

Abstract
Stroke causes significant mortality and morbidity. Currently, there are no treatments which can regenerate brain tissue lost to infarction. Neural progenitor cells (NPCs) are at the forefront of preclinical studies for regenerative stroke therapies. NPCs can differentiate into and replace neurons and promote endogenous recovery mechanisms such as angiogenesis via trophic factor production and release. The stroke core is hypothetically the ideal location for replacement of neural tissue since it is in situ and develops into a potential space where injections may be targeted with minimal compression of healthy peri-infarct tissue. However, the compromised perfusion and tissue degradation following ischemia create an inhospitable environment resistant to cellular therapy. Overcoming these limitations are critical to advancing cellular therapy. In this work, we tested the therapeutic potential of mouse induced pluripotent stem cell derived NPCs encapsulated in a bFGF binding chondroitin sulfate-A (CS-A) hydrogel transplanted into the infarct core in a mouse sensorimotor cortex mini-stroke model. We show that CS-A encapsulation significantly improves vascular remodeling, cortical blood flow, and sensorimotor behavioral outcomes after stroke. We found these improvements were negated by blocking bFGF, suggesting that the sustained trophic signaling endowed by the CS-A hydrogel combined with NPC transplantation can promote tissue repair.
Keywords: Stroke, Neural regeneration, Hydrogel, Neural progenitor cells
Graphical abstract:
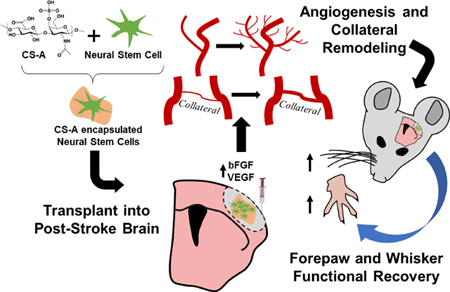
Cellular therapy is a promising avenue for the treatment of ischemic stroke and may be augmented with angiogenic biomaterials. Here, neural progenitor cells were encapsulated with a novel bFGF enriching chondroitin-4-sulfate A hydrogel and transplanted into the infarct core in mice. Treatment with encapsulated cells significantly enhanced angiogenesis, collateral vessel arteriogenesis, blood flow, and functional recovery after ischemic stroke.
Ischemic stroke is one of the leading causes of mortality and morbidity worldwide, and the fifth leading cause of death in the US[1]. Currently there are no therapies that promote recovery after stroke. Cellular therapies hold promise for improving functional recovery after ischemia via trophic support and cell replacement. Neural progenitor cells (NPCs) can differentiate into functional neurons to replace host circuitry lost to stroke. NPCs may also promote endogenous recovery mechanisms such as enhanced vascularization and host neuronal plasticity, immunosuppression, and recruitment of endogenous progenitor cells. For translational cellular therapy purposes, the two main sources of NPCs include embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs). Unlike ESCs, induced pluripotent stem cell derived neural progenitor cells can be generated autologously from the host somatic cells, bypassing potential ethical concerns surrounding the use of cells derived from embryos, and potentially reducing immunogenicity compared to allogeneic cell sources[2]. Our lab has previously used iPSC-derived NPCs for the treatment of ischemic stroke and traumatic brain injury in rodent models[3, 4]. Despite advances in stem cell technologies, the pathophysiology of ischemic stroke makes infarcted brain tissue especially resistant to cellular therapy. Importantly, blood flow to the stroke core region may not recover sufficiently to support newly formed tissue or transplanted cells, thereby limiting the therapeutic efficacy of cellular therapy. Previous studies have circumvented this issue by transplanting into the peri-infarct tissue where the microenvironment is less hostile, or by using methods such as hypoxic preconditioning to increase the resiliency of transplanted cells[4, 5]. However, directing injections to the peri-infarct region requires invasive placement in sensitive brain tissue, which can be damaged simply by delivery approach [6]. Other groups have transplanted on top of the neocortex, however, this approach may limit cellular replacement in situ[7]. Improving transplant retention and enhancing blood flow to the stroke core may be key to overcoming hurdles in cellular therapy. In this work we use a chondroitin-4-sulfate A (CS-A) hydrogel to create a microenvironment in the stroke core that is favorable for transplantation and regeneration. We demonstrate that CS-A encapsulated mouse IPSC-derived NPCs synergistically improve angiogenesis, vascular remodeling, local cerebral blood flow, and behavioral outcomes following ischemic stroke in mice. We found that the bFGF signaling potentiated by enrichment in the CS-A hydrogels mediated the benefits conferred to the NSC-treated stroke tissue.
