

Abstract

Phenotypic whole-cell screening against Mycobacterium tuberculosis (Mtb) in glycerol–alanine–salts supplemented with Tween 80 and iron (GASTE-Fe) media led to the identification of a 2-aminoquinazolinone hit compound, sulfone 1 which was optimized for solubility by replacing the sulfone moiety with a sulfoxide 2. The synthesis and structure–activity relationship (SAR) studies identified several compounds with potent antimycobacterial activity, which were metabolically stable and noncytotoxic. Compound 2 displayed favorable in vitro properties and was therefore selected for in vivo pharmacokinetic (PK) studies where it was found to be extensively metabolized to the sulfone 1. Both derivatives exhibited promising PK parameters; however, when 2 was evaluated for in vivo efficacy in an acute TB infection mouse model, it was found to be inactive. In order to understand the in vitro and in vivo discrepancy, compound 2 was subsequently retested in vitro using different Mtb strains cultured in different media. This revealed that activity was only observed in media containing glycerol and led to the hypothesis that glycerol was not used as a primary carbon source by Mtb in the mouse lungs, as has previously been observed. Support for this hypothesis was provided by spontaneous-resistant mutant generation and whole genome sequencing studies, which revealed mutations mapping to glycerol metabolizing genes indicating that the 2-aminoquinazolinones kill Mtb in vitro via a glycerol-dependent mechanism of action.
Keywords: tuberculosis, Mycobacterium tuberculosis, drug discovery, 2-aminoquinazolinones
Tuberculosis (TB) is an infectious disease, caused by the bacillus Mycobacterium tuberculosis (Mtb), and is the leading cause of death worldwide from a single infectious agent. According to the World Health Organization (WHO), approximately 10 million people acquired TB in 2018 and 1.2 million deaths occurred globally.1 Treatments for drug-susceptible TB have been available for 60 years, with the current regimen comprising an initial two month intensive phase with four frontline drugs (isoniazid, rifampicin, ethambutol, and pyrazinamide), followed by a four month continuation phase using isoniazid and rifampicin. However, resistant strains have emerged over the years possibly as a result of poor patient compliance due to the extensive drug regimen, prolonged treatment time, socioeconomic factors, toxic effects, and the tendency to be asymptomatic before safe completion of the prescribed course.2,3
In the past 15 years, several new classes of anti-TB drugs have emerged in the developmental pipeline including three new agents. Bedaquiline and delamanid received accelerated regulatory approval in 2012 and 2014, respectively, and are being progressed into phase III clinical trials for the treatment of multidrug resistant TB (MDR-TB).2−4 Most recently, pretomanid was approved as part of a six month, three-drug regimen consisting of bedaquiline, pretonamid, and linezolid (BPaL) for the treatment of people with extensively drug resistant TB (XDR-TB).5 Although there are a number of new chemical entities at various phases of clinical development,6 the high attrition rate requires a continuous feed of new chemical matter into the drug discovery pipeline. The development of new TB treatments involves numerous challenges as the TB research community needs to focus on many aspects, e.g., shorter and simpler regimen, less side effects, effectiveness against drug resistant TB, reduced drug–drug interactions for human immunodeficiency virus (HIV)-coinfected patients, and affordable cost for patients in developing countries where the majority of TB cases exist.7,8
2-Aminoquinazolin-4(3H)-ones are derived from the quinazolinone class of compounds, which have been extensively explored in a wide range of therapeutic areas, whereas literature references on 2-aminoquinazolinones are relatively scarce. Secondary and tertiary 2-amino analogues have been found to have pharmacological relevance, with examples of antifungal,9 anti-inflammatory, anticancer,10,11 antibacterial,12,13 antihypertensive,14 antidepressant,15 and antimycobacterial activity.16,17 On the other hand, primary 2-aminoquinazolinone derivatives have not yet been commercially developed but have shown a variety of biological activities as antimalarial,18 antiviral,19,20 and anticancer21 agents. Although there is no known antimycobacterial activity, they appear as a valuable novel scaffold to explore as a potential anti-TB chemotype.
On the basis of structural similarity shared with a previously discovered antimalarial 2-aminopyridine series,22 2-aminoquinazolinones were synthesized for evaluation against the human malaria parasite Plasmodium falciparum. Subsequent phenotypic whole-cell cross-screening against Mtb identified the 2-aminoquinazolinone 1 as an attractive hit compound to follow up on despite its low solubility. During preliminary studies toward improving the solubility of 1, the sulfone substituent was converted to the sulfoxide 2, which displayed improved aqueous solubility while maintaining potency (Figure 1). As a result, medicinal chemistry efforts were subsequently pursued in order to explore the antimycobacterial potential of this 2-aminoquinazolinone series.
Figure 1.

Identification of 2-aminoquinazolinones with in vitro antimycobacterial activity.
Herein, we present the synthesis, structure–activity relationship, in vitro absorption, distribution, metabolism, excretion, and toxicity (ADMET) properties, and in vivo pharmacokinetic (PK) properties of this series. In vivo efficacy and subsequent mechanistic studies are also presented.
Results and Discussion
Chemistry
Structural modifications were explored around the aminoquinazolinone core with the aim of improving potency and aqueous solubility at pH 7.4. Four main points of diversity were established at positions 2, 3, 6, and 7 of the scaffold as indicated in Figure 2. R1 and R2 groups were investigated to identify appropriately substituted ring structures with various electronic, steric, and hydrophobicity properties. The addition of heteroatoms or water solubilizing groups was envisaged to reduce lipophilicity and, therefore, improve aqueous solubility. Additionally, the strategy of increasing sp3 character through saturation of aromatic rings was employed to disrupt the crystal packing of molecules. The position of the substituent on the left-hand side (LHS) from the 6- to the 7-position of the molecule was also explored to determine which of the LHS positions was optimal for activity. Modifications were also made to the 2-position to understand whether or not the 2-amino group was essential for potency.
Figure 2.
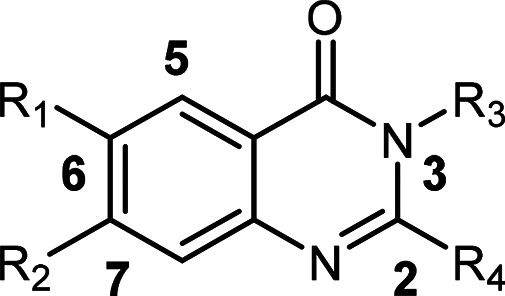
General structure of the quinazolinone core indicating modification sites.
The synthesis of compounds 1, 2, and 7–36 was achieved following a relatively straightforward five-step synthetic route adapted from Leivers et al., starting with the commercially available 2-aminoiodobenzoic acid (Scheme 1).19 Briefly, a cyclization reaction with the appropriately substituted phenyl isothiocyanate afforded intermediate 3. Chlorination of the thioxo intermediate to 4 was obtained using phosphorus oxychloride (POCl3) in the presence of phosphorus pentachloride (PCl5). The 2-amino group was introduced via a 4-methoxybenzylamine intermediate (5) followed by deprotection of the PMB group to afford 6. Finally, a Suzuki cross-coupling reaction with the appropriate boronic acids yielded the desired compounds 1, 2, and 7–36.
Scheme 1. General Synthetic Route for the Preparation of Compounds 1, 2, and 7−36.

Reagents and conditions: (i) dioxane, Et3N, reflux (110 °C), 4 h; (ii) POCl3, PCl5, N2, 110 °C, 14 h; (iii) DMF, 4-methoxybenzylamine, DIPEA, 80 °C, 4 h; (iv) TFA, reflux, 48 h or MW (110 °C) 20 min; (v) dioxane/water, R1/R2B(OH)2, PdCl2(PPh3)2, K2CO3, 80 °C, 1–5 h.
The procedure to obtain the analogue without the 2-amino substituent was adapted from Güngör et al.23 (Scheme 2). This approach involved refluxing 2-amino-5-iodobenzoic acid with 4-(trifluoromethyl)aniline in the presence of triethoxymethane to afford the iodo intermediate 37. A subsequent Suzuki-coupling reaction with (3-(methylsulfonyl)phenyl)boronic acid yielded the desired derivative devoid of the 2-amino group 38.
Scheme 2. Synthesis of Quinazolinone Analogue.

Reagents and conditions: (i) triethoxymethane, dioxane, 100 °C, 16 h; (ii) dioxane, R1B(OH)2, PdCl2(ddpf)2-DCM, Cs2CO3, 90 °C, 5 h.23
Modifications to the 2-amino substituent were achieved using a similar synthetic route to Scheme 1, whereby the appropriate amine was installed by substituting the chloro group of intermediate 4a under basic conditions. Thereafter, the final target compounds 40–43 were obtained using the Suzuki-coupling conditions described previously (Scheme 3).
Scheme 3. Synthetic Route Used to Access Core Modifications.

Reagents and conditions: (i) relevant amine, DMF/THF, base, 80 °C, 3 h; (ii) dioxane/water, R1B(OH)2, PdCl2(PPh3)2, K2CO3, 80 °C, 2 h.
