

Abstract
Generalized epilepsy with febrile seizures plus (GEFS+) is a complex familial epilepsy syndrome. It is mainly caused by mutations in SCN1A gene, encoding type 1 voltage-gated sodium channel α-subunit (NaV1.1), and GABRA1 gene, encoding the α1 subunit of the γ-aminobutyric acid type A (GABAA) receptor, while seldom related with SCN9A gene, encoding the voltage-gated sodium channel NaV1.7. In this study, we investigated a Chinese family with an autosomal dominant form of GEFS+. DNA sequencing of the whole coding region revealed a novel heterozygous nucleotide substitution (c.5873A>G) causing a missense mutation (p.Y1958C). This mutation was predicted to be deleterious by three different bioinformatics programs (The polyphen2, SIFT, and MutationTaster). Our finding reports a novel likely pathogenic SCN9A Y1958C heterozygous mutation in a Chinese family with GEFS+ and provides additional supports that SCN9A variants may be associated with human epilepsies.
Electronic supplementary material
The online version of this article (10.1007/s10072-020-04284-x) contains supplementary material, which is available to authorized users.
Keywords: SCN9A, Mutation, |Epilepsy, Generalized epilepsy with febrile seizures plus
Introduction
SCN9A gene encodes the voltage-gated sodium channel NaV1.7, one of the nine known α members of voltage-gated sodium (Nav) channels [1]. It mainly expresses in dorsal root ganglion neurons, thus its mutations are mainly associated with pain disorders [2]. However, more and more studies have shown that SCN9A mutations in patients are also associated with variable epilepsy phenotypes including febrile seizures (FS) [3], GEFS+ [4], and Dravet syndrome (DS) [5] in recent years. GABRG2 gene (γ-aminobutyric acid receptor subunit gene), SCN1A gene, and GABRA1 gene are most frequently mentioned in GEFS+ [6–8], while the role of SCN9A gene in GEFS+ still remains unknown. Here, we report a novel previously unreported likely pathogenic SCN9A Y1958C heterozygous mutation with no SCN1A mutations in a Chinese family with GEFS+ and explore the possibility of SCN9A contributing to GEFS+.
Material and methods
Family recruitment
A family with 10 living Han members across three generation participated in the study. All the subjects signed the informed consent. Clinical data were collected from all members.
Targeted exon capture and sequencing
After signing the consent, 2~4-ml peripheral blood from the ten members were collected. Blood Genome Column Medium Extraction Kit (Kangweishiji, China) was used to extract genomic DNA from blood samples. Whole-exome enrichment was performed using IDT_xGEN, which targets 39Mb protein-coding region of the human genome and covers 51Mb of partial intron. High-throughput sequencing was performed by Illumina NOVASeq 6000 series sequencer; the sequencing process was performed by Beijing Chigene Translational Medicine Research Center.
Sequence alignment and variant calling
All reads were mapped to the human reference sequence (hg 19) using BWA-MEM (version 0.7.12). Then, a genome analysis tool kit (GATK version 3.4.0) was used to refine the alignments by performing local indel realignment and subsequent base quality recalibration. Single-nucleotide variants (SNVs) and insertions/deletions (indels) were called with the haplotype caller of the GATK. The variant was compared against publicly available databases such as the 1000 Genomes Project and the Exome Aggregation Consortium database (ExAC). At last, protein damage analysis was conducted to qualitatively predict the probability of the results by SIFT, PolyPhen-2, and MutationTaster, and multispecies alignments were performed using Mega 7.0 to determine whether the affected amino acids were conserved.
Results
Case description
The proband (IV1) is a 9-year-old boy with normal spontaneous vaginal delivery and development. He presented the first seizure at 1 year and 4 months when encountering a respiratory tract infection with a fever (41 °C). Since then, he experienced febrile seizures for 9 times in total. The eighth seizure occurred at the age of 4, and he did not have convulsions in the next 4 years. The seizure patterns of the first time to the eighth time were all presented as generalized tonic-clonic seizures (GTCS), while the patient showed GTCS and absences on the ninth seizure when he was 9 years old with a fever. All the seizures last for about 1–2 min and could remit spontaneously. No obvious abnormality was found on neurological examination and brain magnetic resonance imaging (MRI). One week later after the last seizuring, electroencephalogram monitoring showed atypical spike-and-slow waves in the right temporal regions during sleep. In the further pedigree investigation, the proband’s father (III3) experienced febrile seizures at about 1 year old and one of the proband’s aunts (III1) reported febrile seizures at the age of two (Fig. 1a). The seizure patterns were also described as GTCS. Their seizures remitted spontaneously at that time and have not recurred until now. The proband’s grandmother (II4) and grandfather (II3) did not remember whether they had convulsions in their childhood. Besides, the family history of two dead family members (I1, I2) could not be obtained. The other family members denied seizure. None of them were treated with antiepileptic drugs for their seizures.