Hydrogels are biocompatible carrier materials that can be used to promote survival, differentiation, trophic potency, and functional integration of transplanted cells[8, 9, 10–12]. Their ability to emulate the biophysical properties of soft neural tissue makes them ideal for engraftment into brain injuries[13, 14, 15]. Formulating hydrogels from extracellular matrix (ECM) components allows for reduced immunogenicity, preserves bio-inductive properties, and allows for degradation by native mechanisms[14, 16]. Many tissue engineering studies in the stroke field have centered on hyaluronan (HA), which when used as a carrier for cellular therapy, can improve cell survival after transplant, promote angiogenesis and anti-inflammatory signaling, and encourage neuronal differentiation of stem cells[6, 8, 12, 15]. Recently, a HA-based hydrogel was demonstrated to encourage vascular repair and neuronal migration to the stroke core in mice[17]. However, low molecular weight HA is pro-inflammatory, and transplanting HA into the degradatory stroke core could carry an unnecessary risk of exacerbating inflammation[15]. Other matrices have also been tested as scaffolds for cellular therapy in stroke, including poly lactic-co-glycolic acid (PLGA)[18], heparin sulfate[8], collagen[19], and others [12]. Commonly reported outcomes in these studies include histologically observed transplant cell retention and behavioral recovery after stroke, however many studies lack cellular or material controls, and the mechanisms underlying functional improvement remain elusive.
Released proteins such as growth factors play an integral role in regeneration, and ECM binding is a critical step of NPC mediated trophic support[11, 20, 21]. However, materials such as HA and PLGA lack the functional components necessary to enhance trophic factor retention and signaling. Other brain ECM glycosaminoglycans may provide structural elements and functional microdomains that allow for sustained and endogenously regulated trophic signaling, which may improve regenerative potential. Chondroitin sulfate (CS) is a multifunctional sulfated glycosaminoglycan implicated in regeneration and the response to brain injury. The sulfated elements of the CS disaccharide chains attached to native brain CS proteoglycans facilitate growth factor binding, retention, and presentation[22–24]. The various roles of CS can be attributed to their sulfation patterns and pathobiological context. CS-A is involved in endogenous neural stem cell differentiation and migration, neurite/axon growth, and angiogenesis[25, 26, 27]. Other CS moieties such as CS-E and CS-C play a role in neuroprotection as part of the glial scar which limits the spread of inflammation and necrotic debris following cerebral infarction. The homeostatic deposition of these glycosaminoglycans in the peri-infarct region may also assist in regeneration[28]. Regenerative factors such as basic fibroblast growth factor (bFGF) and brain derived neurotrophic factor (BDNF), and the immunomodulatory protein interleukin 10 (IL-10) have higher affinities for CS-A compared to HA and other materials[21, 22, 29]. The secreted mitogen bFGF in particular plays an important role in brain repair after injury by promoting proliferation, migration, and survival of endothelial cells[30]. Furthermore, bFGF also stimulates production of vascular endothelial growth factor (VEGF) which acts in conjunction with bFGF to promote angiogenesis[27, 31]. Arteriogenesis and pial collateral vascular remodeling are also carried out by bFGF signaling after cerebral ischemia[30, 32, 33].
Previous studies have demonstrated that CS-A can support neural stem cells in vitro and transplanted cells in vivo following traumatic brain injury[10, 21]. CS-A was found to reduce progressive cortical degradation induced by controlled cortical impact in a rat model [10]. In a photothrombotic rat stroke model, injection of a combination of chondroitin sulfate, HA, and heparin sulfate gel in addition with growth factors into the stroke region promoted migration of native neural progenitors and angiogenesis in the infarct region[34]. However, CS-A has yet to be tested as a carrier for cellular therapy in ischemic stroke. Given the importance of restoring blood flow to the stroke region for the efficacy of transplanted cells, and the ability of CS-A to facilitate trophic factor potentiation, in particular bFGF enrichment and presentation, we hypothesized that CS-A hydrogels would serve as effective carrier for facilitating NSC-mediated repair in the ischemic stroke core.