Lastly, compounds 47–58 were prepared using a modified synthetic route established by Lecoutey et al. to introduce structural diversity at the R3 position (Scheme 4).24 Initially, methyl 2-amino-5-bromobenzoate was reacted with ethoxycarbonyl isothiocyanate to form the isolated thioureido intermediate 44. Thereafter, cyclization and simultaneous amide coupling with the appropriate amine using the coupling agent 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI) afforded intermediate 45, which was deprotected to give 46. Finally, the methylsulfinylphenyl moiety was installed via a Suzuki-coupling reaction to yield compounds 47–58.
Scheme 4. General Route for the Synthesis of LHS Modifications.

Reagents and conditions: (i) acetonitrile, 25 °C, 10 min; (ii) DCM/DMF, EDCI, R3NH2, 25 °C, 18 h; (iii) TFA, microwave, 110 °C, 20 min; (iv) dioxane/water (4:1), R1B(OH)2, PdCl2(PPh3)2, K2CO3, 80 °C, 5 h.
In Vitro Antimycobacterial Activity
Target compounds were initially evaluated for their in vitro antimycobacterial activity against the drug susceptible Mtb H37Rv strain, under replicating conditions using GASTE-Fe (glycerol–alanine–salts supplemented with Tween 80 and iron) culture media. The minimum inhibitory concentration (MIC90) was read 14 days after incubation. Additionally, compounds were screened for in vitro cytotoxicity against a mammalian cell line, Chinese Hamster Ovarian (CHO). Initial structure–activity relationship (SAR) exploration was performed on the LHS with variations on the 6-position while keeping the 2-aminoquinazolinone core unchanged and the right-hand side (RHS) constant as a para-trifluoromethyl phenyl group (Table 1).
Table 1. SAR and In Vitro ADMET Data for LHS and Core Modifications.


50% inhibitory concentration (IC50) on Chinese Hamster Ovarian (CHO) cell lines.
clogD calculated using StarDrop.
Kinetic solubility assay at pH 7.4 using the HPLC method.
H/R/M: human/rat/mouse. SI: safety index. ND: Not determined.
The R1 phenyl group was first altered to explore the impact of various electronic effects and water solubilizing groups on activity. Changing the methyl sulfone in 1 to the more lipophilic cyclopropyl sulfone (8) led to a loss of activity, as did the introduction of a methyl group at the ortho-position of the biaryl moiety (7). Amide analogues (9, 10, 11) were generally equipotent to the sulfone derivative. Potency was also maintained when an aliphatic chain was extended off the amide functionality as in 13 and 14. Various morpholino-type carboxamides were synthesized to improve aqueous solubility whereby the thiomorpholine analogue, 18, was found to be highly potent (0.656 μM). Amides in the meta-position in combination with polar, ionizable groups, such as pyridyl groups 12 and 17 resulted in a 10- and 15-fold loss in activity, respectively. Transformation to the carboxylic acid group 20 was not tolerated (MIC90 = 112 μM).
Replacements of the phenyl group by heterocycles were also investigated. While the unsubstituted pyridine (22) was equipotent to the frontrunner compounds 1 and 2, the meta-substituted counterparts (23 and 24) displayed a loss in potency (MIC90 >2 μM). Likewise, the pyrimidine analogue (21) was inactive. Exploration of electronic and lipophilic effects identified that electron withdrawing, lipophilic substituents (e.g., CF3 and Cl groups as in 28 and 29, respectively) were highly beneficial for potency, whereas electron donating groups (e.g., OMe, tBu, and NMe2 as in 25, 26, and 27, respectively) were not well-tolerated.
Subsequent SAR explorations focused on moving the LHS substituent from the 6- to the 7-position as well as varying the 2-amino moiety (Table 1). All 7-position derivatives (using side chains in the 6-position, which led to active compounds in order to have matched pairs) maintained potency (<2 μM), thus allowing variation on this position and broadening the SAR scope on the LHS. Thereafter, several analogues were made to explore the scope of the 2-amino moiety while keeping either the phenyl sulfone or sulfoxide group at position 6. Removal of the 2-amino group as in compound 38 led to a 10-fold loss in potency (MIC90 = 2.96 μM) compared to the parent analogue 1. Secondary (40) and tertiary (42) 2-amino analogues interestingly maintained some potency when the 6-position contained the sulfoxide substituent (MIC90 = 1.4 and 2.89 μM, respectively), whereas the sulfone counterparts (41 and 43) were inactive (MIC90 >125 μM). The majority of compounds in this series were found to be noncytotoxic except for the cyclopropyl sulfone (8), tertiary amide (10), and morpholino (16) substituted analogues, although cytotoxicity data always has to be evaluated critically when they are higher than the solubility data.
As far as the SAR exploration on the RHS is concerned, diverse groups were investigated to replace the trifluoromethyl substituent, while keeping the 2-aminoquinazolinone core unchanged and keeping the LHS group as a phenyl sulfoxide at position 6 constant, so that matched pairs could be compared (Table 2). Initially, the exploration of the substitution pattern around the ring highlighted that the CF3 substituent exhibited optimal potency when at the para-position compared to the ortho-position (34) and meta-position (48). Solubility improvement strategies by adding a pyridyl group (47) or introducing a saturated ring (57) maintained moderate potency (MIC90 = 0.922 and 2.75 μM, respectively), whereas the addition of a benzylic methylene linker (58) led to a loss in potency. The replacement of the trifluoromethyl substituent by polar groups (SO2Me, OH, OMe, CN) was detrimental to activity, while lipophilic substituents (F, Me) allowed moderate potency to be retained. The unsubstituted phenyl ring analogue (35) was not tolerated (MIC90 = 33 μM). Overall, the RHS SAR revealed that directly attached phenyl rings bearing lipophilic groups at the para-position were optimal for activity.
Table 2. SAR and in Vitro ADMET Data for RHS Modifications.

14 day readout against the H37Rv Mtb-GFP strain in GASTE-Fe media.
clogD calculated using StarDrop.
Kinetic solubility assay at pH 7.4 using the HPLC method.
H/R/M: human/rat/mouse. ND: Not determined.
Physicochemical and In Vitro Microsomal Metabolic Stability Profiling
As mentioned previously, solubility was established as a critical parameter to optimize. Using the kinetic solubility assay, we aimed to achieve values >100 μM, but compounds with moderate solubility and potency were also progressed. To that effect, we looked at introducing hydrophilic groups as well as reducing overall aromaticity and planarity of the molecules (Table 1). For the latter, a methyl substituent was inserted at the ortho-position to the LHS sulfone, as exemplified by 7, in order to disrupt the planarity of the biaryl scaffold. This resulted in a 10-fold improvement in solubility (53.3 μM) with moderate potency (4.22 μM). Generally, derivatives from the R1 SAR displayed poor to moderate solubility, except for 13 and 14 for which the addition of the aliphatic amide substituents drastically improved solubility (168 and 192 μM, respectively). The addition of heteroatoms as in 22 and 23 did not lead to improvements in solubility, except when the heterocycle contained an additional meta-substituent such as an amide (12) or methoxy (24) moiety (40 μM). Solubility for the morpholine-, thiomorpholine-, and piperazine-type derivatives (16, 18, 19) was moderately improved (46–56 μM).
With regards to microsomal metabolic stability, the LHS modifications led to generally metabolically stable derivatives across all species (human, rat, mouse). A few exceptions though include the morpholino derivative (15), thiomorpholine (18), pyridine (22), and the tertiary amine (27).
Modifications to the 2-position of the core scaffold maintained good metabolic stability, but no significant improvements were made in terms of solubility (Table 1). Removal of the 2-amino group decreased the solubility (38 = <5 μM) of the sulfoxide derivative potentially due to the increase in lipophilicity (clogD = 4.25). The movement of the LHS group from position 6 to position 7 on the scaffold resulted in poor solubility, except for the primary amide derivative (33), which exhibited a 10-fold improvement compared to 9.
RHS modifications were found to be the most favorable to improve aqueous solubility with five analogues (34, 58, 36, 53, and 56) exhibiting moderate to high solubility (>100 μM) (Table 2). The introduction of a pyridyl nitrogen (47) did not improve solubility as seen previously on the LHS; this could be due to the electron withdrawing nature of the trifluoromethyl substituent, which lowers the pKa and reduces the ionizability of the nitrogen. The movement of the trifluoromethyl substituent from the para- to the meta-position was shown to be beneficial for solubility (164 μM), however, a reduction in metabolic stability was observed. Similarly, the incorporation of a benzyl linker improved solubility, albeit detrimental to metabolic stability. Variations of the trifluoromethyl substituent with hydrophilic, electron donating groups such as the methyl (36) and methoxy (53) moieties, resulted in improved solubility (172 and 128 μM, respectively), whereas electron withdrawing groups as in the monofluoro (49) and cyano (54) derivatives were relatively insoluble (37.8 and 16.3 μM). As expected, increasing sp3 character through saturation of the aromatic ring led to improved solubility (56 = 125 μM).
Despite multiple analogues being synthesized and exhibiting favorable in vitro properties, the two frontrunner compounds remained the best overall compounds in terms of combined potency, solubility, metabolic stability, and cytotoxicity properties. Therefore, compound 2 and its active metabolite, 1, were selected for further in vivo PK studies.