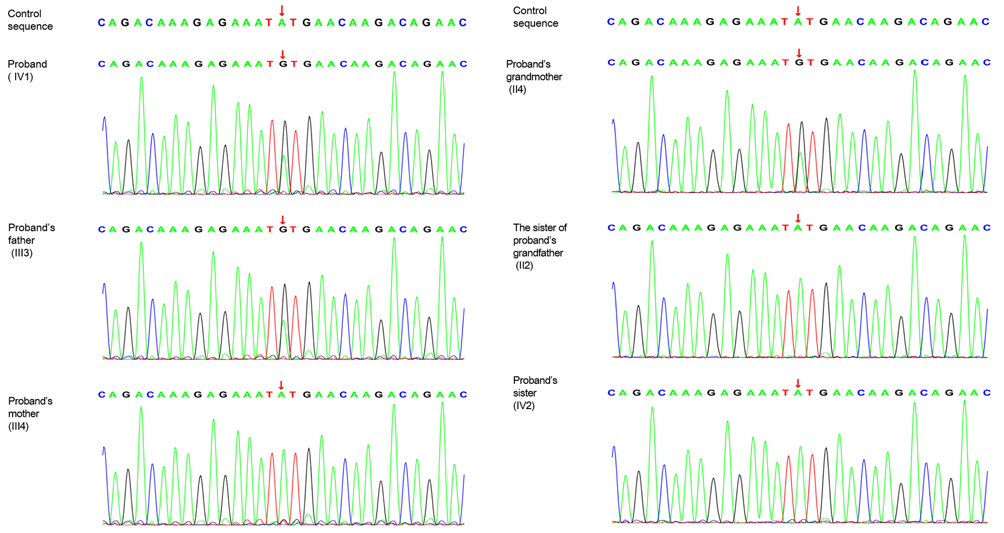
Fig. 1.

a Family pedigree. The black arrow indicates the proband; the legend for the symbols is at the right top of the figure. b Identification of a heterozygous mutation c.5873A>G (p.Y1958C) in the family members: proband (IV1), proband’s father (III3), proband’s aunt (III1), and proband’s grandmother (II4). Additional Sanger sequencing results are given in Online Resource (Figs. 3 and 4) The red arrow shows an A to G transition of nucleotide 5873
Genetic findings
A novel heterozygous SCN9A mutation (c.5873A>G) was detected in the proband (IV1), proband’s father (III3), proband’s aunt (III1), and proband’s grandmother (II4) by using clinical whole-exome sequencing, and Sanger sequencing was used to validate it (Fig. 1b). No mutations in SCN1A were detected. This mutation (c.5873A>G chr2:167055243 p.Y1958C) occurs in the population at a frequency of < 0.5% in the ExAC database (http://exac.broadinstitute.org/variant/2-167055243-T-C) and has not been reported in previous study or presented in dbSNP (http://evs.gs.washington.edu/EVS/) and 1000 Genomes Project (https://www.internationalgenome.org/). Multiple sequence alignment was performed by using Mega 7.0 (https://www.megasoftware.net/), and residue Y1958 is highly conserved (Fig. 2). The results of three bioinformatics programs show that the novel mutation could damage the function of the protein (Table 1). The pathogenicity of this variant is classified likely pathogenic, following the principle of standards and guidelines recommended by ACMG in recent publication [9].
Fig. 2.

Alignment of multiple SCN9A protein sequences across species. The Y1958C affected amino acid locates in the highly conserved amino acid region in different mammals (from Ensembl). Red column shows the Y1958C site
Table 1.
