

Summary
RNA-binding proteins (RBPs) recognize consensus sequences and regulate specific target mRNAs. However, large-scale CLIP-seq revealed loose and broad binding of RBPs to larger proportion of expressed mRNAs: e.g. SFPQ binds to >10,000 pre-mRNAs but distinctly regulates <200 target genes during neuronal development. Identification of crucial binding for regulation and rules of target recognition is highly anticipated for systemic understanding of RBP regulations. For a breakthrough solution, we developed a bioinformatical method for CLIP-seq and transcriptome data by adopting iterative hypothesis testing. Essential binding was successfully identified in C-rich sequences close to the 5′ splice sites of long introns, which further proposed target recognition mechanism via association between SFPQ and splicing factors during spliceosome assembly. The identified features of functional binding enabled us to predict regulons and also target genes in different species. This multilateral bioinformatics approach facilitates the elucidation of functionality, regulatory mechanism, and regulatory networks of RBPs.
Subject Areas: Structural Biology, Protein Structure Aspects, Bioinformatics
Graphical Abstract
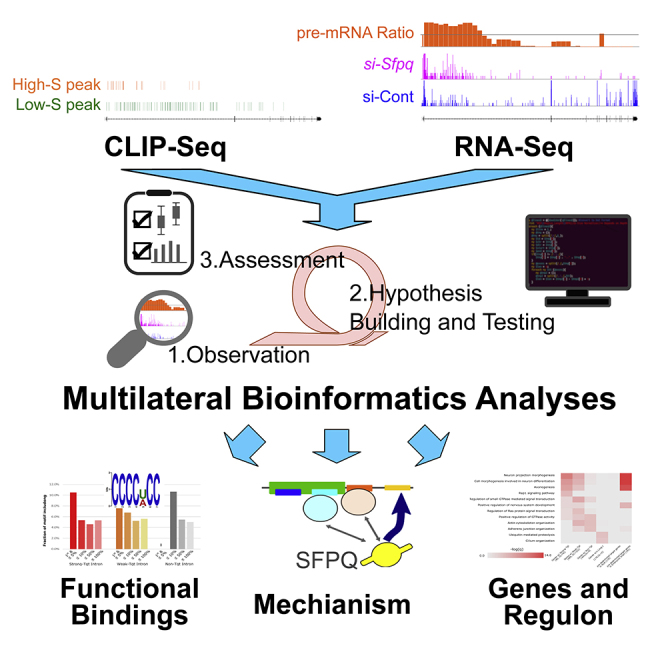
Highlights
-
•
We developed a multilateral bioinformatics (MLBI) analysis for RNA biology study
-
•
MLBI analyses successfully identified functionally essential binding from CLIP-seq
-
•
MLBI analyses revealed the target recognition mechanism of RBP on mRNAs
-
•
MLBI analyses identified RBP regulon and predicted target genes in close species
Structural Biology; Protein Structure Aspects; Bioinformatics
Introduction
In mammals, approximately 1,500 genes encode RNA-binding proteins (RBPs), which directly bind to specific target mRNAs by recognizing consensus sequences and regulate RNA metabolism (Castello et al., 2012; Gerstberger et al., 2014; Hentze et al., 2018; Sundararaman et al., 2016). The consensus sequences of each RBP are called cis-elements; these sequences define the target specificities and functions of RBPs. For example, Rbfox and Nova bind to UGCAUG and YACY motifs close to splice sites, respectively and regulate tissue-specific alternative splicing (Licatalosi et al., 2008; Weyn-Vanhentenryck et al., 2014). Recent studies by the ENCODE project are attempting to reveal the binding properties and functions of entire RBPs by genome-wide identification of target RNAs, binding sites, and consensus sequences using CLIP-seq (ultraviolet [UV]-crosslinking and immunoprecipitation with high-throughput sequencing) (Van Nostrand et al., 2018). However, RBP binding is not necessarily restricted to unique consensus sequences but is broadly observed in a larger proportion of expressed gene transcripts. In their study, totally 26 RBPs were analyzed and more than 30% (8 RBPs including SFPQ) did not showe distinct consensus sequences. For example, SRSF5 (serine and arginine-rich splicing factor 5), PABPN1L (PABPN1-like, cytoplasmic), RBP22, and RBM15B (RNA-binding motif protein 15B) were shown to have more than 10 nonoverlapping motifs in each RBP; however, these motifs accounted for less than 50% of all binding sites (Van Nostrand et al., 2018). These results suggest that RBP binding is not simply defined by consensus sequences but that binding specificities are also regulated by more complex rules. Binding of RBPs is known to be altered by the flexibility of RNA-binding domains to sequence variants of cis-elements, by adjacent sequences and mRNA structures, and by interactions with other RBPs (Fu and Ares, 2014). Furthermore, the recruitment mechanism, binding portion in pre-mRNAs, and co-binding partners allow RBPs to play multiple context-dependent roles, such as roles in transcription, splicing, or stabilization of pre-mRNAs (Hentze et al., 2018). Therefore, an imminent issue is to elucidate the regulatory rules that define the binding specificity and functions of RBPs with regard to distinct subsets of mRNA.
SFPQ (Splicing factor proline and glutamine rich) is a multifunctional RBP that regulates mRNA processing, transcription, and DNA repair (Knott et al., 2016; Yarosh et al., 2015). In a recent study, we found that SFPQ regulates the transcriptional elongation of extra-long genes (≥100 kb) in the brain and muscles (Hosokawa et al., 2019; Takeuchi et al., 2018). Mechanistically, SFPQ co-transcriptionally binds to target pre-mRNAs, recruits cyclin-dependent kinase 9 (CDK9), and activates RNA polymerase II (Pol II). Notably, SFPQ binding has been detected on pre-mRNAs from 11,286 genes in CLIP-seq, and loss of Sfpq disrupts the expression of 141 genes, representing only 1.2% of all SFPQ-bound genes, and 95.6% of these 141 genes have pre-mRNAs > 100 kb in length (Takeuchi et al., 2018). In this study, we aimed to decipher the binding specificity of RBPs linked to unique functions using a newly developed analytical method with SFPQ as a model. Conventional bioinformatic analysis methods for determining RBP functions have adopted pre-settled or so-called “waterfall” analytical designs; however, these approaches have not succeeded in addressing the issues described earlier. Thus, we employed an “agile data science method” (Russell, 2017) as an iterative approach for observation point installation and hypothesis-driven mathematical modeling (Figure S1). We applied this method to elucidate how SFPQ recognizes and regulates specific long pre-mRNAs among the huge spectrum of potential binding target mRNAs and revealed the underlying molecular mechanisms that define the target specificities of SFPQ.