In Vivo Pharmacokinetic Studies
In vivo PK studies were performed on compound 1 (sulfone) and compound 2 (sulfoxide) (Table 3). All studies and procedures were conducted with prior approval of the animal ethics committee of the University of Cape Town (approval numbers 013/028 and 014/028) in accordance with the South African National Standard (SANS 10386:008) for the Care and Use of Animals for Scientific Purposes and guidelines from the Department of Health. The sulfone 1 was poorly absorbed achieving a bioavailability of 18%. Compound 2 had much higher oral bioavailability and, notably, was converted in vivo to the sulfone (1). This resulted in a higher clearance for the sulfoxide 2 (21.8 vs 1.3 mL/min/kg). Higher exposure of the sulfone was achieved when the sulfoxide was dosed and underwent metabolic conversion than when the sulfone was dosed directly (AUC 18430 vs 7249 min·μmol·L–1) as indicated in Figure 3.
Table 3. Pharmacokinetic Parameters for 1 and 2 after Intravenous and Oral Administration.
|
1 |
2 |
1 as metabolite of 2 | |||
|---|---|---|---|---|---|
| parameter | iv | oral | iv | oral | oral |
| dose (mg/kg) | 5 | 20 | 5 | 20 | 20 of compound 2 |
| Cmax (μM) | 5.98 | 3.54 | 10.4 | ||
| Tmax (h) | 3 | 0.5 | 3 | ||
| Vd (L/kg) | 0.81 | 11.1 | |||
| apparent t1/2 (h) | 7.3 | 6.2 | 10.4 | ||
| CLtotal (mL·min–1·kg–1) | 1.3 | 21.8 | |||
| AUC0–∞ (min·μmol·L–1) | 8589 | 7249 | 523 | 2152 | 18430 |
| bioavailability (%) | 18.4 | 93 | |||
Figure 3.

Concentration–time profiles for compound 2 (LHS graph) and its active metabolite 1 (RHS graph) at a 20 mg/kg oral dose (po) and 5 mg/kg intravenous (IV) dose.
In Vivo Tolerability and Efficacy Studies
The high exposure of the sulfone 1 achieved by dosing the sulfoxide 2 suggested that the sulfoxide could be used for in vivo efficacy experiments. Compound 2 was therefore dosed orally at 200 mg/kg to determine the exposure of the compound at the efficacious dose and to confirm that the exposure would be maintained long enough for efficacy. The free concentrations of compound 2 and its sulfone metabolite 1 are represented in Figure 4.
Figure 4.
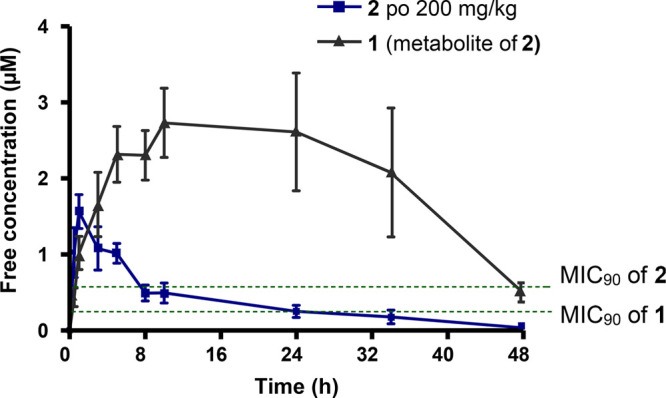
Free concentration–time profile of sulfoxide 2 and its sulfone metabolite 1 at 200 mg/kg dosed orally.
The free exposure was maintained above the MIC90 of compound 1 for more than 24 h, and this dose was therefore projected to give in vivo efficacy.
To test compound 2 for in vivo efficacy, we employed the BALB/c acute TB mouse infection model (see Supporting Information, Section 4.2). In this model, mice are infected by low dose aerosol, resulting in a mean of 2.86 log10 CFU Mtb Erdman-Lux in lungs 1 day following aerosol infection. The lung burdens increased to 4.16 log10 CFU at the start of treatment on day 7. Mice were then dosed once per day with 2 for 12 consecutive days by oral gavage in a 0.2 mL volume at 100 and 200 mg/kg. All mice completed the nonlethal acute TB efficacy trial as scheduled, and there was no evidence of compound tolerability issues. Three days following the last dose, the mean lung burdens were 6.51 log10 CFU for untreated mice compared to 5.72, 4.15, 7.84, and 7.75 log10 CFU for mice treated with rifampin at 10 mg/kg, ethambutol at 100 mg/kg, or 2 at 100 or 200 mg/kg, respectively. Thus, compound 2 was found to lack efficacy in the BALB/c acute infection model, showing higher overall lung burdens relative to the untreated controls. In contrast, rifampin at 10 mg/kg and ethambutol at 100 mg/kg reduced lung burdens as expected by 0.8 and 2.4 log, respectively.
On the basis of the in vivo PK and tolerability studies, the lack of in vivo efficacy is unlikely to be accounted for by low exposure and/or toxicity issues. In addition, plasma concentrations in infected mice at 1 and 24 h were comparable to those in healthy mice at the same time points (Table S1). This suggests that PK was similar between healthy and infected animals. Further investigation into the discrepancy between in vitro and in vivo results was therefore undertaken to understand the in vitro/in vivo disconnect.
Mechanistic Studies
Compound 2 was tested in biology triage assays to assess the activity against promiscuous targets. It did not show hypersensitivity against a cytochrome-bd oxidase knockout mutant strain (Δcyd GASTE-Fe MIC90 = 2.5 μM), which is hypersusceptible to compounds that inhibit the QcrB-containing cytochrome bc1 complex, thereby eliminating QcrB as a potential target of 2.25,26 Additionally, 2 did not show a positive signal in two standard bioluminescence reporter assays: PiniB-LUX27 detects modulation in iniB expression if compounds target Mtb cell-wall biosynthesis, and PrecA-LUX27 detects modulation in recA expression, an indicator of genotoxic compounds. On the basis of this initial triage process, the 2-aminoquinazolinone scaffold was identified as a hit series with a potential novel mechanism of action (MOA).
However, to further understand the discrepancy between in vitro and in vivo results, compound 2 was subsequently retested in vitro using different Mtb strains cultured in different media. As shown in Table 4, 2 was active against both H37Rv and Erdman Mtb strains when grown in media containing glycerol as the carbon source (7H9, glycerol, ADC, and Tween 80). In contrast to these observations, 2 lost activity against both strains when cultured in glycerol-free media (7H12, casitone, palmitic acid, BSA, catalase, and Tween 80).
Table 4. In Vitro Antimycobacterial Activity of 2 in Different Assay Conditions.
| MIC90 (mg/L) |
||||
|---|---|---|---|---|
| strain | media | 2 | rifampin | isoniazid |
| Erdman | 7H9 + glycerol + ADC + Tween 80 | 1 | 0.03 | 0.016 |
| H37Rv | 7H9 + glycerol + ADC + Tween 80 | 0.5 | 0.016 | 0.016 |
| Erdman | 7H12 + casitone + palmitic acid + BSA + catalase + Tween 80 | >16 | <0.008 | 0.016 |
| H37Rv | 7H12 + casitone + palmitic acid + BSA + catalase + Tween 80 | >16 | <0.008 | 0.016 |
Additional in vitro assays using glucose as the carbon source were performed on 15 other aminoquinazolinone analogues. In all cases, no activity was observed in the absence of glycerol in the culture medium, thus suggesting that the glycerol metabolism played a key role in effectively inhibiting Mtb growth. We therefore hypothesized that the in vitro/in vivo disconnect was due to the fact that glycerol was not used as a primary carbon source by Mtb in the mouse lungs, as has previously been observed.28−30
In order to test this hypothesis, MOA studies using spontaneous-resistant mutant generation combined with potential target identification using whole genome sequencing were performed with 2. The spontaneous-resistant mutants were raised on Middlebrook 7H10 containing glycerol and OCDC (oleic acid, casitone, dextrose, and catalse) using the Mtb H37Rv MA strain. The mutants arose with a mutation frequency of 5 × 10–7. Random isolated colonies were picked, grown in compound free medium, and tested for MIC at a compound concentration range of 0.16–160 μM. This led to the isolation of six Mtb spontaneous mutants (MIC90 of compound 2, >160 μM) with full genome sequencing revealing single nucleotide polymorphisms (SNPs) mapping exclusively to glycerol metabolizing genes, specifically glpK and glpD2, as described in Table 5. The GlpK gene (Rv3696) encodes for glycerol kinase, which is the first committing enzyme for glycerol metabolism in most bacteria. The second metabolite in the glycerol dissimilation pathway is dihydroxyacetone phosphate (DHAP), which is formed from glycerol-3-phosphate (G3P) by the enzyme glycerol-3-phosphate dehydrogenase encoded by the glpD1 and glpD2 genes.28
Table 5. Description of Mutations Obtained from Spontaneous -Resistant Mutant Generation.
| no. | MIC90 of 2 (μM) | gene | mutation | descriptor |
|---|---|---|---|---|
| 1 | >160 | glpK | SNP | T96M |
| 2 | >160 | glpK | frameshift mutation | C insertion at a.a. 189 |
| 3 | >160 | glpK | frameshift mutation | C insertion at a.a. 189 |
| 4 | >160 | glpD2 | SNP | F122V |
| 5 | >160 | glpD2 | SNP | F122L |
| 6 | >160 | glpD2 | SNP | F122L |
Previous reports observed that frameshift mutations and SNPs in glpK were linked to resistance to Mtb growth inhibitors.28−30 These studies showed that compounds with glycerol-specific activity promote the accumulation of G3P, resulting in the rapid depletion of adenosine triphosphate (ATP) utilized for phosphorylation of glycerol, ultimately leading to self-poisoning of Mtb.28,29 The activity of these compounds was therefore found to be associated with the metabolism of glycerol contained in 7H9 media.28−30 No such reports have, however, been observed for glpD2 mutants related to in vitro drug screening.
