceptibility to CVB3-induced VM.
miR-192 Overexpression Switches Myocardial-Infiltrating Macrophages to a Predominant M2 Phenotype in AK085865 KO VM Mice
We further examined whether miR-192 overexpression affected the phenotype of heart-infiltrating macrophages in AK085865 KO VM mice. We observed increased proportions of F4/80+ macrophages bearing Arg1 in the hearts of miR-192-overexpression mice on day 7 of VM (Figure 6A). Moreover, we observed strikingly decreased proportions of F4/80+iNOS+ macrophages, which may indicate that M1 macrophages were decreased in the hearts of miR-192-overexpression mice (Figure 6A).
Figure 6.

miR-192 Overexpression Switches Myocardial-Infiltrating Macrophages to a Predominant M2 Phenotype in AK085865 KO VM Mice
AK085865 KO mice received 1 × 105 PFU of CVB3 i.p. on day 0. For in vivo miR-192 treatment, 30 μL Lipofectamine 2000 reagent was mixed with miR-192 agomir or negative control (10 nmol/site), dissolved in a volume of 50 μL PBS, and the liposome complex was injected into the apex of the left ventricle with a 30-gauge needle at six sites per mouse, 3 days prior to CVB3 infection. Hearts were collected on day 7 postinfection, and myocardial-infiltrating mononuclear cells were isolated from the hearts after enzymatic digestion. (A) The percentages of F4/80+iNOS+ or F4/80+Arg1+ cells were analyzed by FACS. (B) The arginase activity of sorted F4/80+ macrophages was assessed by an assay of urea production from the arginine substrate and was normalized to cell counts. (C) qRT-PCR was used to determine the NOS2, Arg1, FIZZ1, and YM-1 expression in sorted F4/80+ macrophages. (D) For colocalization analysis, sections were costained for CD68 (red, macrophage marker) and iNOS (green, M1 marker) or Arg1 (green, M2 marker). DAPI was used for nucleus staining (blue). The arrows just represent that we magnified the zone (arrows pointing) in the right panel images. Scale bars, 100 μm. Experiments were repeated three times in triplicate with 10–15 mice per group. The data represent the mean ± SEM. The results are representative of at least three independent experiments with n ≥ 3. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001.
Consistently, F4/80+ macrophages derived from miR-192 overexpression mice also showed high arginase activity and increased expression of the M2-specific genes Arg1, FIZZ1, and YM-1, compared with the macrophages isolated from the control group mice (Figures 6B and 6C). Similar to the fluorescence-activated cell sorter (FACS) analysis, colocalization demonstrated that a marked increase in the Arg1 signal was detected within the CD68+ macrophages in the heart tissue from the miR-192-overexpression mice, but the iNOS signal was barely detected (Figure 6D). Taken together, these data indicated that miR-192 overexpression switches myocardial-infiltrating macrophages to a predominant M2 phenotype in AK085865 KO VM mice.
Discussion
During CVB3-induced VM, macrophages were shown to be one of the predominant cell types and to display functional heterogeneity with proinflammatory M1 macrophages or anti-inflammatory M2 macrophages. Research by our group and others suggests that the excessive presence of M1 macrophages may cause damage to the host after CVB3 infection, whereas M2 macrophages protect against CVB3-induced VM.10, 11, 12, 13, 14, 15, 16 M1 macrophages express TNF-α, iNOS, and IL-12, and they induce a strong proinflammatory reaction and contribute to VM. In contrast, M2 macrophages express IL-10 and Arg1 instead of iNOS, depleting arginine stores so that NO is not produced and instead, produce polyamine and proline, which are important for cell differentiation and inflammatory response.29,30
Our previous study identified the lncRNA AK085865 as a functionally important regulator during macrophage polarization and demonstrated that AK085865 ablation confers susceptibility to VM by regulating macrophage polarization.10 In the present study, we provide mechanistic insights into how AK085865 regulates macrophage polarization. We used RNA pull-down experiments to identify ILF2 as an important binding partner of AK085865. We found that AK085865 deletion enhanced the binding of ILF2 and ILF3. Our results also identify a sequence in the 3′-466 region that is critical for ILF2 binding. The identification of ILF2 as a functional binding partner of AK085865 adds further support to the role of ILF2 in post-transcriptional regulation, expanding the role of ILF2 beyond its well-known functions. However, further studies will be required to determine whether the secondary structure of this region is involved in mediating AK085865 functions and if all of the effects of AK085865 that we have observed are dependent on its interaction with ILF2.
