

Abstract
Somatic copy-number alterations (SCNAs) drive tumor growth and evolution. However, the functional roles of SCNAs across the genome are still poorly understood. We provide an integrative strategy to characterize the functional roles of driver SCNAs in cancers based on dysregulated competing endogenous RNA (ceRNA) networks. We identified 44 driver SCNAs in lower-grade glioma (LGG). The dysregulated patterns losing all correlation relationships dominated dysregulated ceRNA networks. Homozygous deletion of six genes in 9p21.3 characterized an LGG subtype with poor prognosis and contributed to the dysfunction of cancer-associated pathways in a complementary way. The pan-cancer analysis showed that different cancer types harbored different driver SCNAs through dysregulating the crosstalk with common ceRNAs. The same SCNAs destroyed their ceRNA networks through different miRNA-mediated ceRNA regulations in different cancers. Additionally, some SCNAs performed different functional mechanisms in different cancers, which added another layer of complexity to cancer heterogeneity. Compared with previous methods, our strategy could directly dissect functional roles of SCNAs from the view of ceRNA networks, which not only complemented the functions of protein-coding genes but also provided a new avenue to characterize the functions of noncoding RNAs. Also, our strategy could be applied to more types of cancers to identify pathogenic mechanism driven by the SCNAs.
Graphical Abstract
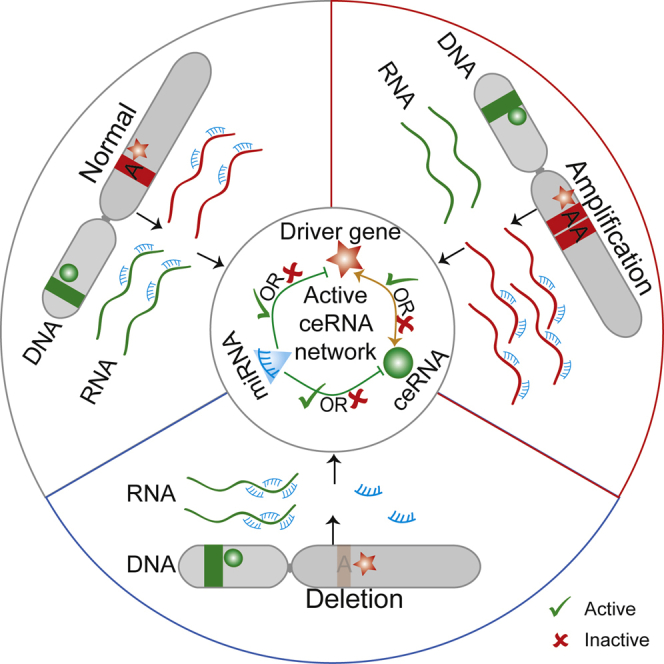
This article provides an integrative strategy to characterize the functional roles of SCNAs based on dysregulated ceRNA networks and identify driver SCNAs in cancers. The driver SCNAs almost completely destroyed the active ceRNA networks in non-alteration conditions to drive cancer development. The functional roles of SCNAs showed multiple cancer-specific manners.
Introduction
The accumulation of driver somatic genetic alterations gradually drives the evolution progression from normal to tumor cells.1 Somatic copy-number alterations (SCNAs), one important type of somatic genetic alterations, have been reported to activate oncogenes and inactivate tumor suppressors, which further made contributions to cancer progression.2 The prevalent focal and arm-level SCNAs affected large cancer genome regions that not only included protein-coding genes (PGs) but also non-coding regions.3,4 How to elucidate the functional roles of SCNAs in cancers and how these SCNAs disorder cellular homeostasis and further promote cancer progression are still unclear.
Many studies have been devoted to explore the functional effects of SCNAs in cancers. It is well known that gene amplifications could increase expression levels of genes in amplified regions, and gene deletions could decrease expression levels of genes in deleted regions.5 SCNAs also influenced expression levels of genes outside the amplified/deleted regions. Some studies investigated the effect of SCNAs on the transcriptional changes by integrating DNA copy number/mutation profiles and gene expression profiles.6,7 Network modeling of global gene expression identified a glioblastoma-associated CNA-driven network in which CNA hubs could explain expression variability of multiple downstream genes.8 The abnormal activities of biological pathways were also caused by driver CNAs through disturbing expression of genes in the pathways.9 Sharma et al.10 showed that the co-amplifications and co-deletions of cancer-causing genes and metabolic genes contributed to reprogram cancer cell metabolism. SCNAs propagated abnormal information along topological structures of biological pathways and contributed to dysfunctional pathways in cancer individuals.11 The driver genetic alterations could induce the dysregulation of multilayer factor-mediated regulatory networks of gene expression and further cause disorder of cancer-associated functions.12 These studies explored the functional roles of driver SCNAs in cancer progression with an indirect way. However, it was limited in understanding direct contributions of SCNAs to tumor progression.
MicroRNAs (miRNAs) are important negative regulators of gene expression. Artificial miRNA sponges can competitively bind and inhibit miRNAs to derepress miRNA targets.13 Recently, the hypothesis of competing endogenous RNAs (ceRNAs) uncovered a new layer of RNA interaction mechanisms.14 ceRNAs can crosstalk with each other through competitively binding to common miRNA response elements. Due to the multiplicity of targets and cooperativity of miRNAs, the ceRNA crosstalking across the transcriptome could form large-scale miRNA-mediated ceRNA networks.15 Endogenous RNA molecules with miRNA target sites have the potential to act as natural miRNA sponges, such as mRNAs, long noncoding RNAs (lncRNAs), pseudogenes, and circular RNAs (circRNAs). The balance of miRNA binding status among ceRNA partners was important for maintaining normal biological functions of cells. Abnormal expression of ceRNAs can break the balance, lead the dysregulated ceRNA networks, and further disorder cell biological functions.16 Pseudogene PTENP1 was targeted by PTEN-targeting miRNAs, and overexpression of the PTENP1 3′ UTR elevated PTEN expression based on miRNA-mediated regulation and performed a tumor suppressive function.17 lncRNA LINCMD1 competitively sponged miR-133 and miR-135 to regulate expression of MAML1 and MEF2C, which could activate muscle-specific gene expression to govern the time of muscle differentiation.18 Exogenous perturbation in expression proved the regulatory role of ceRNAs on expression of their partners. Few studies have focused on the effect of endogenous copy-number alterations on ceRNA networks. DNA copy-number gains induced the high expression of SNHG6, which regulated EZH2 expression by sponging miR-26a/b and miR-214 to promote CRC cell growth and metastasis.19 Copy number loss of both CNOT6LA and VAPA may downregulate PTEN in a miRNA-dependent manner.16 Amplification-caused high EGFR expression downregulated miR-133b to release the control of miR-133b on MET.20 vIRF1 deletion increased miR-218-5p expression level and reduced the level of lnc-OIP5-AS1.21 Xu et al.22 showed that copy number variations can induce the dysregulation of ceRNA-ceRNA interactions. A ceRNA method was proposed to investigate the global transcriptional effect of genomic deletions.23 These studies gave us an inspiration that the view of ceRNA networks associated with SCNAs could provide a more direct manner to explore the functional mechanisms of SCNAs. However, the effect of endogenous expression perturbations of ceRNAs caused by SCNAs on ceRNA networks was less explored.