All considered together, the results from the MOA studies suggest that the aminoquinazolinone series potentially exert its inhibitory effect by dysregulating the glycerol dissimilation pathway,31 leading to the toxic accumulation of sugar phosphates such as G3P and DHAP in Mtb.28 When glycerol is not used as a carbon source by Mtb in vivo, compounds with such MOA do not show efficacy in the mouse model.
These results highlight the importance of validating the relevance of in vitro assay conditions to reproduce the environment encountered by Mtb in the infected host.28,30 The culture media currently used for sufficient Mtb growth were empirically devised several decades ago for optimal propagation of the bacterium in vitro and were not optimized for drug screening efforts. Consequently, these media were not developed to replicate the conditions encountered by Mtb in vivo, thus limiting the predictive value of the in vitro assays. There is no clear consensus thus far as to which in vitro methods best reflect the in vivo biology in order to effectively identify novel candidates that will inhibit Mtb growth in vivo.28,29,32
Conclusion
This study identified a new class of aminoquinazolinone compounds active against Mtb through phenotypic whole-cell assays. Although these compounds exhibited excellent in vitro potency and favorable pharmacological properties, they were found to be inactive in an BALB/c mouse acute TB infection model. The discrepancy between in vitro activity and in vivo efficacy was elucidated through retesting in vitro using different Mtb strains cultured in different media, which revealed that activity was only observed in media containing glycerol. This was supported by spontaneous-resistant mutant generation and whole genome sequencing studies, which revealed mutations mapping to glycerol metabolizing genes, suggesting a major difference in carbon metabolism between bacteria growing in standard TB culture medium containing glycerol compared to those found in TB-infected lungs.
Methods
General Methods
All reagents and solvents were obtained from commercial sources and used as received. All dry solvents were obtained from an SP-1 standalone solvent purification system from LC Technology Solutions Inc. Reactions were monitored by thin layer chromatography (TLC) using Merck TLC silica gel 60 F254 aluminum-backed precoated plates and were visualized by ultraviolet light at 254 nm. Purification of compounds was carried out by either column chromatography on silica gel 60 (Fluka), particle size of 0.063–0.2 mm (70–230 mesh ASTM), as the stationary phase or by Waters’ HPLC using an X-bridge C18 5 μm column (4.6 × 150 mm). Additional details are as follows: organic phase, 10 mM ammonium acetate (pH 3.7) in HPLC grade methanol; aqueous phase, 10 mM ammonium acetate (pH 3.7) in HPLC grade water; flow rate, 15.00 mL/min; detector, photodiode array (PDA). All target compounds and intermediates were characterized by 1H NMR, 13C NMR, and MS. NMR spectra were recorded on either a Varian Mercury-300 (1H 300.1 MHz, 13C 75.5 MHz) or Bruker-400 (1H 400.2 MHz, 13C 100.6 MHz) instrument using CDCl3, MeOH-d4, and DMSO-d6 as solvents. The 1H NMR data are reported as follows: chemical shift in parts per million (δ) downfield of tetramethylsilane (TMS), multiplicity (s = singlet, bs = broad singlet, d = doublet, t = triplet, q = quartet, dd = doublets of doublets, dt = triplets of doublets, and m = multiplet), coupling constant (Hz), and integrated value. The 13C NMR spectra were measured with complete proton decoupling. LC–MS/MS analysis was performed using an Agilent 1260 Infinity Binary Pump, Agilent 1260 Infinity Diode Array Detector (DAD), Agilent 1290 Infinity Column Compartment, Agilent 1260 Infinity Standard Autosampler, and an Agilent 6120 Quadrupole (Single) MS with an APCI/ESI multimode ionization source. The purities were determined by Agilent LCMS/MS or Waters’ HPLC using an X-bridge C18 5 μm column (4.6 × 150 mm). Additional details are as follows: organic phase, 10 mM ammonium acetate (pH 3.7) in HPLC grade methanol; aqueous phase, 10 mM ammonium acetate (pH 3.7) in HPLC grade water; flow rate, 1.20 mL/min; detector, photodiode array (PDA). The purities of all compounds were found to be >95%.
6-Iodo-2-thioxo-3-(4-(trifluoromethyl)phenyl)-2,3-dihydroquinazolin-4(1H)-one (3a)
A solution of 2-amino-5-iodobenzoic acid (5 g, 19 mmol, 1 equiv) and 4-(trifluoromethyl)phenyl isothiocyanate (4 g, 20 mmol, 1.05 equiv) in anhydrous 1,4-dioxane (75 mL) was treated with triethylamine (4 mL, 28.5 mmol, 1.5 equiv). The solution was heated under reflux for 4 h and cooled to room temperature, and the solids that formed were filtered. The solids were resuspended in diethyl ether (Et2O) and filtered again to afford a white solid (7.84 g, 92%); 1H NMR (300 MHz, DMSO-d6) δ 13.18 (s, 1H), 8.20 (d, J = 1.9 Hz, 1H), 8.10 (dd, J = 8.6, 2.0 Hz, 1H), 7.88 (d, J = 8.3 Hz, 2H), 7.56 (d, J = 8.1 Hz, 2H), 7.26 (d, J = 8.6 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 176.14, 159.02, 144.21, 143.32, 139.64, 135.69, 130.72 (2C), 126.60 (2C), 126.56, 118.77, 118.54, 88.18, 66.84. LC-MS (ESI): found m/z = 448.9 [M + H]+, (calcd for C15H8F3IN2OS: 447.0); HPLC purity 99%.
2-Chloro-6-iodo-3-(4-(trifluoromethyl)phenyl)quinazolin-4(3H)-one (4a)
PCl5 (6.37 g, 30.63 mmol, 1.75 equiv) was added in one portion to a mixture of intermediate 3 (7.84 g, 17.5 mmol, 1 equiv) in POCl3 (40 mL, 25 equiv) at room temperature. The mixture was stirred for 15 min at room temperature and was subsequently heated to 110 °C for 14 h under nitrogen. The reaction was cooled to room temperature, concentrated in vacuo, and then diluted with ethyl acetate to be added portionwise to stirred ice-saturated sodium bicarbonate and stirred until all solids dissolved. The organic layer was then separated, washed with brine, dried over magnesium sulfate (Mg2SO4), and concentrated under reduced pressure. The resulting residue was triturated in Et2O and filtered to obtain an off-white solid (6.26 g, 79%); 1H NMR (300 MHz, DMSO-d6) δ 8.39 (d, J = 1.8 Hz, 1H), 8.22 (dd, J = 8.5, 2.1 Hz, 1H), 7.99 (d, J = 8.3 Hz, 2H), 7.81 (d, J = 8.2 Hz, 2H), 7.52 (d, J = 8.7 Hz, 1H); 13C NMR (101 MHz, DMSO-d6) δ 150.20, 143.82, 139.91, 135.86, 135.42, 130.71 (2C), 130.44, 129.22, 127.13 (2C), 126.41, 118.23, 116.99, 85.69. LC-MS (ESI): found m/z = 450.9 [M + H]+, (calcd for C15H7ClF3IN2O: 449.9); HPLC purity 90%.
6-Iodo-2-((4-methoxybenzyl)amino)-3-(4-(trifluoromethyl)phenyl)quinazolin-4(3H)-one (5a)
4-Methoxybenzylamine (2.36 mL, 18 mmol, 1.3 equiv) and N,N-diisopropylethylamine (DIPEA) (4.84 mL, 27.8 mmol, 2 equiv) were dissolved in DMF (45 mL), and the appropriate intermediate 4a (6.26 g, 13.9 mmol, 1.0 equiv) was added portionwise. The reaction was stirred for 4 h at 80 °C and allowed to cool to room temperature. The solvent was concentrated under reduced pressure and diluted with EtOAc. The organic phase was washed with 5% lithium chloride (3 × 20 mL) and brine (20 mL), dried over Mg2SO4, and concentrated in vacuo to yield a brown solid (6.36 g, 83%); 1H NMR (300 MHz, DMSO-d6) δ 8.15 (d, J = 2.0 Hz, 1H), 7.97 (d, J = 8.3 Hz, 2H), 7.87 (dd, J = 8.6, 2.2 Hz, 1H), 7.67 (d, J = 8.1 Hz, 2H), 7.24 (d, J = 8.7 Hz, 2H), 7.11 (d, J = 8.7 Hz, 1H), 6.85 (d, J = 8.7 Hz, 2H), 6.68 (t, J = 5.9 Hz, 1H), 4.44 (d, J = 5.9 Hz, 2H), 3.71 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 161.00, 158.55, 150.59, 149.58, 143.08, 139.05, 134.95, 132.01, 130.97 (2C), 130.70, 130.14 (q, J = 32.0 Hz, 1C, C-CF3), 128.88 (2C), 124.7 (q, J = 272.5 Hz, 1C, CF3), 127.75 (2C), 127.60, 119.49, 114.00 (2C), 84.74, 44.18. LC-MS (ESI): found m/z = 552.0 [M + H]+, (calcd for C23H17F3IN3O2: 551.0); HPLC purity 96%.