The prediction of the identified variant in SCN9A
| Gene name | Position | Transcript | Substitution | ExAC | dbSNP ID | 1000G | Polyphen2 | SIFT | MutationTaster |
|---|---|---|---|---|---|---|---|---|---|
| SCN9A | chr2:167055243 | NM_002977 | c.5873A>G/Y1958C | 0.0001 | Novel | Novel | 0.99 (probably damaging) | 0.01 (damaging) | 0.999993 (disease-causing) |
ExAC, Exome Aggregation Consortium; 1000G, 1000 genomes
Discussion
GEFS+ is a complex autosomal dominant disorder with conspicuous phenotypic heterogeneity [10]. The first and second frequent phenotypes are febrile seizures, where generalized tonic-clonic seizures (GTCS) with fever occur between 3 months and 6 years, and febrile seizures plus (FS+), in which attacks with fever extend beyond 6 years or afebrile GTCS occur, respectively. The other phenotypes include FS/FS+ with absence, myoclonic, atonic, or focal seizures [11]. In our study, the proband experienced FS and FS+ with absence while his father and aunt only had febrile seizures, probably due to the incomplete penetrance and/or the phenotypic heterogeneity. As for the proband’s grandmother (II4), she did not remember whether convulsions had occurred in her childhood.
Several genes have been announced to be associated with GEFS+, in which GABRG2, SCN1A, and GABRA1 account for the major part [6–8], while SCN9A is only reported in a few cases. A potentially pathogenic SCN9A variant, L266M (in exon 7), was discovered in one GEFS+ family [5]. Cen et al. reported a small pedigree diagnosed as GEFS+ with a heterozygous mutation (Q10R) in SCN9A gene without SCN1A mutation [4]. In 2019, a heterozygous mutation in the SCN9A gene, p.(Lys655Arg), in two sisters from a non-consanguineous family who presented GEFS+ was detected [11]. Actually, SCN9A variant is often mentioned as a genetic modifier in SCN1A mutation-associated epilepsy. A heterozygous SCN9A mutation, p.N641Y, was found to be responsible for a large Utah family (K4425) suffering from FS and GEFS+, and the authors identified nine SCN1A mutations with six different SCN9A mutations in this study [3]. However, the exact role of SCN9A mutations without SCN1A variants in GEFS+ has still remained left unclear. Thus, we hope to provide more evidence to illustrate that SCN9A has an important bearing on GEFS+ development in addition to these cases.
SCN9A, encoding sodium channel Nav1.7, contains 27 exons on chromosome 2q24.3 [12]. Nav1.7 is composed of 1977 amino acids and is organized into 4 domains, each with 6 trans-membranes [1]. In our study, a novel SCN9A heterozygous mutation (c.5873A>G) causing a missense mutation (p.Y1958C) was discovered. Regrettably, the mutation didn’t occur in the 4 and the exact cellular mechanism is unclear. However, the mutation in our study was located in highly conserved positions. The SCN9A N641Y mutation in FS proved to reduce thresholds to electrically induced seizures, and increase seizure susceptibility by targeted knock-in mouse model was also in highly conserved positions [3]. So, we speculate that the SCN9A Y1958C mutation might also affect the selectivity of the ion channel. Besides, the bioinformatics programs also demonstrated that the novel mutation could damage the function of the protein. All the evidences confirmed that the SCN9A p.Y1958C mutation should be regarded as pathogenic mutation in this family.
This report further supports that SCN9A mutation without SCN1A mutations is associated with GEFS+ and expands the spectrum of SCN9A gene, but there are limitations in our study that should be addressed. Thus, the functional effect of the mutation should be further studied to strengthen our views.
Electronic supplementary material
{kind=link}
(PNG 199 kb)
{kind=link}
(PNG 329 kb)
Acknowledgments
We would like to thank the patient and his families for their generous participation in this study.
Funding information
The study was supported by The Anhui Natural Science Foundation (grant no.
21608085MH196).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
The patients and his families provided informed consent, and the study was approved by the ethical committees of the First Affiliated Hospital of USTC, Anhui Provincial Hospital. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in the study.