In this study, we provide an integrative strategy to identify the driver SCNAs and characterize the functional roles of SCNAs in cancers based on the dysregulation of ceRNA networks. The integrative strategy includes three steps. First, we integrated the SCNA profiles and expression profiles of protein genes and lncRNAs to identify the candidate driver genes whose SCNAs could concordantly affect their own expression levels. Next, for each candidate driver gene, we constructed a static candidate gene-associated ceRNA network that contained the candidate gene, candidate gene-targeting miRNAs, and the ceRNA partners of the candidate gene. Third, we constructed dynamically activated ceRNA networks under each SCNA status of candidate gene by integrating expression profiles of protein genes, miRNAs, and lncRNAs and the static candidate gene-associated ceRNA network. Then, the dysregulated ceRNA network driven by the SCNA of a candidate driver gene was identified by comparing dynamically activated ceRNA networks (Figure 1). We applied our strategy to lower-grade glioma (LGG). Compared with the normal copy number status, SCNAs almost destroyed the ceRNA networks. Additionally, the major dysregulated pattern was the pattern that all correlation relationships among ceRNAs and miRNAs were lost. Homozygous deletion of six genes in 9p21.3 could characterize a LGG subtype with poor prognosis and contribute to the dysfunction of cancer-associated pathways in a complementary way. We also applied our strategy to 11 other cancer types and dissected the SCNA-induced dysregulated ceRNA networks in common and specific manners across these cancer types.
Figure 1.

The Workflow of the Integrative Strategy for Identifying SCNA-Induced Dysregulated ceRNA Networks
Results
lncRNA SCNAs Were Prevailing across 12 Types of Cancer
We used GISTIC to identify SCNAs (such as homozygous deletion and high-level amplification) of PGs and lncRNAs across 12 cancer types (Figure S1). Through calculating SCNA frequency of PGs and lncRNAs, we found that the SCNA frequency distributions of lncRNAs were comparable to those of PGs (Figure S2A). By combining copy number profiles and gene expression profiles, we identified candidate genes (including PGs and lncRNAs) whose SCNAs concordantly and significantly influenced their expression levels. lncRNAs occupied nearly half of the candidate genes, and especially the fractions were up to approximately 60% in ovarian serous cystadenocarcinoma (OV) and glioblastoma multiforme (GBM) (Figure S2B). Recently, lncRNAs have been reported to act as a class of ceRNAs to regulate expression levels of PGs by sponging miRNAs. Exploring the destructive power of SCNAs of lncRNAs/PGs on the ceRNA networks was helpful to dissect the functions and contributions of lncRNAs/PGs to cancer development. We developed an integrative strategy to identify the lncRNAs/PGs whose SCNAs were able to cause the disorder of their associated ceRNA networks (Figure 1).
The Driver SCNAs in LGG
We applied the integrative strategy to 433 LGG samples that were detected in all of the expression profiles of lncRNAs, PGs, and miRNAs and copy-number profiles, and identified 55 candidate genes (including 8 lncRNAs and 47 PGs) (Figure S3). For each candidate gene, we constructed dynamic ceRNA networks by integrating the expression profiles of lncRNAs, PGs, and miRNAs and a candidate gene-associated static ceRNA network under different copy number status. By comparing dynamic ceRNA networks under different copy number status, 44 driver genes (including 7 lncRNAs and 37 PGs) were identified, whose SCNAs drove differential expression and further caused the unbalance of ceRNA networks. We found that driver gene-associated ceRNA networks worked normally under the non-alteration condition, while these ceRNA networks were extremely destructed by SCNAs of driver genes (Figure 2). For example, MTAP was targeted by 45 miRNAs, which also targeted another 11,848 genes (including 1,783 lncRNAs and 10,065 PGs). An MTAP-associated static ceRNA network consisted of 86,456 ceRNA triples. Under the condition of MTAP non-alteration, 10,149 ceRNA triples were significantly activated while only 206 active ceRNA triples were significantly identified under the condition of MTAP deletion. These results suggested that SCNAs could cause the deregulation of ceRNA networks through increasing or decreasing expression levels of targeting genes.
Figure 2.

The Number of Active ceRNA Triples in Dynamic ceRNA Networks under Different Copy Number Status for 44 Driver Genes in LGG
Among 44 driver genes, 8 genes (including MTAP, CDKN2A, CNTNAP2, CDKN2B, RECQL4, PSCA, CDKN2B-AS1, and LINC-PINT) were recorded in at least one of the four known cancer gene databases (Cancer Gene Census in COSMIC,24 TSGene,25 Bushman [http://www.bushmanlab.org/links/genelists], and Lnc2cancer26). Through literature searches, we also found that many of the rest driver genes were reported to play important cancer-associated functions in vivo or in vitro (Table S2). The knocking down of COMMD9 was reported to inhibit cell proliferation and migration, and it arrested the cell cycle at the G1/S transition.27,28 The depletion of CPSF1 suppressed cell viability and promoted cell apoptosis by inducing cell cycle arrest at the G0/G1 phase.29 HSF1 promoted HCC cell migration and invasion in vitro and in vivo.30,31 HSF1 knockdown reduced cell migration and invasive ability, which were restored by HSF1 overexpression.31 These findings proved the driver roles of genes with SCNAs.
Dysregulated Patterns of ceRNA Triples Driven by SCNAs in LGG
ceRNA triples were elementary units of ceRNA networks. The destructed ceRNA networks driven by SCNAs were composed of dysregulated ceRNA triples, each of which contained a driver gene, a driver gene-targeting miRNA, and a ceRNA of the driver gene. According to the dysregulated status of ceRNA triples, we classified dysregulated ceRNA triples into eight classes (Figure 3A). Class I included triples that were not disturbed; class II included triples that lose inverse correlations between miRNAs and the driver gene; class III included triples that lose inverse correlations between miRNAs and ceRNAs; class IV included triples that lose positive correlations between the driver gene and ceRNAs; class V included triples that lose inverse correlations between miRNAs and both the driver gene and ceRNAs; class VI included triples that lose inverse correlations between miRNAs and the driver gene and positive correlations between the driver gene and ceRNAs; class VII included triples that lose inverse correlations between miRNAs and ceRNAs and positive correlations between the driver gene and ceRNAs; and class VIII included triples that lose all correlation relationships among the driver gene, miRNAs, and ceRNAs.
Figure 3.

The Dysregulated Patterns of ceRNA Triples
(A) Eight types of dysregulated patterns in the dysregulated ceRNA networks. (B) The proportions of eight dysregulated patterns of ceRNA triples in 44 dysregulated ceRNA networks induced by SCNAs of 44 driver genes. (C) The hallmark signatures significantly enriched by the dysregulated ceRNA networks.
For each driver gene, the percentages of eight classes of dysregulated ceRNA triples in the dysregulated ceRNA network were calculated by comparing the active ceRNA networks under the SCNA and non-alteration conditions (Figure 3B). We found that class VIII dominated dysregulated ceRNA networks, with the average percentage of 67.3%. Especially, GRINA amplification completely destroyed the active ceRNA network constructed in the non-alteration condition of GRINA, and class VIII contributed to 99.3% of dysregulated triples. Classes V and VII also occupied major proportions of dysregulated ceRNA networks for some driver genes, such as TNPO3, KLHDC10, MKLN1, RECQL4, and ZNF696. The average percentage of class I was only 0.33% across 44 driver genes. To investigate whether dysregulated ceRNA networks were associated with cancer hallmarks, we downloaded 50 hallmark signatures from MSigDB32 and identified the significantly enriched hallmark signatures by the deregulated ceRNA networks using R package clusterProfiler at a false discovery rate (FDR) of 0.05.33 We found that 37 of 44 deregulated ceRNA networks were significantly enriched in at least one hallmark signature (Figure 3C). For example, the dysregulated ceRNA networks caused by lncRNA ZNF252P-AS1 amplification were significantly enriched in a DNA damage signature (DNA_REPAIR), proliferation signatures (E2F_TARGETS, G2M_CHECKPOINT, MITOTIC_SPINDLE, MYC_TARGETS_V1, and MYC_TARGETS_V2), and a pathway signature (UNFOLDED_PROTEIN_RESPONSE). The most frequently enriched hallmark signatures across 37 dysregulated ceRNA networks were proliferation signatures, including E2F_TARGETS (30/37), G2M_CHECKPOINT (29/37), and MYC_TARGETS_V1 (25/37). These results suggested that driver SCNAs could disorder cancer hallmark signatures by extremely destroying their involved ceRNA networks.