2-Amino-6-iodo-3-(4-(trifluoromethyl)phenyl)quinazolin-4(3H)-one (6a)
Intermediate 5a (6.35 g, 11.5 mmol, 1.0 equiv) was dissolved in TFA (56 mL, 64 equiv) and refluxed at 80 °C for 48 h. The reaction was cooled to room temperature, and the solvent was concentrated. The residue was resuspended in DCM and added portionwise to a stirred ice-saturated sodium bicarbonate solution. The organic phase was separated; the aqueous phase was extracted with DCM, and the combined organic phases were diluted with Et2O to a 1:1 ratio of DCM/Et2O. The solids were filtered, washed with Et2O, and dried under vacuum to afford an off-white solid (4.34 g, 87%); 1H NMR (300 MHz, DMSO-d6) δ 8.14 (d, J = 2.0 Hz, 1H), 7.93 (d, J = 8.3 Hz, 2H), 7.87 (dd, J = 8.7, 2.2 Hz, 1H), 7.63 (d, J = 8.1 Hz, 2H), 7.07 (d, J = 8.7 Hz, 1H), 6.60 (s, 2H); 13C NMR (101 MHz, DMSO-d6) δ 161.06, 152.21, 150.20, 143.04, 139.51, 135.01, 130.59 (2C), 130.03, 127.59, 127.55 (2C), 126.93, 119.24, 84.35. LC-MS (ESI): found m/z = 432.0 [M + H]+, (calcd for C15H9F3IN3O: 431.0); HPLC purity 98%.
General Protocol for the Synthesis of Compounds 1, 2, and 7–36
To a solution of the relevant intermediate 6a–e (1 equiv) and the appropriate arylboronic acid or ester (1.5 equiv) in anhydrous 1,4-dioxane (2 mL) flushed with nitrogen was added bis(triphenylphosphine) palladium(II) dichloride, PdCl2(PPh3)2, (0.1 equiv); cesium carbonate or potassium carbonate (3 equiv) was dissolved in water (0.5 mL) and subsequently added to the reaction mixture. The solution was heated at 80 °C until TLC monitoring showed completion (1–5 h), allowed to cool to room temperature, and diluted with EtOAc to be filtered through Celite. The organic phase was washed with water (2 × 20 mL) and brine (20 mL), dried over Mg2SO4, and concentrated under reduced pressure. The resulting crude residue was purified by silica-gel flash chromatography eluting a gradient of either 0–100% EtOAc in hexane or 0–10% MeOH in DCM. Combined pure fractions were concentrated in vacuo, triturated with Et2O, filtered, and dried under vacuum to give the relevant target compounds.
2-Amino-6-(3-(methylsulfonyl)phenyl)-3-(4-(trifluoromethyl)phenyl)quinazolin-4(3H)-one (1)
Yield 77%. 1H NMR (400 MHz, DMSO-d6) δ 8.22 (d, J = 2.1 Hz, 1H), 8.18 (t, J = 1.7 Hz, 1H), 8.11 (ddd, J = 7.8, 1.8, 1.1 Hz, 1H), 8.09 (dd, J = 8.6, 2.4 Hz, 1H), 7.94 (d, J = 8.3 Hz, 2H), 7.88 (ddd, J = 7.8, 1.8, 1.0 Hz, 1H), 7.73 (t, J = 7.8 Hz, 1H), 7.65 (d, J = 8.1 Hz, 2H), 7.37 (d, J = 8.4 Hz, 1H), 6.57 (br s, 2H), 3.29 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 162.24, 152.18, 150.88, 142.21, 140.96, 139.69, 133.66, 131.86, 131.66, 130.64 (2C), 130.17 (q, J = 32.0 Hz, 1C, C-CF3), 127.61, 127.58 (2C), 125.83, 125.41, 124.96, 124.87, 124.54 (q, J = 272.5 Hz, 1C, CF3), 117.40, 43.95. LC-MS (ESI): found m/z = 460.1 [M + H]+, (calcd for C22H16F3N3O3S: 459.09); HPLC purity 98%.
2-Amino-6-(3-(methylsulfinyl)phenyl)-3-(4-(trifluoromethyl)phenyl)quinazolin-4(3H)-one (2)
Yield 57%. 1H NMR (400 MHz, DMSO-d6) δ 8.20 (d, J = 2.2 Hz, 1H), 8.03 (dd, J = 8.6, 2.2 Hz, 1H), 7.98 (t, J = 2.2 Hz, 1H), 7.95 (d, J = 8.2 Hz, 2H), 7.86 (dt, J = 6.6, 2.2 Hz, 1H), 7.70–7.62 (m, 4H), 7.37 (d, J = 8.6 Hz, 1H), 6.55 (br s, 2H), 2.81 (s, 3H); 13C NMR (101 MHz, DMSO-d6) δ 162.28, 152.08, 150.68, 147.89, 140.82, 139.73, 133.64, 132.55, 130.66 (2C), 130.45, 130.14 (q, J = 32.0 Hz, 1C, C-CF3), 128.86, 127.61, 127.57, 125.34, 124.75, 124.7 (q, J = 272.48 Hz, 1C, CF3), 122.64, 121.49, 117.37, 43.74. LC-MS (ESI): found m/z = 444.1 [M + H]+, (calcd for C22H16F3N3O2S: 443.1); HPLC purity 99%.
6-Iodo-3-(4-(trifluoromethyl)phenyl)quinazolin-4(3H)-one (37)
To a solution of 2-amino-5-iodobenzoic acid (330 mg, 1.25 mmol, 1 equiv) and 4-(trifluoromethyl)aniline (200 mg, 1.25 mmol, 1 equiv) in dioxane (30 mL) was added triethoxymethane (2 g, 12.5 mmol, 10 equiv), and the resulting reaction mixture was heated in a sealed tube at a temperature of 100 °C. After 16 h, a precipitate is formed. The reaction mixture was cooled to room temperature at 25 °C; the precipitate was filtered, washed with Et2O (10 mL), and dried to afford a light yellow solid (150 mg, 0.36 mmol, 29%). LC-MS (ESI): found m/z = 417.0 [M + H]+, (calcd for C15H8F3IN2O: 416.0); HPLC purity 80%.
6-(3-(Methylsulfonyl)phenyl)-3-(4-(trifluoromethyl)phenyl)quinazolin-4(3H)-one (38)
6-Iodo-3-(4-(trifluoromethyl)phenyl)quinazolin-4(3H)-one 37 (150 mg, 0.3 6 mmol, 1 equiv), (3-(methylsulfonyl)phenyl)boronic acid (78 mg, 0.39 mmol, 1.1 equiv), and PdCl2(dppf) (30 mg, 0.041 mmol, 0.11 equiv) were dissolved in dioxane (3 mL). Thereafter, Cs2CO3 (230 mg, 0.706 mmol, 2 equiv) and water (0.3 mL) were added. The mixture was heated in a pressure tube to 85 °C for 2 h. The cooled mixture was diluted with 50 mL of a NaHCO3 solution and extracted with EtOAc (2 × 50 mL). The organic phases were dried over Na2SO4 and evaporated. Flash chromatography eluting a gradient of 30–50% EtOAc in DCM and the evaporation of the product fractions yielded a white solid (80 mg, 50%). 1H NMR (300 MHz, CDCl3) δ (ppm) 8.63 (1H, d, J = 3.0 Hz), 8.30–8.28 (1H, m), 8.18 (1H, s), 8.21 (1H, dd, J = 3.0 and 9.0 Hz), 8.04–7.99 (2H, m), 7.93 (1H, d, J = 9.0 Hz), 7.88 (2H, d, J = 9.0 Hz), 7.77–7.71 (1H, m), 7.64 (2H, d, J = 9.0 Hz), 3.16 (3H, s). LC-MS (ESI): found m/z = 445.1 [M + H]+, (calcd for C22H15F3N2O3S: 444.08); HPLC purity 100%.
6-Iodo-2-(methylamino)-3-(4-(trifluoromethyl)phenyl)quinazolin-4(3H)-one (39a)
2-Chloro-6-iodo-3-(4-(trifluoromethyl)phenyl)quinazolin-4(3H)-one (intermediate 4a) (200 mg, 0.44 mmol, 1.0 equiv) was dissolved in THF (2 mL), and a 33% solution of methylamine in ethanol was added; the mixture was heated in a pressure tube to 60 °C for 3 h. Once cooled, 10 mL of water was added and this mixture was extracted with ethyl acetate (2 × 50 mL). The organic layers were dried over Na2SO4 and evaporated to afford an oily residue (240 mg, 121%). 1H NMR (300 MHz, CDCl3) δ (ppm) 8.44 (1H, d, J = 3.0 Hz), 7.93 (1H, dd, J = 3.0 and 9.0 Hz), 7.92 (2H, d, J = 9.0 Hz), 7.49 (2H, J = 9.0 Hz), 7.46 (1H, d, J = 9.0 Hz), 3.13–3.07 (3H, m). LC-MS (ESI): found m/z = 446.0 [M + H]+, (calcd for C16H11F3IN3O: 444.99).