Our primary goal was to improve the therapeutic potential and brain repair capabilities of NPCs for the treatment of ischemic stroke. Focal ischemic stroke was induced in the right sensorimotor cortex by permanent ligation of distal branches of the middle cerebral artery accompanied with temporary bilateral common carotid occlusions as previously described[41]. CS-A hydrogels were synthesized and tailored to suit brain tissue mechanical properties as previously described[21, 35]. In particular, CS-A gels have pore sizes 10–50 μm and elasticity similar to neural tissue, exhibit minimal swelling, and can be degraded by brain-native enzymes[21]. Mouse induced pluripotent stem cells underwent the 4-/4+ retinoic acid differentiation protocol which produces neuronal lineage committed NPCs[35]. We first tested whether CS-A was conducive to neuronal differentiation of NPCs. NPCs grown in vitro in the CS-A hydrogel expressed the same levels of neuronal markers, including the mature neuron marker NeuN, the pre-synaptic marker SNAP25, and the axonal/dendrite markers MAP2 and TUJ1 as NPCs grown on traditional PDL-laminin coated polystyrene plates (S. Figure 1 A–B). The cells also appeared to have neuronal morphology following MAP2 immunostaining (S. Figure 1C).
We tested the capabilities of the CS-A to promote transplanted cell retention and neuronal replacement following stroke in C57Bl/6 mice. Intracranial transplantations (2 μL, 100k cells/ μL) were performed into the stroke core region 7d after stroke and animals were sacrificed 7 or 14d after transplantation (S. Figure 2). Mice were treated with NPCs, CS-A gel loaded with NPC media, the combined CS-A encapsulated NPCs, or received sham transplantations (Figure 1A). Mice were injected intraperitoneally daily with BrdU following transplantation to label newly formed cells. Significantly more transplanted cells were detectable 2 weeks after transplantation in the CS-A encapsulation group as detected by ex vivo fluorescent imaging and also immunohistochemical cell tracking (S. Figure 3 A–D). In line with the in vitro differentiation studies, CS-A did not alter the % of neuronal differentiation (S. Figure 1E), however, the total number of differentiated neurons was nearly 3 times compared to the non-encapsulated NPC treatment group (S. Figure 1F). This indicates the CS-A hydrogel improved transplant cell retention to facilitate the replacement of neurons in the stroke region.
Figure 1.

A. Schematic illustration of treatment groups following stroke in mice, including sham injection (no treatment), neural progenitor cell (NPC), chondroitin 4 sulfate A hydrogel (CS-A), and CS-A encapsulated NPC (CS-A + NPC) conditions. B. Top row: Fluorescent images of Glut1 and SMA staining showing vessels in the necrotic stroke core across treatments. White dotted line indicates stroke core region. Red dotted line indicates area visualized in part E. Bottom row: Fluorescent images of Glut1 and BrdU staining showing newly formed endothelial cells across treatments. C. Analysis of the vascularity (# Glut1+ vessels) within the stroke core region across treatments. D. Analysis of angiogenesis (Glut1/BrdU colabeled endothelium) across treatments. E. Representative fluorescent image of muscular artery formation (Glut1, SMA, BrdU) within the hydrogel in the CS-A + NPC treatment condition. Scale=50μm. n=8–12/group. *, **, and *** indicate p<0.05, p<0.005, and p<0.0005, respectively (1-way ANOVA, Tukey’s post-hoc).
To study the reparative and regenerative effects of transplantation, we first interrogated the effect of the CS-A encapsulated NPCs (CS-A + NPC) on angiogenesis. Neovascularization in the hypoxic stroke region is essential for the survival and success of both endogenously formed cells and transplanted NPCs and is also prognostic of recovery[30, 33]. Endothelial migration in vitro was significantly enhanced in CS-A gel pre-treated with NPC-conditioned medium compared to the control HA gel (S. Figure 4 A–B). This suggests CS-A can better facilitate endothelial NPC-mediated endothelial haptotaxis. To assess the microvascular and angiogenic effects of treatment, we counted the number of Glut1 (a brain endothelial cell marker[17, 36]) labelled vessels within the stroke core (Figure 1 B–C). We observed a significant increase in the number of microvessels in the stroke core region following treatment with CS-A + NPC. Angiogenesis was also measured by counting the number of Glut1+ and BrdU+ colabeled endothelium. Significantly greater numbers of newly formed endothelial cells in this region were detected, indicating that treatment with CS-A + NPC robustly promotes angiogenesis and improves vascular density (Figure 1D). We also detected the formation of new muscular arteries (SMA/Glut1) within the hydrogel in the CS-A + NPC treatment group (Figure 1E).