Results
SFPQ Regulates Long Intron-Containing Genes
To identify functional SFPQ binding, which is essential for regulating transcriptional elongation of extra-long genes, we integrated data of genome-wide SFPQ-binding mapping (CLIP-seq) with RNA-seq data under Sfpq-KD and si-control (Cont) conditions. We then crosschecked the relationships between SFPQ binding and fold change (FC) of expression. In Neuro2a cells, 176 genes (1.2% of total 11,434 SFPQ-bound genes) were identified as SFPQ regulatory targets that had significant SFPQ-binding peaks, and their expression levels were significantly downregulated under Sfpq-KD condition (FC of Sfpq-KD/Cont ratio <1/3, see Methods for detailed regulatory target gene identification criteria) (Figure 1A, genes indicated by red dots). It is accounted for 13.7% of genes passing the same SFPQ-binding criteria (1,289 genes), whereas 432 (33.5%) were identified as nontarget genes with expression levels not affected under Sfpq-KD condition (0.83 ≤ FC ≤ 1.20; Figure 1A, genes indicated as green dots). These results indicated that total SFPQ binding is not the only factor to identify the SFPQ target genes. Because SFPQ was shown to be required for long gene expression, we next evaluated the gene lengths of SFPQ target genes. The results showed that 85.8% (151/176) were longer than 100 kb, suggesting that only longer genes with significant SFPQ binding were affected by Sfpq-KD. However, the distribution of gene lengths in these two groups overlapped (42 kb–1.9 mb for downregulated genes and 13 kb to 1.1 mb for genes that were not downregulated; Figure 1A), indicating that gene length was informative, but not sufficient, for defining SFPQ target genes.
Figure 1.

SFPQ Regulated Genes Containing Long Introns
(A) Relationships between pre-mRNA lengths and log2 FC in Sfpq-KD and control (Cont-si) Neuro2a cells. The red dots show 176 genes that were significantly downregulated (FC of Sfpq-KD/Cont ratio <1/3) with significant SFPQ binding peaks. The green dots show 432 genes meeting the same SFPQ binding criteria, but without changes in FC values (0.83 < FC < 1.20).
(B) Higher magnification of the graph in A from the range of 20 kb to 1 Mb, showing similar distributions of 120 gene pairs with pre-mRNA lengths (length difference <10 kb).
(C and D) Heatmaps showing pre-mRNA expression profiles for the 120 downregulated genes, captioned as target genes (C) and 120 non-downregulated genes, captioned as non-target genes. (D). Each block shows a 5-kb window for the gene bodies. Down- or upregulation of pre-mRNA expression levels is shown with color according to the Log2 FC of Sfpq-KD/Cont.
(E) Genomic view for Samd4, which was shown to be an SFPQ target gene in a previous study. Smoothed Ribo(−) RNA-seq profiles of Sfpq-KD/si-Cont (orange), Sfpq-KD Ribo(−) RNA-seq profiles (pink), and si-Cont (blue) positions of high-S (red) and low-S peaks (green) are shown.
(F) Genomic view of Ralgapa1, a size-matched gene pair partner of Samd4, among non-target genes.
See also Figures S1, S2, and S7.
To determine the SFPQ-binding characteristic to SFPQ target genes, we selected size-matched gene pairs from SFPQ target and nontarget genes, both of which had substantial SFPQ bindings. We identified 120 gene pairs that exhibited pre-mRNA length differences less than 10 kb and fulfilled the criteria for SFPQ binding (Figure 1B). We next compared the transcriptional elongation statuses in these groups using Ribo(−) RNA-seq and Pol II ChIP-seq. SFPQ target genes showed a gradual decrease in pre-mRNA level and Pol II distribution, particularly beyond 100 kb downstream of transcription start sites (TSSs), which was a symptom of transcriptional elongation defects (Figures 1C and S2A). Although SFPQ nontarget genes had a similar number of SFPQ-binding peak as SFPQ target genes, neither pre-mRNA expression nor Pol II density was decreased for these genes (Figures 1D and S2B), indicating that all but part of SFPQ binding was functionally essential for regulating long pre-mRNA expression.
To identify functionally essential SFPQ binding, we closely examined SFPQ binding in target genes. In typical SFPQ target genes, dense SFPQ binding was detected within extra-long introns. Sfpq-KD caused a gradual decrease of pre-mRNAs expression within these introns, as shown for the Samd4 gene, which possessed a distinctive long intron 2 (131 kb; Figure 1E). A gradual decrease of the pre-mRNA level from the 5′–3′ region in Sfpq-KD was observed as the stepwise decline of the pre-mRNA ratio between Sfpq-KD and control conditions. In contrast, the non-SFPQ target gene Ralgapa1 had a pre-mRNA length similar to that of Samd4 but did not possess extra-long introns (Figure 1F). SFPQ binding on Ralgapa1 pre-mRNA was broadly detected on the entire pre-mRNA, although the density was much lower than that of the Samd4 gene. In addition, the levels of Ralgapa1 pre-mRNA did not gradually decrease, and its KD/Cont pre-mRNA ratio was approximately 1.0 over the entire length. These data suggested that dense SFPQ binding to long introns was functionally essential for long pre-mRNA expression, and regulatory targets of SFPQ showed gradual decrease of their pre-mRNA under Sfpq-KD. To detect target introns of SFPQ, changes in pre-mRNA levels were examined by calculating the FC-FC value as the difference in the KD/Cont pre-mRNA ratio (FC) between the 3′- and 5′-regions. The FC-FC values of SFPQ target introns showed negative values owing to the smaller FC values in 3′-regions than in the 5′-regions. Using this FC-FC value, we analyzed its correlation with intron length for all 422 introns ≥10 kb among the 120 SFPQ target genes. A weakly negative correlation was detected between these two factors (Figure S2C), indicating that intron length was an important feature of SFPQ targets. We further compared the intron lengths of SFPQ target and nontarget genes. The results showed that SFPQ target genes tended to have longer introns than nontarget genes; however, their distribution overlapped (Figure S2D). Together, these results indicated that SFPQ regulated longer introns as its targets and that other factors were expected to delineate target and nontarget genes.