General Procedure for the Synthesis of Compounds 40 and 41
Intermediate 39 (120 mg, 0.22 mmol, 1.0 equiv), the relevant boronic acid (56 mg, 0.28 mmol, 1.3 equiv), and PdCl2(dppf) (20 mg, 0.027 mmol, 0.12 equiv) were dissolved in dioxane (2 mL). Thereafter, cesium carbonate (230 mg, 0.71 mmol, 3 equiv) and water (0.3 mL) were added. The mixture was heated in a pressure tube to 80 °C for 3.5 h. Once complete, the mixture was cooled, diluted with water, and extracted with EtOAc (2 × 50 mL). The organic layers were dried over Na2SO4 and evaporated. Flash chromatography eluting a gradient of 20–40% THF in DCM afforded the target compounds 40 and 41.
2-(Methylamino)-6-(3-(methylsulfinyl)phenyl)-3-(4-(trifluoromethyl)phenyl)quinazolin-4(3H)-one (40)
Yield 75%. 1H NMR (300 MHz, CDCl3) δ (ppm) 8.22 (1H, d, J = 3.0 Hz), 8.06 (1H, dd, J = 3.0 and 9.0 Hz), 8.00–7.94 (3H, m), 7.90–7.86 (1H, m), 7.70–7.68 (4H, m), 7.47 (1H, d, J = 9.0 Hz), 6.08–6.02 (1H, m), 2.81 (6H, “s”). LC-MS (ESI): found m/z = 458.1 [M + H]+, (calcd for C23H18F3N3O2S: 457.11); HPLC purity 99%.
2-(Methylamino)-6-(3-(methylsulfonyl)phenyl)-3-(4-(trifluoromethyl)phenyl)quinazolin-4(3H)-one (41)
Yield 55%. 1H NMR (300 MHz, CDCl3) δ (ppm) 8.40 (1H, d, J = 3.0 hz), 8.24–8.22 (1H, m), 7.98–7.90 (5H, m), 7.72–7.65 (2H, m), 7.53 (2H, d, J = 9.0 Hz), 4.13 (1H, sb), 3.13 (3H, s), 3.08 (3H, d, J = 6.0 Hz). LC-MS (ESI): found m/z = 474.1 [M + H]+, (calcd for C23H18F3N3O3S: 473.10); HPLC purity 98%.
2-(Dimethylamino)-6-iodo-3-(4-(trifluoromethyl)phenyl)quinazolin-4(3H)-one (39b)
2-Chloro-6-iodo-3-(4-(trifluoromethyl)phenyl)quinazolin-4(3H)-one (4a) (220 mg, 0.488 mmol, 1 equiv) was dissolved in DMF (5 mL); dimethylamine hydrochloride (350 mg, 4.8 mmol, 10 equiv), and 1 mL of triethylamine was added, and the mixture was then heated in a pressure tube to 80 °C for 3 h. To the cooled mixture was added 50 mL of water, and this mixture was extracted with ethyl acetate (2 × 50 mL). The organic layers were dried over Na2SO4 and evaporated to afford a yellow solid (270 mg, 120%). 1H NMR (300 MHz, CDCl3) δ (ppm) 8.36 (1H, d, J = 3.0 Hz), 7.83 (1H, dd, J = 3.0 and 9.0 Hz), 7.69 (2H, d, J = 9.0 Hz), 7.42 (2H, J = 9.0 Hz), 7.20 (1H, d, J = 9.0 Hz), 2.61 (6H, s). LC-MS (ESI): found m/z = 460.0 [M + H]+, (calcd for C17H13F3IN3O: 459.0); HPLC purity 98%.
General Procedure for the Synthesis of 42 and 43
Intermediate 39b (135 mg, 0.29 mmol, 1 equiv), the corresponding boronic acid (56 mg, 0.28 mmol, 1 equiv), and PdCl2(dppf) (20 mg, 0.027 mmol, 0.1 equiv) were dissolved in dioxane (2 mL). Thereafter, cesium carbonate (250 mg, 0.77 mmol, 3.0 equiv) and water (0.3 mL) were added. The mixture was heated in a pressure tube to 80 °C for 3.5 h. Once complete, the mixture was cooled, diluted with water, and extracted with EtOAc (2 × 50 mL). The organic layers were dried over Na2SO4 and evaporated. Flash chromatography eluting a gradient of 40–80% EtOAc in hexane afforded the desired final compounds 42 and 43.
2-(Dimethylamino)-6-(3-(methylsulfinyl)phenyl)-3-(4-(trifluoromethyl)phenyl)quinazolin-4(3H)-one (42)
Yield 80%. 1H NMR (300 MHz, CDCl3) δ (ppm) 8.40 (1H, d, J = 3.0 Hz); 8.00 (1H, dd, J = 3.0 and 9.0 Hz); 7.97–7.93 (1H, m); 7.84–7.62 (7H, m); 7.56 (1H, d, J = 9.0 Hz); 2.81 (3H, s); 2.78 (6H, s). LC-MS (ESI): found m/z = 472.1 [M + H]+, (calcd for C24H20F3N3O2S: 471.12); HPLC purity 98%.
2-(Dimethylamino)-6-(3-(methylsulfonyl)phenyl)-3-(4-(trifluoromethyl)phenyl)quinazolin-4(3H)-one (43)
Yield 88%. 1H NMR (300 MHz, CDCl3) δ (ppm) 8.41 (1H, d, J = 3.0 hz), 8.24–8.22 (1H, m), 7.99–7.93 (3H, m), 7.81 (2H, d, J = 9.0 Hz), 7.72–7.65 (2H, m), 7.57 (2H, d, J = 9.0 Hz), 3.13 (3H, s), 2.75 (6H, s). LC-MS (ESI): found m/z = 488.1 [M + H]+, (calcd for C24H20F3N3O3S: 487.11); HPLC purity 98%.
Methyl 5-Bromo-2-(3-(ethoxycarbonyl)thioureido)benzoate (44)
Methyl 2-amino-5-bromobenzoate (3.58 g, 15.5 mmol, 1 equiv) was dissolved in acetonitrile (10 mL), and the yellow solution was treated with ethoxycarbonyl isothiocyanate (2.2 mL, 18.6 mmol, 1.2 equiv). After stirring for 10 min at room temperature, the precipitate that formed was filtered; the resulting mother liquor was concentrated, and solids were refiltered to afford methyl 5-bromo-2-(3-(ethoxycarbonyl)thioureido)benzoate as a white solid (5 g, 90%). 1H NMR (400 MHz, chloroform-d) δ 12.61 (s, 1H), 8.47 (d, J = 8.9 Hz, 1H), 8.18 (s, 1H), 8.16 (d, J = 2.4 Hz, 1H), 7.68 (dd, J = 8.8, 2.5 Hz, 1H), 4.35 (q, J = 7.1 Hz, 2H), 3.97 (s, 3H), 1.37 (t, J = 7.1 Hz, 3H); 13C NMR (101 MHz, chloroform-d) δ 178.10, 165.46, 151.67, 137.95, 135.35, 133.42, 127.86, 123.54, 118.74, 63.07, 52.81, 14.21. LC-MS (ESI): found m/z = 360.9, 362.9 (1:1) [M + H]+, (calcd for C12H13BrN2O4S: 359.98); HPLC purity 99%.
Ethyl(6-bromo-4-oxo-3-(6-(trifluoromethyl)pyridin-3-yl)-3,4-dihydroquinazolin-2-yl)carbamate (45a)
Intermediate 44 (4.94 g, 13.68 mmol, 1 equiv) was dissolved in DCM (60 mL), and 6-(trifluoromethyl)pyridin-3-amine (3.3 g, 20.5 mmol, 1.5 equiv) and EDCI (5.24 g, 27.36 mmol, 2 equiv) were successively added to the solution. The solution was allowed to stir at room temperature for 18 h. Thereafter, the organic layer was washed with 1% of 1 M HCl (2 × 20 mL), water, and brine (20 mL) and dried over magnesium sulfate. The solution was filtered, and the solvent was concentrated under reduced pressure to furnish a pale yellow solid (4.3 g, 69%). 1H NMR (400 MHz, CDCl3) δ 8.65 (d, J = 1.9 Hz, 1H), 8.34 (d, J = 2.2 Hz, 1H), 7.90–7.79 (m, 3H), 7.19 (d, J = 8.6 Hz, 1H), 4.13 (q, J = 7.1 Hz, 2H), 1.27 (t, J = 7.1 Hz, 3H). LC-MS (ESI): found m/z = 4570, 459.0 (1:1) [M + H]+, (calcd for C17H12BrF3N4O3: 456.0); HPLC purity 97%.