ILF2 and ILF3 were first isolated as nuclear factors that bind to a cis element of the IL-2 promoter, known as an antigen receptor response element, in the activated Jurkat T cell line.27,31 ILF2 possesses a zinc-finger nucleic acid binding domain (DZF) and a glutamic acid-rich region. It has been reported that the ILF2-ILF3 complex is involved in mRNA stabilization, translational repression, replication of viral RNA, and transcription in vitro.32, 33, 34, 35 Previous studies demonstrated that the ILF2-ILF3 complex negatively regulates the pri-miRNA processing step, resulting in a reduction of mature miRNA production.25,26
Our results revealed that there was a direct interaction between ILF2 and ILF3 in BMDMs from AK085865−/− and WT mice, and AK085865 deletion enhanced the binding of ILF2 and ILF3. Moreover, miRNA array analysis identified a total of 24 differentially expressed genes in AK085865−/− BMDMs, including 11 upregulated genes and 13 downregulated genes relative to WT BMDMs. Among them, miR-139 and miR-192 were downregulated, whereas the levels of pri-miR-139 and pri-miR-192 were significantly increased in BMDMs from AK085865−/− mice compared with the BMDMs of WT mice. Knockdown of ILF2 increased the levels of mature miR-139 and miR-192, whereas it decreased the expression of pri-miR-139 and pri-miR-192 in BMDMs from AK085865−/− mice. These findings suggest that AK085865 facilitates M2 macrophage polarization by functioning as a negative regulator in the ILF2/ILF3 complex-mediated pri-miR-139 and pri-miR-192 processing pathway, resulting in a reduction of mature miR-139 and miR-192 production.
We next evaluated the effect of miR-139 and miR-192 on macrophage polarization. We found that miR-139 and miR-192 knockdown diminished phenotypical expression of M2 macrophages while promoting polarization to the M1 phenotype in BMDMs from WT mice. We selected miR-192 with higher function for further study. We found that miR-192 overexpression decreased the M1 marker gene and increased M2 marker genes in vitro, suggesting that miR-192 plays a role in the development of macrophage polarization. Our work also demonstrates that miR-192 restrains inflammation in vivo. In response to CVB3 challenge, AK085865−/− mice were more susceptible to CVB3-induced VM compared to their WT counterparts, and miR-192 overexpression could effectively rescue the AK085865 KO mice from enhanced susceptibility to CVB3-induced VM. Furthermore, our study reveals that miR-192 overexpression switches myocardial-infiltrating macrophages to a predominant M2 phenotype in AK085865 KO VM mice. Together, our data suggest that a central role for AK085865 in the modulation of the M1/M2 phenotype may be dependent on miR-192.
For further insight into the function of miR-192 during macrophage polarization, a mRNA target, IRAK1, was predicted to contain a miR-192 3′ UTR binding site and was experimentally validated. IRAK1 is a key cytoplasmic protein kinase induced by TLR signaling. Phosphorylation of these kinases results in the nuclear translocation of the transcription factor NF-κB, which results in the production of proinflammatory cytokines, including TNF-α and IL-6.36, 37, 38 IRAK1 has been shown to regulate the TLR4-mediated inflammatory response, as well as dextran sulfate sodium (DSS)- or LPS-induced colitis and septic shock.39,40 In our current study, the IRAK1 3′ UTR contains an evolutionarily conserved miR-192 binding site, and a mutation of this site was found to abolish miR-192-mediated IRAK1 suppression, as demonstrated by luciferase reporter assays. Moreover, IRAK1 mRNA and protein expression was upregulated following miR-192 inhibition. Altogether, these results indicate that IRAK1 is a functional target of miR-192 and suggest a role for miR-192 in regulating the inhibitory phase of inflammation by suppressing IRAK1 expression.