Identification of Factors Defining SFPQ Target Specificity
Next, we attempted to identify the factors that defined the target specificity of SFPQ, particularly the rules that determined SFPQ binding and functionality in specific introns. We attempted to identify the features of SFPQ binding on SFPQ target introns. We selected introns from SFPQ target genes for which the FC-FC values were significantly negative (≤−1). These introns were designated as “severely downregulated introns.” For comparison, we selected introns from SFPQ target genes for which the length was ≥10 kb and the FC-FC value was between −0.25 and +0.25 as controls as “non-downregulated introns.” With these criteria, 31 and 172 introns were identified as severely downregulated and non-downregulated introns, respectively (Figure 2A). In these two intron groups, the following four potential factors were examined for segregation of target and nontarget introns: (1) intron length; (2) total number of SFPQ binding; (3) density of SFPQ binding using normalized SFPQ binding counts per 10 kb; and (4) position of SFPQ binding closest to the 5′ splice site (termed the primary binding position). Previous studies have demonstrated the significance of SFPQ binding in the 5′ intron region as the seed-binding event mediating the polymerization of SPFQ on mRNA via its coiled-coil domain. This primary binding further allows serial SFPQ binding from 5′ to 3′ on pre-mRNAs and facilitates transcriptional elongation (Huang et al., 2018; Lee et al., 2015; Takeuchi et al., 2018). Therefore, the position of 5′ SFPQ binding was examined. In these analyses, we considered the stringency of SFPQ binding, which indicates the specificity and exclusiveness of SPFQ binding at certain RNA loci compared with size-matched input controls (SMI) (Van Nostrand et al., 2016). Among all SFPQ binding peaks with p values <0.01, those with CLIP-tag/SMI-tag FC ≥ 2 were designated as “High-S peaks” (high-stringent binding peaks compared with SMI), and the remaining peaks were termed as “Low-S peaks” (low-stringent binding peaks compared with SMI), as defined in our previous study (Takeuchi et al., 2018). These peaks accounted for 27,732 High-S peaks and 208,828 Low-S peaks. Then, factors (1)–(4) were analyzed and compared between severely downregulated and non-downregulated intron considering High-S- and Low-S SFPQ-binding peaks, except factor (1) (seven factors were investigated in total). The seven factors described earlier were examined to determine whether these factors could delineate severely downregulated and non-downregulated introns by drawing cumulative curves (Figures 2B–2H). Six features clearly segregated the 2 groups: (1) intron length, (2) High-S peak count, (3) Low-S peak count, (4) High-S peak density, (5) Low-S peak density, and (6) primary High-S peak binding position. Next, we checked whether the combination of these six features could be used for identifying functional SFPQ binding. For this purpose, we selected the following two thresholds for each feature: (1) a threshold giving the maximum difference ratio between two intron groups (designated as “high-stringent criteria,” indicated with black arrows), and (2) a threshold including 90% of highly downregulated introns (termed as “low-stringent criteria,” indicated with gray arrows; Figures 2B–2H). The distribution of severely downregulated and non-downregulated introns separated by these criteria was summarized in Table 1. Combinations of the six features were tested using these two thresholds with 31 downregulated and 172 non-downregulated introns in SFPQ target genes as following. Total 203 introns were classified into three groups: (1) introns fulfilling high-stringent criteria for all six features (designated as “strong-target introns”); (2) introns not meeting at least one high-stringent criterion but fulfilling low-stringent criteria for all six features (termed as “weak-target introns”); and (3) the remaining introns (termed as “nontarget introns”; Tables S1 and S2). We checked whether these criteria could clearly identify downregulated introns by plotting the FC-FC value for each intron in these three groups (Figure 2I). Fourteen of 17 strong-target introns (82%) were included in the severely downregulated intron group (defined above and in Figure 2A), indicating that the high-stringent criteria successfully enriched downregulated introns under Sfpq-KD conditions. Moreover, 13 of 56 weak-target introns (23%) were included in the severely downregulated intron group. Enrichment of SFPQ target introns in the weak-target intron group was lower than that in the strong-target intron group, but some severely downregulated introns that did not meet the strong-target criteria were included. The nontarget introns contained only four severely downregulated introns (3%) out of 130, indicating that the low-stringent criteria efficiently segregated SFPQ nontarget introns (Table S1). We also assessed the relationships between SFPQ binding close to the 5′-end of the intron and total SFPQ binding within introns. The relative High-S binding peaks position close to 5′ introns and total SFPQ binding density within introns were examined for all 2,509 introns ≥30 kb in length within the expressed genes (Figure S3). The presence of upstream SFPQ binding, with relative position in the total intron length within 5% from its 5′ end, was significantly correlated with elevated SFPQ-binding density within entire introns, consistent with a model showing that primary SFPQ binding close to the 5′ end of the intron causes serial SFPQ binding on entire introns by polymerization (Huang et al., 2018, Lee et al., 2015, Takeuchi et al., 2018).
Figure 2.

Identification of Factors Defining SFPQ Target Specificity
(A) Histogram of FC-FC values of introns in downregulated genes and having lengths ≥20 kb. In total, 31 severely downregulated introns (FC-FC values ≤ −1, indicated with red) and 172 non-downregulated introns (−0.25 < FC-FC < +0.25, indicated with blue) were compared.
(B–H) Cumulative plots for intron length (B), number of high-S peaks (C), density of high-S peaks (counts per 10 kb) (D), relative position of primary high-S peaks (E), number of low-S peaks (F), density of low-S peaks (counts per 10 kb) (G), and relative position of low-S peaks (H). Black and gray arrows indicate the values for the high-stringent and low-stringent criteria, respectively.
(I) Boxplot of FC-FC values of introns fulfilling all of the high-stringent criteria, introns fulfilling all of the low-stringent criteria, and the remaining introns. Upper, middle, and lower lines indicate the first quartile, median, and third quartile, respectively. Whiskers show the furthest point within 1.5 interquartile ranges (IQRs) from the upper and lower quartiles.
(J) Graphic abstract summarizing the identified features for strong SFPQ target introns. Detailed criteria are shown in Table 1.
Table 1.