2-Amino-6-bromo-3-(6-(trifluoromethyl)pyridin-3-yl)quinazolin-4(3H)-one (46a)
The 2-carbamate-based intermediate 45a (4.17 g, 9.1 mmol, 1 equiv) was dissolved in TFA (14 mL, 20 equiv). This solution was heated under microwave conditions at 110 °C for 20 min. After cooling to room temperature and evaporating the solvent, the residue was dissolved in DCM/MeOH (1:1). Amberlyst A21 free base was then added to this solution; the mixture was stirred at ambient temperature for 45 min, followed by filtration and rinsing of the resin with MeOH. The filtrate was then concentrated under reduced pressure to give a residue that was triturated in Et2O, filtered, and dried to afford an off-white solid (2.9 g, 64%). 1H NMR (400 MHz, DMSO-d6) δ 8.83 (d, J = 2.3 Hz, 1H), 8.22 (dd, J = 7.7, 2.3 Hz, 1H), 8.13 (d, J = 7.6 Hz, 1H), 7.97 (d, J = 2.4 Hz, 1H), 7.77 (dd, J = 8.8, 2.5 Hz, 1H), 7.23 (d, J = 8.7 Hz, 1H), 6.80 (br s, 2H). LC-MS (ESI): found m/z = 385.0, 387.0 (1:1) [M + H]+, (calcd for C14H8BrF3N4O: 384.0); HPLC purity 99%.
2-Amino-6-(3-(methylsulfinyl)phenyl)-3-(6-(trifluoromethyl)pyridin-3-yl)quinazolin-4(3H)-one (47)
(3-(Methylsulfinyl)phenyl)boronic acid (1.2 equiv) and the corresponding 2-amino-6-bromoquinazolinone-based intermediate 46a (1 equiv) were dissolved in DMF (5 mL). The mixture was flushed with nitrogen gas for about 10 min at room temperature, after which Pd(PPh3)2Cl2 (0.05 equiv) and K2CO3 (3 equiv) were added successively, and the mixture was heated at 100 °C for 1 to 4 h. After cooling to room temperature, the mixture was diluted with EtOAc (10 mL) and water (30 mL) and then extracted with EtOAc (3 × 20 mL). The combined organic fractions were filtered through a pad of Celite, washed with 5% LiCl (3 × 30 mL) and brine (2 × 30 mL), and dried over anhydrous MgSO4. The solvents were then removed in vacuo to yield a residue that was purified by flash chromatography eluting a gradient of 0–15% MeOH in DCM. Product fractions were combined, and the solvents were rotary evaporated to give a solid that was then triturated with Et2O, filtered, or recrystallized in an appropriate solvent (ethanol was used in most cases) and dried to furnish the desired final target compounds. Yield 56%. 1H NMR (400 MHz, DMSO-d6) δ 8.86 (d, J = 2.3 Hz, 1H), 8.25 (dd, J = 8.3, 2.3 Hz, 1H), 8.22 (d, J = 2.3 Hz, 1H), 8.14 (d, J = 8.3 Hz, 1H), 8.05 (dd, J = 8.6, 2.4 Hz, 1H), 7.98 (dd, J = 2.2, 1.2 Hz, 1H), 7.91–7.84 (m, 1H), 7.71–7.64 (m, 2H), 7.39 (d, J = 8.6 Hz, 1H), 6.78 (br s, 2H), 2.82 (s, 3H). LC-MS (ESI): found: m/z = 445.1 [M + H]+, (calcd for C21H15F3N4O2S: 444.09); HPLC purity 99%.
Acknowledgments
The authors wish to acknowledge Helena I. M. Boshoff from the National Institute of Allergy and Infectious Diseases (NIAID) for technical assistance and advice with the in vitro antimycobacterial assays. We thank Véronique Dartois and Firat Kaya from Rutgers University for their PK analysis on infected mice from the in vivo efficacy study. From H3D, Ronett Seldon is acknowledged for the routine screening of MIC90; Nesia Barnes, Warren Oilifant, and Duane Knowles are acknowledged for their work on the ADMET assays; Sumaya Salie and Virgil Verhoog are thanked for their cytotoxicity work. This work was supported by grants from the Novartis Research Foundation, the Bill & Melinda Gates Foundation (Global Health Grant OPP1066878), and the South African Medical Research Council (SAMRC). The University of Cape Town, SAMRC, and South African Research Chairs Initiative of the Department of Science and Technology, administered through the South African National Research Foundation, are gratefully acknowledged for support (K.C.).
Glossary
Abbreviations
- TB
tuberculosis
- Mtb
Mycobacterium tuberculosis
- INH
isoniazid
- RIF
rifampicin
- MDR
multidrug resistant
- EDC
N-(3-(dimethylamino)propyl)-N′-ethylcarbodiimide hydrochloride
- ADMET
absorption, distribution, metabolism, excretion, and toxicity
- HPLC
high-performance liquid chromatography
- LC-MS
liquid chromatography–mass spectrometry
- MIC90
concentration of drug that inhibits growth of 90% of the bacterial population
- PPB
plasma protein binding
- SRM
spontaneous-resistant mutants
- SAR
structure–activity relationship
- PK
pharmacokinetics
- WHO
World Health Organization
- LHS
left-hand side
- RHS
right-hand side
- GASTE-Fe
glycerol–alanine–salts supplemented with Tween 80 and iron
- AUC
area under curve
- MOA
mechanism of action
- SNP
single nucleotide polymorphism
- G3P
glycerol-3-phosohate
- ATP
adenosine triphosphate
- DCM
dichloromethane
- DMF
dimethylformamide
- THF
tetrahydrofuran
- TFA
trifluoracetic acid
- TLC
thin layer chromatography
- NMR
nuclear magnetic resonance
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.0c00252.
Additional details of the synthetic protocols and characterization of all intermediates and final compounds and the procedures used for the in vitro biological assays, ADMET, and in vivo studies (PDF)
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Author Status
† A. Myrick: Deceased.
Dedication
This paper is dedicated to Alissa Myrick.
Supplementary Material
References
- WHO (2019) Global tuberculosis report 2019, WHO, Geneva. [Google Scholar]
- Horsburgh C. R.; Barry C. E.; Lange C. (2015) Treatment of Tuberculosis. N. Engl. J. Med. 373, 2149–2160. 10.1056/NEJMra1413919. [DOI] [PubMed] [Google Scholar]
- Dover L. G.; Coxon G. D. (2011) Current Status and Research Strategies in Tuberculosis Drug Development. J. Med. Chem. 54, 6157–6165. 10.1021/jm200305q. [DOI] [PubMed] [Google Scholar]
- Liu B.; Li F.; Zhou T.; Tang X. Q.; Hu G. W. (2018) Quinoline Derivatives with Potential Activity Against Multidrug-Resistant Tuberculosis. J. Heterocycl. Chem. 55, 1863–1873. 10.1002/jhet.3241. [DOI] [Google Scholar]
- TB Alliance (2019) FDA Approves New Treatment for Highly Drug-Resistant Forms of Tuberculosis, https://www.tballiance.org/news/fda-approves-new-treatment-highly-drug-resistant-forms-tuberculosis (accessed 2020-02-24).
- Working Group for New TB Drugs Clinical Pipeline, https://www.newtbdrugs.org/pipeline/clinical (accessed 2020-01-16).