Collectively, the present study demonstrated that AK085865 promotes macrophage M2 polarization in CVB3-induced VM by regulating ILF2-ILF3 complex-mediated miR-192 biogenesis, which rescues AK085865 KO mice from enhanced susceptibility to CVB3-induced VM (Figure 7). Our findings uncover a critical mechanism of AK085865 in the regulation of macrophage polarization in vitro and in vivo and further advance our understanding of the physiological roles of lncRNAs in general and the growing importance of these molecules in inflammatory heart disease.
Figure 7.

Summary of the Role of lncRNA AK085865 on CVB3-Induced VM
The lncRNA AK085865 facilitates M2 macrophage polarization by functioning as a negative regulator in the ILF2-ILF3 complex-mediated pri-miR-192 processing, which results in an increase in mature miR-192 biogenesis, attenuating the susceptibility to CVB3-induced VM.
Materials and Methods
Ethics Statement
All of the animal experimental procedures were approved by the Animal Ethics Committee of Wannan Medical College (Wuhu, China) and were performed according to the guidelines for the Care and Use of Laboratory Animals (Ministry of Health, China, 1998). The animals were euthanized by cervical dislocation following anesthetization with a mixture of isoflurane and oxygen (3% v/v). All efforts were made to minimize suffering of the animals.
Cell Culture and Reagents
BMDMs were isolated from the femurs and tibias of adult mice, as previously described.41 Macrophages were cultured in Dulbecco’s modified Eagle’s medium (DMEM) complete medium (20% fetal bovine serum [FBS], 20% L929 cell supernatant) at 37°C and 5% CO2 for 7 days. M1 and M2 macrophage polarization was obtained by removing the culture medium and culturing cells for an additional 48 h in DMEM, supplemented with 10% FBS and 100 ng/mL LPS (Sigma) plus 20 ng/mL IFN-γ (PeproTech) (for M1 polarization) or 20 ng/mL IL-4 (PeproTech) (for M2 polarization).
RNA Pull-Down Assay and MS
Biotin-labeled RNAs were in vitro transcribed using the Biotin RNA Labeling Mix and T7 RNA polymerase (Ambion) and purified with the RNeasy Mini Kit (QIAGEN) on-column digestion of DNA. The biotinylated sense, antisense, or truncated AK085865 was incubated with cell lysates (containing RNasin) overnight at 4°C. The interacting complexes were purified with streptavidin beads for 3 h at room temperature and visualized by silver staining (Pierce silver stain kit; Thermo Scientific) for further analysis by MS or by immunoblotting using a specific antibody to ILF2. For MS, a specific band present in the experimental lane was extracted (the corresponding region in the control lane was also extracted) for further analysis.
RIP
RIP was performed using a Magna RIP RNA-Binding Protein Immunoprecipitation Kit (Millipore). Briefly, BMDMs were harvested by adding RIP lysis buffer. Clear supernatant containing ILF2 protein, Protein G beads, and ILF2 antibody (or immunoglobulin G [IgG] control; Millipore) was mixed to perform the immunoprecipitation. After washing, RNAs bound to ILF2 were eluted and quantified. qRT-PCR was performed to examine whether certain RNAs were coimmunoprecipitated with the ILF2 antibody.
Animals and Myocarditis Model
lncRNA AK085865−/− mice and littermate WT mice with a C57BL/6 background were generated from the Nanjing Biomedical Research Institute of Nanjing University (Nanjing, China), and they were fed a standard diet and housed in pathogen-free mouse colonies. The Animal Care and Use Committee of Wannan Medical College approved all of the housing and surgical procedures.
The original stock of CVB3 (Nancy strain) was a kind gift from Professor Wei Hou (School of Basic Medical Sciences, Wuhan University) and was maintained by passage through HeLa cells (ATCC number: CCL-2). Virus titer was routinely determined prior to infection by a 50% tissue-culture infectious dose (TCID50) assay of HeLa cell monolayers, according to previously published procedures.42 Mice were infected by an intraperitoneal (i.p.) injection of 0.1 mL of phosphate-buffered saline (PBS) containing approximately 1 × 105 plaque-forming units (PFUs) of the virus on day 0. For in vivo miR-192 overexpression, 6-week-old C57BL/6 AK085865−/− mice were anesthetized, intubated, and mechanically ventilated. Next, 30 μL Lipofectamine 2000 reagent was mixed with miR-192 agomir or negative control (10 nmol/site; RiboBio) in a volume of 50 μL that was injected into the apex of the left ventricle with a 30-gauge needle at six sites per mouse, 3 days prior to CVB3 infection. Mice were euthanized, and tissues or cells were collected on day 7.