Definition of High- and Low-Stringent Criteria Used for Defining Strong- and Weak-Target Introns of SFPQ and the Number of Introns Passing Each Criterion
| High Stringent Criteria | # Of Intron Passed | Fraction of Severely Downregulated Intron | Fraction of Non-downregulated Intron | Difference of Fractions | Low Stringent Criteria | # Of Intron Passed | Fraction of Severely Downregulated Introns |
|---|---|---|---|---|---|---|---|
| Intron length ≥45kb | 25 | 0.807 | 0.291 | 0.516 | Intron length ≥30kb | 28 | 0.903 |
| Count of High-S peaks ≥20 | 18 | 0.581 | 0.076 | 0.505 | Count of High-S peaks ≥1 | 28 | 0.903 |
| Density of High-S peaks (counts/10kb) ≥ 0.5 | 26 | 0.839 | 0.326 | 0.513 | Density of High-S peaks (counts/10kb) ≥ 0.03 | 28 | 0.903 |
| Relative position of primary High-S peak ≤0.0128 | 15 | 0.484 | 0.052 | 0.432 | Relative position of primary High-S peak ≤0.6 | 28 | 0.903 |
| Count of Low-S peaks ≥25 | 25 | 0.807 | 0.204 | 0.603 | Count of Low-S peaks ≥10 | 29 | 0.936 |
| Density of Low-S peaks (counts/10kb) ≥ 5 | 24 | 0.774 | 0.302 | 0.472 | Density of Low-S peaks (counts/10kb) ≥ 1.5 | 29 | 0.936 |
Introns fulfilling all six high-stringent criteria were designated as “strong-target introns” and introns not classified into strong-target introns but fulfilling low-stringent criteria for all six features were termed as “weak-target introns.”
In summary, the identified features that defined SFPQ target introns by high-stringent criteria were as follows: long introns ≥45 kb, total High-S SFPQ binding peaks in introns ≥20 with a density ≥0.5 per 10 kb intron length, total Low-S binding peaks ≥25 with a density ≥5 per 10 kb intron length, and primary High-S SFPQ binding within 1.28% of the relative position in the total intron length. These identified criteria are schematically summarized in Figure 2J. A typical example of SFPQ target introns could be observed in the shorter gene Cbx7. The length of Cbx7 pre-mRNA was 55 kb, which was much shorter than 100 kb; however, this gene had a strong-target intron with a length of 47 kb (>45 kb). Loss of Sfpq caused a gradual decrease of Cbx7 pre-mRNA within this intronic region and subsequently decreased its mature mRNA expression (Figure S4). Thus, our multimodal analysis combining Ribo(−) RNA-seq and CLIP-seq data successfully identified the features of SFPQ target introns.
The Identified Features Could Segregate Significant SFPQ Binding to Regulate the Transcriptional Elongation of Long Genes
The features of functional SFPQ binding in target introns were identified using a limited number of target and nontarget genes by integrating CLIP-seq and transcriptome data. Therefore, we next tested whether the identified features could be expanded to detect target introns from entire transcripts using CLIP-seq data. According to the identified seven features with high and low stringencies, we sorted all introns (n = 2,509) in all expressed genes into strong-target, weak-target, and nontarget intron groups. When the FC-FC value was examined, strong-target introns showed significantly lower FC-FC values than weak-target or nontarget introns, indicating successful separation of SFPQ target introns from all expressed genes (Figure 3A). Then, we further examined whether identification of the SFPQ target intron could be expanded to predict SFPQ target genes. First, we classified the expressed long genes (≥100 kb) according to the presence of long introns (≥30 kb) as a minimum criterion for SFPQ target introns. Among 1,196 expressed long genes (≥100 kb), 1,018 genes (85.1%) possessed long introns (≥30 kb), indicating a positive correlation between total gene length and long introns. Genes with more than one long intron were further classified into three groups according to the presence of strong-target, weak-target, and nontarget introns (Figures 3B and Table S3). We examined changes in the expression levels of these gene groups under Sfpq-KD conditions. As expected, genes possessing strong-target introns showed the lowest mean FCs under Sfpq-KD conditions (Figure 3C, red plot), and the mean FCs of genes possessing weak-target and nontarget introns increased stepwise (Figure 3C, orange and gray plots, respectively). In addition, the mean FC of long genes without long introns was 0.13 (linear value: 1.09), indicating that these genes were not downregulated under Sfpq-KD conditions (Figure 3C, green plot). Taken together, these results indicated that multilateral bioinformatics analyses could successfully identify SFPQ target genes that were defined by SFPQ target introns.
Figure 3.

Identified Features Segregated Functional SFPQ Binding to Regulate Transcriptional Elongation of Long Genes
(A) Violin plots for FC-FC values of all expressed genes ≥30 kb, which were classified according to the identified SFPQ target intron criteria. Numbers in brackets shows intron counts in long genes (≥100 kb).
(B) Flowchart for long gene classification results by intron length and SFPQ target intron criteria. Genes for which pre-mRNA lengths ≥100 kb were classified into four groups: genes with strong-target introns (Genes w/Strong-Tgt), genes with weak-target introns (Genes w/Weak-Tgt), genes with non-target introns (Genes w/Non-Tgt), and genes without long introns (Genes w/o Long-Intron).
(C) Violin plots for fold changes (si-Sfpq/si-Cont) of classified long genes.
See also Figures S1 and S4 and Table S3.
Sequence Feature Analysis of SFPQ-Binding Sites Suggested an Association between SFPQ Binding and Upstream Exon Recognition
Next, we attempted to identify the molecular mechanisms through which functional SFPQ binding formed to regulate its specific target introns. Our results indicated that SFPQ binding close to the 5′ intronic region was essential for subsequent serial binding of SFPQ toward the 3′ end (Figures 2E and S3). We expected that 5′ regions of SFPQ target introns, particularly primary binding sites close to the 5′ end, should have stronger and/or unique consensus SFPQ-binding sequences; therefore, we attempted to identify specific features of primary binding sites. SFPQ-binding sites in strong-target, weak-target, and nontarget introns were classified according to their relative binding portions in introns as follows. The first binding site located within the upstream region (5% of the total intron length from the 5′ intron end) was classified into the first (≤5%) group. The remaining sites were classified according to relative position within the introns, i.e., within 10% region (≤10%), within 10%–50% region (≤50%), and the remaining regions (≤100%). This classification is schematically outlined in Figure S5A.