- Barry C. E.; Boshoff H. I.; Dartois V.; Dick T.; Ehrt S.; Flynn J.; Schnappinger D.; Wilkinson R. J.; Young D. (2009) The Spectrum of Latent Tuberculosis: Rethinking the Biology and Intervention Strategies. Nat. Rev. Microbiol. 7, 845–855. 10.1038/nrmicro2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koul A.; Arnoult E.; Lounis N.; Guillemont J.; Andries K. (2011) The Challenge of New Drug Discovery for Tuberculosis. Nature 469, 483–490. 10.1038/nature09657. [DOI] [PubMed] [Google Scholar]
- El-Hashash M. A.; Elshahawi M. M.; Ragab E. A.; Nagdy S. (2015) Synthesis and Antifungal Activity of Novel Quinazolin-4(3H)-One Derivatives. Synth. Commun. 45, 2240–2250. 10.1080/00397911.2015.1074697. [DOI] [Google Scholar]
- Radwan A. A.; Alanazi F. K.; Al-Dhfyan A. (2013) Synthesis, and Docking Studies of Some Fused-Quinazolines and Quinazolines Carrying Biological Active Isatin Moiety as Cell-Cycle Inhibitors of Breast Cancer Cell Lines. Drug Res. (Stuttgart, Ger.) 63, 129–136. 10.1055/s-0032-1333306. [DOI] [PubMed] [Google Scholar]
- Rizza P.; Pellegrino M.; Caruso A.; Iacopetta D.; Sinicropi M. S.; Rault S.; Lancelot J. C.; El-Kashef H.; Lesnard A.; Rochais C.; Dallemagne P.; Saturnino C.; Giordano F.; Catalano S.; Ando S. (2016) 3-(Dipropylamino)-5-Hydroxybenzofuro[2,3-f]Quinazolin-1(2H)-One (DPA-HBFQ-1) Plays an Inhibitory Role on Breast Cancer Cell Growth and Progression. Eur. J. Med. Chem. 107, 275–287. 10.1016/j.ejmech.2015.11.004. [DOI] [PubMed] [Google Scholar]
- Alanazi A. M.; Abdel-Aziz A. A.-M.; Shawer T. Z.; Ayyad R. R.; Al-Obaid A. M.; Al-Agamy M. H. M.; Maarouf A. R.; El-Azab A. S. (2016) Synthesis, Antitumor and Antimicrobial Activity of Some New 6-Methyl-3-Phenyl-4(3H)-Quinazolinone Analogues: In Silico Studies. J. Enzyme Inhib. Med. Chem. 31, 721–735. 10.3109/14756366.2015.1060482. [DOI] [PubMed] [Google Scholar]
- Azab M. E.; Kassab E. A.; El-Hashash M. A.; Ali R. S. (2009) Synthesis and Antibacterial Activity of Some New 4(3H)Quinazolin-4-One Derivatives. Phosphorus, Sulfur Silicon Relat. Elem. 184, 610–625. 10.1080/10426500802243182. [DOI] [Google Scholar]
- Hess H. J.; Cronin T. H.; Scriabine A. (1968) Antihypertensive 2-Amino-4(3H)-Quinazolinones. J. Med. Chem. 11, 130–136. 10.1021/jm00307a028. [DOI] [PubMed] [Google Scholar]
- Gupta C. M.; Bhaduri A. P.; Khanna N. M. (1968) Drugs Acting on the Central Nervous System. Syntheses of Substituted Quinazolinones and Quinazolines and Triazepino- and Triazocinoquinazolinones. J. Med. Chem. 11, 392–395. 10.1021/jm00308a057. [DOI] [PubMed] [Google Scholar]
- Couturier C.; Lair C.; Pellet A.; Upton A.; Kaneko T.; Perron C.; Cogo E.; Menegotto J.; Bauer A.; Scheiper B.; Lagrange S.; Bacqué E. (2016) Identification and Optimization of a New Series of Anti-Tubercular Quinazolinones. Bioorg. Med. Chem. Lett. 26, 5290–5299. 10.1016/j.bmcl.2016.09.043. [DOI] [PubMed] [Google Scholar]
- Alagarsamy V.; Anjana G. V.; Sulthana M. T.; Parthiban P.; Solomon V. R. (2016) Antimicrobial Activities of Some Synthesized 1-(3-(2-Methylphenyl)-4-Oxo-3H-Quinazolin-2-Yl-4-(Substituted)Thiosemicarbazide Derivatives. Russ. J. Bioorg. Chem. 42, 332–339. 10.1134/S106816201603002X. [DOI] [Google Scholar]
- Jirgensons A., Domraceva I., Lapsa I., Rasina D., Jaudzems K., and Otikovs M. (2015) Novel Substituted 2-Aminoquinazolin-4(3H)-One Derivatives as Malarial Aspartic Protease Inhibitors, WO/2015/063544.
- Leivers A. L.; Tallant M.; Shotwell J. B.; Dickerson S.; Leivers M. R.; McDonald O. B.; Gobel J.; Creech K. L.; Strum S. L.; Mathis A.; Rogers S.; Moore C. B.; Botyanszki J. (2014) Discovery of Selective Small Molecule Type III Phosphatidylinositol 4-Kinase Alpha (PI4KIIIα) Inhibitors as Anti Hepatitis C (HCV) Agents. J. Med. Chem. 57, 2091–2106. 10.1021/jm400781h. [DOI] [PubMed] [Google Scholar]
- Banka A. L., Botyanszki J., Duan M., Leivers M. R., Shotwell J. B., Tallant M. D., Dickerson S. H., Tai V. W.-F., Mcfadyen R. B., Redman A. M., Yu J., Li X., Garrido D. M., Catalano J. G., and Adjabeng G. (2012) Quinazolinone Derivatives as Antiviral Agents, WO2012087938A1.
- Hughes A. N.; Rafi I.; Griffin M. J.; Calvert A. H.; Newell D. R.; Calvete J. A.; Johnston A.; Clendeninn N.; Boddy A. V. (1999) Phase I Studies with the Nonclassical Antifolate Nolatrexed Dihydrochloride (AG337, THYMITAQ) Administered Orally for 5 Days. Clin. Cancer Res. 5, 111–118. [PubMed] [Google Scholar]
- Younis Y.; Douelle F.; Feng T.-S.; Cabrera D. G.; Le Manach C.; Nchinda A. T.; Duffy S.; White K. L.; Shackleford D. M.; Morizzi J.; Mannila J.; Katneni K.; Bhamidipati R.; Zabiulla K. M.; Joseph J. T.; Bashyam S.; Waterson D.; Witty M. J.; Hardick D.; Wittlin S.; Avery V.; Charman S. A.; Chibale K. (2012) 3,5-Diaryl-2-Aminopyridines as a Novel Class of Orally Active Antimalarials Demonstrating Single Dose Cure in Mice and Clinical Candidate Potential. J. Med. Chem. 55, 3479–3487. 10.1021/jm3001373. [DOI] [PubMed] [Google Scholar]
- Güngör T.; Chen Y.; Golla R.; Ma Z.; Corte J. R.; Northrop J. P.; Bin; Dickson J. K.; Stouch T.; Zhou R.; Johnson S. E.; Seethala R.; Feyen J. H. M. (2006) Synthesis and Characterization of 3-Arylquinazolinone and 3-Arylquinazolinethione Derivatives as Selective Estrogen Receptor Beta Modulators. J. Med. Chem. 49, 2440–2455. 10.1021/jm0509389. [DOI] [PubMed] [Google Scholar]
- Lecoutey C.; Fossey C.; Rault S.; Fabis F. (2011) Efficient Room-Temperature One-Pot Synthesis of 2-Amino-3-Alkyl(3-Aryl)Quinazolin-4(3H)-Ones. Eur. J. Org. Chem. 2011, 2785–2788. 10.1002/ejoc.201100200. [DOI] [Google Scholar]
- Arora K.; Ochoa-Montaño B.; Tsang P. S.; Blundell T. L.; Dawes S. S.; Mizrahi V.; Bayliss T.; Mackenzie C. J.; Cleghorn L. A. T.; Ray P. C.; Wyatt P. G.; Uh E.; Lee J.; Barry C. E.; Boshoff H. I. (2014) Respiratory Flexibility in Response to Inhibition of Cytochrome c Oxidase in Mycobacterium Tuberculosis. Antimicrob. Agents Chemother. 58, 6962–6965. 10.1128/AAC.03486-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moosa A.; Lamprecht D. A.; Arora K.; Barry C. E.; Boshoff H. I. M.; Ioerger T. R.; Steyn A. J. C.; Mizrahi V.; Warner D. F. (2017) Susceptibility of Mycobacterium Tuberculosis Cytochrome Bd Oxidase Mutants to Compounds Targeting the Terminal Respiratory Oxidase, Cytochrome C. Antimicrob. Agents Chemother. 61, e01338-17 10.1128/AAC.01338-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naran K.; Moosa A.; Barry C. E.; Boshoff H. I. M.; Mizrahi V.; Warner D. F. (2016) Bioluminescent Reporters for Rapid Mechanism of Action Assessment in Tuberculosis Drug Discovery. Antimicrob. Agents Chemother. 60, 6748–6757. 10.1128/AAC.01178-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pethe K.; Sequeira P. C.; Agarwalla S.; Rhee K.; Kuhen K.; Phong W. Y.; Patel V.; Beer D.; Walker J. R.; Duraiswamy J.; Jiricek J.; Keller T. H.; Chatterjee A.; Tan M. P.; Ujjini M.; Rao S. P. S.; Camacho L.; Bifani P.; Mak P. A.; Ma I.; Barnes S. W.; Chen Z.; Plouffe D.; Thayalan P.; Ng S. H.; Au M.; Lee B. H.; Tan B. H.; Ravindran S.; Nanjundappa M.; Lin X.; Goh A.; Lakshminarayana S. B.; Shoen C.; Cynamon M.; Kreiswirth B.; Dartois V.; Peters E. C.; Glynne R.; Brenner S.; Dick T. A. (2010) Chemical Genetic Screen in Mycobacterium Tuberculosis Identifies Carbon-Source-Dependent Growth Inhibitors Devoid of in Vivo Efficacy. Nat. Commun. 1, 57. 10.1038/ncomms1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley S. A.; Grant S. S.; Kawate T.; Iwase N.; Shimizu M.; Wivagg C.; Silvis M.; Kazyanskaya E.; Aquadro J.; Golas A.; Fitzgerald M.; Dai H.; Zhang L.; Hung D. T. (2012) Identification of Novel Inhibitors of M. Tuberculosis Growth Using Whole Cell Based High-Throughput Screening. ACS Chem. Biol. 7, 1377–1384. 10.1021/cb300151m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manjunatha U. H.; Smith P. W. (2015) Perspective: Challenges and Opportunities in TB Drug Discovery from Phenotypic Screening. Bioorg. Med. Chem. 23, 5087–5097. 10.1016/j.bmc.2014.12.031. [DOI] [PubMed] [Google Scholar]
- Baughn A. D.; Rhee K. Y. (2014) Metabolomics of Central Carbon Metabolism in Mycobacterium Tuberculosis. Microbiol. Spectrum 2, 1–16. 10.1128/microbiolspec.MGM2-0026-2013. [DOI] [PubMed] [Google Scholar]
- Yuan T.; Sampson N. S. (2018) Hit Generation in TB Drug Discovery: From Genome to Granuloma. Chem. Rev. 118, 1887–1916. 10.1021/acs.chemrev.7b00602. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.