Western Blotting
Cells or tissues were harvested at the indicated times and lysed in radioimmunoprecipitation assay (RIPA) extraction solution (15 mM Tris, pH 7.5, 120 mM NaCl, 25 mM KCl, 2 mM EGTA, 2 mM EDTA, 0.1 mM dithiothreitol, 0.5% Triton X-100, and protease inhibitor cocktail [Sigma]). The protein concentration was assessed by bicinchoninic acid (BCA) assay. Total protein lysates (25 μg/lane) were subjected to SDS-PAGE and transferred onto an Immobilon polyvinylidene difluoride (PVDF) membrane (Millipore). The antibodies used were the following: anti-ILF2 (Abcam), anti-ILF3 (Abcam), anti-EEF1A1 (Novus), anti-Rbmxl1 (Novus), and anti-β-actin antibody as the loading control (Santa Cruz Biotechnology). Western blots were quantified by Quantity One Analysis software (Bio-Rad).
Microarray Analysis
The miRNA microarray work was performed by Shanghai Biotechnology (Shanghai, China). In brief, total RNA from each sample was amplified and labeled by a Low Input Quick Amp WT Labeling Kit (Agilent Technologies), following the manufacturer’s instructions. The labeled complementary RNA (cRNA) was purified using an RNeasy mini kit (QIAGEN). The concentration and specific activity of the labeled cRNAs (pmol Cy3/μg cRNA) were measured with a NanoDrop 2000 spectrophotometer. Each microarray slide was hybridized with 1.65 μg Cy3-labeled cRNA using a Gene Expression Hybridization Kit and a hybridization oven, according to the manufacturer’s instructions. After 17 h of hybridization, slides were washed in staining dishes using the Gene Expression Wash Buffer Kit following the manufacturer’s instructions. Next, the slides were scanned using an Agilent Microarray Scanner G2565C (Agilent Technologies) with the following default settings: dye channel = green, scan resolution = 3 μm, photomultiplier tube (PMT) 100%, 20 bit. Agilent Feature Extraction software (version [v.]10.7) was used to analyze the acquired array images. Quantile normalization and subsequent data processing were performed using GeneSpring Software 11.0 (Agilent Technologies).
Isolation of RNA and qRT-PCR
Total RNAs were extracted using TRIzol reagent (Invitrogen), and cDNA was generated using a RevertAid First Strand cDNA Synthesis Kit (Thermo Fisher Scientific) with oligo(dT) and random primers, according to the manufacturer’s instructions. SYBR Green dye-based quantitative real-time PCR was performed using the SYBR Green PCR Master Mix and CFX96 Real-Time PCR System from Bio-Rad. For miRNA real-time PCR, a commercial Hairpin-it miRNA qPCR Quantification Kit (RiboBio) was used. Briefly, 1 μg RNA was used as a template and then reverse transcribed using a miRNA-specific RT primer. The resulting cDNA was further amplified with a universal reverse primer and a specific forward primer. Calculations of mRNA or miRNA expression levels were performed using the comparative Cycle Threshold (ΔΔCT) method and normalized against GAPDH or U6 small nuclear RNA (snRNA) levels. All of the reactions were run in triplicate. The primers used are shown in Table S2.
Cytokine Enzyme-Linked Immunosorbent Assay (ELISA)
Cytokine levels of TNF-α and IL-12 were measured in cell supernatants using respective cytokine ELISA kits (R&D Systems), according to the manufacturer’s instructions.
Transfections
BMDMs were transfected with micrON miR-192 mimic (100 nM), micrOFF miR-192 inhibitor (100 nM), micrOFF miR-139 inhibitor (100 nM), and control RNAs using the Lipofectamine 3000 (Invitrogen, USA) transfection reagent, according to the manufacturer’s instructions. The efficiency and function assay were confirmed 48 h after small interfering RNA (siRNA) transfection.