The Homozygous Deletion of 9p21.3 Characterized an LGG Subtype with Poor Prognosis
To test whether SCNAs of driver lncRNAs/PGs destroying their ceRNA networks were associated with LGG prognosis, we used Kaplan-Meier analysis and a log-rank test to perform survival analysis of LGG patients based on the SCNA profiles of 44 driver genes. For each driver gene, we divided the LGG patients into three subgroups based on SCNA status of the driver gene as follows: one subgroup of patients with heterozygous deletion (−2) of the driver gene, one subgroup of patients with high-level amplification (2) of the driver gene, and one subgroup of patients with non-alteration (0) of the driver gene. The survival times among the subgroups were compared. The results showed that homozygous deletion of six genes was associated with poor prognosis of LGG, including CDKN2B-AS1 (p = 8.71e−08, log-rank test), CDKN2B (p = 1.84e−08, log-rank test), CDKN2A (p = 1.84e−08, log-rank test), MTAP (p = 3.35e−09, log-rank test), KLHL9 (p = 0.000494, log-rank test), and ELAVL2 (p = 0.00316, log-rank test) (Figure 4A; Figure S4). The expression levels of the six genes were significantly decreased by homozygous deletion (Figure 4B). We investigated the SCNA distributions of these six genes across LGG samples and found that homozygous deletion of the six genes showed co-occurrence patterns and obvious mutual exclusive patterns with alterations of the rest driver genes (Figure 4C). We also found that the genome locations of these six genes were in 9p21.3. To further analyze the association of 9p21.3 deletions with LGG poor prognosis in spite of alterations in the rest of the driver genes, we classified the LGG patients into three subgroups based on the SCNA status of the 44 driver genes as follows: subgroup I, including patients without any SCNAs of the 44 driver genes; subgroup II, including patients with homozygous deletions of at least one of the six genes in 9p21.3; and subgroup III, including patients without homozygous deletions of the six genes but with homozygous deletions or high-level amplifications of the rest of the 38 driver genes. Survival analysis of three subgroups showed that survival times of subgroup II were significantly shorter than those of subgroups I and III (p = 4.67e−09, log-rank test), and there was no significant difference in survival times between subgroups I and III (Figure 4D). To explore how homozygous deletions of these six genes contribute to poor prognosis, we comprehensively analyzed the dysregulated ceRNA networks. We found that homozygous deletions of these driver genes destroyed the ceRNA networks through different miRNAs (Figure 4E). Additionally, 79.4% of ceRNAs appeared in only one of six deregulated ceRNA networks. By preforming function enrichment analysis, we found that these six dysregulated ceRNA networks were consistently and significantly enriched in the proliferation hallmark signatures, including E2F_TARGETS, G2M_CHECKPOINT and MYC_TARGETS_V1 (Figure 4F). Pathway analysis showed that cancer-associated pathways such as spliceosome, apoptosis, and cell cycle were also significantly enriched (Figure 4G).These results suggested that homozygous deletion of these six driver genes contributed to the LGG poor prognosis in a complementary way, characterizing a new LGG subtype.
Figure 4.

The Homozygous Deletion of 9p21.3 Characterized a LGG Subtype with Poor Prognosis
(A) The homozygous deletions of six genes, including CDKN2B-AS, CDKN2B, CDKN2A, MTAP, KLHL9, and ELAVL2 were associated with poor prognosis in LGG. (B) The homozygous deletions of six genes significantly decreased their own gene expression. (C) The heatmap for SCNAs of 44 driver genes across LGG patients. (D) 9p21.3 deletions characterized a new LGG subtype with poor prognosis. (E) Comprehensive skeleton of the dysregulated ceRNA networks induced by homozygous deletions of six driver genes. (F) Hallmark signatures significantly enriched by the six dysregulated ceRNA networks. (G) Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways significantly enriched by the six dysregulated ceRNA networks.
The Dysregulated ceRNA Networks Driven by SCNAs across 12 Cancer Types
To investigate the generality and specificity of driver SCNAs and their driving dysregulated ceRNA networks across multiple cancers, we applied the integrative strategy to the other 11 cancer types to identify the driver SCNAs and their induced dysregulated ceRNA networks. For each cancer, a comprehensive dysregulated ceRNA network was constructed through assembling all SCNA-driven dysregulated ceRNA networks. We compared the comprehensive ceRNA networks across 12 cancer types from four aspects, including driver genes, miRNAs, ceRNAs, and dysregulated ceRNA triples. The number of driver genes ranged from 6 to 497, with a median of 109 (Figure 5A). We found that the SCNAs in breast invasive carcinoma (BRCA) and OV were more likely to destroy the ceRNA networks. The number of miRNAs was significantly associated with the number of driver genes in the networks (Pearson Correlation Coefficient [PCC] = 0.76, p = 0.004), while the number of ceRNAs and ceRNA triples did not show similar correlations (PCC = 0.38, p = 0.2 for ceRNAs; PCC = 0.49, p = 0.1 for triples). By comparing these comprehensive ceRNA networks, we found that 12 cancer types shared very few driver genes, and the average share proportion was only 1.9%. Thus, the share proportions of dysregulated ceRNA triples were very low, ranging from 0% to 1.1% (Figure 5B). Interestingly, we found that the share proportions of miRNAs and ceRNAs were clearly high. The average share proportions of miRNA and ceRNAs were 31.4% and 32.9%, respectively. In total, we identified 1,655 driver genes with SCNAs including 203 lncRNAs and 1,452 PGs for 12 cancer types. To further investigate the distributions of driver lncRNAs and PGs across 12 cancer types, we found that most of the driver genes were specific to specific cancer type, and few driver genes were shared by two or more cancer types (Figure 5C for lncRNAs and Figure 5D for PGs). These results suggested that different cancer types harbored different driver genomic alterations contributing to cancer carcinogenesis through dysregulating the crosstalks with common ceRNAs.
Figure 5.

Comparison Analysis of the Comprehensive ceRNA Networks across 12 Cancer Types
(A) The numbers of driver genes, miRNAs, ceRNAs, and triples in the comprehensive ceRNA networks in 12 cancer types. (B) The overlap ratio of driver genes, miRNAs, ceRNAs, and triples across 12 cancer types. (C) Heatmap of driver lncRNAs across 12 cancer types. (D) Heatmap of driver PGs across 12 cancer types.