Footnotes
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Klugbauer N, Lacinova L, Flockerzi V, Hofmann F. Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. EMBO J. 1995;14(6):1084–1090. doi: 10.1002/j.1460-2075.1995.tb07091.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Drenth JP, Waxman SG. Mutations in sodium-channel gene SCN9A cause a spectrum of human genetic pain disorders. J Clin Invest. 2007;117(12):3603–3609. doi: 10.1172/JCI33297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Singh NA, Pappas C, Dahle EJ, Claes LR, Pruess TH, De Jonghe P, Thompson J, Dixon M, Gurnett C, Peiffer A, White HS, Filloux F, Leppert MF. A role of SCN9A in human epilepsies, as a cause of febrile seizures and as a potential modifier of Dravet syndrome. PLoS Genet. 2009;5(9):e1000649. doi: 10.1371/journal.pgen.1000649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cen Z, Lou Y, Guo Y, Wang J, Feng J. Q10R mutation in SCN9A gene is associated with generalized epilepsy with febrile seizures plus. Seizure. 2017;50:186–188. doi: 10.1016/j.seizure.2017.06.023. [DOI] [PubMed] [Google Scholar]
- 5.Mulley JC, Bree H, Mcmahon JM, Xenia I, Susannah B, Mullen SA, Kevin F, Mark M, Lynette S, Andrew B. Role of the sodium channel SCN9A in genetic epilepsy with febrile seizures plus and Dravet syndrome. Epilepsia. 2014;54(9):e122–e126. doi: 10.1111/epi.12323. [DOI] [PubMed] [Google Scholar]
- 6.Audenaert D, Schwartz E, Claeys KG, Claes L, Deprez L, Suls A, Van Dyck T, Lagae L, Van Broeckhoven C, Macdonald RL, De Jonghe P. A novel GABRG2 mutation associated with febrile seizures. Neurology. 2006;67(4):687–690. doi: 10.1212/01.wnl.0000230145.73496.a2. [DOI] [PubMed] [Google Scholar]
- 7.Wallace RH, Scheffer IE, Barnett S, Richards M, Dibbens L, Desai RR, Lerman-Sagie T, Lev D, Mazarib A, Brand N, Ben-Zeev B, Goikhman I, Singh R, Kremmidiotis G, Gardner A, Sutherland GR, George AL, Jr, Mulley JC, Berkovic SF. Neuronal sodium-channel alpha1-subunit mutations in generalized epilepsy with febrile seizures plus. Am J Hum Genet. 2001;68(4):859–865. doi: 10.1086/319516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Johannesen K, Marini C, Pfeffer S, Møller RS, Maljevic S (2016) Phenotypic spectrum of GABRA1: from generalized epilepsies to severe epileptic encephalopathies. Neurology 87(11). 10.1212/WNL.0000000000003087 [DOI] [PubMed]
- 9.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, Voelkerding K, Rehm HL, Comm ALQA. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang YH, Burgess R, Malone JP, Glubb GC, Helbig KL, Vadlamudi L, Kivity S, Afawi Z, Bleasel A, Grattan-Smith P, Grinton BE, Bellows ST, Vears DF, Damiano JA, Goldberg-Stern H, Korczyn AD, Dibbens LM, Ruzzo EK, Hildebrand MS, Berkovic SF, Scheffer IE. Genetic epilepsy with febrile seizures plus refining the spectrum. Neurology. 2017;89(12):1210–1219. doi: 10.1212/Wnl.0000000000004384. [DOI] [PubMed] [Google Scholar]
- 11.Alves RM, Uva P, Veiga MF, Oppo M, Zschaber FCR, Porcu G, Porto HP, Persico I, Onano S, Cuccuru G, Atzeni R, Vieira LCN, Pires MVA, Cucca F, Toralles MBP, Angius A, Crisponi L (2019) Novel ANKRD11 gene mutation in an individual with a mild phenotype of KBG syndrome associated to a GEFS plus phenotypic spectrum: a case report. BMC Med Genet 20. 10.1186/s12881-019-0745-7 [DOI] [PMC free article] [PubMed]
- 12.Ding JZJ, GUO Y, Zhang Y, Chen Z. Novel mutations in SCN9A occurring with fever-associated seizures or epilepsy. Seizure. 2019;71:214–218. doi: 10.1016/j.seizure.2019.06.005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(PNG 199 kb)
(PNG 329 kb)