Plasmid Construction and Luciferase Assay
The entire mouse IRAK1 3′ UTR segment was amplified by PCR using genomic DNA as a template. The PCR product was subcloned into the PLUC vector following the manufacturer’s protocol (Promega). Plasmid DNA was sequenced to ensure its authenticity. Then, 293T cells were cultured in a 96-well plate, and each well was transfected with 0.2 μg luciferase reporter constructs (described above) and at the same time with micrON miR-192 mimic (100 nM), micrOFF miR-192 inhibitor (100 nM), or control RNAs using Lipofectamine 3000 (Invitrogen, USA). Cells were assayed using luciferase assay kits (Promega), 48 h after transfection.
Histopathology
Hearts were cut longitudinally, fixed in 10% phosphate-buffered formalin, and then embedded in paraffin. Tissue sections (5 μm thick) were cut at various depths and stained with hematoxylin and eosin (H&E) to determine the level of inflammation. The sections were examined by two independent investigators blinded to the disease state of the mice.
Serological Index of Myocarditis
Yijishan Hospital performed the serum cTnI measurement with a DXI800 (Beckman Coulter) immunology analyzer.
FACS Analysis
Single-cell suspensions were pooled from mouse heart tissues. Surface marker F4/80 (eBioscience) was stained with fluorochrome-conjugated monoclonal antibodies (mAbs) diluted in 1% FBS in PBS. For intracellular Arg1 and iNOS (BD Pharmingen; BioLegend) staining, cells were fixed and permeabilized using fixation buffer and permeabilization solution (eBioscience). Cell fluorescence was measured using FACS (Beckman Coulter), and the data were analyzed using FlowJo software (Tree Star).
Immunofluorescence
Frozen heart sections were fixed in 4°C acetone for 10 min and permeabilized with 0.3% Triton X-100. Then, cells was washed three times with PBS and treated with the first antibody at 37°C; 2 h later, cells were washed three times with PBS and treated with fluorescent-labeled secondary antibody for 1 h at 37°C. Nuclei were labeled with 4′,6-diamidino-2-phenylindole (DAPI). We continued with several rounds of washing and finished with mounting the coverslip onto a microscope slide using an anti-fade mounting medium. All of the immunofluorescence staining was photographed under a confocal microscope. The antibodies used were as follows: CD68 antibody (diluted at 1:200; Abcam), iNOS antibody (diluted at 1:100; Abcam), arginase antibody (diluted at 1:1,000; Abcam), and goat anti-mouse IgG (ab150115; Abcam).
Data Availability
The GEO database accession number for the miRNA profile data from BMDMs of the WT and lncRNA AK085865 KO mice reported in this study is GEO: GSE125574.
Statistical Analysis
The data are shown as the mean ± SEM. Statistical analysis of the data was performed with the two-tailed independent Student’s t test. Analysis of variance (ANOVA) was conducted using SPSS software, and the Kaplan-Meier survival curves were drawn using GraphPad Prism v.5.0 (GraphPad Software). p < 0.05 was considered to be statistically significant.
Author Contributions
Y.Z., X.L., and C.W. designed the experiments, performed most of the experiments, and analyzed the data. M.Z. and H.Y. assisted with cell culture and western blot. Y.Z. and K.L. conceived the project, analyzed the data, and wrote the manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
We thank LetPub (https://www.letpub.com) for its linguistic assistance during the preparation of this manuscript. This work was supported by the National Natural Science Foundation of China (81472017, 81772180, 81870017, 81701557, and 81802503) and Key Projects of Natural Science Research of Universities in Anhui Province (KJ2019A0413 and KJ2018A0265), and funding from “Peak” Training Program for Scientific Research of Yijishan Hospital, Wannan Medical College (GF2019T01, GF2019G09, and GF2019G15).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.06.017.