Amplification of lncRNA PVT1 Disturbed Distinct ceRNA Triples in Different Cancers
Among the 203 driver lncRNAs, lncRNA PVT1 was most frequently identified in 12 cancer types. We found that amplification of lncRNA PVT1 could destroy its ceRNA networks in six cancer types, including OV, head and neck squamous cell carcinoma (HNSC), lung adenocarcinoma (LUAD), stomach adenocarcinoma (STAD), BRCA, and bladder urothelial carcinoma (BLCA) (Figures S5–S10). PVT1 has been reported as an oncogene. The high protein level of oncogene MYC was dependent on the gain of PVT1 expression to promote tumorigenesis.34 Amplification of PVT1 could contribute to the development ovarian and breast cancer.35 Knockdown of PVT1 could inhibit cell migration and proliferation.36 PVT1 showed frequent amplification across cancer types, and the frequencies were above 10% in six cancers (47.3% in OV, 12.8% in HNSC, 12.6% in LUAD, 13.4% in STAD, 20.6% in BRCA, and 11.2% in BLCA; Figure 6A). Compared with non-alteration status, amplification significantly elevated the expression levels of PVT1 (Figure 6B). In the PVT1-associated static ceRNA network, 33,741 ceRNA triples were formed by PVT1, 38 miRNAs, and 5,522 ceRNAs. By constructing the PVT1-associated dynamic ceRNA networks under PVT1 non-alteration, we found that the numbers of active ceRNA triples varied over three orders of magnitude, with from 0 to 1,501 (Figure 6C). The amplification of PVT1 completely destroyed the active ceRNA networks, and the numbers of active ceRNA triples dropped to 0. Surprisingly, there were no active ceRNA triples under PVT1 non-alteration status while PVT1 amplification caused six active ceRNA triples in OV. By analyzing the dysregulated ceRNA networks, one to six miRNAs mediated the ceRNA relationships between PVT1 and its ceRNAs (Figure 6D). Additionally, the ceRNAs of PVT1 also exhibited a clear difference (Figure 6E). lncRNAs occupied the major part of PVT1 ceRNAs in OV, HNSC, and LUAD, while PGs dominated ceRNAs of PVT1 in STAD. In both BLCA and BRCA, lncRNAs and PGs had equal shares of PVT1 ceRNAs. The results of genomic locations showed that the active ceRNAs of PVT1 spread universally across the whole genome (Figure 6F). To further investigate the generality and specificity of miRNAs and ceRNAs, we compared the six dysregulated ceRNA networks driven by PVT1 amplification and found that miRNAs and ceRNAs showed low generality across six cancer types. Obvious mutual exclusivity was observed in both miRNAs and ceRNAs (Figures 6G and 6H). The most frequent driver PG was MTAP. The homozygous deletion of MTAP was identified to induce dysregulated ceRNA networks in five cancer types, including LGG, lung squamous cell carcinoma (LUSC), LUAD, STAD, and BLCA. Comparative analysis showed that MTAP deletion also disturbed distinct ceRNA triples in different cancers (Figure S11).
Figure 6.

Amplification of lncRNA PVT1 Disturbed Distinct Different ceRNA Networks in Six Types of Cancers
(A) The amplification frequencies of PVT1 in 12 cancer types. (B) PVT1 amplification elevated the expression levels of PVT1. (C) The number of active ceRNA triples participated by PVT1 under the conditions of PVT1 non-alteration and amplification. (D) The numbers of miRNAs in PVT1 amplification-induced dysregulated ceRNA networks in cancers. (E) The numbers of PGs and lncRNAs in PVT1-induced dysregulated ceRNA networks in different cancers. (F) Genome-wide view of ceRNA partners of PVT1 in different cancers. Blue indicates lncRNA; red indicates PGs. (G) Heatmap for miRNAs in PVT1-mediated dysregulated ceRNA networks in different cancers. (H) Heatmap for ceRNAs of PVT1 in PVT1-mediated dysregulated ceRNA networks in different cancers.
We also found that ceRNA triples exhibited different active states under different conditions across cancer types. For example, PVT1 and lncRNA NEAT1 shared a miRNA hsa-miR-326 and formed a static ceRNA triple. This ceRNA triple was activated under non-alteration of PVT1 in HNSC. PVT1 and NEAT1 showed significant positive expression correlation (PCC = 0.38, p = 2.2e−12), and hsa-miR-326 and both PVT1 and NEAT1 showed significant negative expression correlations (PCC = −0.19, p = 0.00059; PCC = −0.26, p = 3.3e−6; Figure 7). However, the significant inverse relationships between hsa-miR-326 and both PVT1 and NEAT1 were destroyed by PVT1 amplification in HNSC. In other cancer types, this active ceRNA tripe was not identified in spite of the alteration status of PVT1. These results suggested that SCNAs of lncRNAs/PGs could contribute to abnormal transformation of cancer by destroying their ceRNA networks through different miRNA-mediated ceRNAs.
Figure 7.

An Active ceRNA Triple Was Specific under the Condition of PVT1 Non-alteration in HNSC, which Included PVT1, NEAT1, and has-miR-326
The green checkmark represents active, and the red cross represents inactive.
Cancer-Specific Driver Genes
We found that the major identified driver genes were specific to one type of cancer. Several driver genes were cancer-specific due to the fact that their frequent alterations only occurred in a specific cancer. For example, the amplification frequency of lncRNA GAS5 was above 0.1 only in BRCA (Figure 8A). lncRNA GAS5 induced growth arrest and apoptosis in breast cancer.37 The overexpression of GAS5 enhanced cell sensitivity to tamoxifen in breast cells both in vitro and in vivo.38 Compared with non-alteration, GAS5 amplification significantly elevated expression level (Figure 8A) and completely destroyed the GAS5-associated active ceRNA network in the non-alteration condition, which contained 3 miRNAs and 28 ceRNAs (26 lncRNAs and 2 PGs; Figures 8B–8D; Figure S12). In another situation, although the highly frequent alterations of driver genes occurred in multiple cancer types, the SCNA-driven dysregulated ceRNA network was specific to a specific cancer. For example, EGFR showed frequent amplification in GBM and HNSC; especially, amplification frequency was up to 43.9% in GBM (Figure 8E). EGFR amplification was associated with poor prognosis in both GBM and HNSC (p = 0.0242 for GBM, p = 0.0314 for HNSC; Figure S13). Compared with non-alteration, amplification significantly elevated EGFR expression levels in both GBM and HNSC (p = 3.6e−16 for GBM and p = 1.3e−9 for HNSC; Figure 8F). However, EGFR did not participate in the ceRNA regulatory mechanism in spite of non-alteration or amplification in GBM (FDR = 0.05, PCC < 0; Figure 8G). Under non-alteration of EGFR in HNSC, four miRNAs showed significant inverse correlations with EGFR while no inverse correlations were identified between any miRNA and EGFR under EGFR amplifications (FDR = 0.05, PCC < 0; Figure 8H; Figure S14). These results suggested that the same SCNA of driver genes may contribute to cancer development by different functional mechanisms, which added another layer of complexity on cancer heterogeneity.
Figure 8.

Cancer-Specific Driver Genes
(A) The amplification frequency of GAS5 was greater than 0.1 in only BRCA. GAS5 amplification significantly elevated its expression. (B) Volcano plot for expression correlations between GAS5 and miRNAs in the static GAS5-associated ceRNA networks. (C) Volcano plot for expression correlations between ceRNA partners of GAS5 and miRNAs in the static GAS5 associated ceRNA networks. (D) Volcano plot for expression correlations between GAS5 and its ceRNA partners in the static GAS5-associated ceRNA networks. (E) The amplification frequencies of EGFR were greater than 0.1 in both GBM and HNSC. (F) EGFR amplification significantly elevated its expression. (G) Volcano plot for expression correlations between EGFR and miRNAs from the static EGFR associated ceRNA networks in GBM. (H) Volcano plot for expression correlations between EGFR and miRNAs from the static EGFR-associated ceRNA networks in HNSC.