Our previous study identified C-rich and AG-rich sequences as cis-elements of SFPQ (Takeuchi et al., 2018). Accordingly, we next examined the presence of these motifs. CU-rich sequences with a core UCC sequence had significantly higher occupancy exclusively in the first and ≤5% SFPQ binding sites of strong-target introns compared with remaining regions of strong-target introns and compared with the same regions in weak-target and nontarget introns (Figure 4A), whereas AG-rich sequences were not (Figure S5B). Next, we searched for another consensus sequence that was specific to the first position of strong- or weak-target introns. Then, we identified the C-rich sequence that was significantly enriched in the first and ≤10% SFPQ-binding sites of strong-target introns compared with the remaining regions of strong-target introns and the same regions of weak-target introns (Figure 4B). These results indicated that CU- or C-rich sequences contributed to the formation of SFPQ binding close to the 5′ intronic region. This and previous studies have demonstrated that SFPQ binds to target mRNAs using rather non-strict-binding sequences. Accordingly, we then attempted to identify additional factors that regulate the target specificities of SFPQ. Because previous studies have demonstrated the interactions between SFPQ and splicing factors (Patton et al., 1993; Snijders et al., 2015; Talukdar et al., 2011), we performed quantitative analyses focusing on sequences related to splice site recognition, i.e., intronic splicing enhancer/silencer sequences (ESEs, ISEs, ESSs, and ISSs). SFPQ target introns tended to possess significantly more ESEs, ESSs, and ISEs but fewer ISSs per 100 bases (Figures 4C–4F). Significant numbers of ESE, ESS, and ISE sequences adjacent to the 5′ splice sites in strong-target introns suggested that exon recognition of upstream target introns was regulated by several splicing factors and that target recognition of SFPQ close to 5′ splice sites was conducted via association. The mechanistic insight of SFPQ target recognition at 5′ splice sites and sequential binding on target introns is schematically summarized in Figure 4G.
Figure 4.

Sequence feature analysis of SFPQ-binding sites suggested an association between SFPQ binding and upstream exon recognition
(A and B) Bar graphs showing the fraction of SFPQ target introns possessing SFPQ binding motifs. The C-rich motif found in our previous study (A) and the CU-rich motif newly identified in primary SFPQ-binding sites among strong- or weak-target introns (B).
(C–F) Box plots showing the numbers of splicing enhancer and silencer sequences in upstream exons of SFPQ target introns. Counts of ESE (C), ESS (D), ISE (E), and ISS (F) were determined. ∗p < 0.05, ∗∗p < 0.01. The box plot format is similar to that in Figure 2I.
(G) Working model for SFPQ binding to its target intron.
See also Figures S1 and S5.
Prediction of SFPQ Regulatory Target Genes by Identifying Functional SFPQ Binding
Our previous study indicated that SFPQ target genes in mouse embryonic brains are highly enriched in gene ontology (GO) categories related to nervous system development, specifically axonal guidance, dendrite projections, and neuronal projections (Takeuchi et al., 2018). This previous result supported the idea that RBPs comprehensively regulate clusters of genes having specific functions and that these functional gene clusters are called regulons. In this study, using all expressed genes sorted according to the presence of strong-target, weak-target, and nontarget introns (Figure 3B and Table S3), we conducted GO analysis. As a result, GO categories related to neural development, including neuron projection morphogenesis, cell morphology involved in neuro differentiation, and axonogenesis, were identified as enriched terms in genes with strong- and weak-target introns (Figure 5 and Table S4). In our previous study, 176 genes were identified as SFPQ targets in differentiated Neuro2a cells (Figure 5, predetermined target genes in N2a), and GO categories related to neural development were not identified, presumably because brain-specific gene expression was much weaker in vitro than in vivo, which hindered SFPQ target gene identification. However, using the combined criteria, we detected only 85 genes harboring strong-target introns of SFPQ and 489 genes harboring weak-target introns, and these genes were highly enriched in GO categories related to neural development. These results indicated that SFPQ comprehensively regulated essential genes involved in neural development and that prediction of SFPQ target genes from the identified functional SFPQ-binding events detected unique regulons.
Figure 5.

Prediction of SFPQ Regulatory Target Genes via Identification of Functional SFPQ Binding in Neuro2a Cells
Heatmap showing q-values of enriched GO terms for SFPQ target long genes and predetermined SFPQ target genes in Neuro2a (N2a) cells and embryonic mouse brain tissues. Numbers in parentheses show the count of genes belonging to each gene group and the counts of genes showing overlap (OL) with predetermined SFPQ target genes in Neuro2a cells. See also Table S4.
SFPQ expression is spatially and temporally controlled in developing brains (Takeuchi et al., 2018). This observation implies that the presence or absence of SFPQ can modulate SFPQ target gene expression. Thus, we examined the correlations between SFPQ and predicted SFPQ target genes using public proteome data in human brains (Carlyle et al., 2017). First, we compared SFPQ protein expression in different brain regions, including the dorsolateral prefrontal cortex (DFC), primary visual cortex (V1C), hippocampus (HIP), amygdala (AMY), mediodorsal thalamic nucleus (MD), striatum (STR), and cerebellar cortex (CBC), in brain samples from patients aged 0–60 years. The CBC showed the highest expression of SFPQ, and an age-dependent decrease in SFPQ expression was observed (Figure 6A). Among different brain regions, the expression levels of 492 proteins were positively correlated with SFPQ protein levels (Pearson correlation coefficient ≥0.5, Table S5). We further analyzed whether these positively correlated genes possessed SFPQ target introns in mouse homologous genes. Genes possessing strong-target introns showed the highest enrichment in positively correlated genes, followed by genes with weak-target introns, although these enrichments were not statistically significant (Figure 6B). Genes with nontarget introns and genes without long introns did not show enrichment.
Figure 6.

Prediction of SFPQ Regulatory Target Genes in Human Brains
(A) Protein expression levels of SFPQ in the indicated brain regions at different ages (peptides per 30 million peptides [PP30M]). Abbreviations of postmortem brain regions: dorsolateral prefrontal cortex (DFC), primary visual cortex (V1C), hippocampus (HIP), amygdala (AMY), mediodorsal thalamic nucleus (MD), striatum (STR), and cerebellar cortex (CBC).
(B) Over-representation analysis between classified long genes and genes positively correlated with SFPQ (PCC ≥0.5). Long genes (≥100 kb) were classified into four groups according to the presence of SFPQ target introns, similar to that in Figure 3B.
(C) Violin plots for protein expression levels from long genes. Five postmortem brain samples with low SFPQ expression and another five samples with high SFPQ expression were analyzed.
(D) Scatterplot showing the correlation between SFPQ (x) and FTO (y) protein expression levels. Abbreviations: DFC (D); V1C (V); HIP (H); AMY (A); MD (M); STR (S); CBC (C).