Supplemental Information
References
- 1.Steinberger J., Lucas R.V., Jr., Edwards J.E., Titus J.L. Causes of sudden unexpected cardiac death in the first two decades of life. Am. J. Cardiol. 1996;77:992–995. doi: 10.1016/s0002-9149(96)00035-5. [DOI] [PubMed] [Google Scholar]
- 2.Fairweather D., Stafford K.A., Sung Y.K. Update on coxsackievirus B3 myocarditis. Curr. Opin. Rheumatol. 2012;24:401–407. doi: 10.1097/BOR.0b013e328353372d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Luo H., Yanagawa B., Zhang J., Luo Z., Zhang M., Esfandiarei M., Carthy C., Wilson J.E., Yang D., McManus B.M. Coxsackievirus B3 replication is reduced by inhibition of the extracellular signal-regulated kinase (ERK) signaling pathway. J. Virol. 2002;76:3365–3373. doi: 10.1128/JVI.76.7.3365-3373.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhang H., Yue Y., Sun T., Wu X., Xiong S. Transmissible endoplasmic reticulum stress from myocardiocytes to macrophages is pivotal for the pathogenesis of CVB3-induced viral myocarditis. Sci. Rep. 2017;7:42162. doi: 10.1038/srep42162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fairweather D., Rose N.R. Coxsackievirus-induced myocarditis in mice: a model of autoimmune disease for studying immunotoxicity. Methods. 2007;41:118–122. doi: 10.1016/j.ymeth.2006.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gordon S. Alternative activation of macrophages. Nat. Rev. Immunol. 2003;3:23–35. doi: 10.1038/nri978. [DOI] [PubMed] [Google Scholar]
- 7.Martinez F.O., Helming L., Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu. Rev. Immunol. 2009;27:451–483. doi: 10.1146/annurev.immunol.021908.132532. [DOI] [PubMed] [Google Scholar]
- 8.Mosser D.M., Edwards J.P. Exploring the full spectrum of macrophage activation. Nat. Rev. Immunol. 2008;8:958–969. doi: 10.1038/nri2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Martinez F.O., Sica A., Mantovani A., Locati M. Macrophage activation and polarization. Front. Biosci. 2008;13:453–461. doi: 10.2741/2692. [DOI] [PubMed] [Google Scholar]
- 10.Zhang Y., Li X., Kong X., Zhang M., Wang D., Liu Y., Lv K. Long non-coding RNA AK085865 ablation confers susceptibility to viral myocarditis by regulating macrophage polarization. J. Cell. Mol. Med. 2020;24:5542–5554. doi: 10.1111/jcmm.15210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang Y., Zhang M., Li X., Tang Z., Wang X., Zhong M., Suo Q., Zhang Y., Lv K. Silencing MicroRNA-155 Attenuates Cardiac Injury and Dysfunction in Viral Myocarditis via Promotion of M2 Phenotype Polarization of Macrophages. Sci. Rep. 2016;6:22613. doi: 10.1038/srep22613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li K., Xu W., Guo Q., Jiang Z., Wang P., Yue Y., Xiong S. Differential macrophage polarization in male and female BALB/c mice infected with coxsackievirus B3 defines susceptibility to viral myocarditis. Circ. Res. 2009;105:353–364. doi: 10.1161/CIRCRESAHA.109.195230. [DOI] [PubMed] [Google Scholar]
- 13.Yang X., Yue Y., Xiong S. Dpep2 Emerging as a Modulator of Macrophage Inflammation Confers Protection Against CVB3-Induced Viral Myocarditis. Front. Cell. Infect. Microbiol. 2019;9:57. doi: 10.3389/fcimb.2019.00057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang C., Dong C., Xiong S. IL-33 enhances macrophage M2 polarization and protects mice from CVB3-induced viral myocarditis. J. Mol. Cell. Cardiol. 2017;103:22–30. doi: 10.1016/j.yjmcc.2016.12.010. [DOI] [PubMed] [Google Scholar]
- 15.Gou W., Zhang Z., Yang C., Li Y. MiR-223/Pknox1 axis protects mice from CVB3-induced viral myocarditis by modulating macrophage polarization. Exp. Cell Res. 2018;366:41–48. doi: 10.1016/j.yexcr.2018.03.004. [DOI] [PubMed] [Google Scholar]