The Performance Evaluation of the Method
Three gene sets of known cancer genes were collected from the Cancer Gene Census of COSMIC, TSGene, and Bushman, respectively. We found that the driver genes in LGG identified by our method significantly overlapped with these known cancer genes (p = 0 for Cancer Gene Census, p = 0.02 for TSGene, and p = 0.02 for Bushman at the threshold of 0.1 in alteration frequency; Figure 9). Due to the less frequent SCNAs of known cancer genes, we re-identified the driver genes in LGG for the threshold of alteration frequencies at 0.08, 0.06, 0.04, and 0.02, which also could significantly capture known cancer genes. We also compared our method with two previous methods which identified driver copy-number alterations (Zhou et al.39 and DriverDBv340). Compared with these methods, our method could identify more known cancer genes from the Cancer Gene Census of COSMIC, TSGene, and Bushman (Figures 10A–10C). Due to the need for a certain sample size, our method had the limits in identifying the driver genes with very low SCNA frequencies or without SCNAs in the cancer population (Figure 10D). The driver genes identified by our method significantly overlapped those identified by Zhou et al.,39 but showed no significant overlap with those from DriverDBv3 (Figure 10E). These results suggested that our method was useful for identifying the driver genes by dissecting the functional mechanisms of SCNAs from the view of dysregulated ceRNAs.
Figure 9.

The Significant Overlaps between the Driver Genes Identified by Our Method and Cancer Genes Recorded in Three Known Cancer Gene Databases, Including the Cancer Gene Census of COSMIC, TSGene, and Bushman, at Different Thresholds of SCNA Frequency
Figure 10.

Comparison of Our Method with Other Methods
(A) The number of cancer genes in the Cancer Gene Census of COSMIC identified by three methods at different thresholds of SCNA frequencies. (B) The number of cancer genes in TSGene identified by three methods at different thresholds of SCNA frequencies. (C) The number of cancer genes in Bushman identified by three methods at different thresholds of SCNA frequencies. (D) The cumulative distributions of SCNA frequencies of driver genes identified by three methods. (E) The overlaps between driver genes identified by our method and the other two methods at different thresholds of SCNA frequencies.
Discussion
In this study, we developed an integrative strategy to identify driver SCNAs and characterize functional roles of SCNAs in multiple cancer types. By integrating copy number profiles, expression profiles of PGs, lncRNAs, and miRNAs, and ceRNA networks, we identified SCNA-induced dysregulated ceRNA networks. We analyzed the dysregulated patterns of dysregulated ceRNA networks and dissected the cooperative functional roles of SCNAs contributing to cancer subtypes. Comparative analysis of SCNA-induced dysregulated ceRNA networks across multiple cancer types showed that the functional roles of SCNAs were cancer-specific.
It is well accepted that non-coding regions occupy major parts of the genome. The SCNA frequency distributions of lncRNAs were comparable to those of PGs across multiple cancer types. However, few studies focused on the functional roles of lncRNA SCNAs. Enhanced characterization of the functional effects of lncRNA SCNAs in cancer could help understand cancer etiology and guide cancer diagnosis and therapy. Our strategy provided a direct way to characterize lncRNA functions through identifying the lncRNA SCNA-induced dysregulated ceRNA network, in which lncRNAs directly participated in each of the ceRNA triples. lncRNA SCNAs directly destroyed the miRNA-mediated co-regulated patterns between them and their ceRNA partners by influencing their own expression levels. Meanwhile, our strategy could be applied to dissect other types of noncoding RNAs along with the advent of corresponding biological profiles.
Due to the cooperativity of miRNAs and multiplicity of targets, ceRNA networks were extremely complicated, which contained not only protein-coding mRNAs and the known non-coding RNAs (such as lncRNAs, pseudogenes, and circRNAs), but also the unknown RNAs with miRNA target sites. Constructing comprehensive ceRNA networks was necessary for understanding the molecular mechanism in cancer cells. Analysis of ceRNA networks in previous studies explored ceRNA partners of PGs (PTEN) based on mRNA-mRNA ceRNA networks or predicted lncRNA functions based on lncRNA-mRNA ceRNA networks. These ceRNA networks were not enough to capture ceRNA-mediated functions of lncRNAs or PGs. We further expanded ceRNA networks including mRNA-mRNA, lncRNA-mRNA, and lncRNA-lncRNA ceRNA partnerships, which provided us a more comprehensive view to explore functional effects of SCNAs in cancers. Analysis of SCNA-induced dysregulated ceRNA networks showed that lncRNAs contributed important parts to dysregulated ceRNA triples induced by driver SCNAs. However, the constructed ceRNA networks were still limited due to lacking matched biological profiles of other RNAs. With the development of functional studies of lncRNAs and data accumulation of multi-omics, our strategy could comprehensively characterize functional roles of SCNAs.
The analysis of SCNAs across multiple cancers showed that cancer specificities of SCNAs were varied. We found that 68.8% of genes and lncRNAs with high SCNA frequency were present in only one cancer type. Exploring the functional roles of these cancer-specific SCNAs was useful for understanding cancer-specific pathogenesis. For example, lncRNA GAS5 showed high amplification frequency only in BRCA. GAS5 amplification destroyed the dynamic ceRNA network that was constructed under GAS5 non-alteration. Although some genes and lncRNAs (such as EGFR) showed high SCNA frequency in multiple cancer types, their SCNAs performed different functional effects in different cancer types. Amplification of EGFR could elevate EGFR expression levels in both HNSC and GBM. EGFR amplification dysregulated the EGFR-associated ceRNA network in HNSC, while EGFR did not participate in the ceRNA regulatory mechanism in GBM. In other case, SCNAs of genes or lncRNAs could disturb their associated ceRNA networks through different miRNAs and different ceRNA partners in different cancer types. The functional diversity of SCNAs led us to investigate the cancer-specific functional roles of SCNAs from different perspectives.
The driver genes identified by our method could be affected by the input data, including copy number profiles and expression profiles of lncRNAs, miRNAs, and PGs. With the threshold level of undetected frequency decreasing or the threshold of mean expression level increasing, the numbers of lncRNAs, miRNAs, and PGs in the expression profiles as input decreased. The potential driver genes with a low expression level could not be input into our method. Additionally, the number of identified driver genes would decreased slightly (Figure S15). Meanwhile, the threshold of SCNA frequency also determined the copy number profiles as input. With the threshold of SCNA frequency decreasing, more lncRNAs/PGs were input into our method, and more potential driver genes with higher SCNA frequencies than threshold could be identified (Figure S16). Due to the need for a certain sample size, our method had limits in identifying the driver genes with very low SCNA frequencies in the cancer population.
In conclusion, we proposed an integrative strategy to investigate the functional roles of driver SCNAs based on dysregulated ceRNA networks, which was complementary with previous methods. Our method had good extensibility with accumulation of multi-omics.
Materials and Methods
Materials
We collected four types of datasets, that is, copy number profiles and gene expression profiles of PGs, lncRNAs, and miRNAs for 12 cancer types, including LGG, GBM, HNSC, BLCA, OV, BRCA, LUAD, LUSC, STAD, kidney renal clear cell carcinoma (KIRC), cervical squamous cell carcinoma (CESC), and prostate adenocarcinoma (PRAD). The copy number profiles and gene expression profiles of PGs and miRNAs were downloaded from The Cancer Genome Atlas (TCGA) (https://www.cancer.gov/about-nci/organization/ccg/research/structural-genomics/tcga). The copy number profiles of 12 cancer types involved 5,814 cancer samples in TCGA. Additionally, the expression profiles of PGs and miRNA expression profiles contained 5,701 and 5,069 cancer samples in TCGA, respectively. We collected the lncRNA expression profiles of 12 cancer types from TANRIC,41 which contained 4,578 cancer samples from TCGA. There were 3,508 common cancer samples from which all of the copy number profiles, expression profiles of PGs, miRNA expression profiles, and lncRNA expression profiles were detected (Table S1; Figure S1). The datasets from these 3,508 common cancer samples were used for subsequent analysis.