To further evaluate the effect of CS-A + NPC transplantation on vascular dynamics, we examined arteriogenesis and collateralization of pial arteries in genetically modified alpha smooth muscle actin-GFP (αSMA-GFP) mice. Pial collateral arteries are major regulators of blood flow to the ischemic region following brain infarction, and their recruitment is a primary mechanism of recovery and tissue support[30, 32, 33]. Following focal ischemia, pial arteries near the stroke region increase in diameter to encourage perfusion of the ischemic tissue. Both the extent of increase in diameter as well as the proportion of collateral channels that are recruited locally correlates with blood flow recovery to the infarct region. Since our stroke model surgery targets the middle cerebral artery (MCA), we measured the number and widths of MCA anastomoses from whole brain images taken of αSMA-GFP mice in all treatment groups (S. Figure 5). The average total number of pial collateral arteries were not significantly different between groups (S. Figure 5 A). A recruited collateral was defined as having a diameter greater than 25% than the average collateral in the non-injured (sham surgery) mouse. Mice treated with CS-A + NPC had both significantly greater widths and a greater percentage of collaterals with increased widths compared to other study arms (Figure 2 A–B, S. Figure 5 B). This indicated that the CS-A + NPC treatment enhanced pial collateral arteriogenesis.
Figure 2.
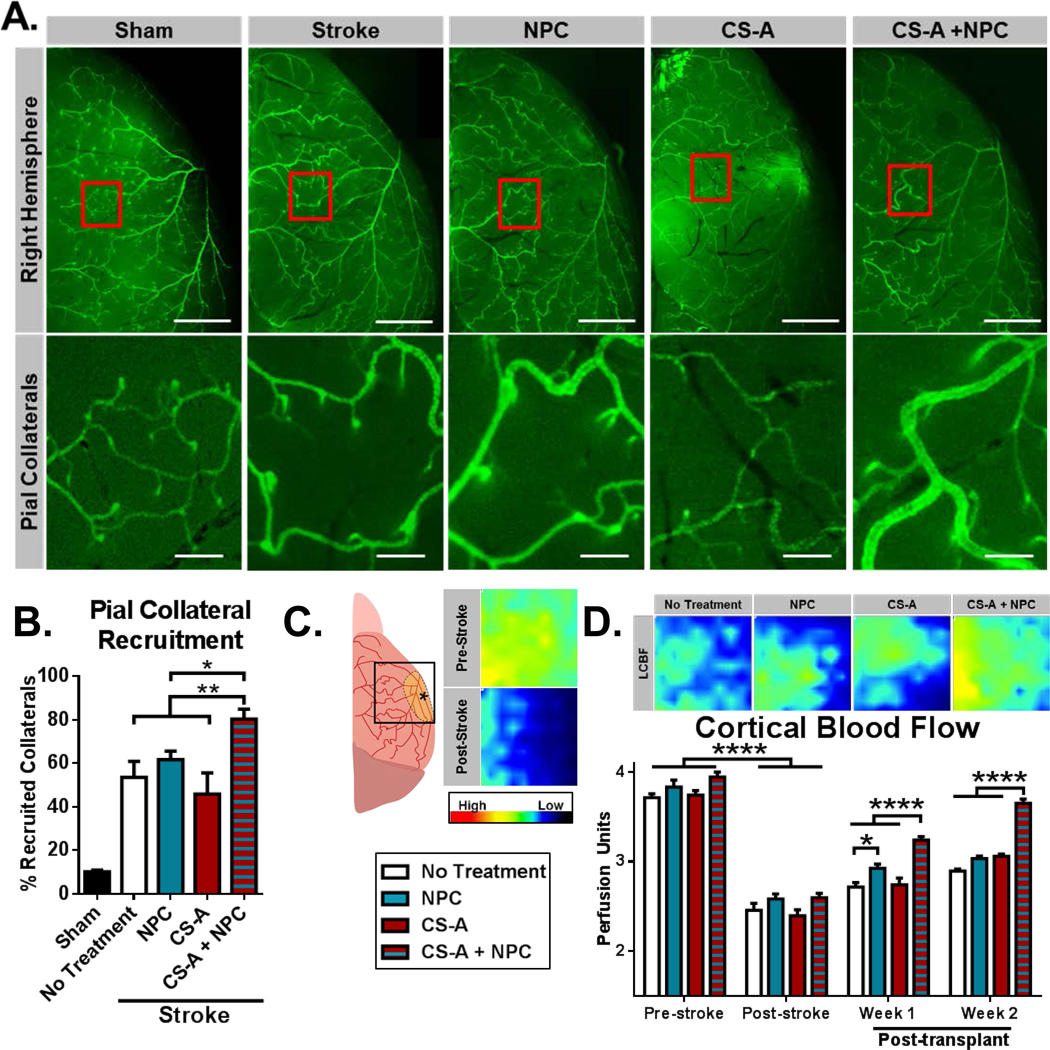
A. Top panel: Representative fluorescent images of right hemisphere of brains isolated from SMA-GFP mice 2 weeks after respective treatment (scale=1mm). Red boxes indicate area magnified for bottom panel. Bottom panel: Fluorescent images of pial collateral vessels. B. Analysis of pial collateral vessel recruitment (scale=0.2mm). n=5–6/group. * and ** indicate p<0.05 and p<0.005, respectively (1 way ANOVA, Holm-Sidak post-hoc vs CS-A + NPC; all treatment groups significantly greater than sham stroke). C. Schematic illustration of laser Doppler flowmetry test region (3×3mm) and representative pre-stroke and post-stroke (7 days) flowmetry heatmaps. D. Representative laser Doppler flowmetry heatmaps indicating local cerebral blood flow (LCBF) 2 weeks after respective treatments and analysis of longitudinal quantification of LCBF across treatments. n=8–12/group. * and **** indicates p<0.05 and p<0.0001, respectively (2-way ANOVA, Bonferroni’s correction).