To further identify relationships between SFPQ protein levels and global long gene expression, we selected brain regions in which SFPQ protein levels were relatively high (SFPQ-high) or low (SFPQ-low). In total, 7 samples had higher SFPQ expression (>25 PP30K), and 14 have lower SFPQ expression (7 PP30K). As expected, genes possessing strong-target introns showed elevated expression in SFPQ-high samples compared with that in SFPQ-low samples (Figure 6C). Protein expression from genes with weak-target introns was not different between these two groups, and genes with nontarget introns showed slightly decreased protein levels in SFPQ-high samples. Fto is a long gene with one strong-target intron (intron 8) and one weak-target intron (intron 1; Figure S6). SFPQ and FTO protein levels were highly positively correlated (PCC = 0.9; Figure 6D). These results indicated that the expression of genes possessing strong-target introns in the human brain was significantly correlated with SFPQ expression. Taken together, our multilateral bioinformatics analyses identified functional SFPQ binding and facilitated prediction of SFPQ target genes.
Discussion
By integrating CLIP-seq with transcriptome data and carrying out multilateral bioinformatics analyses, we successfully segregated functional SFPQ binding from total SFPQ deposition on a large proportion of expressed pre-mRNAs. We found that SFPQ binding close to 5′ splice sites of introns was a crucial factor distinguishing target and nontarget introns and could regulate mRNA expression of long introns (≥30 kb) (Figures 2 and 3). The presence of High-S SFPQ binding close to the 5′ intron within 1.2% of the total intron length provided clear segregation between severely downregulated introns and non-downregulated introns (Figure 2E) In addition, High-S SFPQ binding close to 5′ intron within ∼5% of the total intron length increased total SFPQ binding in the entire intron (Figure S3), indicating that SFPQ binding close to exon-intron junctions was critical for regulating downstream introns. These results were consistent with findings in a structural study of SFPQ showing that SFPQ deposition at high-affinity binding sites acted as the seed for multimerization of SFPQ via SFPQ-SFPQ binding on target nucleic acids using its coiled-coil domains (Lee et al., 2015). From these observations, we proposed a model in which High-S SPFQ binding peaks close to the 5′ intron end facilitated SFPQ recruitment toward downstream pre-mRNAs, which caused co-transcriptional SFPQ binding to entire long introns and serially activated transcriptional elongation (Takeuchi et al., 2018). In addition to High-S peaks, we also assessed the effects of Low-S SFPQ binding peaks near 5′ splice sites. However, these peaks did not segregate between severely downregulated and non-downregulated introns (Figure 2H). These results indicated that selective SFPQ binding in the 5′-end of introns detected as high-S peaks were significantly involved in the formation of functional binding of SFPQ.
SFPQ binding sites close to the 5′ splice sites were enriched with nonstrict CU- or C-rich sequences (Figures 4A, 4B, and S5B). We found that enrichment of ESEs, ESSs, and ISEs within 300 bp of 5′ splice sites in SFPQ target introns was compared with that in nontarget introns (Figures 4C–4F). These results indicated that functional binding of SFPQ and target recognition was facilitated by splicing factors through exon recognition (Figure 4G). To unravel the mechanism of target recognition by SFPQ, we analyzed the presence of consensus sequences of different RBPs in the SFPQ target introns and found that binding sites of hnRNP-H1/2/3, -F, and -K were enriched (data not shown). Because these hnRNPs have been reported to interact with SFPQ (Talukdar et al., 2011; Yamamoto et al., 2016), it is possible that these RBPs facilitate SFPQ recruitment to 5′ splice sites. For proteins that interact with SFPQ, it is necessary to consider its family members NONO (non-POU domain-containing octamer binding protein) and PSPC1 (paraspeckle component 1). SFPQ and these proteins belong to the Drosophila behavior/human splicing (DBHS) family (Knott et al., 2016), and NONO and PSPC1 are known to interact with SFPQ and each other to form heterodimers (Huang et al., 2018, 2020; Knott et al., 2016; Lee et al., 2015). Thus, it is also possible that DBHS family members contribute to the target specification and co-transcriptional binding of SFPQ. Further studies to identify RBPs that share target RNAs or binding sites could also improve our understanding of cooperative regulation of RBPs for target specification. Although our study successfully identified and extracted functional bindings of SFPQ among entire deposition, mechanism of the sequence and structure dependent-recognition of RBPs to target RNAs remained to be elucidated. Several studies provided useful computational methods. For example, CapR surveyed specific secondary structures of target RNAs based on CLIP-seq data (Fukunaga et al., 2014). A web-tool beRPB estimates target RNAs based on single or multiple cis-elements (Yu et al., 2018). Further studies using existing computational method would help our understanding about the molecular mechanism for target RNA recognition of extracted functional bindings of RBPs.
In a previous study, SFPQ target genes were identified by the combination of substantial SFPQ binding on pre-mRNAs and downregulation of genes under SFPQ-KO or KD conditions (FC < 1/3). In this study, we integrated CLIP-seq and Ribo(−) RNA-seq data, identified functional SFPQ binding from SFPQ target introns, and used criteria to determine SFPQ target genes (Figures 3A and 3C). Indeed, 45 of 85 strong-SFPQ target genes were newly identified based on the current criteria and were subjected to GO enrichment evaluation. For example, the GO term “neuron projection morphogenesis” contained 22 SFPQ strong-target genes, 15 of which, including Kif5c, Auts, and Kalrn, were newly identified (Figure 5 and Table S4). The downregulation of these genes in Sfpq-KD Neuro2A cells was moderate (their log2(FC) values ranged from 0.92 to 0.03), and this could explain the difference in target genes. Because SFPQ target genes possessing strong-target introns showed high enrichment in nervous system development in GO analysis (Figure 5), we concluded that RBPs regulated specific functional gene clusters and that the methods developed in this study could successfully capture SFPQ regulons. In proteome analysis in human brains, SFPQ protein expression and the protein expression levels of genes possessing SFPQ target introns were significantly correlated (Figure 6), demonstrating that functional SFPQ binding enabled prediction of SFPQ target genes. We identified the m6A eraser gene FTO as the gene with the highest PCC among SFPQ strong-target genes (Figure 6D). Moreover, because FTO has been reported to bind SFPQ and work together with SFPQ to eliminate m6A modification (Song et al., 2020), identification of SFPQ regulatory target genes may further enable identification of SFPQ interacting partners. Furthermore, SFPQ showed relatively high expression levels in spatiotemporally limited regions, such as the young cerebral cortex (Figures 6B and 6C), and SFPQ strong-target genes, i.e., CAMK1D, KALRN, and FMNL2, which are essential genes for neuron development, showed co-expression patterns with SFPQ, with high PCC values of ≥0.5 (Table S5). Thus, our results indicated that such a specific transcriptome would be formed via RBPs, including SFPQ, and that prediction of RBP target genes could greatly improve our understanding of gene regulatory networks, as demonstrated for nervous system development.