- 16.Li Y., Huang Y., Wu W., Wei B., Qin L. B Cells Increase Myocardial Inflammation by Suppressing M2 Macrophage Polarization in Coxsackie Virus B3-Induced Acute Myocarditis. Inflammation. 2019;42:953–960. doi: 10.1007/s10753-018-0950-0. [DOI] [PubMed] [Google Scholar]
- 17.Ponting C.P., Oliver P.L., Reik W. Evolution and functions of long noncoding RNAs. Cell. 2009;136:629–641. doi: 10.1016/j.cell.2009.02.006. [DOI] [PubMed] [Google Scholar]
- 18.Goodrich J.A., Kugel J.F. Non-coding-RNA regulators of RNA polymerase II transcription. Nat. Rev. Mol. Cell Biol. 2006;7:612–616. doi: 10.1038/nrm1946. [DOI] [PubMed] [Google Scholar]
- 19.Bartolomei M.S., Zemel S., Tilghman S.M. Parental imprinting of the mouse H19 gene. Nature. 1991;351:153–155. doi: 10.1038/351153a0. [DOI] [PubMed] [Google Scholar]
- 20.Zheng J., Huang X., Tan W., Yu D., Du Z., Chang J., Wei L., Han Y., Wang C., Che X. Pancreatic cancer risk variant in LINC00673 creates a miR-1231 binding site and interferes with PTPN11 degradation. Nat. Genet. 2016;48:747–757. doi: 10.1038/ng.3568. [DOI] [PubMed] [Google Scholar]
- 21.Li Z., Chao T.C., Chang K.Y., Lin N., Patil V.S., Shimizu C., Head S.R., Burns J.C., Rana T.M. The long noncoding RNA THRIL regulates TNFα expression through its interaction with hnRNPL. Proc. Natl. Acad. Sci. USA. 2014;111:1002–1007. doi: 10.1073/pnas.1313768111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Carpenter S., Aiello D., Atianand M.K., Ricci E.P., Gandhi P., Hall L.L., Byron M., Monks B., Henry-Bezy M., Lawrence J.B. A long noncoding RNA mediates both activation and repression of immune response genes. Science. 2013;341:789–792. doi: 10.1126/science.1240925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hu G., Tang Q., Sharma S., Yu F., Escobar T.M., Muljo S.A., Zhu J., Zhao K. Expression and regulation of intergenic long noncoding RNAs during T cell development and differentiation. Nat. Immunol. 2013;14:1190–1198. doi: 10.1038/ni.2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Du M., Yuan L., Tan X., Huang D., Wang X., Zheng Z., Mao X., Li X., Yang L., Huang K. The LPS-inducible lncRNA Mirt2 is a negative regulator of inflammation. Nat. Commun. 2017;8:2049. doi: 10.1038/s41467-017-02229-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sakamoto S., Aoki K., Higuchi T., Todaka H., Morisawa K., Tamaki N., Hatano E., Fukushima A., Taniguchi T., Agata Y. The NF90-NF45 complex functions as a negative regulator in the microRNA processing pathway. Mol. Cell. Biol. 2009;29:3754–3769. doi: 10.1128/MCB.01836-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Todaka H., Higuchi T., Yagyu K., Sugiyama Y., Yamaguchi F., Morisawa K., Ono M., Fukushima A., Tsuda M., Taniguchi T., Sakamoto S. Overexpression of NF90-NF45 Represses Myogenic MicroRNA Biogenesis, Resulting in Development of Skeletal Muscle Atrophy and Centronuclear Muscle Fibers. Mol. Cell. Biol. 2015;35:2295–2308. doi: 10.1128/MCB.01297-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kim B.R., Cho Y.C., Cho S. Anti-inflammatory effects of a novel compound, MPQP, through the inhibition of IRAK1 signaling pathways in LPS-stimulated RAW 264.7 macrophages. BMB Rep. 2018;51:308–313. doi: 10.5483/BMBRep.2018.51.6.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Giriwono P.E., Shirakawa H., Ohsaki Y., Sato S., Aoyama Y., Ho H.J., Goto T., Komai M. Geranylgeraniol Suppresses the Expression of IRAK1 and TRAF6 to Inhibit NFκB Activation in Lipopolysaccharide-Induced Inflammatory Responses in Human Macrophage-Like Cells. Int. J. Mol. Sci. 2019;20:E2320. doi: 10.3390/ijms20092320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Timmers L., Pasterkamp G., de Hoog V.C., Arslan F., Appelman Y., de Kleijn D.P. The innate immune response in reperfused myocardium. Cardiovasc. Res. 2012;94:276–283. doi: 10.1093/cvr/cvs018. [DOI] [PubMed] [Google Scholar]