The Copy Number Profiles of Protein Genes and lncRNAs
Segmentation data (level 3) of copy numbers in 12 cancer types were collected. For each cancer type, we applied GISTIC (version 2)42 to copy number data and identified the SCNAs of PGs and lncRNAs. Five values were used to measure the extent of copy-number alterations, including homozygous deletion, heterozygous deletion, and diploid, gain, and high-level amplification. Only homozygous deletion and high-level amplification were considered as reliable SCNAs, and then the discrete SCNA profiles of PGs and lncRNAs were established, in which 2 (high-level amplification) and −2 (heterozygous deletion) represented SCNAs and 0 represented non-alterations.
The Expression Profiles of PGs
The gene expressions of PGs in GBM and OV were detected by microarray technology. The gene expression of PGs for the remaining cancer types were detected using RNA sequencing (RNA-seq) technology. The expression levels of genes were calculated by multiplying the tau value calculated using RSEM and 106.43 For subsequent calculation, the expression levels were transformed using log2.
The Expression Profiles of miRNAs
The gene expression levels of miRNAs in GBM and OV were also detected using microarray technology, and the miRNA IDs from microarrays were transferred into miRNA IDs in miRBase version 20.44 For the rest of the cancer types, miRNA expression profiles were detected using miRNA sequencing (miRNA-seq) technology. Expression levels of miRNAs were measured using RPM. For subsequent calculation, the expression levels were transformed using log2.
The Expression Profiles of lncRNAs
lncRNA expression profiles of TCGA samples for 12 cancer types were collected from TANRIC,41 which were frequently used to explore the functional roles of lncRNAs (Table S3). The expression levels of lncRNAs were quantified as RPKM (reads per kilobase transcript per million mapped reads), which were calculated based on the paired-end RNA-seq BAM files of TCGA samples. The expression levels of lncRNAs were transformed by log2 for subsequent calculation.
For each cancer, the common samples across the above four levels were selected (Figure S1). Any PG or lncRNA or miRNA was deleted when the undetected frequency was greater than 30% or the mean expression level was lower than 0.1.
The miRNA-PG/miRNA-lncRNA Target Interactions
We downloaded the miRNA-PG/miRNA-lncRNA target interactions from starBase v2.045 and the miRNA-lncRNA target interactions from lnCeDB.46 These target relationships between miRNAs and PGs/ncRNAs were supported from the view of sequence matching, which provided a comprehensive view of static miRNA-PGs/lncRNAs target interactions. To identify the activated miRNA-PGs/lncRNAs target interactions in a specific cancer, we integrated the static miRNA-PGs/lncRNAs target interactions and expression profiles of PGs, miRNAs, and lncRNAs from the specific cancer samples. Therefore, only the miRNA-PGs/lncRNAs target relations, in which both miRNAs and PGs/lncRNAs were detected in expression profiles of PGs, miRNAs, and lncRNAs from the specific cancer, were kept for subsequent analysis.
Methods
We defined driver genes as those genes whose SCNAs could significantly affect their own expression levels and further destroy the activated ceRNA networks under the condition of non-alterations. Through integrating the discrete SCNA profiles, expression profiles of PGs, miRNAs, and lncRNAs, and miRNA target interactions, we developed an integrative strategy to characterize the functional roles of SCNAs and identify the driver genes in cancers by constructing SCNA-induced dysregulation of ceRNA networks (Figure 1). The integrative strategy included three steps: identifying the candidate PGs/lncRNAs with frequent SCNAs, constructing the static ceRNA networks for candidate genes, and constructing dysfunctional ceRNA networks driven by SCNAs. The detailed information is presented below.
Identifying the Candidate PGs/lncRNAs with Frequent SCNAs
The candidate genes should meet two criteria: the SCNAs of candidate genes should recur frequently in cancer populations, and the SCNAs could affect the expression of candidate genes. We calculated the SCNA frequencies of PGs/lncRNAs from the discrete SCNA profiles and selected the PGs/lncRNAs with SCNA frequencies greater than 0.1. By combining expression profiles of PGs/lncRNAs and the discrete SCNA profiles, we identified PGs/lncRNAs whose SCNAs could significantly and concordantly influence their expression levels (t test, p ≤ 0.05). Finally, these identified PGs/lncRNAs with miRNA-target interactions were considered as candidate genes.
Constructing the Static ceRNA Networks for Candidate Genes
For each candidate gene, we identified miRNAs that targeted this candidate gene from miRNA-PGs/lncRNAs interactions. Then, PGs/lncRNAs targeted by these miRNAs were considered as ceRNA competitors of the candidate gene. We defined a ceRNA triple as one containing the candidate gene, a ceRNA, and a miRNA, in which both the candidate gene and the ceRNA were targeted by the miRNA. Then, we enumerated all candidate gene-associated ceRNA triples and assembled these ceRNA triples into a static ceRNA network of the candidate gene.
Constructing Dysfunctional ceRNA Networks Driven by SCNAs
Candidate genes may show the status of three copy numbers across cancer samples, that is, SCNAs (homozygous deletion and high-level amplification) and non-alteration. Differential expression levels of candidate genes driven by SCNAs may destroy the homeostasis of the ceRNA networks activated in the condition of non-alteration.
According to copy number status of each candidate gene, we grouped cancer samples into subgroups and deleted the subgroups with fewer than five samples. For each copy number status, we constructed the dynamic ceRNA networks by integrating expression profiles of PGs, lncRNAs, and miRNAs of the corresponding subgroup and the static ceRNA network associated with the candidate gene. The correlation coefficients were calculated among the candidate gene, miRNAs, and ceRNA competitors of ceRNA triples in the static ceRNA network. We identified the activated ceRNA triples in which correlation coefficients between the candidate gene and miRNAs and those between miRNAs and ceRNA competitors were significantly negative at a FDR of 0.05, and the correlation coefficients between candidate gene and ceRNA competitors were significantly positive at a FDR of 0.05. These activated ceRNA triples were assembled into the dynamic ceRNA network under the condition of specific copy number status.
To characterize the SCNA influence of the candidate gene on the ceRNA network, the dynamic ceRNA networks constructed under the SCNA of the candidate gene were compared with the dynamic ceRNA network from non-alteration status. We identified the dysregulated ceRNA triples as the ceRNA triples that were only activated in only one of the conditions of the SCNAs and non-alteration of the candidate gene. Then, all dysregulated ceRNA triples constituted the dysregulated ceRNA networks driven by the SCNA of the candidate gene. The driver genes were considered as the candidate genes whose SCNAs could drive the dysregulated ceRNA networks.
Survival Analysis
For a given PG or lncRNA, three SCNA status levels may be present across cancer populations: heterozygous deletion (−2), high-level amplification (2), and non-alterations (0). According to SCNA status of the PG or lncRNA, the cancer patients were divided into subgroups as follows: one subgroup of patients with heterozygous deletion (−2) of the PG or lncRNA, one subgroup of patients with high-level amplification (2) of the PG or lncRNA, and one subgroup of patients with non-alteration (0) of the PG or lncRNA. To test whether the SCNA status of the PG or lncRNA was associated with prognosis, we used Kaplan-Meier analysis and a log-rank test to compare the survival times among the subgroups.