Past studies demonstrated SFPQ's multi-functions to regulate mRNA splicing, microRNA targeting via 3′ UTR, or DNA repair (Knott et al., 2016; Yarosh et al., 2015). Although we had assessed these functions in brains and differentiating neuronal cells, it was unlikely that loss of Sfpq directly caused alternative splicing changes, modulated microRNA (miRNA) targeting of mRNAs with long 3′ UTRs, or disrupted DNA repair (Takeuchi et al., 2018). Thus, our observation indicates that a major function of SFPQ in developing brains is to facilitate transcriptional elongation and loss of Sfpq caused downregulation of extra-long genes. As the supportive evidence, among genes having ≥1 High-S peak and ≥32 Low-S peaks, only one gene was upregulated in Sfpq-KD Neuro2A cells. The gene, Cdk19 (Cdk11 in old name), had 134 kb length with two long introns >40 kb but these long introns did not pass the criteria of SFPQ target introns (Figure S7).
In conclusion, using an integrated data science approach, we provided a solution for identifying the features of functionally essential binding of RBPs among entire deposition on expressed mRNAs. In addition, we showed that these features could be used for predicting regulatory target genes. This approach also provided insights into the molecular mechanisms through which target recognition of RBPs could be formed, i.e., not merely depending on consensus sequences but also through interactions with other RBPs during mRNA processing. Thus, we propose that the multilateral bioinformatics approach employed in this study could be applied as a standard analytical approach in RNA biology to identify functional RBP binding and determine the biological roles of these proteins that lead to understand complicated regulative nature of RBPs on RNA molecules and regulatory network of each RBPs; regulon.
Limitation of the Study
Elucidating precise molecular mechanisms of RBP's target recognition further required determination of the binding and working co-partners. For this purpose, it is highly anticipated to accumulate the information of functional bindings of other RBPs.
Resource Availability
Lead Contact
Further information and requests for resources should be addressed to the Lead Contact, Akihide Takeuchi (takeuchi.akihide.8r@kyoto-u.ac.jp)
Materials Availability
N/A.
Data and Code Availability
The raw data for CLIP-seq, RNA-seq, pol II ChIP-seq were deposited and available in the NCBI Gene Expression Omnibus (GEO) with accession number GSE60246. Computer programs used in this study can be available at GitHub; https://github.com/keiiida.
Methods
All methods can be found in the accompanying Transparent Methods supplemental file.
Acknowledgments
We thank Dr. Motoyasu Hosokawa for helpful comments and discussions.
This work was supported in part by the Japan Society for the Promotion of Science (JSPS) Grants-in-Aid for Scientific Research (KAKENHI) from the Ministry of Education, Culture, Sports, Science and Technology of Japan [MEXT, JSPS KAKENHI 19500269, 25500288, 21249013, 15H05721, 19K06907, 20K07310 to M.H., A.T., and K.I.]; Innovative Cell Biology by Innovating Technology [Cell Innovation to M.H. and A.T.]; a Core Research for Evolutional Science and Technology grant from the Japan Science and Technology Agency [to M.H.]; a grant from the Japan Agency for Medical Research and Development [to M.H.]; the Asian CORE Program of JSPS [to M.H.]; iCeMS Cross-Disciplinary Research Promotion Project of Kyoto University [to A.T.]; and the Fujiwara Memorial Foundation [to A.T.].
Author Contributions
K.I. conceived the project, developed the bioinformatics analytical method, and performed analysis. A.T. conceived the project and contributed to the bioinformatics analyses. All authors contributed to the writing of the final manuscript.
Declaration of Interests
The authors declare no competing interests.
Published: July 24, 2020
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.isci.2020.101325.
Supplemental Information
Location of long introns (≥10kb), the number of SFPQ-binding peaks, relative position of primary SFPQ-binding sites, and other related information are shown
All expressed long genes were classified using our criteria and mRNA length, presence of target introns, expression levels in poly-A RNA-seq, and FC between Control and Sfpq-KD conditions are shown
Detailed information of GO Enrichment Analysis which provided gene list included in identified GO terms and their classification according to target specificities of SFPQ are shown
Full list of proteins which PCC is ≥0.5 to SFPQ and their classification according to SFPQ's target specificities identified in mouse genes are shown
References
- Carlyle B.C., Kitchen R.R., Kanyo J.E., Voss E.Z., Pletikos M., Sousa A.M.M., Lam T.T., Gerstein M.B., Sestan N., Nairn A.C. A multiregional proteomic survey of the postnatal human brain. Nat. Neurosci. 2017;20:1787–1795. doi: 10.1038/s41593-017-0011-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castello A., Fischer B., Eichelbaum K., Horos R., Beckmann B.M., Strein C., Davey N.E., Humphreys D.T., Preiss T., Steinmetz L.M. Insights into RNA biology from an Atlas of Mammalian mRNA-binding proteins. Cell. 2012;149:1393–1406. doi: 10.1016/j.cell.2012.04.031. [DOI] [PubMed] [Google Scholar]
- Fu X.-D., Ares M. Context-dependent control of alternative splicing by RNA-binding proteins. Nat. Rev. Genet. 2014;15:689–701. doi: 10.1038/nrg3778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukunaga T., Ozaki H., Terai G., Asai K., Iwasaki W., Kiryu H. CapR: revealing structural specificities of RNA-binding protein target recognition using CLIP-seq data. Genome Biol. 2014;15:R16. doi: 10.1186/gb-2014-15-1-r16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerstberger S., Hafner M., Tuschl T. A census of human RNA-binding proteins. Nat. Rev. Genet. 2014;15:829–845. doi: 10.1038/nrg3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze M.W., Castello A., Schwarzl T., Preiss T. A brave new world of RNA-binding proteins. Nat. Rev. Mol. Cell Biol. 2018;19:327–341. doi: 10.1038/nrm.2017.130. [DOI] [PubMed] [Google Scholar]