- 30.Zhang Y., Zhang M., Li X., Tang Z., He L., Lv K. Expansion of CD11b+Ly-6C+ myeloid-derived suppressor cells (MDSCs) driven by galectin-9 attenuates CVB3-induced myocarditis. Mol. Immunol. 2017;83:62–71. doi: 10.1016/j.molimm.2017.01.013. [DOI] [PubMed] [Google Scholar]
- 31.Kao P.N., Chen L., Brock G., Ng J., Kenny J., Smith A.J., Corthésy B. Cloning and expression of cyclosporin A- and FK506-sensitive nuclear factor of activated T-cells: NF45 and NF90. J. Biol. Chem. 1994;269:20691–20699. [PubMed] [Google Scholar]
- 32.Shim J., Lim H., Yates J.R., Karin M. Nuclear export of NF90 is required for interleukin-2 mRNA stabilization. Mol. Cell. 2002;10:1331–1344. doi: 10.1016/s1097-2765(02)00730-x. [DOI] [PubMed] [Google Scholar]
- 33.Pfeifer I., Elsby R., Fernandez M., Faria P.A., Nussenzveig D.R., Lossos I.S., Fontoura B.M., Martin W.D., Barber G.N. NFAR-1 and -2 modulate translation and are required for efficient host defense. Proc. Natl. Acad. Sci. USA. 2008;105:4173–4178. doi: 10.1073/pnas.0711222105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tominaga-Yamanaka K., Abdelmohsen K., Martindale J.L., Yang X., Taub D.D., Gorospe M. NF90 coordinately represses the senescence-associated secretory phenotype. Aging (Albany NY) 2012;4:695–708. doi: 10.18632/aging.100497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kiesler P., Haynes P.A., Shi L., Kao P.N., Wysocki V.H., Vercelli D. NF45 and NF90 regulate HS4-dependent interleukin-13 transcription in T cells. J. Biol. Chem. 2010;285:8256–8267. doi: 10.1074/jbc.M109.041004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ellett J.D., Evans Z.P., Atkinson C., Schmidt M.G., Schnellmann R.G., Chavin K.D. Toll-like receptor 4 is a key mediator of murine steatotic liver warm ischemia/reperfusion injury. Liver Transpl. 2009;15:1101–1109. doi: 10.1002/lt.21782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Takeda K., Akira S. TLR signaling pathways. Semin. Immunol. 2004;16:3–9. doi: 10.1016/j.smim.2003.10.003. [DOI] [PubMed] [Google Scholar]
- 38.Seki E., Brenner D.A. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 2008;48:322–335. doi: 10.1002/hep.22306. [DOI] [PubMed] [Google Scholar]
- 39.Jeong J.J., Jang S.E., Hyam S.R., Han M.J., Kim D.H. The Rhizome Mixture of Anemarrhena asphodeloides and Coptidis chinensis Ameliorates Acute and Chronic Colitis in Mice by Inhibiting the Binding of Lipopolysaccharide to TLR4 and IRAK1 Phosphorylation. Evid. Based Complement. Alternat. Med. 2014;2014:809083. doi: 10.1155/2014/809083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Toubiana J., Courtine E., Pène F., Viallon V., Asfar P., Daubin C., Rousseau C., Chenot C., Ouaaz F., Grimaldi D. IRAK1 functional genetic variant affects severity of septic shock. Crit. Care Med. 2010;38:2287–2294. doi: 10.1097/CCM.0b013e3181f9f9c7. [DOI] [PubMed] [Google Scholar]
- 41.Zhang Y., Zhang Y., Li X., Zhang M., Lv K. Microarray analysis of circular RNA expression patterns in polarized macrophages. Int. J. Mol. Med. 2017;39:373–379. doi: 10.3892/ijmm.2017.2852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Henke A., Huber S., Stelzner A., Whitton J.L. The role of CD8+ T lymphocytes in coxsackievirus B3-induced myocarditis. J. Virol. 1995;69:6720–6728. doi: 10.1128/jvi.69.11.6720-6728.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The GEO database accession number for the miRNA profile data from BMDMs of the WT and lncRNA AK085865 KO mice reported in this study is GEO: GSE125574.