Functional recovery is the ultimate goal of therapy. We examined the effects of treatment with CS-A + NPCs on local cerebral blood flow following ischemic stroke. Increasing blood flow to the ischemic region is critical for sustained tissue regeneration and is indicative of recovery. Local cerebral blood flow (LCBF) was measured using laser Doppler flowmetry as previous described. Blood flow was reduced from ~3.8 to ~2.5 perfusion units 1 week following ischemic stroke to the sensorimotor cortex (Figure 2C). Following treatment with CS-A + NPCs, LCBF was significantly enhanced 1 and 2 weeks after transplantation compared to all other treatment groups (Figure 2D). Our ischemic stroke model targets the sensorimotor cortex responsible for rodent whisker and forelimb sensation and motor function[37]. We evaluated whisker sensation using the corner test, which detects unilateral deficits that involve damage to rodent whisking pathways[38]. After walking into a corner within a monitored arena, mice tend to rear then turn towards the side which they whisked. Following ischemic stroke in the right sensorimotor cortex, mice turn right more frequently due to a lack of sensation of the left whiskers, resulting in an elevated right/left turn ratio[38]. Following transplantation of CS-A + NPC, the turn ratio was significantly restored 1 week after treatment (Figure 3A). Forepaw sensorimotor function was evaluated using the adhesive removal test, which is a detailed task that requires the mouse to sense and remove a sticker placed on the paw[39]. Mice treated with CS-A + NPCs showed a significantly improved time to sticker removal 1 week after transplantation (Figure 3B). Due to high rates of functional recovery in the corner and adhesive removal tests by 3 weeks after induction of mini-strokes to the sensorimotor cortex (Figure 3 A–B), we also tested forepaw motor function using the volitional isometric pull test, which more readily detects subtle defects in forepaw motor function and dexterity[40]. Two weeks after transplantation, mice treated with CS-A + NPC showed improved pull strength compared to the sham treatment group and superior hit rates compared to the cellular control group (Figure 3 C–D). Together, these blood flow and behavior results indicate functional recovery of the sensorimotor cortex and suggest that CS-A encapsulation can improve the therapeutic potential of cellular therapy for stroke.
Figure 3.

A. Analysis of corner test. B. Analysis of forepaw adhesive removal test. n=8–12/group for corner tests and sticky dot. C. Analysis of pull strength. D. Analysis of hit rate in the pull test. n=3–4/group for pull tests. *, ***, and **** indicate p<0.05, p<0.0005, and p<0.0001, respectively (2-way ANOVA, Bonferroni’s multiple comparison test).