- Hosokawa M., Takeuchi A., Tanihata J., Iida K., Takeda S., Hagiwara M. Loss of RNA-binding protein Sfpq causes long-gene transcriptopathy in skeletal muscle and severe muscle mass reduction with metabolic myopathy. iScience. 2019;13:229–242. doi: 10.1016/j.isci.2019.02.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Casas Garcia G.P., Perugini M.A., Fox A.H., Bond C.S., Lee M. Crystal structure of a SFPQ/PSPC1 heterodimer provides insights into preferential heterodimerization of human DBHS family proteins. J. Biol. Chem. 2018;293:6593–6602. doi: 10.1074/jbc.RA117.001451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang J., Ringuet M., Whitten A.E., Caria S., Lim Y.W., Badhan R., Anggono V., Lee M. Structural basis of the zinc-induced cytoplasmic aggregation of the RNA-binding protein SFPQ. Nucleic Acids Res. 2020;48:3356–3365. doi: 10.1093/nar/gkaa076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott G.J., Bond C.S., Fox A.H. The DBHS proteins SFPQ, NONO and PSPC1: a multipurpose molecular scaffold. Nucleic Acids Res. 2016;44:3989–4004. doi: 10.1093/nar/gkw271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee M., Sadowska A., Bekere I., Ho D., Gully B.S., Lu Y., Iyer K.S., Trewhella J., Fox A.H., Bond C.S. The structure of human SFPQ reveals a coiled-coil mediated polymer essential for functional aggregation in gene regulation. Nucleic Acids Res. 2015;43:3826–3840. doi: 10.1093/nar/gkv156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Licatalosi D.D., Mele A., Fak J.J., Ule J., Kayikci M., Chi S.W., Clark T.A., Schweitzer A.C., Blume J.E., Wang X. HITS-CLIP yields genome-wide insights into brain alternative RNA processing. Nature. 2008;456:464–469. doi: 10.1038/nature07488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand E.L., Pratt G.A., Shishkin A.A., Gelboin-Burkhart C., Fang M.Y., Sundararaman B., Blue S.M., Nguyen T.B., Surka C., Elkins K. Robust transcriptome-wide discovery of RNA-binding protein binding sites with enhanced CLIP (eCLIP) Nat. Methods. 2016;13:508–514. doi: 10.1038/nmeth.3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Nostrand E.L., Freese P., Pratt G.A., Wang X., Wei X., Xiao R., Blue S.M., Chen J.-Y., Cody N.A.L., Dominguez D. A large-scale binding and functional map of human RNA binding proteins. BioRxiv. 2018:179648. doi: 10.1038/s41586-020-2077-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patton J.G., Porro E.B., Galceran J., Tempst P., Nadal-Ginard B. Cloning and characterization of PSF, a novel pre-mRNA splicing factor. Genes Dev. 1993;7:393–406. doi: 10.1101/gad.7.3.393. [DOI] [PubMed] [Google Scholar]
- Russell J. 2017. Agile Data Science 2.0: Building Full-Stack Data Analytics Applications with Spark. [Google Scholar]
- Snijders A.P., Hautbergue G.M., Bloom A., Williamson J.C., Minshull T.C., Phillips H.L., Mihaylov S.R., Gjerde D.T., Hornby D.P., Wilson S.A. Arginine methylation and citrullination of splicing factor proline- and glutamine-rich (SFPQ/PSF) regulates its association with mRNA. RNA. 2015;21:347–359. doi: 10.1261/rna.045138.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H., Wang Y., Wang R., Zhang X., Liu Y., Jia G., Chen P.R. SFPQ is an FTO-binding protein that facilitates the demethylation substrate preference. Cell Chem. Biol. 2020;27:283–291.e6. doi: 10.1016/j.chembiol.2020.01.002. [DOI] [PubMed] [Google Scholar]
- Sundararaman B., Zhan L., Blue S.M., Stanton R., Elkins K., Olson S., Wei X., Van Nostrand E.L., Pratt G.A., Huelga S.C. Resources for the comprehensive discovery of functional RNA elements. Mol. Cell. 2016;61:903–913. doi: 10.1016/j.molcel.2016.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi A., Iida K., Tsubota T., Hosokawa M., Denawa M., Brown J.B., Ninomiya K., Ito M., Kimura H., Abe T. Loss of Sfpq causes long-gene transcriptopathy in the brain. Cell Rep. 2018;23:1326–1341. doi: 10.1016/j.celrep.2018.03.141. [DOI] [PubMed] [Google Scholar]
- Talukdar I., Sen S., Urbano R., Thompson J., Yates J.R., Webster N.J.G. hnRNP A1 and hnRNP F modulate the alternative splicing of exon 11 of the insulin receptor gene. PLoS One. 2011;6:e27869. doi: 10.1371/journal.pone.0027869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weyn-Vanhentenryck S.M., Mele A., Yan Q., Sun S., Farny N., Zhang Z., Xue C., Herre M., Silver P.A., Zhang M.Q. HITS-CLIP and integrative modeling define the Rbfox splicing-regulatory network linked to brain development and autism. Cell Rep. 2014;6:1139–1152. doi: 10.1016/j.celrep.2014.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto K., Furukawa M.T., Fukumura K., Kawamura A., Yamada T., Suzuki H., Hirose T., Sakamoto H., Inoue K. Control of the heat stress-induced alternative splicing of a subset of genes by hnRNP K. Genes Cells. 2016;21:1006–1014. doi: 10.1111/gtc.12400. [DOI] [PubMed] [Google Scholar]
- Yarosh C.A., Iacona J.R., Lutz C.S., Lynch K.W. PSF.: Nuclear busy-body or nuclear facilitator? Wiley Interdiscip. Rev. RNA. 2015;6:351–367. doi: 10.1002/wrna.1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H., Wang J., Sheng Q., Liu Q., Shyr Y. beRBP: binding estimation for human RNA-binding proteins. Nucleic Acids Res. 2018;47:e26. doi: 10.1093/nar/gky1294. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Location of long introns (≥10kb), the number of SFPQ-binding peaks, relative position of primary SFPQ-binding sites, and other related information are shown
All expressed long genes were classified using our criteria and mRNA length, presence of target introns, expression levels in poly-A RNA-seq, and FC between Control and Sfpq-KD conditions are shown
Detailed information of GO Enrichment Analysis which provided gene list included in identified GO terms and their classification according to target specificities of SFPQ are shown
Full list of proteins which PCC is ≥0.5 to SFPQ and their classification according to SFPQ's target specificities identified in mouse genes are shown
Data Availability Statement
The raw data for CLIP-seq, RNA-seq, pol II ChIP-seq were deposited and available in the NCBI Gene Expression Omnibus (GEO) with accession number GSE60246. Computer programs used in this study can be available at GitHub; https://github.com/keiiida.