For a specific SCNA of the genome region, we divided the cancer patients based on the SCNA profiles of driver genes as follows: one subgroup of patients without SCNAs of driver genes, one subgroup of patients with the specific SCNA of at least one driver gene in the genome region, and one subgroup that consist of the rest of the patients. The significance of a survival difference between the three subgroups was estimated using Kaplan-Meier analysis and log-rank test.
The Code for the Integrative Strategy
The code for the integrative strategy is available at https://github.com/pingyanyan/Driver-SCNAs.
Author Contributions
Y.X. and C.X. designed the idea of this work; Y.P., L.P., and Y.Z. processed the data and performed bioinformatics analysis; Y.P. and J.H. wrote the manuscript. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported in part by the National Key R&D Program of China (2018YFC2000100); the National Natural Science Foundation of China (31601076, 61873075, 31900478); the China Postdoctoral Science Foundation (2016M601444); Wu lien-teh Youth Science Fund Project of Harbin Medical University (WLD-QN1407); Special Funds for the Construction of Higher Education in Heilongjiang Province (grant UNPYSCT-2018068); and by the Hei Long Jiang Postdoctoral Foundation (LBH-Z16119).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtn.2020.06.012.
Contributor Information
Chaohan Xu, Email: chaohanxu@hrbmu.edu.cn.
Yun Xiao, Email: xiaoyun@ems.hrbmu.edu.cn.
Supplemental Information
References
- 1.Martincorena I., Campbell P.J. Somatic mutation in cancer and normal cells. Science. 2015;349:1483–1489. doi: 10.1126/science.aab4082. [DOI] [PubMed] [Google Scholar]
- 2.Zack T.I., Schumacher S.E., Carter S.L., Cherniack A.D., Saksena G., Tabak B., Lawrence M.S., Zhsng C.Z., Wala J., Mermel C.H. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013;45:1134–1140. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Yan X., Hu Z., Feng Y., Hu X., Yuan J., Zhao S.D., Zhang Y., Yang L., Shan W., He Q. Comprehensive genomic characterization of long non-coding RNAs across human cancers. Cancer Cell. 2015;28:529–540. doi: 10.1016/j.ccell.2015.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Deng Y., Luo S., Zhang X., Zou C., Yuan H., Liao G., Xu L., Deng C., Lan Y., Zhao T. A pan-cancer atlas of cancer hallmark-associated candidate driver lncRNAs. Mol. Oncol. 2018;12:1980–2005. doi: 10.1002/1878-0261.12381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fehrmann R.S., Karjalainen J.M., Krajewska M., Westra H.J., Maloney D., Simeonov A., Pers T.H., Hirschhorn J.N., Jansen R.C., Schultes E.A. Gene expression analysis identifies global gene dosage sensitivity in cancer. Nat. Genet. 2015;47:115–125. doi: 10.1038/ng.3173. [DOI] [PubMed] [Google Scholar]
- 6.Akavia U.D., Litvin O., Kim J., Sanchez-Garcia F., Kotliar D., Causton H.C., Pochanard P., Mozes E., Garraway L.A., Pe’er D. An integrated approach to uncover drivers of cancer. Cell. 2010;143:1005–1017. doi: 10.1016/j.cell.2010.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bashashati A., Haffari G., Ding J., Ha G., Lui K., Rosner J., Huntsman D.G., Caldas C., Aparicio S.A., Shah S.P. DriverNet: uncovering the impact of somatic driver mutations on transcriptional networks in cancer. Genome Biol. 2012;13:R124. doi: 10.1186/gb-2012-13-12-r124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jörnsten R., Abenius T., Kling T., Schmidt L., Johansson E., Nordling T.E., Nordlander B., Sander C., Gennemark P., Funa K. Network modeling of the transcriptional effects of copy number aberrations in glioblastoma. Mol. Syst. Biol. 2011;7:486. doi: 10.1038/msb.2011.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Albertson D.G., Collins C., McCormick F., Gray J.W. Chromosome aberrations in solid tumors. Nat. Genet. 2003;34:369–376. doi: 10.1038/ng1215. [DOI] [PubMed] [Google Scholar]
- 10.Sharma A.K., Eils R., König R. Copy number alterations in enzyme-coding and cancer-causing genes reprogram tumor metabolism. Cancer Res. 2016;76:4058–4067. doi: 10.1158/0008-5472.CAN-15-2350. [DOI] [PubMed] [Google Scholar]
- 11.Ping Y., Zhang H., Deng Y., Wang L., Zhao H., Pang L., Fan H., Xu C., Li F., Zhang Y. IndividualizedPath: identifying genetic alterations contributing to the dysfunctional pathways in glioblastoma individuals. Mol. Biosyst. 2014;10:2031–2042. doi: 10.1039/c4mb00289j. [DOI] [PubMed] [Google Scholar]
- 12.Ping Y., Deng Y., Wang L., Zhang H., Zhang Y., Xu C., Zhao H., Fan H., Yu F., Xiao Y., Li X. Identifying core gene modules in glioblastoma based on multilayer factor-mediated dysfunctional regulatory networks through integrating multi-dimensional genomic data. Nucleic Acids Res. 2015;43:1997–2007. doi: 10.1093/nar/gkv074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ebert M.S., Neilson J.R., Sharp P.A. MicroRNA sponges: competitive inhibitors of small RNAs in mammalian cells. Nat. Methods. 2007;4:721–726. doi: 10.1038/nmeth1079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Salmena L., Poliseno L., Tay Y., Kats L., Pandolfi P.P. A ceRNA hypothesis: the Rosetta Stone of a hidden RNA language? Cell. 2011;146:353–358. doi: 10.1016/j.cell.2011.07.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tay Y., Rinn J., Pandolfi P.P. The multilayered complexity of ceRNA crosstalk and competition. Nature. 2014;505:344–352. doi: 10.1038/nature12986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tay Y., Kats L., Salmena L., Weiss D., Tan S.M., Ala U., Karreth F., Poliseno L., Provero P., Di Cunto F. Coding-independent regulation of the tumor suppressor PTEN by competing endogenous mRNAs. Cell. 2011;147:344–357. doi: 10.1016/j.cell.2011.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Poliseno L., Salmena L., Zhang J., Carver B., Haveman W.J., Pandolfi P.P. A coding-independent function of gene and pseudogene mRNAs regulates tumour biology. Nature. 2010;465:1033–1038. doi: 10.1038/nature09144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cesana M., Cacchiarelli D., Legnini I., Santini T., Sthandier O., Chinappi M., Tramontano A., Bozzoni I. A long noncoding RNA controls muscle differentiation by functioning as a competing endogenous RNA. Cell. 2011;147:358–369. doi: 10.1016/j.cell.2011.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu M., Chen X., Lin K., Zeng K., Liu X., Xu X., Pan B., Xu T., Sun L., He B. lncRNA SNHG6 regulates EZH2 expression by sponging miR-26a/b and miR-214 in colorectal cancer. J. Hematol. Oncol. 2019;12:3. doi: 10.1186/s13045-018-0690-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kabat H., Tunkle L., Lee I. ceRNA search method identified a MET-activated subgroup among EGFR DNA amplified lung adenocarcinoma patients. Pac. Symp. Biocomput. 2017;22:438–448. doi: 10.1142/9789813207813_0041. [DOI] [PubMed] [Google Scholar]