We next sought to determine a potential mechanism for the enhancement of NPC-mediated regeneration via CS-A encapsulation. Given the robust augmentation of angiogenesis and arteriogenesis following the combined treatment, we hypothesized that bFGF may play a central role in mediating these effects since CS-A is known to regulate bFGF enrichment, presentation, and signaling in vitro and in the brain[24, 26, 27, 29, 33, 34, 41]. We confirmed that bFGF is enriched in the stroke area 1 week following transplantation of CS-A + NPC using Western blotting (Figure 4 A–B). Furthermore, VEGF was also increased in the combined treatment group. Consistent with increased bFGF and VEGF, we saw that levels of p-STAT3 and p-AKT, primary downstream signaling molecules involved in endothelial activation, were also increased[41]. To test whether bFGF was driving the angiogenesis, pial collateral arteriogenesis, and resulting cortical blood flow and sensorimotor behavior recovery in mice treated with CS-A + NPCs, we compared the combined treatment group with a neutralizing antibody towards bFGF (Figure 4C). We found that angiogenesis and pial collateral artery recruitment were significantly reduced 2 weeks following bFGF inhibition (Figure 4 D–F). Accordingly, LCBF was also reduced (Figure 4G). Mice treated with the CS-A encapsulated NPCs and the bFGF antibody showed a higher rightwards deviation in the corner test, indicating that behavioral recovery was negated by bFGF inhibition (Figure 4H). Together, these results suggest that CS-A acts via bFGF to potentiate NPC-mediated angiogenesis and vascular remodeling, blood flow recovery, and sensorimotor behavior recovery after stroke.
Figure 4.

A. Representative Western blotting for bFGF, p-STAT3, STAT3, p-AKT, AKT, VEGF, and Tubulin across treatment groups. B. Quantifications of Western blot intensity. n=4–5/group. *, **, and *** indicate p<0.05, p<0.005, and p<0.0005, respectively (1 way ANOVA, Tukey’s post-hoc). C. Schematic illustration of treatment groups, including CS-A encapsulated neural progenitor cells (CS-A + NPCs) and CS-A + NPCs co-administered with a neutralizing antibody towards bFGF (CS-A + NPCs + anti-bFGF). D. Representative images of Glut1/BrdU staining (top; scale=50μm), pial collateral arteries from SMA-GFP mice (middle; scale=0.2mm), and laser Doppler flowmetry heatmaps indicating local cerebral blood flow in the stroke region 2 weeks after treatment E. Analysis of angiogenesis (Glut1/BrdU). F. Analysis of pial collateral recruitment. G. Analysis of cortical blood flow. H) Analysis of corner test. n=4–5/group. *, **, and *** indicates p<0.05, **p<0.005, ***p<0.0005 respectively; two-tailed T-test.
In summary, encapsulation with CS-A hydrogel can improve neural stem cell transplantation for stroke. We transplanted iPSC-derived neural progenitor cells into the stroke core in mice 7 days after surgically induced infarction and compared CS-A versus non-encapsulated treatment groups. We found that CS-A encapsulation of NPCs significantly improved angiogenesis and vascular density in the ischemic tissue. Pial collateral vessels in the mice treated CS-A + NPCs had a greater average diameter and were recruited to a greater extent compared to other treatments. These effects led to functional recovery of cortical blood flow and sensorimotor behavior. Furthermore, we showed that many of these outcomes were dependent on bFGF, an important mediator of angiogenesis and arteriogenesis that is regulated and augmented by CS-A. We showed that CS-A can extend the trophic potential of NPCs for the treatment of ischemic stroke. This strategy of combining sustained trophic potentiation with cellular therapy may be a viable solution to overcoming the limitations of both approaches for regenerative medicine.
Supplementary Material
Acknowledgements
We greatly appreciate Chao Qu’s assistance in the work presented above.
LW acknowledges NIH R01 funding (R01NS085568, R01NS091585), SPY acknowledges VA National Merit Grants RX000666 and RX001473, and LK acknowledges NIH R01 funding (R01NS099596).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Conflict of Interest
The authors declare no conflicts of interest.
Citations
- [1].Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de Ferranti S, Després J-P, Fullerton HJ, Circulation 2016, 133, 447. [DOI] [PubMed] [Google Scholar]
- [2].Itakura G, Ozaki M, Nagoshi N, Kawabata S, Nishiyama Y, Sugai K, Iida T, Kashiwagi R, Ookubo T, Yastake K, Matsubayashi K, Kohyama J, Iwanami A, Matsumoto M, Nakamura M, Okano H, Sci Rep 2017, 7, 12996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Chau MJ, Deveau TC, Song M, Gu X, Chen D, Wei L, Stem Cells 2014, 32, 3075; [DOI] [PubMed] [Google Scholar]; Wei L, Wei ZZ, Jiang MQ, Mohamad O, Yu SP, Prog Neurobiol 2017, 157, 49; [DOI] [PMC free article] [PubMed] [Google Scholar]; Wei ZZ, Lee JH, Zhang Y, Zhu YB, Deveau TC, Gu X, Winter MM, Li J, Wei L, Yu SP, Cell Transplant 2016, 25, 797. [DOI] [PubMed] [Google Scholar]
- [4].Chau M, Deveau TC, Song M, Wei ZZ, Gu X, Yu SP, Wei L, Oncotarget 2017, 8, 97537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Francis KR, Wei L, Cell Death Dis 2010, 1, e22; [DOI] [PMC free article] [PubMed] [Google Scholar]; Wei N, Yu SP, Gu X, Taylor TM, Song D, Liu XF, Wei L, Cell Transplant 2013, 22, 977; [DOI] [PubMed] [Google Scholar]; Sun J, Wei ZZ, Gu X, Zhang JY, Zhang Y, Li J, Wei L, Exp Neurol 2015, 272, 78. [DOI] [PubMed] [Google Scholar]
- [6].Moshayedi P, Nih LR, Llorente IL, Berg AR, Cinkornpumin J, Lowry WE, Segura T, Carmichael ST, Biomaterials 2016, 105, 145; [DOI] [PMC free article] [PubMed] [Google Scholar]; Nih LR, Sideris E, Carmichael ST, Segura T, Adv Mater 2017, 29; [DOI] [PMC free article] [PubMed] [Google Scholar]; Cook DJ, Nguyen C, Chun HN, I LL, Chiu AS, Machnicki M, Zarembinski TI, Carmichael ST, J Cereb Blood Flow Metab 2017, 37, 1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Osanai T, Kuroda S, Yasuda H, Chiba Y, Maruichi K, Hokari M, Sugiyama T, Shichinohe H, Iwasaki Y, Neurosurgery 2010, 66, 1140. [DOI] [PubMed] [Google Scholar]
- [8].Zhong J, Chan A, Morad L, Kornblum HI, Fan G, Carmichael ST, Neurorehabil Neural Repair 2010, 24, 636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Jendelova P, Kubinova S, Sandvig I, Erceg S, Sandvig A, Sykova E, Expert Opin Biol Ther 2016, 16, 43; [DOI] [PubMed] [Google Scholar]; Lim TC, Spector M, Transl Stroke Res 2017, 8, 57. [DOI] [PubMed] [Google Scholar]
- [10].Betancur MI, Mason HD, Alvarado-Velez M, Holmes PV, Bellamkonda RV, Karumbaiah L, ACS Biomater Sci Eng 2017, 3, 420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nih LR, Carmichael ST, Segura T, Curr Opin Biotechnol 2016, 40, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Cohen LK, Jensen MB, Arch Neurosci 2015, 2, e25364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Banerjee A, Arha M, Choudhary S, Ashton RS, Bhatia SR, Schaffer DV, Kane RS, Biomaterials 2009, 30, 4695. [DOI] [PMC free article] [PubMed] [Google Scholar]; Seidlits SK, Khaing ZZ, Petersen RR, Nickels JD, Vanscoy JE, Shear JB, Schmidt CE, Biomaterials 2010, 31, 3930. [DOI] [PubMed] [Google Scholar]
- [14].Saldin LT, Cramer MC, Velankar SS, White LJ, Badylak SF, Acta Biomater 2017, 49, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Moshayedi P, Carmichael ST, Biomatter 2013, 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ghuman H, Massensini AR, Donnelly J, Kim SM, Medberry CJ, Badylak SF, Modo M, Biomaterials 2016, 91, 166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Nih LR, Gojgini S, Carmichael ST, Segura T, Nat Mater 2018, 17, 642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ju R, Wen Y, Gou R, Wang Y, Xu Q, Cell Transplant 2014, 23 Suppl 1, S83; [DOI] [PubMed] [Google Scholar]; Bible E, Qutachi O, Chau DY, Alexander MR, Shakesheff KM, Modo M, Biomaterials 2012, 33, 7435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Yu H, Cao B, Feng M, Zhou Q, Sun X, Wu S, Jin S, Liu H, Lianhong J, Anat Rec (Hoboken) 2010, 293, 911. [DOI] [PubMed] [Google Scholar]