- 21.Li W., Wang Q., Feng Q., Wang F., Yan Q., Gao S.J., Lu C. Oncogenic KSHV-encoded interferon regulatory factor upregulates HMGB2 and CMPK1 expression to promote cell invasion by disrupting a complex lncRNA-OIP5-AS1/miR-218-5p network. PLoS Pathog. 2019;15:e1007578. doi: 10.1371/journal.ppat.1007578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu J., Hou X., Pang L., Sun S., He S., Yang Y., Liu K., Xu L., Yin W., Xu C., Xiao Y. Identification of dysregulated competitive endogenous RNA networks driven by copy number variations in malignant gliomas. Front. Genet. 2019;10:1055. doi: 10.3389/fgene.2019.01055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Arancio W., Genovese S.I., Bongiovanni L., Tripodo C. A ceRNA approach may unveil unexpected contributors to deletion syndromes, the model of 5q- syndrome. Oncoscience. 2015;2:872–879. doi: 10.18632/oncoscience.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sondka Z., Bamford S., Cole C.G., Ward S.A., Dunham I., Forbes S.A. The COSMIC Cancer Gene Census: describing genetic dysfunction across all human cancers. Nat. Rev. Cancer. 2018;18:696–705. doi: 10.1038/s41568-018-0060-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhao M., Kim P., Mitra R., Zhao J., Zhao Z. TSGene 2.0: an updated literature-based knowledgebase for tumor suppressor genes. Nucleic Acids Res. 2016;44(D1):D1023–D1031. doi: 10.1093/nar/gkv1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gao Y., Wang P., Wang Y., Ma X., Zhi H., Zhou D., Li X., Fang Y., Shen W., Xu Y. Lnc2Cancer v2.0: updated database of experimentally supported long non-coding RNAs in human cancers. Nucleic Acids Res. 2019;47(D1):D1028–D1033. doi: 10.1093/nar/gky1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhan W., Wang W., Han T., Xie C., Zhang T., Gan M., Wang J.B. COMMD9 promotes TFDP1/E2F1 transcriptional activity via interaction with TFDP1 in non-small cell lung cancer. Cell. Signal. 2017;30:59–66. doi: 10.1016/j.cellsig.2016.11.016. [DOI] [PubMed] [Google Scholar]
- 28.Solban N., Jia H.P., Richard S., Tremblay S., Devlin A.M., Peng J., Gossard F., Guo D.F., Morel G., Hamet P. HCaRG, a novel calcium-regulated gene coding for a nuclear protein, is potentially involved in the regulation of cell proliferation. J. Biol. Chem. 2000;275:32234–32243. doi: 10.1074/jbc.M001352200. [DOI] [PubMed] [Google Scholar]
- 29.Zhang B., Liu Y., Liu D., Yang L. Targeting cleavage and polyadenylation specific factor 1 via shRNA inhibits cell proliferation in human ovarian cancer. J. Biosci. 2017;42:417–425. doi: 10.1007/s12038-017-9701-x. [DOI] [PubMed] [Google Scholar]
- 30.Fang F., Chang R., Yang L. Heat shock factor 1 promotes invasion and metastasis of hepatocellular carcinoma in vitro and in vivo. Cancer. 2012;118:1782–1794. doi: 10.1002/cncr.26482. [DOI] [PubMed] [Google Scholar]
- 31.Nakamura Y., Fujimoto M., Fukushima S., Nakamura A., Hayashida N., Takii R., Takaki E., Nakai A., Muto M. Heat shock factor 1 is required for migration and invasion of human melanoma in vitro and in vivo. Cancer Lett. 2014;354:329–335. doi: 10.1016/j.canlet.2014.08.029. [DOI] [PubMed] [Google Scholar]
- 32.Liberzon A., Birger C., Thorvaldsdóttir H., Ghandi M., Mesirov J.P., Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. doi: 10.1016/j.cels.2015.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yu G., Wang L.G., Han Y., He Q.Y. clusterProfiler: an R package for comparing biological themes among gene clusters. OMICS. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tseng Y.Y., Moriarity B.S., Gong W., Akiyama R., Tiwari A., Kawakami H., Ronning P., Reuland B., Guenther K., Beadnell T.C. PVT1 dependence in cancer with MYC copy-number increase. Nature. 2014;512:82–86. doi: 10.1038/nature13311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Guan Y., Kuo W.L., Stilwell J.L., Takano H., Lapuk A.V., Fridlyand J., Mao J.-H., Yu M., Miller M.A., Santos J.L. Amplification of PVT1 contributes to the pathophysiology of ovarian and breast cancer. Clin. Cancer Res. 2007;13:5745–5755. doi: 10.1158/1078-0432.CCR-06-2882. [DOI] [PubMed] [Google Scholar]
- 36.Yu Y., Zhang M., Liu J., Xu B., Yang J., Wang N., Yan S., Wang F., He X., Ji G. Long non-coding RNA PVT1 promotes cell proliferation and migration by silencing ANGPTL4 expression in cholangiocarcinoma. Mol. Ther. Nucleic Acids. 2018;13:503–513. doi: 10.1016/j.omtn.2018.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mourtada-Maarabouni M., Pickard M.R., Hedge V.L., Farzaneh F., Williams G.T. GAS5, a non-protein-coding RNA, controls apoptosis and is downregulated in breast cancer. Oncogene. 2009;28:195–208. doi: 10.1038/onc.2008.373. [DOI] [PubMed] [Google Scholar]
- 38.Gu J., Wang Y., Wang X., Zhou D., Shao C., Zhou M., He Z. Downregulation of lncRNA GAS5 confers tamoxifen resistance by activating miR-222 in breast cancer. Cancer Lett. 2018;434:1–10. doi: 10.1016/j.canlet.2018.06.039. [DOI] [PubMed] [Google Scholar]
- 39.Zhou W., Zhao Z., Wang R., Han Y., Wang C., Yang F., Han Y., Liang H., Qi L., Wang C. Identification of driver copy number alterations in diverse cancer types and application in drug repositioning. Mol. Oncol. 2017;11:1459–1474. doi: 10.1002/1878-0261.12112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu S.H., Shen P.C., Chen C.Y., Hsu A.N., Cho Y.C., Lai Y.L., Chen F.H., Li C.Y., Wang S.C., Chen M. DriverDBv3: a multi-omics database for cancer driver gene research. Nucleic Acids Res. 2020;48:D863–D870. doi: 10.1093/nar/gkz964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li J., Han L., Roebuck P., Diao L., Liu L., Yuan Y., Weinstein J.N., Liang H. TANRIC: an interactive open platform to explore the function of lncRNAs in cancer. Cancer Res. 2015;75:3728–3737. doi: 10.1158/0008-5472.CAN-15-0273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mermel C.H., Schumacher S.E., Hill B., Meyerson M.L., Beroukhim R., Getz G. GISTIC2.0 facilitates sensitive and confident localization of the targets of focal somatic copy-number alteration in human cancers. Genome Biol. 2011;12:R41. doi: 10.1186/gb-2011-12-4-r41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kozomara A., Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic Acids Res. 2014;42:D68–D73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li J.H., Liu S., Zhou H., Qu L.H., Yang J.H. starBase v2.0: decoding miRNA-ceRNA, miRNA-ncRNA and protein-RNA interaction networks from large-scale CLIP-seq data. Nucleic Acids Res. 2014;42:D92–D97. doi: 10.1093/nar/gkt1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Das S., Ghosal S., Sen R., Chakrabarti J. lnCeDB: database of human long noncoding RNA acting as competing endogenous RNA. PLoS ONE. 2014;9:e98965. doi: 10.1371/journal.pone.0098965. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.