- [20].Bliss T, Guzman R, Daadi M, Steinberg GK, Stroke 2007, 38, 817; [DOI] [PubMed] [Google Scholar]; Machalinski B, Expert Rev Neurother 2014, 14, 959; [DOI] [PubMed] [Google Scholar]; Macri L, Silverstein D, Clark RA, Adv Drug Deliv Rev 2007, 59, 1366. [DOI] [PubMed] [Google Scholar]
- [21].Karumbaiah L, Enam SF, Brown AC, Saxena T, Betancur MI, Barker TH, Bellamkonda RV, Bioconjug Chem 2015, 26, 2336. [DOI] [PubMed] [Google Scholar]
- [22].Karumbaiah L, Saxena T, Betancur M, Bellamkonda RV, Curr Med Chem 2014, 21, 4257. [DOI] [PubMed] [Google Scholar]
- [23].Sirko S, von Holst A, Wizenmann A, Gotz M, Faissner A, Development 2007, 134, 2727. [DOI] [PubMed] [Google Scholar]
- [24].Sirko S, von Holst A, Weber A, Wizenmann A, Theocharidis U, Gotz M, Faissner A, Stem Cells 2010, 28, 775. [DOI] [PubMed] [Google Scholar]
- [25].Purushothaman A, Sugahara K, Faissner A, J Biol Chem 2012, 287, 2935; [DOI] [PMC free article] [PubMed] [Google Scholar]; Nishimura K, Ishii M, Kuraoka M, Kamimura K, Maeda N, Neuroscience 2010, 169, 1535; [DOI] [PubMed] [Google Scholar]; Maeda N, Cent Nerv Syst Agents Med Chem 2010, 10, 22. [DOI] [PubMed] [Google Scholar]; Ryan E, Shen D, Wang X, FEBS J 2016, 283, 1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Rauvala H, Paveliev M, Kuja-Panula J, Kulesskaya N, Neural Regen Res 2017, 12, 687; [DOI] [PMC free article] [PubMed] [Google Scholar]; Kwok JC, Warren P, Fawcett JW, Int J Biochem Cell Biol 2012, 44, 582. [DOI] [PubMed] [Google Scholar]
- [27].Le Jan S, Hayashi M, Kasza Z, Eriksson I, Bishop JR, Weibrecht I, Heldin J, Holmborn K, Jakobsson L, Soderberg O, Spillmann D, Esko JD, Claesson-Welsh L, Kjellen L, Kreuger J, Arterioscler Thromb Vasc Biol 2012, 32, 1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Liu Z, Chopp M, Prog Neurobiol 2016, 144, 103; [DOI] [PMC free article] [PubMed] [Google Scholar]; Cekanaviciute E, Buckwalter MS, Neurotherapeutics 2016, 13, 685; [DOI] [PMC free article] [PubMed] [Google Scholar]; Sofroniew MV, Nat Rev Neurosci 2015, 16, 249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Smith SM, West LA, Govindraj P, Zhang X, Ornitz DM, Hassell JR, Matrix Biol 2007, 26, 175. [DOI] [PubMed] [Google Scholar]
- [30].Lapi D, Colantuoni A, J Vasc Res 2015, 52, 22. [DOI] [PubMed] [Google Scholar]
- [31].Salehi A, Zhang JH, Obenaus A, J Cereb Blood Flow Metab 2017, 37, 2320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Nishijima Y, Akamatsu Y, Weinstein PR, Liu J, Brain Res 2015, 1623, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Liu J, Wang Y, Akamatsu Y, Lee CC, Stetler RA, Lawton MT, Yang GY, Prog Neurobiol 2014, 115, 138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Jian WH, Wang HC, Kuan CH, Chen MH, Wu HC, Sun JS, Wang TW, Biomaterials 2018, 174, 17. [DOI] [PubMed] [Google Scholar]
- [35].Bain G, Ray WJ, Yao M, Gottlieb DI, Biochem Biophys Res Commun 1996, 223, 691. [DOI] [PubMed] [Google Scholar]
- [36].Cornford EM, Hyman S, Swartz BE, J Cereb Blood Flow Metab 1994, 14, 106; [DOI] [PubMed] [Google Scholar]; Wei L, Fraser JL, Lu ZY, Hu X, Yu SP, Neurobiol Dis 2012, 46, 635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Wei L, Rovainen CM, Woolsey TA, Stroke 1995, 26, 1459. [DOI] [PubMed] [Google Scholar]
- [38].Zhang L, Schallert T, Zhang ZG, Jiang Q, Arniego P, Li Q, Lu M, Chopp M, J Neurosci Methods 2002, 117, 207. [DOI] [PubMed] [Google Scholar]
- [39].Bouet V, Boulouard M, Toutain J, Divoux D, Bernaudin M, Schumann-Bard P, Freret T, Nat Protoc 2009, 4, 1560. [DOI] [PubMed] [Google Scholar]
- [40].Becker AM, Meyers E, Sloan A, Rennaker R, Kilgard M, Goldberg MP, J Neurosci Methods 2016, 258, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chen SH, Murphy DA, Lassoued W, Thurston G, Feldman MD, Lee WM, Cancer Biol Ther 2008, 7, 1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.